

Abstract
Genes which confer a relative longevity advantage may be regulated at the level of transcription or translation. Alternatively, pro-longevity genes may mediate their effects at the level of protein structure-functional relationships that are beneficially optimized in long-lived species. Longevity associated genes (LAGs) may be operationally defined as genes that confer beneficial effects and are relatively more conserved among long-lived species. Global and local protein sequence alignments of over 10,000 genes across at least 30 mammalian species were examined to identify LAGs. Known LAGs, including growth hormone receptor (GHR), and breast cancer 1, early onset (BRCA1), have strong associations with maximum lifespan by our analysis. Several common categories of protein function were observed among genes ranked with the strongest associations with MLS identified by all regression models. These genes included those that function in the immune system, cell cycle regulation, and DNA damage response. We provide a ranking of genes with the strongest associations with species maximum lifespan (MLS) by several phylogenetic generalized least squares regression models, including adjustment for confounding variables such as body weight and gestation length.
Keywords: Aging, longevity, genetics, genes, homology
1. Introduction
Although phenotypic changes related to aging have been abundantly described in both human and other mammalian species, genetic effectors of the aging process are difficult to determine. Current approaches to determining the role of a gene in the aging process include animal models in which either gain or loss of function mutations result in changes in lifespan and comparative human studies of long-lived cohorts [1-4]. With respect to human studies, age-related genes have been identified by a variety of approaches including comparison of differential gene expression in older individuals (particularly centenarians) with younger individuals, longitudinal studies of octogenarians and nonagenarians to identify genes that are differentially expressed in those that reach 100 years of age, and determination of genes in which naturally occurring polymorphisms or mutations result in a change in the aging process [5].
A novel approach for identification of potentially age-related genes is to examine the conservation of genes in multiple mammalian species and correlate this with the maximum lifespan (MLS) across species. Genetic conservation of DNA or protein sequences can be quantified by alignment methods including the Needleman-Wunsch and Smith-Waterman algorithms, which account for base pair or amino acid matches as well as gaps created to improve overall score and substitution of amino acids with similar functional groups [6, 7]. Based on standard homology algorithms, the bit score (hereafter referred to as alignment score) for two sequences increases as similarity between the sequences increases. Highly evolutionarily conserved genes are those genes with sequences that are almost identical across species. On the other hand, genes that have a beneficial effect on lifespan may be functionally optimized in longer-lived species based on conservation of DNA or protein sequence information. In this case, it would be expected that these genes and the proteins they encode would be more conserved in a comparison between two longer-lived species and less conserved when comparing between longer-lived and shorter-lived species.
Alignment scores for sequence similarity may thus be used to determine if a gene is longevity-related. By comparing a reference sequence – the genetic sequence in a reference organism – with the homologous sequence in each of the comparison species, a score for the gene in each species may be obtained and compared with the species' maximum lifespan (MLS). As shown in Figure 1a, three relations can arise when correlating alignment scores with MLS: (1) the gene or protein sequence is near completely or completely conserved across species and independent of MLS, i.e. a Highly Evolutionarily Conserved Gene (HECG), (2) the gene or protein is more similar among longer lived species compared to shorter lived species, and (3) conversely, the gene or protein may be more similar among shorter lived species and less similar among longer lived species. The latter two relationships, at the extremes, represent the cases for longevity associated genes (LAGs).
Figure 1. Relationships of alignment scores for homologous genes and maximum lifespan.
