

Abstract
Adult hippocampal neurogenesis is the process whereby adult neural precursor cells (aNPCs) in the subgranular zone (SGZ) of the dentate gyrus (DG) generate adult-born, functional neurons in the hippocampus. This process is modulated by various extracellular and intracellular stimuli, and the adult-born neurons have been implicated in hippocampus-dependent learning and memory. However, studies on how neurotoxic agents affect this process and the underlying mechanisms are limited. The goal of this study was to determine whether lead, a heavy metal, directly impairs critical processes in adult neurogenesis and to characterize the underlying signaling pathways using primary cultured SGZ-aNPCs isolated from adult mice. We report here that lead significantly increases apoptosis and inhibits proliferation in SGZ-aNPCs. In addition, lead significantly impairs spontaneous neuronal differentiation and maturation. Furthermore, we found that activation of the c-Jun NH2-terminal kinase (JNK) and p38 mitogen activated protein (MAP) kinase signaling pathways are important for lead cytotoxicity. Our data suggest that lead can directly act on adult neural stem cells and impair critical processes in adult hippocampal neurogenesis, which may contribute to its neurotoxicity and adverse effects on cognition in adults.
Keywords: Lead, adult neurogenesis, neurotoxicity, apoptosis, proliferation, differentiation
Introduction
Adult hippocampal neurogenesis is the process whereby adult neural precursor cells (aNPCs) in the subgranular zone (SGZ) of the dentate gyrus (DG) lead to the generation and functional integration of adult-born neurons in the hippocampus (Ming and Song, 2011). These adult-born neurons can influence certain forms of hippocampus-dependent learning and memory formation (Clelland et al., 2009; Deng et al., 2009; Garthe et al., 2009; Pan et al., 2012a; Pan et al., 2012b; Wang et al., 2014). Importantly, the various stages and cell types involved in adult hippocampal neurogenesis can be modulated by various physiological and pathological factors, including other cell types in the neurogenic niche, growth factors, cytokines, neurotrophins, and processes such as mating, aging, stress, and exercise (Ming and Song, 2011; Pan et al., 2012a,b; Pan et al., 2013b; Wang et al., 2014). However, the effects of neurotoxicant exposure on adult hippocampal neurogenesis have not been studied extensively.
The heavy metal lead is a ubiquitous environmental contaminant and a major public health concern. The combustion of leaded gas in the U.S. in the 20th century released approximately 4 million metric tons of lead into the environment (Toscano and Guilarte, 2005b). The phasing out of leaded paint and gasoline has contributed to a significant decline in ambient lead levels as well as mean blood lead levels (12.8 to 1.6 μg/dL from 1976 to 2002) in the U.S. population (Toscano and Guilarte, 2005b; ATSDR, 2007). However, lead can persist in the soil for decades and no level of lead is considered safe (White et al., 2007). In addition to its well-characterized developmental neurotoxicity, cumulative lead exposure can also cause neurological impairment in adults (van Wijngaarden et al., 2009). Monkeys and rats exposed to low concentrations of lead have increased cognitive decline and Alzheimer’s disease-associated neuropathology later in life (Basha et al., 2005; Wu et al., 2008; Bihaqi et al., 2013; Grossman, 2014). Furthermore, longitudinal studies from a cohort of non-occupationally exposed elderly men found an association between relatively low blood (mean 5.5 μg/dL) and/or patella lead levels and increased cognitive decline (Payton et al., 1998; Weisskopf et al., 2004). These blood lead levels are comparable to background blood lead levels in the adult U.S. population (1.5 and 2.2 μg/dL among 20-59 and ≥ 60 year-olds, respectively) (ATSDR, 2007). Thus, lead may contribute to increased or accelerated cognitive decline at environmentally relevant exposure levels.
