

Abstract
Though defective genome maintenance and DNA repair have long been know to promote phenotypes of premature aging, the role protein methylation plays in these processes is only now emerging. We have recently identified the first N-terminal methyltransferase, NRMT1, which regulates protein-DNA interactions and is necessary for both accurate mitotic division and nucleotide excision repair. To demonstrate if complete loss of NRMT1 subsequently resulted in developmental or aging phenotypes, we constructed the first NRMT1 knockout (Nrmt1−/−) mouse. The majority of these mice die shortly after birth. However, the ones that survive exhibit decreased body size, female-specific infertility, kyphosis, decreased mitochondrial function, and early-onset liver degeneration; phenotypes characteristic of other mouse models deficient in DNA repair. The livers from Nrmt1−/− mice produce less reactive oxygen species (ROS) than wild type controls, and Nrmt1−/− mouse embryonic fibroblasts show a decreased capacity for handling oxidative damage. This indicates that decreased mitochondrial function may benefit Nrmt1−/− mice and protect them from excess internal ROS and subsequent DNA damage. These studies position the NRMT1 knockout mouse as a useful new system for studying the effects of genomic instability and defective DNA damage repair on organismal and tissue-specific aging.
Keywords: N-terminal methylation, DNA repair, aging, mitochondria
1. Introduction
There is an intricate relationship between genome maintenance and the onset of aging. Cells that fail to strictly monitor the integrity of their chromosomes or adequately repair DNA damage accumulate lesions that promote accelerated aging both in mice and in humans (Hasty et al., 2003). Understanding how loss of genomic maintenance and accumulation of mutations drives accelerated aging will help clarify how these phenomenon are also driving normal aging and how their loss can be counteracted to slow the aging process.
The most common human progeroid syndromes have deficiencies in at least one form of genome maintenance. Hutchinson-Gilford Progeria Syndrome (HGPS) is caused by a mutation in the nuclear lamin protein lamin A (LMNA). Though not directly involved in regulating DNA repair, LMNA controls the nuclear/cytoplasmic Ran-GTP gradient and thereby regulates nuclear transport (Kelley et al., 2011). Disruption of the Ran-GTP gradient in HGPS models results in decreased nuclear import of Ubc9, the E2 for SUMOylation, and increased production of reactive oxygen species (ROS) (Datta et al., 2014). As Ubc9 is an important enzyme for both nucleotide excision repair (NER) (Silver et al., 2011) and conservative non-homologous end joining (NHEJ) (Hu and Parvin, 2014), this results in both increased DNA damage from the ROS and a decreased capacity to repair it. Cockayne syndrome (CS), Werner's Syndrome, Rothmund-Thompson Syndrome, and Trichothiodystrophy (TTD) are all caused by mutations in DNA helicases necessary for nucleotide excision repair or double-strand break repair (Hasty et al., 2003). Ataxia telangiectasia results from homozygous mutations in the ATM kinase, another major player in the repair of double-strand DNA breaks (DSBs) (Savitsky et al., 1995).
A variety of mouse models have been generated in an attempt to replicate these human accelerated aging syndromes and understand the underlying molecular mechanisms. TTD mice harbor the same mutation in the DNA helicase subunit Xeroderma pigmentosum group D (XPD) as human patients and exhibit kyphosis, osteoporosis, female-specific infertility, hyperkeratotic epidermis, gray hair, and reduced life span (de Boer et al., 2002). Embryonic fibroblasts derived from these mice (MEFs) are also defective in nucleotide excision repair and sensitive to oxidative damage (de Boer et al., 2002). In general, CS mouse models are defective in transcription-coupled repair, and they develop tremors, cachexia, and retinal degeneration, though these phenotypes are less severe than those seen in humans (van der Horst et al., 1997). The CSB mouse model (mutation in the in the Cockayne syndrome group B protein) also shows an increase in metabolism and oxygen consumption both at the whole organism and cellular level (Scheibye-Knudsen et al., 2012). This phenotype is due to an increase in mitochondrial content and resultant reactive oxygen species as a result of failed mitochondrial degradation (Scheibye-Knudsen et al., 2012). Current mouse models defective in homologous recombination (HR) do not exhibit premature aging phenotypes, though many of them are embryonic lethal (van Gent et al., 2001). However, the embryonic lethality of a Brca1 hypomorphic mutation can be rescued by haploid loss of p53, and these Brca1Δ11/Δ11p53+/− mice exhibit premature aging phenotypes that include reduced size, decreased life span, kyphosis, and osteoporosis (Cao et al., 2003). Knockout mice for the NHEJ protein Ku86 (Ku86−/−) exhibit osteoporosis, atrophic skin, liver pathology, sepsis, cancer, and reduced life span (Vogel et al., 1999). Additionally, mice deplete of the endonuclease ERCC1 (Ercc1−/−) are also defective for repair of double-strand lesions and develop cachexia, neuronal degeneration, and skin atrophy, and most die before three weeks of age from liver or kidney failure (Weeda et al., 1997). These models have all provided valuable insight into how faulty genome maintenance contributes to the aging process. However, each represents a mutation in a single aspect of the genome maintenance machinery, making it hard to completely mimic normal human aging (Hasty et al., 2003).
There is mounting evidence that protein methylation plays an important role in many modes of genome maintenance and DNA damage repair (Escargueil et al., 2008; Li, 2012), and accordingly, animals with altered methylation patterns are showing promise as novel aging models (McCauley and Dang, 2014). Knockdown of the ASH-2/WDR-5/SET-2 complex that trimethylates H3K4 (a mark corresponding to transcriptional activation) in C. elegans significantly extends lifespan, while knockdown of the H3K4 demethylase RBR-2 decreases lifespan, indicating that excess H3K4 methylation promotes aging (Greer et al., 2010). Interestingly, Drosophilia strains with mutations in E (Z), the H3K27 methyltransferase (a mark associated with repressed transcription), are also long-lived, and reintroducing H3K27 methylation restores normal lifespan (Siebold et al., 2010). Senescence accelerated mice (SAMP8) also have increased levels of H3K27me3 and demonstrate premature neurodegeneration (Wang et al., 2010). The similar aging phenotypes resulting from the misregulation of these functionally opposing histone methylation marks suggest methylation can regulate lifespan in a mode independent of transcriptional activation or repression, perhaps through their DNA repair functions. However, additional model systems are needed to understand the interconnecting roles between protein methylation, DNA damage repair, and the aging process.
We have recently discovered the first eukaryotic N-terminal methyltransferase, NRMT1 (Tooley et al., 2010). NRMT1 is a ubiquitously expressed nuclear trimethylase that methylates the α-amino group of the first N-terminal amino acid after cleavage of the initiating methionine (Tooley et al., 2010). NRMT1 methylates a three amino acid N-terminal consensus sequence. The first amino acid of the sequence is most commonly alanine, proline, serine, glycine or methionine (Petkowski et al., 2012). The second position accepts most uncharged polar or nonpolar amino acids, and only lysine or arginine are permissive in the third position (Petkowski et al., 2012). Database searches using this consensus predicts over 300 NRMT1 targets, including proteins involved in chromatin structure (CENP-A, CENP-B, HP1γ, SET), DNA repair (RB, DDB2, PARP3, BAP1), and metabolism (NDUFA11, UGT1A10, ARSB, FMO3). We, and others, have subsequently verified dozens of these substrates, and currently, all tested proteins containing this consensus have been found to be N-terminally methylated by NRMT1 (Cai et al., 2014; Dai et al., 2013; Petkowski et al., 2012).
N-terminal methylation was originally found to protect proteins against cellular proteases (Pettigrew and Smith, 1977; Stock et al., 1987). However, we have been able to demonstrate that N-terminal methylation also regulates protein-DNA interactions (Chen et al., 2007; Tooley et al., 2010). Loss of N-terminal methylation of RCC1 reduces its affinity for DNA and causes its mislocalization from chromatin, multi-polar spindle formation, aberrant mitotic division, and aneuploidy (Chen et al., 2007; Tooley et al., 2010). N-terminal methylation of Centromere protein B (CENP-B) regulates its binding to centromeric DNA and is enriched in response to cellular stress (Dai et al., 2013). N-terminal methylation of Damaged DNA-Binding Protein 2 (DDB2) regulates its binding to DNA and is necessary for effective NER (Cai et al., 2014). We have also found that loss of NRMT1 sensitizes cells to agents that produce DSBs (etoposide and γ-irradiation) and propose that this is due to decreased binding of the NRMT1 substrates PARP3 and BAP1 to sites of damage (LB unpublished data). Given these phenotypes in cell culture and the variety of NRMT1 substrates involved in chromatin structure, DNA repair, and metabolism we generated the first NRMT1 knockout (Nrmt1−/−) mouse to determine if global loss of NRMT1 affects development and the aging process.