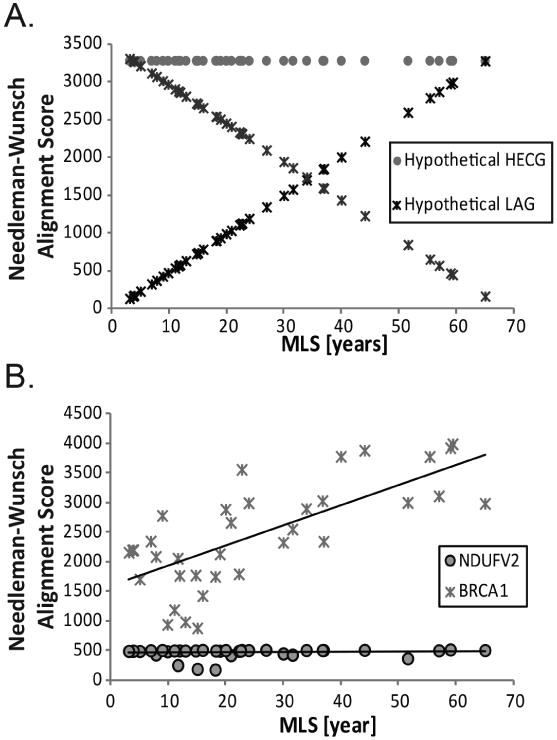
(a) Alignment scores for three hypothetical genes were plotted against the maximum lifespan (MLS) of the species. Hypothetical LAGs may have a steep, positive or negative slope and a correlation approaching ± 1. The hypothetical highly evolutionarily conserved gene (HECG) is a horizontal line indicating that the gene is completely conserved across all species. (b) The alignment scores for the known LAG, BRCA1, and the HECG, NADH dehydrogenase (NDUFV2), are plotted against species MLS. As can be seen from the figure BRCA1 has a steeper slope compared to NDUFV2.
Here we describe an approach to identify genes related to a trait of interest based on an analysis of their sequence similarity in species where the trait is expressed compared to species where the trait is less favored. By examining the relationship between the extent of conservation of genes across species and the MLS of each species, we report the identification of genes potentially related to longevity and aging (LAGs).
2. Methods
2.1. Data Collection
Protein sequences for genes conserved across at least 30 mammalian species were obtained from the OrthoMaM database as FASTA files [11]. Using MATLAB, orthologous sequences were compared using both the Smith-Waterman and Needleman-Wunsch algorithms with BLOSUM62 matrix, gap initiation penalty of 11 and gap extend penalty of 1. Both algorithms generate an alignment score as a measure of sequence alignment and similarity. For each algorithm higher scores indicate a greater degree of conservation and lower scores indicate more divergent sequences. The Needleman-Wunsch algorithm performs a global alignment of sequences and is more sensitive for more closely related sequences [7] while the Smith-Waterman algorithm performs local alignments and is more sensitive for more distantly related proteins [6]. Initial analysis indicated ∼95% similarity in rankings of genes by the two algorithms. Orthologous genes are closely related by definition so alignment scores produced by the Needleman-Wunsch algorithm were used for analyses due to the greater sensitivity for more closely related sequences. For all comparisons, the human protein sequence was used as the reference sequence and any gene for which there was no human sequence was excluded.
2.2. Phylogenetic Generalized Least Squares (PGLS)
The caper package in R [23, 24] was used to apply the PGLS model to each gene with the similarity score as the dependent variable and the species maximum lifespan as the independent variable (univariable analysis). The PGLS model uses a phylogenetic tree to correct the generalized least squares model for evolutionary relatedness. The mammalian phylogenetic supertree was used for the PGLS model [25]. The p-value for the slope of the regression (pMLS) of each gene was determined. A second PGLS model was applied including gestation length (GL) and body weight (BW) as potential confounding variables (multivariable analysis) and pMLS was determined.
2.3. Residual Analysis with Phylogenetic Correction
For each gene, residuals were calculated from the regression of the alignment scores with a confounding variable (ResASvCV). Residuals were also calculated from the regression of MLS with a confounding variable (ResMLSvCV). The PGLS model was then applied with ResASvCV and ResMLSvCV as the dependent and independent variables, respectively, to determine pMLS [8]. This method was applied using BW (SpeakmanBW) and GL (SpeakmanGL) as confounding variables separately. These p-values were not adjusted for multiple comparisons but used primarily to compile a ranking of genes of interest.
2.4. Identification of Genes of Interest
For each model, genes with a pMLS of <0.01 were identified. The genes with the smallest p-values are those orthologous genes that are most divergent when compared between longer and shorter lived species, and thus more conserved among long-lived species. We defined consensus genes as those genes with a pMLS <0.05 identified by all models. The known functions and mutant phenotypes of these consensus genes was determined from the GeneCard gene encyclopedia which compiles various information on genes from multiple sources [26].