Lead may facilitate and accelerate cognitive decline through impaired adult hippocampal neurogenesis. Several studies have examined the effect of early life lead exposure on adult hippocampal neurogenesis and found that developmental lead exposure is associated with altered proliferation, survival, and dendritic morphology of adult-born neurons in the hippocampus and altered hippocampus learning and memory in rats (Verina et al., 2007) (Jaako-Movits et al., 2005). However, results have not been entirely consistent among various studies. Gilbert et al. (2005) found that lead-treated rats have reduced adult-born cell (BrdU+) survival but no change in cell proliferation, while a similar study reported that lead decreases adult-born cell survival as well as proliferation (Verina et al., 2007). Furthermore, Jaako-Movits et al. (2005) found that lead impaired adult-born cell proliferation and neuronal maturation, while Verina et al. found no effect of lead on neuronal differentiation (Verina et al., 2007). Although these studies are very interesting, the inconsistent results warrant further investigation. Moreover, the early life exposure paradigms used in these in vivo studies may have introduced potential confounding factors due to the adverse effects of lead during development (Gilbert et al., 2005; Jaako-Movits et al., 2005; Verina et al., 2007). Only one study to date has assessed the effect of postnatal lead exposure alone on adult neurogenesis (Schneider et al., 2005). Schneider et al. (2005) exposed male rats to 1500 ppm lead acetate for 30-35 days starting at postnatal day 25 and found that lead impaired adult-born cell proliferation in the SGZ. However, they did not assess the effect of lead on other stages of adult neurogenesis or cognitive behavior. Thus, additional research is needed to determine whether adult-only lead exposure is sufficient to impair adult hippocampal neurogenesis and to characterize the signaling mechanisms underlying lead-induced impairment in adult neurogenesis. In this study, we used primary cultured aNPCs isolated from the hippocampus (SGZ-aNPCs) of adult mice as an in vitro model system to test the hypothesis that lead exposure impairs adult hippocampal neurogenesis and to elucidate the underlying signaling mechanisms.
Materials and Methods
Reagents
The preparation, use, and disposal of hazardous agents were carried out according to the Environmental Health and Safety Office at the University of Washington. Lead (II) acetate trihydrate (Cat. 316512, Sigma-Aldrich, St. Louis, MO) was dissolved in deionized distilled water (H2O) to make a 5 mM stock solution and stored at −20°C. Z-VAD-FMK (Cat. FMK001, R&D Systems, Minneapolis, MN) was dissolved in dimethyl sulfoxide (DMSO) to make a 20 mM stock and used according the manufacturer’s specifications. The p38 (Cat. SB2021990, EMD Millipore Calbiochem, Billerica, MA) and JNK (Cat. SP600125, EMD Millipore Calbiochem) inhibitors were dissolved in DMSO to yield 3 mM stock solutions and stored at −20°C. 5-bromo-2'-deoxyuridine (BrdU) was from Sigma (Cat. B9285) and stored as a 65 mM stock solution. The primary antibodies and dilutions used in immunocytochemistry were rat anti-BrdU (1:500, Bio-Rad Laboratories AbD Serotec, Raleigh, NC), mouse anti-βIII-tubulin (1:500, Promega, Madison, WI), and mouse anti-SOX2 (1:500, R&D Systems). Goat anti-rat and goat anti-mouse Alexa Fluor-conjugated secondary antibodies as well as Hoechst 33342 were from Invitrogen (Carlsbad, CA). For Western Blot analysis, the following rabbit primary antibodies from Cell Signaling (Beverly, MA) were used at a 1:1000 dilution unless otherwise specified: monoclonal anti-phospho-Akt (Cat. 4060, 1:2000), polyclonal anti-phospho-p38 (Cat. 9211), monoclonal anti-phospho JNK (Cat. 4668), monoclonal anti-JNK (Cat. 9258), polyclonal anti-phospho-c-Jun (Cat. 9164), and monoclonal anti-GAPDH (Cat. 2118). Horseradish peroxidase-conjugated secondary antibodies were from EMD Millipore (Billerica, MA). All of the primary and secondary antibodies were diluted into the appropriate blocking buffer.