Here we show that Nrmt1−/− mice are phenotypically similar to other transgenic mice deficient in DNA repair and most closely resemble the TTD mice deficient in NER. Most die before reaching three weeks of age, and the ones that survive are smaller, exhibit female-specific infertility, kyphosis, decreased mitochondrial function, and liver pathogenesis. Similar to TTD mice (de Boer et al., 2002), the liver pathogenesis is accompanied by altered expression of genes involved in apoptosis, oxidative phosphorylation, fatty acid metabolism, and steroid biosynthesis, with the levels of these transcripts in Nrmt1−/− mice more closely correlated to those in aged mouse livers. Nrmt1−/− livers also show increased aconitase activity (indicating decreased ROS production), while Nrmt1−/− MEFs have a 30% increase in sensitivity to hydrogen peroxide treatment. These results indicate the decreased mitochondrial function seen in Nrmt1−/− mice may be beneficial and serve to protect from excess ROS production and subsequent DNA damage. We propose that having both NER and double strand break repair simultaneously compromised (but not eliminated) better recapitulates the gradual pace of normal mammalian aging. Therefore, the surviving Nrmt1−/− mice can be utilized to study how failure to maintain genetic integrity and adequately protect DNA from extracellular insult accelerates both tissue and organismal aging.
2. Materials and Methods
2.1 Animals
The experimental protocol was approved by the University of Louisville School of Medicine Institutional Animal Care and Use Committee. C57BL/6J embryonic stem (ES) cells expressing a cassette that replaces the third exon of NRMT1 with an E. coli lac Z reporter construct were obtained from the North American Conditional Mouse Mutagenesis Project (NorCOMM). NRMT1 knockout mice (Nrmt1−/−) were generated by injection of the ES cells into blastocysts obtained post fertilization of albino B6 mice and transplanted into pseudopregnant females at the University of Cincinnati Transgenic Mouse Facility. Chimeric males were mated to albino B6 females and the resultant black progeny were bred to homozygosity at the University of Louisville. Genotyping was performed with the following primers: P1 - TGATTGGGTGGGACTTGTGAC, P2 - GAGCTGCATGAAAAGGAGTCAC, and P3 – GAAGAATAGGAACTTCGTCGAC. P1 and P2 produce a 164 bp band from wild type DNA. P1 and P3 produce a 129 bp band from Nrmt1−/− DNA. All other C57BL/6J wild type mice were obtained from Jackson Laboratories. All mating pairs consisted of one male and one female mouse unless otherwise noted. Mice were weighed at indicated time points with a standard laboratory scale.
2.2 Western Blots and Real Time PCR analysis
Mice were sacrificed, and tissue was extracted, snap-frozen, and stored at −80°C until needed. Tissue was homogenized in protein lysis buffer (0.1% NP40, 0.25 M NaCl, 5 mM EDTA, 50 mM Hepes pH 7.5, 0.1% protease inhibitor cocktail, 0.5 mM DTT) for western blot or in Trizol (Life Technologies) for RNA isolation. Primary antibodies used for western blot analysis include: 1:2000 dilution rabbit anti-NRMT1 (Tooley et al., 2010), 1:3000 dilution rabbit anti-GAPDH (Trevigen, Gaithersburg, MD), and 1:10,000 dilution rabbit anti-trimethylated RCC1 (me3-SPK: though made against N-terminally trimethylated RCC1, this antibody recognizes most N-terminally trimethylated proteins with an XPK consensus) (Chen et al., 2007; Tooley et al., 2010). Samples for RNA isolation were mixed with chloroform to extract RNA, the RNA was precipitated using isopropanol and washed with ethanol. cDNA was synthesized using the SuperScript First-Strand Synthesis System (Life Technologies). Quantitative RT-PCR was performed with SYBR green PCR Master Mix and the CFX96 Touch™ Real-Time PCR Detection System and Sequence Detection Software (BioRad, Hercules, CA). Real time RT-PCR parameters were as follows: 50°C for 10 minutes, 95°C for 5 minutes, then 39 cycles at 95°C for 10 seconds then 60°C for 30 seconds. The melting curve parameters were 95°C for 10 seconds and 65°C for 5 seconds. Primer sequences (Integrated DNA Technologies, Coralville, IA) are indicated in Supplementary Table 1. All real-time PCR assays included analysis of melting curves and agarose gel electrophoresis to confirm the presence of single PCR reaction products.
2.3 Histology
Mice were sacrificed, and tissue was extracted, fixed in 10% formalin, and washed in 70% ethanol. Histological preparation was then performed in the University of Louisville Pathology Core Research Facility. Sections were embedded in paraffin, sectioned, and stained with hematoxylin and eosin. Images were taken on a Axio Observer microscope (Zeiss Microscopy, Jena, Germany).
2.4 Liver Mitochondria Isolation
Livers were excised and placed immediately in ice-cold isolation buffer 1 (250 mM sucrose, 0.5 mM Na2EDTA, and 10 mM Tris, pH 7.4). Livers were minced, washed 3 times and homogenized using the GKH- Homogenizer (Glas-Col). The homogenate was centrifuged and supernatant was filtered through gauze followed by another round of centrifugation. The mitochondrial pellet was washed two times using isolation buffer 2 (250 mM sucrose, 0.5 mM Na2EDTA, 10 mM Tris, and 1g/L BSA, pH 7.4) followed by centrifugation. The mitochondrial pellet was then resuspended in respiration buffer (0.5 mM EGTA, 3 mM MgCl2, 60 mM K-lactobionate, 20 mM taurine, 10 mM KH2PO4, 20 mM HEPES, 110 mM sucrose, and 1 g/L BSA). Mitochondria concentration was determined using the Bradford Protein Assay and samples were used immediately for respiration assays.
2.5. Respiration Assays
Mitochondria (0.025 mg/ml) was added to closed Oxygraph-2k (Oroboros Instruments) chambers containing respiration buffer. LEAK (proton leak) respiration was measured following the addition of 10 mM glutamate and 2 mM malate. This was followed by addition of 1 mM ADP to measure State 3 respiration. After induction of State 3 respiration, cytochrome c (2.5 μM) was added as a control to evaluate the integrity of the outer mitochondrial membrane. State 4 respiration was then induced by adding Oligomycin A (2.5 μM), followed by addition of 3.5 μM carbonyl cyanide p-trifluoro-methoxyphenyl hydrazone (FCCP) to obtain maximum mitochondrial respiration in the non-coupled state of electron transfer system (ETS) capacity. A complex II inhibitor, antimycin A, was then added after depleting the oxygen in the chamber. This was followed by re-oxygenation to measure residual oxygen consumption due to oxidative side reactions.
2.6 Aconitase Assay
Livers were excised and homogenized in stabilization buffer (2 mM sodium citrate, 0.6 mM manganese chloride, 50 mM Tris HCl pH 7.4). Protein concentration was measured and samples were immediately frozen. At the time of the assay 25 ug protein, 50 mM Tris-HCl pH 7.4, 0.2 mM NADP, 0.6 mM manganese chloride, 5 mM sodium citrate, 18.75 μl/mg isocitrate dehydrogenase was added to a 1.5 ml cuvette (Starna Cells). Change in absorbance at 340 nm was measured every thirty seconds for one hour using a Synergy 2 (BioTek) spectrophotometer. The mU aconitase activity per mg protein was then calculated using the molar extinction coefficient 6220 M−1cm−1 for NADP.
2.7 Mouse Embryonic Fibroblasts (MEFs)
Pregnant wild type or Nrmt1+/− heterozygous females were euthanized by CO2 asphyxiation between embryonic days 13.5 to 14.5. Uterine horns containing embryos were removed from mice and placed in a dish of sterile phosphate buffered saline. Embryos were then taken out of the embryonic sac and the head and body cavity were removed. Wild type embryos were processed together. Embryos from the Nrmt1+/− mothers were processed individually, and the resultant MEF lines genotyped to distinguish Nrmt1−/− from Nrmt1+/− lines. The remaining body tissue from the embryos was minced, placed in a tube with 2X trypsin, and incubated at 37°C for 5 minutes. Supernatant was then transferred to a tube containing pre-warmed Dulbecco's Modified Eagle Medium (DMEM) with 10% fetal bovine serum (FBS, Atlanta Biologicals, Atlanta, GA) and 1% penicillin-streptomycin (P/S, CellGro, Manassas, VA). This was repeated a total of four times before cells were centrifuged and resuspended in fresh media. Cells were plated on standard adherent tissue culture plates and the media was changed the next day to remove non-adherent cells.