3. Results
Figure 1b shows the relationship between alignment score and MLS for BRCA1, a known LAG, and NADH dehydrogenase (NDUFV2), a known HECG. As can be seen from the figure, the LAG has a steeper slope than the HECG [34.39 for BRCA1; 0.58 for NDUFV2]. Based on the four regression analyses, BRCA1 has an average pMLS of 0.004 while NDUFV2 has an average pMLS of 0.53 where pMLS is the p-value for the slope of the regression model with MLS as the variable of interest.
For all analyses, smaller p-values are over-represented compared to the uniform distribution expected for the case where the null hypothesis (H0), no relation between MLS and alignment score, is true (figure 2). There is a small subset of 167 genes (out of over 10,000) which demonstrated pMLS ≤ 0.05 by all four methods and among these are 11 genes which have a pMLS ≤ 0.01 by each method applied. The gene symbols, average p-values, names, accession numbers, and functions of the genes which have a pMLS ≤ 0.01 by all regression analyses are listed in table 1 and the genes which have a pMLS ≤ 0.05 are listed in additional file 1.
Figure 2. Distribution of p-values (pMLS) suggests de facto relationship between protein homology and MLS.
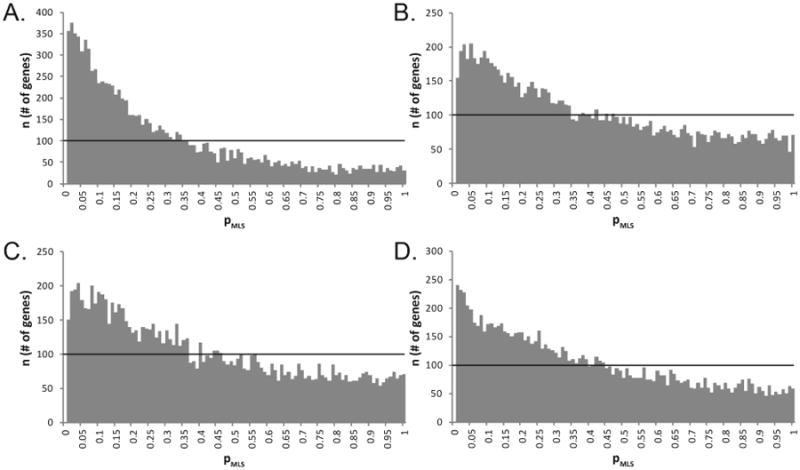
The distribution of p values for the slope of the regression model with MLS as the independent variable of interest for univariable (a), multivariable including confounders (b), SpeakmanGL (c), and SpeakmanBW (d) models is shown. For each model, smaller p-values are over-represented compared to the uniform distribution expected for the case where alignment score and MLS are truly unrelated. The uniform distribution for this data is represented by the horizontal line.
Table 1. Summary of Consensus Genes (pMLS ≤ 0.01) by all methods ranked by average p-value.
| Symbol | Avg p-value | Name | Accession # | Summary |
|---|---|---|---|---|
| CRNN | 7.51E-04 | Cornulin | GC01M152381 | Heat shock protein and survival factor involved in cell cycle regulation |
| TEX101 | 1.82E-03 | testis expressed 101 | GC19P043892 | May play a role in signal transduction |
| HYLS1 | 2.15E-03 | hydrolethalus syndrome 1 | GC11P125753 | Component of centrioles and required for cilia formation |
| CCDC30 | 2.18E-03 | coiled-coil domain containing 30 | GC01P042929 | Unknown function |
| ANUBL1 | 2.67E-03 | zinc finger, AN1-type domain 4 | GC10M046112 | zinc finger protein |
| CD3EAP | 2.70E-03 | CD3e Molecule, Epsilon Associated Protein | GC19P045909 | Component of t-cell receptor complex and RNA polymerase I |
| CD8B | 2.97E-03 | CD8 beta | GC02M087042 | part of coreceptor on cytotoxic T-cell |
| BRCA1 | 3.93E-03 | Breast cancer 1, early onset | GC17M041197 | Involved in transcription, DNA repair, cell cycle regulation and DDR |
| BPI | 5.00E-03 | bactericidal/permeability increasing protein | GC20P036888 | Bactericidal activity |
| MRPL49 | 5.45E-03 | mitochondrial ribosomal protein L49 | GC11P064889 | mitochondrial ribosomal protein |
| IL7R | 6.74E-03 | interleukin 7 receptor | GC05P035892 | Regulator of immune system function |
All methods incorporate a correction for phylogeny in order to account for the greater sequence similarity expected for more closely related species. The first approach of applying the phylogenetic generalized least squares (PGLS) method to the regression of alignment score versus MLS assumes that correlation with confounding variables, in addition to MLS, does not exclude genes from being considered LAGs. The second approach, applying the PGLS method to the multiple regression of alignment score versus MLS, gestation length (GL), and body weight (BW), accounts for both confounding variables in one model. The third and fourth approaches remove the effect of a single confounding variable (GL and BW, respectively) as previously described by Speakman [8]. Because pMLS is necessarily positive, any gene for which pMLS is sufficiently small is considered a potential LAG, regardless of slope.