Cell culture
The University of Washington Institutional Animal Care and Use Committee approved all experimental procedures. The primary aNPCs were prepared as previously described (Guo et al., 2012; Pan et al., 2013) from the SGZ of the DG from 6-7 week-old male C57BL/6J mice (Taconic, Hudson, NY). The solutions and media used during the aNPC isolation were filter sterilized. Briefly, the whole brain from four adult male mice was harvested and placed in HBSS (Invitrogen). Each brain was then sliced into 1 mm sections using an adult mouse brain matrix (Kent Scientific, Torrington, CT), and then the SGZ was isolated from these sections via microdissection under a dissection microscope. The SGZ tissue was placed in Solution A (30 mM Glucose, 26 mM NaCO3, 2mM HEPES pH 7.4 (Invitrogen) in HBSS (Invitrogen)) and spun down for 10 min at 1,000 rpm. The pelleted tissue was then resuspended and a combination of mechanical and enzymatic digestion (MACS Neural Tissue Dissociation Kit, Miltenyi Biotec, San Diego, CA) was used to dissociate the tissue. To stop the digestion, DMEM/F-12 medium (Invitrogen) with 10% Fetal Bovine Serum (FBS, Invitrogen) was added and the SGZ tissue was then filtered through a cell strainer (70 μm cell strainer, Fisher Scientific, Waltham, MA) and spun down for 3 min at 1,000 rpm. The pellet was washed once with DMEM/F-12 medium with 10% FBS and once with DMEM/F-12 medium with 10% FBS plus Percoll (GE Healthcare Life Sciences, Pittsburgh, PA) solution (1:10 Percoll in PBS) followed by spins at 1,000 rpm for 3 and 15 min, respectively. The pellet was washed once with Solution A and once with initial proliferation medium (Neurobasal medium (Invitrogen); 1X B27 supplement without Vitamin A (Invitrogen); 2mM L-Glutamine (Invitrogen); 100 U/ml penicillin/streptomycin (Invitrogen), 20 ng/ml of epidermal growth factor (EGF; EMD Chemicals) and 10 ng/ml basic fibroblast growth factor (bFGF; EMD Chemicals) followed by 5 min spins at 1,500 rpm. The cells were then plated in a petri dish with initial proliferation medium and cultured at 37°C and 6.5% CO2. Growth factors (EGF and bFGF) were refreshed every 3-4 d unless noted otherwise. Primary neurospheres formed after 7-14 d, at which time, the neurospheres were collected, enzymatically and mechanically dissociated, and then resuspended in growth media (Advanced DMEM/F-12, 1X N2 Supplement (Invitrogen), 1X B27 Supplement, 100 U/ml Penicillin/streptomycin, 2 mM L-Glutamine, 2 μg/ml Heparin sodium salt (Sigma), 20 ng/ml EGF, and 10 ng/ml bFGF). The neurospheres were maintained in petri dishes in the growth media and passaged ≤ 10 times.
Drug treatment
For experiments, the neurospheres were dissociated (0.125% trypsin-EDTA (Invitrogen) for 5 min; 0.014% soybean trypsin inhibitor (Sigma) for 5 min) and seeded as a monolayer culture on poly-L-ornithine- (15 μg/ml) and fibronectin (1 mg/ml) (BD Biosciences, San Jose, CA)-coated ACLAR coverslips or culture plates. To assess cell number, proliferation, and apoptosis, cells were seeded at a cell density of 5 × 103 cells per well (48-well plate) and allowed to attach overnight. The next day, the media was changed (Advanced DMEM/F-12, 1X N2 Supplement, Penicillin/streptomycin, 2 mM L-Glutamine, 2 μg/ml Heparin sodium salt, 20 ng/ml EGF, and 10 ng/ml bFGF) and the cells were treated with agents for 0-48 h as described in the figures and figure legends. For a given experiment, each agent was administered once (not replenished). Lead was dissolved in H2O while the p38 and JNK inhibitors were dissolved in DMSO, so either an equal volume of H2O or an equal concentration of DMSO was used as a vehicle control. To assess cell proliferation, BrdU was added to each well (final concentration of 10 μM) for the last 2 h of the experiment.
To assess spontaneous differentiation, the cells were seeded at a cell density of 5 × 103 cells per well (48-well plate) overnight. The next day, the media was replaced with EGF/bFGF-free growth media supplemented with 1 mg/ml bovine serum albumin (BSA) (Equitech Bio, Kerrville, TX), and then the cells were treated with lead or vehicle control and cultured for 5 d (each agent was administered once).
Immunocytochemistry
For immunocytochemistry, the cells were fixed by removing half of the media from each well and replacing it with an equal volume of 8% paraformaldehyde (PFA)/8% sucrose in PBS for 30 min at room temperature (RT). The fixed cells were washed for 3X5 min in PBS, permeabilized by 1X5 min in 1% SDS in PBS, and washed 3X5 min in PBS. The cells were blocked with 5% BSA in PBST (0.1% Triton-X 100 in PBS) for 30 min at RT, and then incubated with primary antibodies at 4°C overnight. For BrdU staining, the cells underwent additional processing prior to blocking: 5 min in H2O at RT, 10 min in 1 N HCl at 4°C, 30 min in 2 N HCl at 37°C, 2X15 min in 0.1 M borate buffer (pH 8.5). Following incubation with primary antibodies, the cells were washed 3X10 min with PBST and then incubated with secondary antibodies for 2 h at RT. The cells were washed 3X10 min with PBST, incubated with 2.5 μg/ml Hoechst 33342 for 30 min, washed 1X with PBST, and then mounted onto slides using anti-fade Aqua Poly/Mount (Polysciences) solution.