2.8 Cell Viability Assays
For hydrogen peroxide treatments, MEFs were plated at 10,000 cells in triplicate in a 96-well plate. The next day and once a day throughout the experiment cells were treated with 50 μM hydrogen peroxide (Sigma). Cell viability was assayed using the CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega, Madison, WI) by addition of 20 μl of CellTiter AQueous One Solution and measurement of absorbance at 490 nm. Relative fold increase was calculated by dividing by absorbance measurements at day zero.
2.9 Statistical analysis
All statistical analysis was performed using GraphPad Prism Software (La Jolla, CA). The specific statistical test used is noted in the respective figure caption. Results are shown as mean ± standard error unless otherwise noted.
3. Results
3.1 Generation of an Nrmt1−/− knockout mouse strain
As many NRMT1 targets are involved in maintaining genome integrity, and NRMT1 itself is required for accurate mitotic division and nucleotide excision repair, we were interested in determining the developmental defects associated with complete NRMT1 loss (Cai et al., 2014; Tooley et al., 2010). We purchased C57B/L6 (B6) derived mouse embryonic stem (ES) cells generated by the North American Conditional Mouse Mutagenesis Project (NorCOMM) expressing a cassette that replaces the third exon of NRMT1 with an E. coli lac Z reporter construct (Fig. 1a). The ES cells were injected into blastocysts obtained post fertilization of albino B6 mice and transplanted into pseudopregnant females. Chimeric males were mated to albino B6 females and monitored for germline transmission/black progeny. The resultant Nrmt1+/− heterozygotes were bred and offspring were genotyped to identify Nrmt1−/− homozygotes (Fig. 1b).
Fig. 1. Generation of the Nrmt1−/− knockout mouse line.
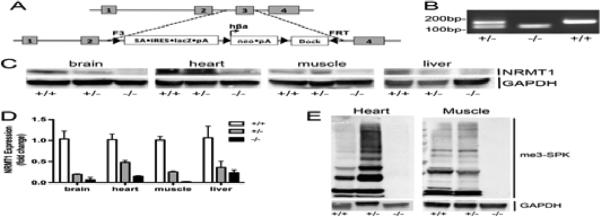
(a) Diagram of the reporter construct used to replace the third exon of NRMT1 and generate the Nrmt1−/− mice. (b) PCR genotyping of Nrmt1+/− heterozygotes (+/−), Nrmt1−/− homozygotes (−/−), and wild type C56BL/6J (+/+) mice. (c) Western blot illustrating loss of NRMT1 protein expression in Nrmt1−/− brain, heart, muscle, and liver tissue as compared to wild type and heterozygotes. GAPDH is used as a loading control. (d) qRT-PCR analysis of NRMT1 mRNA levels in wild type, heterozygous, and homozygous Nrmt1−/− mouse tissues. Levels given as fold change from wild type. (e) Western blot illustrating that along with NRMT1 protein levels, there is a loss of N-terminally methylated proteins (me3-SPK) in the Nrmt1−/− heart and muscle tissue as compared to wild type and heterozygotes. GAPDH was used as a loading control.
Tissues were isolated from the homozygous animals and NRMT1 protein levels were analyzed in the Nrmt1−/− tissues by western blot. No NRMT1 protein was visible in any of the Nrmt1−/− tissues examined (Fig. 1c). NRMT1 mRNA levels were analyzed by quantitative real-time PCR (qRT-PCR). Minimal NRMT1 expression was seen in the Nrmt1−/− mice as compared to wild type B6 and Nrmt1+/− heterozygotes (Fig. 1d). There were also no visible N-terminally trimethylated proteins in the knockout tissues (Fig. 1e). We concluded from these data that the knockout construct produced a functionally null allele of NRMT1, and these mice represent a viable knockout strain.
3.2 Nrmt1−/− mice exhibit developmental, age-related, and premature aging defects
Upon generation of the first Nrmt1−/− homozygotes from the breeding of heterozygous parents, a few striking phenotypes were apparent. First, homozygous knockout pups were represented at less than the expected 25% associated with normal Mendelian inheritance. Second, the knockout pups that did survive were considerably smaller than their wild type and heterozygous littermates (Fig. 2a and b). To quantitate the size difference between wild type mice, Nrmt1+/− heterozygous, and Nrmt1−/− homozygous littermates, mice were weighed at 3 weeks and 3 months of age. There was no statistical difference between wild type males or females and heterozygous males and females at any age (Fig. 2b). However, at 3 weeks and 3 months of age, the Nrmt1−/− males and females weighed significantly less than the wild type and heterozygotes (Fig. 2b). To better quantitate the lifespan of the knockout mice, we documented the age of death of each Nrmt1−/− homozygote. As compared to wild type B6 mice whose average life span is approximately 26 months (Rowlatt et al., 1976), approximately 40% of the Nrmt1−/− mice died before they reached 1 month of age (Fig. 2c). Of those that survived past one month, about half died before 3 months of age, and the other half survived past 6 months (Fig. 2c). The lifespan curves for males and females that survived past one month showed similar patterns, though the percent of female mice surviving past 6 months was lower than that of the males (Fig. 2c). There were also far fewer total female mice that made it past 1 month of age (23 males vs. 14 females), suggesting developmental defects may be more pronounced in the females. Interestingly, the males that survived to six months exhibited phenotypes associated with much older mice, including hair loss, occasional premature graying, and kyphosis (Fig. 2d). The kyphosis was also seen in younger mice (Fig. 2a) and may actually be the result of a developmental defect. Upon necropsy of the animals that died, it was also noted that the main gross anatomical abnormalities were dark amorphous livers that often consisted of only one lobe (Fig. 2e).
Fig. 2. Initial phenotypes of Nrmt1−/− mice.
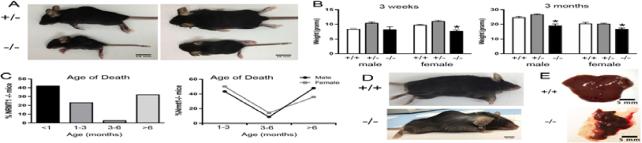
(a and b) Nrmt1−/− homozygotes (−/−) are significantly smaller then their heterozygous (+/−) littermates. Shown are 3 week-old male littermates, side and top view. The weights of Nrmt1−/− males and females differ significantly from wild type mice (+/+) and Nrmt1+/− heterozygotes at 3 weeks and 3 months of age. Each group contained at least 3 mice and statistical analysis was by Student's t-test, * denotes p<0.05. (c) Nrmt1−/− mice die prematurely. Most do not survive past weaning (< 1 month-old), ~20% live 1-3 months, and just over 30% survive past 6 months. The lifespan curves for males and females that survived past one month showed similar patterns. However, of these mice, the percent of female mice surviving past 6 months (~35%) was lower than that of the males (~50%). (d) Of those that survive past 6-months old, some develop premature graying and hair loss. Shown are 9 month-old WT (+/+) and 9 month-old Nrmt−/− males. (e) Nrmt1−/− mice that die before 3 months of age, exhibit severe liver degeneration. The Nrmt1−/− (−/−) livers are much smaller and darker than wild type livers (+/+) of matched age. Shown are 3 month-old male livers.
We also observed fertility problems in the Nrmt1−/− mice. To increase the number of Nrmt1−/− homozygous pups produced by each litter, we initially attempted to mate homozygous Nrmt1−/− males with homozygous females. None of the three breeding pairs produced any progeny even after two months of being housed together. To maintain the line and determine whether the Nrmt1−/− males or females were infertile, each breeding pair was separated and paired with an Nrmt1+/− heterozygote of the opposite sex. The mating pairs of Nrmt1−/− homozygous females and heterozygous males did not produce any progeny. However, the mating pairs of Nrmt1−/− homozygous males paired with heterozygous females produced litters that averaged approximately seven pups (data not shown). Nrmt1−/− females paired with wild type B6 males also produced no progeny. From these data, we concluded that Nrmt1−/− females are infertile, while the males are fertile and produce litters similar in size to those produced by wild type B6 mice (Green and Seyfried, 1991).