Of the 167 consensus genes, those genes with a pMLS <0.05 identified by all models, 36 (22 %) were related to the immune system and 24 (14 %) were related to cell cycle regulation (Table 2). Additionally, 64 genes were identified from the Mouse Genome Database (MGD) as having known phenotypes when mutated in mice [9]. Table 3 shows the categories of phenotypes displayed when these genes are mutated and the numbers of genes with mutant phenotypes in those categories. Many genes resulted in more than one phenotype when mutated. Of the 64 genes identified, 35 genes resulted in an altered immune phenotype when mutated and 29 genes resulted in a phenotype with a known effect on mortality and aging in mice [9].
Table 2. Summary of consensus gene (pMLS ≤ 0.05) function.
| System | Class | Symbol |
|---|---|---|
| Immune system and inflammation | Cytokine/Chemokine | CCL1, CXCL17, CYTL1, IFNG, IL17A, IL17A, IL6 |
| Immune Receptor | CD226, CD3EAP, CD4, CD8B, HLA-DOA, IL28RA, IL4R, IL7R, SLAMF1, SLAMF9, FAIM3 | |
| Modulation | ADAMDEC1, GAB3, GC, GPR18, HIVEP1, IFNG, IL7R, PTPRJ, STAT2, TAPBP, TNFRSF11A, TNIP3, TRAFD1, ZC3H12A | |
| Innate immunity | BPI, IL28RA, LBP, LPO, MAVS, RNASEL | |
| Inflammation | F2, IL17A, IL6, ZC3H12A | |
| Cell Cycle | Regulation | BRCA1, C11orf82, CDC25C, CENPC1, CENPT, CEP55, CRNN, MAP4, MCC, DLEC1 |
| DNA maintenance | BRCA1, EME1, FANCB, MBD4, NEIL2 | |
| DDR | BRCA1, FBXO6 | |
| Apoptosis | C11orf82, HIVEP1, PPP1R15A, FAIM3 | |
| Growth | ARHGAP31, PTPRJ | |
| Chromosome | CDCA2, CENPT, NCAPH, OFD1, AUNIP | |
| Centromere | MLF1IP, SGOL2 |
Table 3. Known mutant phenotypes of consensus genes (pMLS ≤ 0.05).
| Phenotype | n |
|---|---|
| Immune system phenotype | 35 |
| Cellular phenotype | 34 |
| Homeostasis/metabolism phenotype | 29 |
| Mortality/aging | 29 |
| Growth/size phenotype | 25 |
| Hematopoietic system phenotype | 25 |
| Nervous system phenotype | 17 |
| Behavior/neurological phenotype | 13 |
| Digestive/alimentary phenotype | 13 |
| Endocrine/exocrine gland phenotype | 13 |
| Skeleton phenotype | 13 |
| Muscle phenotype | 12 |
| Respiratory system phenotype | 12 |
| Cardiovascular system phenotype | 11 |
| Embryogenesis phenotype | 11 |
| Integument phenotype | 11 |
| Reproductive system phenotype | 11 |
| Liver/biliary system phenotype | 9 |
| Renal/urinary system phenotype | 9 |
| Taste/olfaction phenotype | 8 |
| Tumorigenesis | 8 |
| Craniofacial phenotype | 6 |
| Limbs/digits/tail phenotype | 5 |
| Vision/eye phenotype | 5 |
| Adipose tissue phenotype | 4 |
4. Discussion
We have presented a novel approach for identification of potential LAGs through the use of multiple regression models comparing global alignment scores of protein sequences with species maximum lifespan. This approach is complementary to, but much less expensive and faster than traditional approaches that are based on animal models or human studies. We have applied this approach to 10,411 genes conserved across more than 30 mammalian species. Many of the consensus genes function in pathways of the immune system, inflammation, DNA maintenance, and DNA damage response -- pathways that have been implicated in multiple pathological processes related to aging.