Imaging and quantification of immunostained cells
All images were captured using a fluorescence microscope (Zeiss) equipped with a camera with a 10x or 20x objective (Zeiss). Images were uniformly adjusted for color, brightness, and contrast with Adobe Photoshop CS4 (Adobe Systems Inc.). A cell with nuclear condensation or fragmentation was scored as apoptotic. A cell was scored as marker+ if the cell had a uniformly stained Hoechst+ nucleus as well as marker expression in the nucleus (BrdU) or cell body and neurites (βIII-tubulin+ neurites ≥ length of soma). All quantification was conducted by an experimenter blinded to treatment. At least 250 cells per coverslip per treatment were quantified from at least 10 randomly selected fields on each coverslip.
Western Blot analysis
The cells were seeded at 2 × 105 cells per well in poly-L-ornithine and fibronectin-coated 12-well plates for 24 h. Cells were then treated as described in the figure legends and then washed with ice-cold PBS followed by Triton-X cell lysis buffer with protease inhibitors. The cell lysates were clarified by centrifugation and stored at −80°C. The protein concentration was measured using the BCA protein assay (Thermo Scientific, Waltham, MA). Samples containing 5 μg protein were separated by gel electrophoresis on a 12.5% SDS-PAGE gel and transferred to a PVDF membrane (EMD Millipore). Following antibody incubation, the protein of interest was detected with ECL Prime (GE Healthcare Life Sciences) using a ChemiDoc XRS Imaging System (BioRad). ImageJ (NIH) was used for the densitometry analysis and determination of fold induction normalized to a loading control (total protein or GAPDH).
Statistical Analysis
Statistical analyses were conducted using GraphPad Prism software (version 5.0c for Mac, GraphPad Software Inc., San Diego, CA, USA). All of the immunocytochemical data are from at least two independent experiments with triplicates or quadruplicates for each data point (n ≥ 2 independent experiments for each data point). The Western blot data are representative data from two independent experiments with duplicates. For dose-response experiments, a Student’s t test with two-tailed analysis (α = 0.05) was used for pair-wise comparison of the means. One-way ANOVA with a Bonferroni post-hoc analysis (α = 0.05) was used to analyze all of the drug treatment data (Z-VAD-FMK, MAPK inhibitors). Data represent mean ± SEM., n.s. not significant, * p < 0.05; ** p < 0.01; *** p < 0.001.
Results
SGZ-aNPCs are maintained as neurospheres in vitro and retain their stem cell characteristics
After isolation from the dentate gyrus of the hippocampus of adult C57Bl/6J male mice, the SGZ-aNPCs are maintained as neurospheres in culture (Fig. 1A). Upon dissociation and seeding as a monolayer in growth media, at least 98.5% of cells continue to express the stem cell marker, SOX2 (Fig. 1B), after more than six passages. Thus, these SGZ-aNPCs are an appropriate in vitro model for studying the effects of lead on the stem/progenitor pool in the hippocampus because these cells proliferate and retain their stem cell characteristics after multiple passages.
Fig. 1. The SGZ-aNPCs are maintained as neurospheres and retain their stem cell characteristics, in vitro.
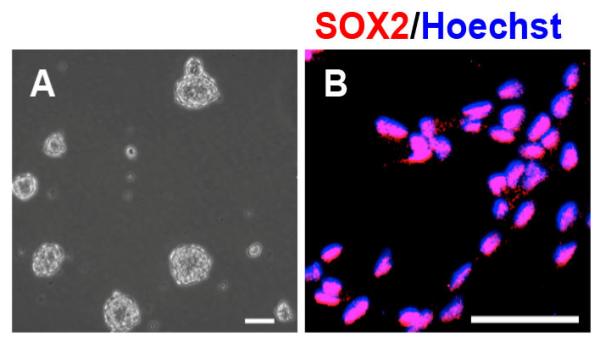
A, SGZ-aNPCs continue to proliferate as neurospheres after six passages. B, SGZ-aNPCs continue to express SOX2 (red), a stem cell marker, when dissociated and seeded as a monolayer in proliferative conditions. Scale bars. 50 μm.
Lead significantly decreases the total cell number and increases apoptosis in SGZ–aNPCs
To determine whether lead is cytotoxic and induces apoptosis in aNPCs, cells were treated with 0 to 2 μM lead for 48 h. Lead significantly decreased the total cell number in a dose-dependent manner (Fig. 2A-D) starting at 0.1 μM. Furthermore, lead significantly increased the percent of apoptotic cells, quantified by nuclear condensation and/or fragmentation (Fig. 2E), beginning at 0.1 μM, the lowest concentration tested. A 2 h pretreatment with 5 μM Z-VAD-FMK, a pan-caspase inhibitor, almost completely blocked lead (0.5 μM)-induced cell loss (Fig. 2F) and apoptosis (Fig. 2G). These data suggest that lead is toxic to SGZ-aNPCs and induces caspase-dependent apoptosis in these cells.