3.3 Histological analysis of Nrmt1−/− tissue
The combination of reduced size, reduced life span, premature graying and hair loss, kyphosis, liver defects, and female-specific infertility were reminiscent of phenotypes seen in the TTD mouse model of trichothiodystrophy (de Boer et al., 2002). TTD mice have a mutation in the XPD gene, which encodes a DNA helicase necessary for transcription-coupled NER. They are a mouse model of premature aging that exhibit osteoporosis, osteosclerosis, kyphosis, early graying, cachexia, female-specific infertility, hyperkeratotic epidermis, loss of subcutaneous fat, and reduced lifespan (de Boer et al., 2002). They also have altered liver metabolism, increased levels of liver apoptosis, and increased sensitivity to UV-irradiation and oxidative stress (de Boer et al., 2002; Park et al., 2008; Wijnhoven et al., 2005). Though XPD is not a direct target of NRMT1, DNA damage-binding protein 2 (DDB2), another protein essential for NER, is methylated by NRMT1 (Cai et al., 2014). Given the phenotypic similarities between Nrmt1−/− knockout and TTD mice, and the role for both proteins in NER, we aimed to determine if Nrmt1−/− mice have similar histological, metabolic, and repair deficiencies.
The female infertility seen in TTD mice differs somewhat from what is observed in Nrmt1−/− females. Some TTD females are capable of completing a full-term pregnancy, though they never produced more than one litter and become progressively infertile with age (de Boer et al., 2002). Histological examination of TTD ovaries divided the females into two groups, one group demonstrated immature follicles and no active oestrus, and the other group failed to accumulate corpora lutea, indicating infrequent ovulation (de Boer et al., 2002). Similarly, ovaries were harvested from 3 month-old wild type B6 and Nrmt1−/− homozygous females. Histology showed that Nrmt1−/− ovaries were very small compared to wild type ovaries (Fig. 3a). In addition, Nrmt1−/− ovaries contained preantral and small antral follicles but no corpus luteum indicating incomplete ovulation (Fig. 3a). The ovaries appeared cystic and resembled those seen in mouse models of polycystic ovary syndrome (van Houten and Visser, 2014). This deficiency could account for the infertility of the Nrmt1−/− females.
Fig. 3. Nrmt1−/− organ histology.

(a) Ovaries isolated from 3 month-old wild type (+/+) and Nrmt1−/− homozygous (−/−) females and stained with hematoxylin and eosin (H&E). Nrmt1−/− ovaries are much smaller and contain preantral and small antral follicles but few corpus luteum, indicating incomplete ovulation. The Nrmt1−/− ovaries also appear cystic and resemble those seen in mouse models of polycystic ovary syndrome. (b) Skin isolated from 3 and 6 month-old wild type and Nrmt1−/− males stained with H&E. At 3 months-old there are no drastic differences in skin morphology, though some mice exhibited a thinner epidermal layer (arrows). At 6 months, there were smaller and fewer blood vessels in the Nrmt1−/− mice, the epidermis was consistently thinner, and there was a dramatic increase in collagen bundles (dark pink staining), reminiscent of skin fibrosis. (c) Livers isolated from 3 and 6 month-old wild type and Nrmt1−/− males stained with H&E. There were no apparent histological differences at either time point, all tissues were highly cellular and relatively devoid of fat.
The skin of TTD mice shows severe dilation of hair follicles, massive hyperkeratosis and absence of subcutaneous fat (de Boer et al., 2002). Therefore, we examined the skin of 3 and 6 month-old wild type B6 and Nrmt1−/− homozygous males. Skin from 3 month-old Nrmt1−/− mice appeared relatively normal with a slight thinning of the epidermis (Fig 3b). However, at 6 months-old Nrmt1−/− skin exhibited a substantial decrease in blood vessel size and an increase in narrowly spaced, thick collagen bundles that are reminiscent of skin fibrosis (Fig. 3b). Despite the gross liver degeneration seen at time of death of Nrmt1−/− mice that die prematurely, we found no difference in histology of the livers of wild type and Nrmt1−/− homozygous males at 3 or 6 months of age (Fig. 3c). However, these livers were harvested from Nrmt1−/− mice that lived to 3 or 6 months (and by definition had the least severe phenotype) that were euthanized specifically for tissue histology. Histological analysis of tissues from animals that die naturally is complicated by amount of time lapsing between time of death and tissue harvest, and it was not possible to predict which of the Nrmt1−/− mice were going to die prematurely. We speculate that, had we been able to isolate fresh livers from the Nrmt1−/− mice that died prematurely, they would have exhibited more severe histological phenotypes.
3.4 Characterization of Nrmt1−/− liver defects
As the liver histology of the Nrmt1−/− mice did not clearly indicate a cause for the observed degeneration, we wanted to determine if the liver mitochondria had impaired function or the livers themselves were becoming apoptotic and/or altering their metabolic states. Mitochondrial dysfunction can lead to accumulated mitochondria oxidative damage or promote cellular senescence, both drivers of aging phenotypes (Ziegler et al., 2014). We first tested the respiration of liver mitochondria of 3 month-old wild type and Nrmt1−/− homozygous males. We found that Nrmt1−/− liver mitochondria had significantly decreased proton leak and State 3 respiration as compared to aged-matched wild type controls (Fig. 4a and b). In addition, Nrmt1−/− livers had significantly reduced maximum mitochondrial respiration in the non-coupled state of the electron transfer system (ETS) (Fig. 4c). This data indicates that Nrmt1−/− livers have decreased mitochondria respiration and therefore decreased ability to generate energy for optimal liver function. In addition to mitochondria respiration, we also measured aconitase activity of the liver of 3 month-old wild type and Nrmt1−/− males. Aconitase activity is very sensitive to reactive oxygen species (ROS) generated by the mitochondria (Cantu et al., 2009). We found that aconitase activity of Nrmt1−/− livers was increased compared to wild type livers (Fig. 4d). This data supports the observation of reduced mitochondrial liver function in Nrmt1−/− mice, as increased aconitase activity indicates less ROS generation.
Fig. 4. Nrmt1−/− liver has decreased mitochondrial function.
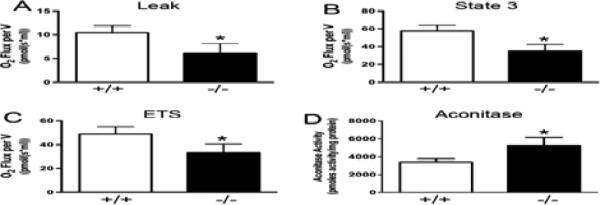
(a, b, and c) Mitochondrial respiration measurements of proton leak, State 3 respiration, and electron transfer system of isolated mitochondria from 3 month-old Nrmt1−/− and wild type males. (d) Aconitase activity of liver from 3 month-old Nrmt1−/− and wild type males indicating less reactive oxygen species generation. Each bar represents the mean ± SEM of three mice. Statistical analysis was by Student's t-test, * denotes p<0.05.
TTD mice exhibit increased liver apoptosis and decreased expression of genes involved in lipid metabolism, steroid biosynthesis, and oxidative phosphorylation (Park et al., 2008). In this DNA repair-defective background, reducing liver metabolism is a potential mechanism for lessening the production of damage-inducing ROS and attempting to extend lifespan (Wijnhoven et al., 2005). To see if a similar mechanism was being employed by Nrmt1−/− mice, qRT-PCR analysis was performed on the livers of 3 and 6 month-old wild type and Nrmt1−/− homozygous males for genes involved in apoptosis (Fas), lipid metabolism (FABP1, ACAA1, ELOVL5), steroid biosynthesis (TM7SF2, SC5D, APOE), and oxidative phosphorylation (COX5B, NDUFC1, ATP6VOC).
At 3 months of age, there were few differences between wild type and homozygous livers. There was no increase in Fas levels and only one gene in each of the other three categories showed any significant change (Fig. 5a-d). Of the three markers for lipid metabolism, fatty acid binding protein 1 (FABP1) was significantly reduced in the Nrmt1−/− homozygotes (Fig. 5b). Of the three markers for steroid biosynthesis, the sterol reductase TM7SF2 showed increased expression in homozygotes (Fig. 5c). Of the oxidative phosphorylation genes, the ATPase subunit ATP6VOC, also showed a small but significant increase in homozygotes (Fig. 5d).