Some known LAGs may have been excluded from analysis based on the availability of sequence data. Of the 25 genes in the GenAge database [10] that are known to be related to aging through direct evidence in humans or mammalian models, 12 were excluded from analysis (5 due to sequence data being available for fewer than 30 species, 7 because sequence data was not available in the OrthoMaM database[11]). Of the remaining 13, BRCA1 and IL6 were identified as consensus genes (pMLS<0.05 among all methods) while PLAU, GHR, EPS8, and SLC13A1 were identified with a pMLS<0.05 by at least one method. For some LAGs, their primary protein sequence may be so essential that even minor alterations would adversely affect function or even be lethal. These genes may still impact longevity through expression level, which is regulated by other proteins (e.g., transcription factors), as well as adjacent or distant cis-acting sequences, or epigenetic factors, and thus not captured by analysis of the primary protein sequence alone. Our approach does not identify those genes which exert their impact on aging or longevity primarily through expression level, and thus some LAGs may be not identified by this method.
Among the consensus genes, BRCA1 is a known LAG which has been shown to have different genotype frequencies in centenarian populations compared to controls [3]. BRCA1 functions in transcription, DNA repair, cell cycle regulation, and DNA damage response (DDR). Mutations in BRCA1 are associated with early onset breast cancer. In addition to BRCA1, the FANCB and FBXO6 genes are among our identified consensus genes involved in DDR. FANCB encodes a core component of the Fanconi Anemia core complex which is involved in a DNA repair and maintenance pathway with BRCA1 and BRCA2 [12]. FBXO6 is involved in the regulation of the ubiquitination and degradation of CHK1 which phosphorylates and accumulates in response to DDR resulting in cell cycle arrest [13]. Growing evidence suggests that accumulation of DNA damage contributes to degenerative changes associated with aging. This is supported by the facts that inhibition of DNA damage can delay cellular senescence [14], while DNA damage that generates a DDR can result in cellular senescence [15].
The immune system has been shown to exhibit age-related changes termed immunosenescence and resulting in a persistent state of low-grade inflammation [16]. Conversely, immune function and lower levels of inflammatory cytokines are preserved in extremely long lived mice compared to old and very old [17]. We provide 3 examples from our consensus genes with important roles in the immune system: CD3EAP, CD8B, and IL7R. CD3EAP encodes a binding protein associated with the epsilon subunit of the T cell receptor (TCR) CD3 complex and is involved in signal transduction through the TCR pathway regulating activation, differentiation, and function of T cells [18]. CD8B is a consensus gene which encodes the precursor to the beta subunit of the CD8 coreceptor. CD8 functions in T cell activation through interaction with the major histocompatibility complex (MHC) during MHC/TCR-peptide interaction [19]. The gene IL7R encodes the IL7 receptor subunit alpha precursor, part of the IL7 receptor heterodimer, and mutations in IL7R result in a form of severe combined immunodeficiency characterized by the absence of T-cells in the presence of B cells and natural killer (NK) cells [20, 21]. Our finding, of immune related genes among the consensus LAGs, provides further support of a role for the immune system in aging.
Semeiks & Grishin used a similar rational to look at longevity selected positions within proteins [22]. In contrast, our method examines specific genes and based on the entire sequence determines if there is a correlation to longevity. While the two approaches may have similar rationales, our approach identifies specific genes based on the global sequence and alignment rather than motifs within those genes [22]. Of note, both methods identify a subset of genes with similar functions in development, inflammation, and immune fidelity. Additionally, both methods identify BRCA1 as a LAG.