Fig. 2. Lead significantly decreases the total cell number and increases apoptosis in SGZ-aNPCs.

A-E, SGZ-aNPCs were treated with 0, 0.1, 0.2, 0.5, 1, or 2 μM lead for 48 h. Representative Hoechst nuclei staining from (A) untreated, (B) 0.2 μM, and (C) 1 μM lead-treated SGZ-aNPCs. Quantification of (D) the total cell number and (E) the percent apoptotic cells. F-G, SGZ-aNPCs were pretreated with 5 μM Z-VAD-FMK, a pan-caspase inhibitor, for 2 h followed by 0.5 μM lead for 48 hand (F) the total cell number and (G) the percent apoptotic cells were quantified. Arrows: nuclear condensat ion and/or fragmentation. Hoechst: nuclei staining. Scale bar: 50 μm. n = 2-3 independent experiments for a total of 4-8 coverslips per data point. Data represent mean ± SEM., n.s. not significant, * p < 0.05; ** p < 0.01; *** p < 0.001.
Lead decreases proliferation in SGZ–aNPCs
To determine whether lead decreases aNPC cell proliferation, we pulsed the cells with BrdU during the final 2 h of a 48 h lead treatment. BrdU is a thymidine analog and it is incorporated into the DNA of actively replicating cells during S phase of the cell cycle. Starting at 0.5 μM lead, we observed a significant decrease in the number of BrdU+ cells after a 48 h-treatment (Fig. 3). Combined with data shown in Fig. 2, these results suggest that lead inhibits cell proliferation of aNPCs at higher concentrations. However, at lower concentrations (< 0.5 μM), lead causes cell loss primarily through apoptosis.
Fig. 3. Lead decreases proliferation in SGZ-aNPCs.

A-D, SGZ-aNPCs were treated with 0, 0.1, 0.2, 0.5, 1, or 2 μM lead for 48 h. Representative BrdU and Hoechst co-staining from (A) untreated, (B) 0.2 μM, and (C) 0.5 μM lead-treated SGZ-aNPCs, and quantification of (D) the percent BrdU+ cells. BrdU: a marker for cells in S phase of the cell cycle. Scale bars: 50 μm. n = 2 independent, experiments for a total of 8 coverslips per data point. Data represent mean ± SEM., n.s. not significant, * p < 0.05; ** p < 0.01.
Lead decreases spontaneous neuronal differentiation and maturation of SGZ-aNPCs
We also examined whether lead disrupts neuronal differentiation and maturation, another critical step in adult neurogenesis. SGZ-aNPCs were seeded overnight, and then the cells were cultured in EGF/bFGF-free growth media containing vehicle or lead for 5 d to allow spontaneous neuronal differentiation in the absence of mitogens. We assessed neuronal differentiation by immunostaining for βIII-tubulin (also known as Tuj-1), a marker of immature neurons. Treatment with low concentrations (≥ 0.1 μM) of lead significantly decreased the percent βIII-tubulin+ cells (βIII-tubulin+ soma and neurites ≥ length of soma) (Fig. 4A-G).
Fig. 4. Lead decreases spontaneous neuronal differentiation of SGZ-aNPCs.

A-H, SGZ-aNPCs were treated with 0, 0.1, 0.2, 0.5, 1, and 2 μM lead for 5 d in EGF/bFGF-free growth media. Representative βIII-tubulin and Hoechst co-staining from (A,D) untreated, (B,E) 0.2 μM, and (C,F) 0.5 μM lead-treated SGZ-aNPCs. Images D-F correspond to the boxed regions in images A-C, respectively. Quantification of (G) the percent βIII-tubulin+ cells and (H). βIII-tubulin+: immature neuron marker. Scale bars for A-C: 50 μm; D-F: 25 μm. n = 2 independent experiments for a total of 8 coverslips per data point. Data represent mean ± SEM., n.s. not significant,* p < 0.05; ** p < 0.01; *** p < 0.001.
To determine whether lead may also impair neuronal maturation, we assessed the effect of lead on neurite morphology and complexity. Using the ImageJ Simple Neurite Tracer plug-in, we traced all of the neurites from 12-16 randomly chosen fields (20X magnification) per treatment and calculated the average neurite length and average number of branching points per βIII-tubulin+ cell. Lead significantly decreased the mean neurite length (Fig. 5A-E) and the mean number of branching points (Fig. 5A-D, F) per βIII-tubulin+ cell. These data suggest that lead impairs neuronal differentiation and maturation of SGZ-aNPCs.