Fig. 5. qRT-PCR analysis of liver mRNA expression for genes involved in apoptosis, fatty acid metabolism, steroid biosynthesis and oxidative phosphorylation.

(a) Expression of Fas receptor (Fas) is significantly increased in 6 month-old Nrmt1−/− males (as compared to wild type control), indicating their livers are becoming more apoptotic. (b) Expression of fatty acid-binding protein 1 (FABP1) is significantly decreased at both 3 and 6 months, indicating a decrease in the amount of fatty acids being metabolized. Expression of acetyl-Coenzyme A acyltransferase 1 (ACAA1) is significantly increased in the Nrmt1−/− males at 6 months, which could be an attempt to compensate for decreased mitochondrial function. (c and d) Similarly, the sterol reductase TM7SF2, the sterol desaturase SC5D, the apolipoprotein APOE (all three involved in sterol biosynthesis), the cytochrome c oxidase subunit COX5B, the mitochondrial Complex I subunit NDUFC1, and the ATPase subunit ATP6VOC (all three involved in oxidative phosphorylation) showed significant increases in the Nrmt1−/− males at 6 months. We propose these increases are also an attempt to compensate for the inherently low mitochondrial function of Nrmt1−/− mice. Each bar represents the mean ± SEM of three mice. Statistical analysis was by Student's t-test, * denotes p<0.05.
It is unclear why FABP1 levels have a slight decrease in 3-month old Nrmt1−/− homozygotes, but the increase in the other markers seems to mark the start of a change in liver metabolism for Nrmt1−/− mice. In contrast to the TTD mice, by 6 months of age Nrmt1−/− homozygotes showed an increase in almost all markers tested (FABP1 increased slightly compared to 3-month but still remained significantly decreased from wild type) (Fig. 5a-d). There was a greater than 3-fold increase in Fas expression in Nrmt1−/− livers, indicating an increase in apoptotic rates (Fig. 5a). There was also a greater than 5-fold increase in expression of the acetyl-Coenzyme A acyltransferase ACAA1, the mitochondrial Complex I subunit NDUFC1, TM7SF2, and ATP6VOC and a greater than 2-fold increase in the apolipoprotein APOE, the sterol desaturase SC5D, and the cytochrome c oxidase subunit COX5B (Fig. 5b-d). These data indicate, that contrary to TTD mice, which dampen liver metabolism in attempt to decrease potential DNA damage and extend lifespan (Wijnhoven et al., 2005), Nrmt1−/− mice have upregulated liver metabolism. This could represent an attempt to compensate for decreased mitochondrial liver respiration and normally N-terminally methylated metabolic enzymes that are now working at suboptimal activity. Again these livers were taken from Nrmt1−/− mice with the least severe phenotypes, and we predict these differences in mitochondrial function and gene expression are even more pronounced in the mice that die prematurely.
We have previously noted that at 6 weeks of age, NRMT1 expression is quite low in the liver (Petkowski et al., 2013). To ascertain if NRMT1 function in the liver may be more critical as an animal ages, we obtained livers from wild type C57B/L6 mice aged 3, 10, 18, 24, and 33 months. Interestingly, NRMT1 levels increased significantly from 3 to 10 months and remained elevated through 24 months of age (Fig. 6a). Levels began to decrease at 33 months, as mice approached the end of their normal lifespan (Fig. 6a). To determine if 6 month-old Nrmt1−/− livers had similar gene expression patterns to aged wild type livers, qRT-PCR was performed on the aged livers for Fas, FABP1, NDUFC1, ATP6VOC, and TM7SF2. 6 month-old Nrmt1−/− livers have higher Fas expression than wild type livers at 6, 24, or 33 months, indicating these livers are abnormally apoptotic (Fig. 6b). FABP1 expression decreases as animals age and liver function decreases, and 6 month Nrmt1−/− livers have levels similar to those seen in 10 to 24 month wild type mice (Fig. 6c). The other three markers, NDUFC1, ATP6VOC, and TM7SF2 demonstrated significantly increased levels as the livers aged. Expression of the 6 month Nrmt1−/− livers most closely resembled the levels seen in 10 month wild type mice (Fig. 6d and e). These data suggest NRMT1 expression is increasingly necessary in the liver as animals age, and these altered metabolic markers are signs of premature aging, as the expression pattern of 6 month Nrmt1−/− livers more closely resembles the expression pattern of older wild type mice than matched 6 month controls.
Fig. 6. qRT-PCR analysis of mRNA expression levels in aged mouse livers.
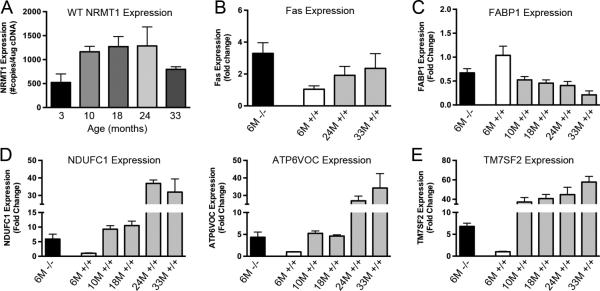
(a) NRMT1 mRNA expression in wild type C57BL/6J males is lowest at 3 months of age. These levels increase by 10 months of age and remain constant until they decrease with extreme old age (33 months). This indicates NRMT1 expression is important during the aging process. (b) Fas Receptor (Fas) mRNA expression is higher in 6 month-old Nrmt1−/− males than even 33 month-old wild type males, indicating their livers are highly apoptotic. (c) Expression of Fatty acid-binding protein 1 (FABP1) generally decreases with age, and levels of FABP1 mRNA in Nrmt1−/− males most closely resemble the levels seen in 10-24 month-old wild type mice. (d and e) Conversely, expression of NDUFC1, ATP6VOC, and TM7SF2 increase with age, and levels of these transcripts in Nrmt1−/− males most closely resemble the levels seen in 10-18 month-old wild type mice. These data indicate the livers in Nrmt1−/− males may be aging at an accelerated rate, and this could contribute to the liver degeneration seen at death. Each bar represents the mean ± SEM of three mice.
3.5 Nrmt1−/− MEFs are sensitive to oxidative damage
Finally, as NRMT1 loss has been shown to increase sensitivity to agents that produce UV-induced damage (Cai et al., 2014) and double-strand DNA breaks (LB unpublished data), and results in less ROS production (Fig. 4d), we generated wild type and Nrmt1−/− knockout MEFs (Fig. 7a and b) and assayed their sensitivity to the ROS producing and DNA damaging agent hydrogen peroxide. Though basal growth rates are similar between wild type and Nrmt1−/− knockout MEFs (Fig. 7c), treatment with hydrogen peroxide significantly decreased the viability of Nrmt1−/− MEFs at 24, 48 and 72 hours post-treatment as compared to wild type MEFs (Fig. 7d). These data suggest that loss of NRMT1 sensitizes cells to ROS production, in addition to other DNA damaging agents, and the decreased mitochondrial function of Nrmt1−/− mice might provide beneficial protection from excessive internal ROS production. Whether this decreased mitochondrial function is in response to defective DNA repair (as hypothesized with TTD mice (Park et al., 2008)) or an unrelated advantageous phenotype remains to be determined.
Fig. 7. Nrmt1−/− mouse embryonic fibroblasts have increased sensitivity to hydrogen peroxide.
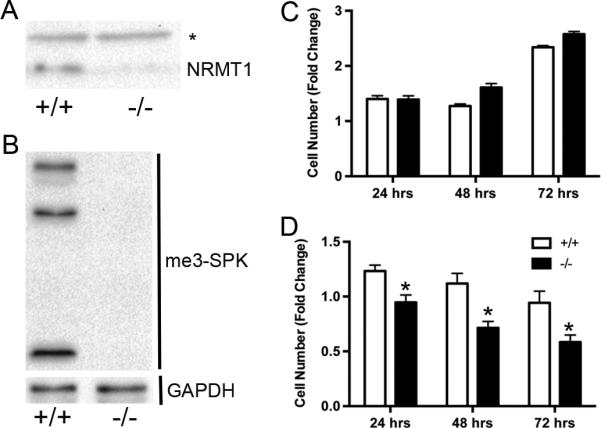
(a) Similar to other Nrmt1−/− tissues, no NRMT1 protein expression is detectable in the Nrmt1−/− mouse embryonic fibroblasts (MEFs). * is non-specific band detected by the anti-NRMT1 antibody, which is used as a loading control. The level of NRMT1 in wild type MEFs (+/+) is shown as a control. (b) No N-terminal trimethylation (me3-SPK) is detectable in the Nrmt1−/− MEFs. GAPDH is used as a loading control. (c) Cell proliferation assay measuring the basal growth rates of wild type and Nrmt1−/− MEFs. No significant difference was detected at 24, 48, or 72 hours. (d) Cell proliferation assay measuring the decreased growth rate of Nrmt1−/− MEFs in response to 50 μM hydrogen peroxide as compared to wild type MEFs. Each bar represents the mean ± SEM of three to six independent experiments. Statistical analysis was by Two-Way Anova, * denotes p<0.05.