We note that using the unadjusted p-value levels of 0.05 and 0.01 to identify those genes for further investigation regarding mechanisms is quite liberal, in the sense of identifying more genes than would be selected based on a stricter criterion of statistical significance. For example, a simple Bonferroni correction would identify statistically significant associations if p<0.000005, which did not occur for any of our candidate genes. We recognize that this is a limitation of the methods used. A second limitation is the small number of available species used, which affects the statistical power to detect true associations. We have partially addressed these limitations by focusing on ranking and selection.
5. Conclusions
We provided a ranking of genes with the strongest associations with species MLS by several phylogenetic generalized least squares regression models. Using our models, we observed non-uniform distributions of p-values for the correlation of alignment score and MLS. Among the consensus genes identified by our method we found subsets of genes with roles in immune fidelity, inflammation, development, and DDR.
Supplementary Material
Highlights.
We identify longevity associated genes (LAGs) by multiple regression analyses.
We evaluated over 10,000 homologous genes across 30-39 mammalian species.
We provide a ranking of genes with the strongest associations with species maximum lifespan (MLS).
Known LAGs have strong associations with species maximum MLS.
LAGs were commonly related to immune function, cell cycle, and DNA damage response.
Acknowledgments
This work was supported by the National Institute of Health, National Institute on Aging grant R01A6028873 (RJP) and the National Center for Advancing Translational Science grant UL1TR000003 (KJP).
Grant Support: National Institute of Health, National Institute on Aging R01A6028873
Footnotes
Author Contributions: Carter M. Lindborg: collection, assembly, analysis, and interpretation of data, writing of manuscript, final approval of manuscript
Kathleen J. Propert: analysis and interpretation of data, writing of manuscript, final approval of manuscript
Robert J. Pignolo: conception, design, collection, assembly, analysis, and interpretation of data, writing of manuscript, final approval of manuscript
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Park SK. Genomic approaches for the understanding of aging in model organisms. BMB Rep. 2011;44:291–297. doi: 10.5483/BMBRep.2011.44.5.291. [DOI] [PubMed] [Google Scholar]
- 2.Hekimi S. How genetic analysis tests theories of animal aging. Nat Genet. 2006;38:985–991. doi: 10.1038/ng1881. [DOI] [PubMed] [Google Scholar]
- 3.Vijg J, Perls T, Franceschi C, van Orsouw NJ. BRCA1 gene sequence variation in centenarians. Ann N Y Acad Sci. 2001;928:85–96. doi: 10.1111/j.1749-6632.2001.tb05639.x. [DOI] [PubMed] [Google Scholar]
- 4.Cao L, Li W, Kim S, Brodie SG, Deng CX. Senescence, aging, and malignant transformation mediated by p53 in mice lacking the Brca1 full-length isoform. Genes Dev. 2003;17:201–213. doi: 10.1101/gad.1050003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jylhava J, Hurme M. Gene variants as determinants of longevity: focus on the inflammatory factors. Pflugers Arch. 2010;459:239–246. doi: 10.1007/s00424-009-0726-3. [DOI] [PubMed] [Google Scholar]
- 6.Smith TF, Waterman MS. Identification of common molecular subsequences. J Mol Biol. 1981;147:195–197. doi: 10.1016/0022-2836(81)90087-5. [DOI] [PubMed] [Google Scholar]
- 7.Needleman SB, Wunsch CD. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J Mol Biol. 1970;48:443–453. doi: 10.1016/0022-2836(70)90057-4. [DOI] [PubMed] [Google Scholar]
- 8.Speakman JR. Correlations between physiology and lifespan – two widely ignored problems with comparative studies. Aging Cell. 2005:167–175. doi: 10.1111/j.1474-9726.2005.00162.x. [DOI] [PubMed] [Google Scholar]