Fig. 5. Lead decreases the neuronal maturation of SGZ-aNPCs.

A-F, SGZ-aNPCs were treated with 0, 0.1, 0.2, 0.5, 1, and 2 μM lead for 5 d in EGF/bFGF-free growth media. Representat ive (A, C) Hoechst staining and (B, D) neurite traces from untreated and 0.2 μM lead-treated SGZ-aNPCs, respectively. Quantification of (E) the mean neurite length per βIII-tubulin+ cell and (F) the average number of branch points per βIII-tubulin+ cell. Scale bar: 25 μm. n = 2 independent experiments for a total of 8 coverslips per data point. Data represent mean ± SEM.; * p < 0.05; ** p < 0.01; *** p < 0.001.
Lead inhibits activation of the Akt signaling pathway
To begin to elucidate the molecular mechanisms underlying lead toxicity, we determined if lead inhibits antiapoptotic signaling pathways, such as the Akt pathway. Akt plays a critical role in regulating cell survival and growth, and Akt activation can prevent stress-induced apoptosis (Song et al., 2005). Lead significantly decreased Akt phosphorylation starting at 4 h (Fig. 6), indicative of reduced Akt activation. Thus, lead-induced cell death may involve inhibition of the prosurvival Akt pathway.
Fig. 6. Lead inhibits activation of the Akt signaling pathway.
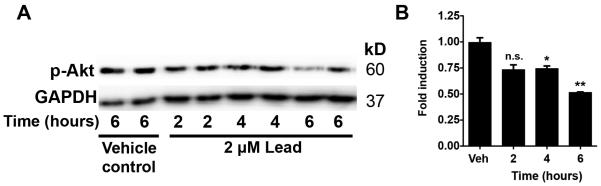
A-B, SGZ-aNPCs were treated with 2 μM lead for the indicated amount of time, and the cell lysates were subjected to Western blot analysis for (A) phosphorylated-Akt. (B) The fold induction of Akt normalized to GAPDH compared to DMSO controls. n = 2 independent experiments with duplicates. Data represent mean ± SEM., n.s. not significant, * p < 0.05; ** p < 0.01.
Activation of the JNK signaling pathway contributes to lead-induced cytotoxicity
The c-Jun NH2-terminal kinase (JNK) is a member of the mitogen activated protein kinase (MAPK) family, and it couples various external stimuli, such as stress and growth factors, to a variety of biological responses, including apoptosis and cell proliferation, respectively (Davis, 2000; Dhanasekaran and Reddy, 2008). Thus, to begin elucidating signaling mechanisms underlying lead-induced apoptosis, we first determined the effect of lead on JNK and c-Jun phosphorylation. SGZ–aNPCs were treated with 2 μM lead for 0-12 h, and the cell lysates were subjected to Western blot analysis. Treatment with lead for 8 h or longer significantly increased JNK phosphorylation, indicative of JNK activation (Fig. 7A, B). Furthermore, lead treatment for 12 h caused a significant increase in the phosphorylation of c-Jun, the prototypical JNK substrate (Fig. 7C, D). To determine whether inhibition of JNK is sufficient to attenuate the effect of lead on cell number and apoptosis, SGZ–aNPCs were pretreated with 0.5 μM of the ATP-competitive, pan-JNK inhibitor SP600125 for 1 h and then treated with 0.5 μM lead for an additional 48 h. Pretreatment with the JNK inhibitor without lead co-treatment had no effect on total cell number (Fig. 7E) or the number of BrdU+ cells (data not shown), suggesting that the endogenous basal JNK activity does not play a major role in cell proliferation under normal proliferation conditions in the presence of growth factors. However, pretreatment with the JNK inhibitor almost completely reversed lead-induced cell loss and apoptosis (Fig. 7E, F). These data suggest that activation of the JNK signaling pathway contributes to lead-induced apoptosis.
Fig. 7. Activation of the JNK signaling pathway contributes to lead-induced cytotoxicity.

A-D, SGZ-aNPCs were treated with 2 μM lead for the indicated amount of time. The cell lysates were subjected to Western blot analysis for (A) phosphorylated-JNK and (C) phosphorylated c-Jun, and the fold induction of (B) p-JNK (normalized to total JNK) and (D) p-c-Jun (normalized to GAPDH) in lead-treated cells compared to DMSO controls was quantified. E-F, SGZ-aNPCs were pretreated with 0.5 μM of the ATP-competitive, pan-JNK inhibitor SP600125 for 1 h and then treated with 0.5 μM lead for an additional 48 h. Quantification of (E) the total cell number and (F) the percent apoptot ic cells. A-D: n = 2 independent experiments with duplicates . E-F: n = 3 independent experiments for a total of 9 coverslips per data point. Data represent mean ± SEM., n.s. not significant,* p < 0.05; ** p < 0.01; *** p < 0.001.