4. Discussion
The generation of Nrmt1−/− knockout mice described here demonstrates for the first time the importance of N-terminal methylation in mammalian development and aging. While it had previously been shown that N-terminal methylation of DNA damage-binding protein 2 (DDB2) is necessary for efficient NER (Cai et al., 2014) and loss of NRMT1 sensitizes cells to agents that cause double-strand DNA breaks (LB unpublished data), we wanted to demonstrate that these deficiencies in DNA repair have phenotypic consequences in vivo. We found that Nrmt1−/− mice exhibit developmental defects (decreased size, kyphosis, female specific infertility), age-related defects (skin pathology), and premature aging phenotypes (early greying, hair loss, and altered liver metabolic markers). The observed phenotypes of the Nrmt1−/− knockout mice complement the in vitro data, and we propose a model where NRMT1 loss results in a global decrease in the affinity of its targets for DNA. In regards to DNA repair, this prevents accumulation of important NER and double stranded break repair proteins at sites of damage and results in inefficient resolution of lesions. It remains to be determined if additional modes of the DNA damage response are also deficient in Nrmt1−/− knockout mice, as RB1 and HP1γ are also NRMT1 substrates (proven and predicted, respectively) and are implicated in not just NER and DSB repair, but base excision repair (BER) as well (Carr et al., 2014; Lin et al., 2009; Luijsterburg et al., 2009; Sudhakar et al., 2014).
The majority of the work studying post-translational modification during DNA repair has focused on phosphorylation, especially the role of the signaling kinases ATM and ATR (ataxia telangiectasia mutated and ataxia telangiectasia and Rad3 related, respectively) (Savitsky et al., 1995 and Stiff et al., 2006). However, there is a growing appreciation for the existence of significant crosstalk between phosphorylation and other post-translational modifications at sites of DNA damage, including methylation, acetylation, ubiquitination, and SUMOylation (Zhao et al., 2014). Phosphorylation is a rapid and reversible way to regulate protein function in response to DNA damage (Mariotti et al., 2013), and it is possible other modifying enzymes are recruited by phosphoproteins to provide a longer-lived, yet still reversible, response. It is still highly debated whether N-terminal post-translational modifications can be removed, as there are currently no known N-terminal deacetylases, demethylases, or deubiquitinating enzymes (Tooley and Schaner Tooley, 2014). However, levels of both N-terminal acetylation and methylation can vary according to metabolic state or cell cycle stage (Chen et al., 2007; Yi et al., 2011), so it will be interesting to determine if they are being enzymatically removed or simply lost due to N-terminal cleavage.
The observed liver phenotypes in the Nrmt1−/− knockout mice could result directly from an accumulation of DNA damage, or alternatively, a decrease in inherent liver function due to loss of NRMT1. The liver is a slowly proliferating tissue with high oxygen metabolism, and thereby especially sensitive and prone to accumulating DNA damage (Osterod et al., 2001). Though all organs in the Nrmt1−/− knockout mouse would theoretically be equally deficient in DNA repair, the liver could potentially be the first to exhibit phenotypes due to its increased burden of damage-inducing ROS. However, the significantly increased aconitase activity in Nrmt1−/− livers signifies a general decrease in ROS production and indicates this is not the case. The corresponding decrease in mitochondrial function and increase in expression of genes involved in lipid metabolism, steroid biosynthesis, and oxidative phosphorylation imply Nrmt1−/− livers may be trying to compensate unsuccessfully for an inherent functional impairment. TTD mice attempt to compensate for their inefficient DNA repair by decreasing liver metabolism and its corresponding ROS production (Wijnhoven et al., 2005). Nrmt1−/− mice may already have inherent metabolic problems due to lack of N-terminal methylation of key metabolic enzymes.
The mitochondrial Complex I component NDUFA11 contains the consensus sequence for N-terminal methylation, as do the UDP-glucuronosyltransferase UGT1A10, the arylsulfatase ARSB, the flavin containing monooxygenase FMO3, and a variety of other metabolic enzymes (Petkowski et al., 2012). Though it is unlikely N-terminal methylation is regulating the interaction of these enzymes with DNA, N-terminal methylation is often found on proteins in large multi-subunit complexes, is predicted to regulate protein/protein interactions, and may prevent proper formation of these large enzymatic complexes (Stock et al., 1987). If N-terminal methylation of NDUFA11 helps maintain the structural integrity of Complex I, overexpression of other genes involved in oxidative phosphorylation could help balance this destabilized enzyme complex. Similarly, N-terminal methylation of UGT1A10 could regulate its homo- or heterodimerization (Operana and Tukey, 2007), and loss of this modification could disrupt enzyme function and steroid excretion. Impaired UGT1A10 function has been associated with bilirubin accumulation (Milton et al., 2012), and the Nrmt1−/− mice exhibiting liver degeneration also appear jaundiced (LB unpublished data). We are currently looking at Complex I function in the mitochondria of Nrmt1−/− livers and assaying plasma bilirubin levels in Nrmt1−/− mice. We are also interested in looking at the kidney function of Nrmt1−/− mice, as this organ is also metabolically quite active (Forbes et al., 2008) and may be especially susceptible to loss of NRMT1.
In light of the DNA repair deficiencies and altered liver function, it is not surprising that the Nrmt1−/− knockout mice also exhibit some phenotypes associated with premature aging. However, approximately 40% of Nrmt1−/− knockout mice also die before reaching 3 weeks of age. This indicates additional developmental defects or variable penetrance of the DNA repair defects. TTD mice develop normally to adulthood and only then begin to exhibit phenotypes, indicating normal development followed by genuine aging defects (de Boer et al., 2002). However, complete absence of NER or transcription couple repair can result in death within 3 weeks, while low levels of residual repair activity can mitigate the severity and rate of aging phenotypes (de Boer et al., 2002). We hypothesize the variable lifespan seen in Nrmt1−/− knockout mice could result from a maternal effect of NRMT1 expression and an unequal distribution of maternal NRMT1 mRNA between oocytes.
NRMT1 would not be the first protein involved in DNA repair to exhibit maternal effect phenotypes. The Drosophila paralogs of Rad51 and Rad54 were discovered in a maternal effect screen for abnormal meiosis (Morris and Lehmann, 1999). These proteins are required to resolve the double stranded DNA breaks that occur during meiotic recombination and their loss results in female-specific infertility (Staeva-Vieira et al., 2003), which is also seen in the Nrmt−/− knockout mice. In mice, DNA mismatch repair (DMR) also exhibits a maternal effect. When mated to wild type males, female mice deficient in Pms2, the mammalian homolog of mutL, can pass DMR defects to their progeny even though these progeny are technically DMR proficient (Gurtu et al., 2002). Maternal Pms2 deficiency in one-cell embryos seems to promote unrepaired replication errors during early cell division (Gurtu et al., 2002). We propose a similar mechanism may be taking place in NRMT1 deficient embryos, and the amount of genomic lesions acquired early is determining later phenotypes. In completely null Nrmt1−/− females, abnormal chromosomal segregation and unrepaired damage could accumulate and no viable oocytes would be produced. In Nrmt1+/− heterozygous females, the maternal load of NRMT1 could be unequally distributed, leading to embryos with no expression and extensive instability that die, embryos with moderate expression and damage that are born live but cannot survive past 3 weeks, and embryos with higher expression and few lesions that make it through to adulthood but eventually accumulate additional damage and exhibit aging phenotypes prematurely.