- 9.Blake JA, Bult CJ, Eppig JT, Kadin JA, Richardson JE Mouse Genome Database G. The Mouse Genome Database: integration of and access to knowledge about the laboratory mouse. Nucleic Acids Res. 2014;42:D810–817. doi: 10.1093/nar/gkt1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tacutu R, Craig T, Budovsky A, Wuttke D, Lehmann G, Taranukha D, Costa J, Fraifeld VE, de Magalhaes JP. Human Ageing Genomic Resources: integrated databases and tools for the biology and genetics of ageing. Nucleic Acids Res. 2013;41:D1027–1033. doi: 10.1093/nar/gks1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ranwez V, Delsuc F, Ranwez S, Belkhir K, Tilak MK, Douzery EJ. OrthoMaM: a database of orthologous genomic markers for placental mammal phylogenetics. BMC Evol Biol. 2007;7:241. doi: 10.1186/1471-2148-7-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meetei AR, Levitus M, Xue Y, Medhurst AL, Zwaan M, Ling C, Rooimans MA, Bier P, Hoatlin M, Pals G, et al. X-linked inheritance of Fanconi anemia complementation group B. Nat Genet. 2004;36:1219–1224. doi: 10.1038/ng1458. [DOI] [PubMed] [Google Scholar]
- 13.Zhang YW, Brognard J, Coughlin C, You Z, Dolled-Filhart M, Aslanian A, Manning G, Abraham RT, Hunter T. The F box protein Fbx6 regulates Chk1 stability and cellular sensitivity to replication stress. Mol Cell. 2009;35:442–453. doi: 10.1016/j.molcel.2009.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tilstra JS, Robinson AR, Wang J, Gregg SQ, Clauson CL, Reay DP, Nasto LA, St Croix CM, Usas A, Vo N, et al. NF-kappaB inhibition delays DNA damage-induced senescence and aging in mice. J Clin Invest. 2012 doi: 10.1172/JCI45785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192:547–556. doi: 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ostan R, Bucci L, Capri M, Salvioli S, Scurti M, Pini E, Monti D, Franceschi C. Immunosenescence and immunogenetics of human longevity. Neuroimmunomodulation. 2008;15:224–240. doi: 10.1159/000156466. [DOI] [PubMed] [Google Scholar]
- 17.Arranz L, Lord JM, De la Fuente M. Preserved ex vivo inflammatory status and cytokine responses in naturally long-lived mice. Age (Dordr) 2010;32:451–466. doi: 10.1007/s11357-010-9151-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamazaki T, Hamano Y, Tashiro H, Itoh K, Nakano H, Miyatake S, Saito T. CAST, a novel CD3epsilon-binding protein transducing activation signal for interleukin-2 production in T cells. J Biol Chem. 1999;274:18173–18180. doi: 10.1074/jbc.274.26.18173. [DOI] [PubMed] [Google Scholar]
- 19.Garcia KC, Scott CA, Brunmark A, Carbone FR, Peterson PA, Wilson IA, Teyton L. CD8 enhances formation of stable T-cell receptor/MHC class I molecule complexes. Nature. 1996;384:577–581. doi: 10.1038/384577a0. [DOI] [PubMed] [Google Scholar]
- 20.Roifman CM, Zhang J, Chitayat D, Sharfe N. A partial deficiency of interleukin-7R alpha is sufficient to abrogate T-cell development and cause severe combined immunodeficiency. Blood. 2000;96:2803–2807. [PubMed] [Google Scholar]
- 21.Puel A, Ziegler SF, Buckley RH, Leonard WJ. Defective IL7R expression in T(-)B(+)NK(+) severe combined immunodeficiency. Nat Genet. 1998;20:394–397. doi: 10.1038/3877. [DOI] [PubMed] [Google Scholar]
- 22.Semeiks J, Grishin NV. A method to find longevity-selected positions in the mammalian proteome. PLoS One. 2012;7:e38595. doi: 10.1371/journal.pone.0038595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.David Orme RF, Thomas Gavin, Petzoldt Thomas, Fritz Susanne, Isaac Nick, Pearse Will. caper: Comparative Analyses of Phylogenetics and Evolution in R. 0.5. 2012. [Google Scholar]
- 24.Team RC. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2013. [Google Scholar]
- 25.Bininda-Emonds OR, Cardillo M, Jones KE, MacPhee RD, Beck RM, Grenyer R, Price SA, Vos RA, Gittleman JL, Purvis A. The delayed rise of present-day mammals. Nature. 2007;446:507–512. doi: 10.1038/nature05634. [DOI] [PubMed] [Google Scholar]
- 26.Safran M, Dalah I, Alexander J, Rosen N, Iny Stein T, Shmoish M, Nativ N, Bahir I, Doniger T, Krug H, et al. GeneCards Version 3: the human gene integrator. Database (Oxford) 2010;2010:baq020. doi: 10.1093/database/baq020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.