Activation of the p38 MAP kinase signaling pathway is also important for lead cytotoxicity
p38 is another member of the stress activated MAP kinases (Kyriakis and Avruch, 2012). Lead significantly increased p38 phosphorylation [6.25 ± 0.14 (SEM)-fold increase] after treatment for 8 h (Fig. 8A, B), indicative of p38 activation. Pretreatment with 0.5 μM SB2021990, a p38 inhibitor, had no effect by itself on total cell number (Fig. 8C), apoptosis (Fig. 8D), or the number of BrdU+ cells (data not shown), but completely blocked the adverse effect of lead on cell number and apoptosis. Together, the data in Fig. 7 and Fig. 8 suggest that activation of both JNK and p38 are critical for lead-induced apoptosis.
Fig. 8. Activation of the p38 MAP kinase signaling pathway is also important for lead cytotoxicity.

A-B, SGZ-aNPCs were treated with 2 μM lead for the indicated amount of time, and the cell lysates were subjected to Western blot analysis for (A) phosphorylated-p38. (B) The fold induction of p-38 normalized to GAPDH compared to DMSO controls. C-D, SGZ-aNPCs were pretreated with 0.5 μM of a p38 inhibitor for 1 hand then treated with 0.5 μM lead for 48 h. Quantification of (C) the total cell number and (D) apoptosis. A-B : n = 2 independent experiments with duplicates . C-D: n = 3 independent experiments for a total of 9 coverslips per data point. Data represent mean ± SEM., n.s. not significant, * p < 0.05; ** p < 0.01; *** p < 0.001.
Discussion
Various extracellular and intracellular stimuli have been shown to modulate the survival, proliferation, and differentiation of adult-born cells in the hippocampus (Clelland et al., 2009; Deng et al., 2009; Garthe et al., 2009; Ming and Song, 2011; Pan et al., 2013b; Wang et al., 2013). However, the effects of toxicants on adult neurogenesis are not well understood. A recent study from our lab found that a hydroxylated metabolite of PBDE-47, a brominated flame retardant, interferes with the differentiation, proliferation, and survival of primary cultured aNPCs isolated from the subventricular zone of the lateral ventricle (Li et al., 2013). In addition, several studies have examined the effect of developmental lead exposure on adult hippocampal neurogenesis, although the results have been inconsistent (Gilbert et al., 2005; Jaako-Movits et al., 2005; Verina et al., 2007). In addition, their use of early life exposure paradigms makes it difficult to conclude that these observations are not due to the cumulative effects of lead on both developmental and adult neurogenesis (Gilbert et al., 2005; Jaako-Movits et al., 2005; Verina et al., 2007).Thus, additional studies are needed to determine whether adult-only lead exposure is sufficient to impair adult hippocampal neurogenesis and learning and memory. Most importantly, the signaling mechanisms underlying lead-induced impairment in adult neurogenesis have not been investigated. Thus, the goal of this study was to determine whether lead directly impairs critical processes in adult neurogenesis using primary cultured SGZ-aNPCs as a model system and to characterize the underlying signaling pathways.
Our data demonstrate that lead impairs several key processes in adult neurogenesis. Under proliferation conditions, lead significantly reduced aNPC total cell number and increased apoptosis at concentrations as low as 0.1 μM, the lowest tested concentration. It also significantly decreased BrdU incorporation, a measure of cell proliferation, starting at 0.5 μM. Under differentiation conditions when cells were cultured in media lacking the mitogenic growth factors EGF/bFGF, lead significantly impaired spontaneous neuronal differentiation and neuronal maturation, starting at 0.1 μM. These data are consistent with the notion that adult lead exposure, alone, may be sufficient to directly impair multiple aspects of adult neurogenesis (Schneider et al., 2005), with apoptosis and neuronal differentiation being most sensitive.