Given the abundance of NRMT1 targets, it is likely DNA repair is not the only biological process misregulated in the Nrmt1−/− mice. However, our work with these mice and with NRMT1 loss in cell culture indicates it is a predominant factor in driving the resultant phenotypes. In cell culture, NRMT1 loss also causes a multi-polar spindle phenotype and unequal cell division (Chen et al., 2007; Tooley et al., 2010), so it will be interesting to determine if aneuploid cells are found in Nrmt1−/− tissues. This could be an additional driving force of genomic instability and contribute to the observed phenotypes (Pratt et al., 2011). As future work is done to comprehensively understand these Nrmt1−/− knockout mice, we believe they will become a novel and useful model for studying both organismal and tissue-specific development and aging. Contrary to current aging models, which are predominantly depleted of one mode of genomic repair, surviving Nrmt1−/− mice are impaired in multiple mechanisms of genome maintenance and may more closely mimic the gradual accumulation of different types of damage seen with normal aging.
Supplementary Material
Highlights.
Knockout of NRMT1 phenocopies transgenic mice deficient in DNA repair
Nrmt1−/− mice have a short life span, decreased body size, and female infertility
They have kyphosis, decreased mitochondrial function, and altered liver metabolism
Useful for studying how weakening of multiple DNA repair pathways accelerates aging
Acknowledgements
This research was supported by grants awarded to C.E.S.T. from the National Cancer Institute [CA158009] and M.P.C. from the National Heart, Lung, and Blood Institute [HL95769].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Cai Q, Fu L, Wang Z, Gan N, Dai X, Wang Y. alpha-N-Methylation of Damaged DNA-binding Protein 2 (DDB2) and Its Function in Nucleotide Excision Repair. J Biol Chem. 2014;289:16046–56. doi: 10.1074/jbc.M114.558510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantu D, Schaack J, Patel M. Oxidative inactivation of mitochondrial aconitase results in iron and H2O2-mediated neurotoxicity in rat primary mesencephalic cultures. PLoS One. 2009;4:e7095. doi: 10.1371/journal.pone.0007095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Li W, Kim S, Brodie SG, Deng CX. Senescence, aging, and malignant transformation mediated by p53 in mice lacking the Brca1 full-length isoform. Genes Dev. 2003;17:201–13. doi: 10.1101/gad.1050003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr SM, Munro S, Zalmas LP, Fedorov O, Johansson C, Krojer T, Sagum CA, Bedford MT, Oppermann U, La Thangue NB. Lysine methylation-dependent binding of 53BP1 to the pRb tumor suppressor. Proc Natl Acad Sci U S A. 2014;111:11341–6. doi: 10.1073/pnas.1403737111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T, Muratore TL, Schaner-Tooley CE, Shabanowitz J, Hunt DF, Macara IG. N-terminal alpha-methylation of RCC1 is necessary for stable chromatin association and normal mitosis. Nat Cell Biol. 2007;9:596–603. doi: 10.1038/ncb1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai X, Otake K, You C, Cai Q, Wang Z, Masumoto H, Wang Y. Identification of novel alpha-n-methylation of CENP-B that regulates its binding to the centromeric DNA. J Proteome Res. 2013;12:4167–75. doi: 10.1021/pr400498y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S, Snow CJ, Paschal BM. A pathway linking oxidative stress and the Ran GTPase system in progeria. Mol Biol Cell. 2014;25:1202–15. doi: 10.1091/mbc.E13-07-0430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer J, Andressoo JO, de Wit J, Huijmans J, Beems RB, van Steeg H, Weeda G, van der Horst GT, van Leeuwen W, Themmen AP, Meradji M, Hoeijmakers JH. Premature aging in mice deficient in DNA repair and transcription. Science. 2002;296:1276–9. doi: 10.1126/science.1070174. [DOI] [PubMed] [Google Scholar]
- Escargueil AE, Soares DG, Salvador M, Larsen AK, Henriques JA. What histone code for DNA repair? Mutat Res. 2008;658:259–70. doi: 10.1016/j.mrrev.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Forbes JM, Coughlan MT, Cooper ME. Oxidative stress as a major culprit in kidney disease in diabetes. Diabetes. 2008;57:1446–54. doi: 10.2337/db08-0057. [DOI] [PubMed] [Google Scholar]
- Green RC, Seyfried TN. Kindling susceptibility and genetic seizure predisposition in inbred mice. Epilepsia. 1991;32:22–6. doi: 10.1111/j.1528-1157.1991.tb05605.x. [DOI] [PubMed] [Google Scholar]
- Greer EL, Maures TJ, Hauswirth AG, Green EM, Leeman DS, Maro GS, Han S, Banko MR, Gozani O, Brunet A. Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans. Nature. 2010;466:383–7. doi: 10.1038/nature09195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurtu VE, Verma S, Grossmann AH, Liskay RM, Skarnes WC, Baker SM. Maternal effect for DNA mismatch repair in the mouse. Genetics. 2002;160:271–7. doi: 10.1093/genetics/160.1.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasty P, Campisi J, Hoeijmakers J, van Steeg H, Vijg J. Aging and genome maintenance: lessons from the mouse? Science. 2003;299:1355–9. doi: 10.1126/science.1079161. [DOI] [PubMed] [Google Scholar]
- Hu Y, Parvin JD. SUMO isoforms and conjugation-independent function in DNA double-strand break repair pathways. J Biol Chem. 2014;289:21289–95. doi: 10.1074/jbc.C114.582122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley JB, Datta S, Snow CJ, Chatterjee M, Ni L, Spencer A, Yang CS, Cubenas-Potts C, Matunis MJ, Paschal BM. The defective nuclear lamina in Hutchinson-gilford progeria syndrome disrupts the nucleocytoplasmic Ran gradient and inhibits nuclear localization of Ubc9. Mol Cell Biol. 2011;31:3378–95. doi: 10.1128/MCB.05087-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S. Implication of posttranslational histone modifications in nucleotide excision repair. Int J Mol Sci. 2012;13:12461–86. doi: 10.3390/ijms131012461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin PS, McPherson LA, Chen AY, Sage J, Ford JM. The role of the retinoblastoma/E2F1 tumor suppressor pathway in the lesion recognition step of nucleotide excision repair. DNA Repair (Amst) 2009;8:795–802. doi: 10.1016/j.dnarep.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luijsterburg MS, Dinant C, Lans H, Stap J, Wiernasz E, Lagerwerf S, Warmerdam DO, Lindh M, Brink MC, Dobrucki JW, Aten JA, Fousteri MI, Jansen G, Dantuma NP, Vermeulen W, Mullenders LH, Houtsmuller AB, Verschure PJ, van Driel R. Heterochromatin protein 1 is recruited to various types of DNA damage. J Cell Biol. 2009;185:577–86. doi: 10.1083/jcb.200810035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariotti LG, Pirovano G, Savage KI, Ghita M, Ottolenghi A, Prise KM, Schettino G. Use of the gamma-H2AX assay to investigate DNA repair dynamics following multiple radiation exposures. PLoS One. 2013;8:e79541. doi: 10.1371/journal.pone.0079541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCauley BS, Dang W. Histone methylation and aging: Lessons learned from model systems. Biochim Biophys Acta. 2014;1839:1454–62. doi: 10.1016/j.bbagrm.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton JN, Sebastiani P, Solovieff N, Hartley SW, Bhatnagar P, Arking DE, Dworkis DA, Casella JF, Barron-Casella E, Bean CJ, Hooper WC, DeBaun MR, Garrett ME, Soldano K, Telen MJ, Ashley-Koch A, Gladwin MT, Baldwin CT, Steinberg MH, Klings ES. A genome-wide association study of total bilirubin and cholelithiasis risk in sickle cell anemia. PLoS One. 2012;7:e34741. doi: 10.1371/journal.pone.0034741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris J, Lehmann R. Drosophila oogenesis: versatile spn doctors. Curr Biol. 1999;9:R55–8. doi: 10.1016/s0960-9822(99)80010-4. [DOI] [PubMed] [Google Scholar]
- Operana TN, Tukey RH. Oligomerization of the UDP-glucuronosyltransferase 1A proteins: homo- and heterodimerization analysis by fluorescence resonance energy transfer and co-immunoprecipitation. J Biol Chem. 2007;282:4821–9. doi: 10.1074/jbc.M609417200. [DOI] [PubMed] [Google Scholar]
- Osterod M, Hollenbach S, Hengstler JG, Barnes DE, Lindahl T, Epe B. Age-related and tissue-specific accumulation of oxidative DNA base damage in 7,8-dihydro-8-oxoguanine-DNA glycosylase (Ogg1) deficient mice. Carcinogenesis. 2001;22:1459–63. doi: 10.1093/carcin/22.9.1459. [DOI] [PubMed] [Google Scholar]
- Park JY, Cho MO, Leonard S, Calder B, Mian IS, Kim WH, Wijnhoven S, van Steeg H, Mitchell J, van der Horst GT, Hoeijmakers J, Cohen P, Vijg J, Suh Y. Homeostatic imbalance between apoptosis and cell renewal in the liver of premature aging Xpd mice. PLoS One. 2008;3:e2346. doi: 10.1371/journal.pone.0002346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkowski JJ, Bonsignore LA, Tooley JG, Wilkey DW, Merchant ML, Macara IG, Schaner Tooley CE. NRMT2 is an N-terminal monomethylase that primes for its homologue NRMT1. Biochem J. 2013;456:453–62. doi: 10.1042/BJ20131163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petkowski JJ, Schaner Tooley CE, Anderson LC, Shumilin IA, Balsbaugh JL, Shabanowitz J, Hunt DF, Minor W, Macara IG. Substrate specificity of mammalian N-terminal alpha-amino methyltransferase NRMT. Biochemistry. 2012;51:5942–50. doi: 10.1021/bi300278f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettigrew GW, Smith GM. Novel N-terminal protein blocking group identified as dimethylproline. Nature. 1977;265:661–2. doi: 10.1038/265661a0. [DOI] [PubMed] [Google Scholar]
- Pratt CH, Curtain M, Donahue LR, Shopland LS. Mitotic defects lead to pervasive aneuploidy and accompany loss of RB1 activity in mouse LmnaDhe dermal fibroblasts. PLoS One. 2011;6:e18065. doi: 10.1371/journal.pone.0018065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowlatt C, Chesterman FC, Sheriff MU. Lifespan, age changes and tumour incidence in an ageing C57BL mouse colony. Lab Anim. 1976;10:419–42. doi: 10.1258/002367776780956917. [DOI] [PubMed] [Google Scholar]
- Savitsky K, Bar-Shira A, Gilad S, Rotman G, Ziv Y, Vanagaite L, Tagle DA, Smith S, Uziel T, Sfez S, Ashkenazi M, Pecker I, Frydman M, Harnik R, Patanjali SR, Simmons A, Clines GA, Sartiel A, Gatti RA, Chessa L, Sanal O, Lavin MF, Jaspers NG, Taylor AM, Arlett CF, Miki T, Weissman SM, Lovett M, Collins FS, Shiloh Y. A single ataxia telangiectasia gene with a product similar to PI-3 kinase. Science. 1995;268:1749–53. doi: 10.1126/science.7792600. [DOI] [PubMed] [Google Scholar]
- Scheibye-Knudsen M, Ramamoorthy M, Sykora P, Maynard S, Lin PC, Minor RK, Wilson DM, 3rd, Cooper M, Spencer R, de Cabo R, Croteau DL, Bohr VA. Cockayne syndrome group B protein prevents the accumulation of damaged mitochondria by promoting mitochondrial autophagy. J Exp Med. 2012;209:855–69. doi: 10.1084/jem.20111721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siebold AP, Banerjee R, Tie F, Kiss DL, Moskowitz J, Harte PJ. Polycomb Repressive Complex 2 and Trithorax modulate Drosophila longevity and stress resistance. Proc Natl Acad Sci U S A. 2010;107:169–74. doi: 10.1073/pnas.0907739107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver HR, Nissley JA, Reed SH, Hou YM, Johnson ES. A role for SUMO in nucleotide excision repair. DNA Repair (Amst) 2011;10:1243–51. doi: 10.1016/j.dnarep.2011.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staeva-Vieira E, Yoo S, Lehmann R. An essential role of DmRad51/SpnA in DNA repair and meiotic checkpoint control. EMBO J. 2003;22:5863–74. doi: 10.1093/emboj/cdg564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stock A, Clarke S, Clarke C, Stock J. N-terminal methylation of proteins: structure, function and specificity. FEBS Lett. 1987;220:8–14. doi: 10.1016/0014-5793(87)80866-9. [DOI] [PubMed] [Google Scholar]
- Sudhakar J, Khetan V, Madhusudan S, Krishnakumar S. Dysregulation of human apurinic/apyrimidinic endonuclease 1 (APE1) expression in advanced retinoblastoma. Br J Ophthalmol. 2014;98:402–7. doi: 10.1136/bjophthalmol-2013-304166. [DOI] [PubMed] [Google Scholar]
- Stiff T, Walker SA, Cerosaletti K, Goodarzi AA, Petermann E, Concannon P, O’Driscoll M, Jeggo PA. ATR-dependent phosphorylation and activation of ATM in response to UV treatment or replication fork stalling. EMBO J. 2006;25:5775–5782. doi: 10.1038/sj.emboj.7601446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooley CE, Petkowski JJ, Muratore-Schroeder TL, Balsbaugh JL, Shabanowitz J, Sabat M, Minor W, Hunt DF, Macara IG. NRMT is an alpha-N-methyltransferase that methylates RCC1 and retinoblastoma protein. Nature. 2010;466:1125–8. doi: 10.1038/nature09343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tooley JG, Schaner Tooley CE. New roles for old modifications: Emerging roles of N-terminal post-translational modifications in development and disease. Protein Sci. 2014;23:1641–9. doi: 10.1002/pro.2547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Horst GT, van Steeg H, Berg RJ, van Gool AJ, de Wit J, Weeda G, Morreau H, Beems RB, van Kreijl CF, de Gruijl FR, Bootsma D, Hoeijmakers JH. Defective transcription-coupled repair in Cockayne syndrome B mice is associated with skin cancer predisposition. Cell. 1997;89:425–35. doi: 10.1016/s0092-8674(00)80223-8. [DOI] [PubMed] [Google Scholar]
- van Gent DC, Hoeijmakers JH, Kanaar R. Chromosomal stability and the DNA double-stranded break connection. Nat Rev Genet. 2001;2:196–206. doi: 10.1038/35056049. [DOI] [PubMed] [Google Scholar]
- van Houten EL, Visser JA. Mouse models to study polycystic ovary syndrome: a possible link between metabolism and ovarian function? Reprod Biol. 2014;14:32–43. doi: 10.1016/j.repbio.2013.09.007. [DOI] [PubMed] [Google Scholar]
- Vogel H, Lim DS, Karsenty G, Finegold M, Hasty P. Deletion of Ku86 causes early onset of senescence in mice. Proc Natl Acad Sci U S A. 1999;96:10770–5. doi: 10.1073/pnas.96.19.10770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CM, Tsai SN, Yew TW, Kwan YW, Ngai SM. Identification of histone methylation multiplicities patterns in the brain of senescence-accelerated prone mouse 8. Biogerontology. 2010;11:87–102. doi: 10.1007/s10522-009-9231-5. [DOI] [PubMed] [Google Scholar]
- Weeda G, Donker I, de Wit J, Morreau H, Janssens R, Vissers CJ, Nigg A, van Steeg H, Bootsma D, Hoeijmakers JH. Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Curr Biol. 1997;7:427–39. doi: 10.1016/s0960-9822(06)00190-4. [DOI] [PubMed] [Google Scholar]
- Wijnhoven SW, Beems RB, Roodbergen M, van den Berg J, Lohman PH, Diderich K, van der Horst GT, Vijg J, Hoeijmakers JH, van Steeg H. Accelerated aging pathology in ad libitum fed Xpd(TTD) mice is accompanied by features suggestive of caloric restriction. DNA Repair (Amst) 2005;4:1314–24. doi: 10.1016/j.dnarep.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Yi CH, Pan H, Seebacher J, Jang IH, Hyberts SG, Heffron GJ, Vander Heiden MG, Yang R, Li F, Locasale JW, Sharfi H, Zhai B, Rodriguez-Mias R, Luithardt H, Cantley LC, Daley GQ, Asara JM, Gygi SP, Wagner G, Liu CF, Yuan J. Metabolic regulation of protein N-alpha-acetylation by Bcl-xL promotes cell survival. Cell. 2011;146:607–20. doi: 10.1016/j.cell.2011.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Brickner JR, Majid MC, Mosammaparast N. Crosstalk between ubiquitin and other post-translational modifications on chromatin during double-strand break repair. Trends Cell Biol. 2014;24:426–434. doi: 10.1016/j.tcb.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler DV, Wiley CD, Velarde MC. Mitochondrial effectors of cellular senescence: beyond the free radical theory of aging. Aging Cell. 2015;14:1–7. doi: 10.1111/acel.12287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.