The underlying mechanisms for the effects of lead on adult neurogenesis have not been well characterized. JNK and p38 are stress-activated MAP kinases (Barger et al., 1993; Davis, 2000; Dhanasekaran and Reddy, 2008), and their activation has been implicated in toxicant-induced neuronal apoptosis (Xia et al., 1995; Newhouse et al., 2004; Giordano et al., 2007; Klintworth et al., 2007; Choi et al., 2010). JNK and p38 are also activated by growth factors and mediate cell proliferation (Davis, 2000; Kyriakis and Avruch, 2012). Here we show that pharmacological inhibition of JNK or p38 had no effect on total cell number or BrdU incorporation, suggesting that JNK or p38 do not play a major role in the cell proliferation of cultured SGZ-aNPCs. However, lead increases JNK and p38 phosphorylation as well as phosphorylation of the transcription factor c-Jun, a downstream target of JNK. These results are consistent with the findings from another study in which acute treatment with 1-10 μM lead acetate increased p38 phosphorylation in a human derived cell line (SH-SY5Y cells), although cell survival and apoptosis were not assessed (Leal et al., 2002). Furthermore, in our study, we found that pharmacological inhibition of either JNK or p38 was sufficient to prevent lead-induced cell loss and apoptosis. Thus, lead-induced cell loss and apoptosis may require activation of both JNK and p38 MAP kinase signal transduction pathways.
Akt is important for the regulation of cell growth and survival, and Akt activation can prevent stress-induced apoptosis (Kim et al., 2001; Tobiume et al., 2001; Yoon et al., 2002; Song et al., 2005). We found that lead treatment significantly decreased Akt phosphorylation starting at 4 h. Interestingly, Akt phosphorylates and negatively regulates the MAPKKK apoptosis signal-regulating kinase 1 (Ask1) (Tobiume et al., 2001). Inhibition of Akt results in the dissociation of Ask1 and the subsequent activation of downstream targets, including p38 and JNK (Yoon et al., 2002). Thus, lead-induced cell death may also be mediated through the inhibition of the pro-survival Akt pathway.
Endoplasmic reticulum (ER) stress, oxidative stress, and disrupted calcium homeostasis have also been implicated in lead-induced neurotoxicity and other target organ toxicities (Toscano and Guilarte, 2005a; White et al., 2007; Baranowska-Bosiacka et al., 2013; Liu et al., 2013a, b; Akande et al., 2014). For example, lead may bind to sulfhydryl groups, leading to a reduction in cellular antioxidant capacity through the depletion of the cellular thiol status and inhibition of antioxidant enzymes, inducing oxidative stress (Ercal et al., 2001). Increased oxidative stress is associated with reduced cell viability and cell proliferation in embryonic and adult neural progenitor cells (Sava et al., 2007; Choi et al., 2014). In our study, we found that lead increased JNK and p38 activation in SGZ-aNPCs. Importantly, both of these MAP kinases can be activated by oxidative stress (Davis, 2000), and JNK may be activated via the IRE1/JNK pathway in response to lead-induced ER stress (Qian et al., 2001; Qian and Tiffany-Castiglioni, 2003). Thus, additional research is needed to determine whether oxidative and ER stress may underlie lead toxicity in SGZ-aNPCs.
Both animal and epidemiological studies have found an association between low blood lead levels (~ 10 μg/dL) and increased cognitive decline in older adults (Stewart et al., 2002; Weisskopf et al., 2004; Stewart et al., 2006; Wu et al., 2008). Although we cannot directly compare the lead concentrations we used to human blood lead levels, we observed lead toxicity in SGZ-aNPCs at 0.1 μM lead, which is equivalent to 2.05 μg lead/dL media. Thus, lead may exert toxic effects on adult neurogenesis under environmentally relevant exposure conditions.
In summary, using an in vitro model system, we provide evidence that lead significantly impairs aNPC survival, proliferation, and differentiation in a dose-dependent manner. In addition, we show that the activation of proapoptotic JNK and p38 MAP kinase and inhibition of the prosurvival Akt signaling may mediate lead toxicity. Because adult hippocampal neurogenesis plays an important role for hippocampus-dependent learning and memory, impairment in adult neurogenesis may underlie cognitive decline in adults exposed to lead.
Highlights.
Lead decreases aNPC survival via increased apoptosis and decreased proliferation
Lead impairs the neuronal differentiation and maturation of aNPCs in vitro
Lead aNPC cytotoxicity is mediated by JNK and p38 activation and Akt inhibition
Acknowledgements
We thank members of the Xia lab for their technical assistance and critical reading of the manuscript.
Funding Information
This work was supported by the National Institutes of Health (R01 MH95840 to Z.X., T32ES015459 to A.E.).
Abbreviations
- aNPC
adult neural precursor cells
- SGZ
subgranular zone
- DG
dentate gyrus
- JNK
c-Jun NH2-terminal kinase
- MAP
mitogen activated protein
- DMSO
dimethyl sulfoxide
- BrdU
5-bromo-2'-deoxyuridine
- EGF
epidermal growth factor
- bFGF
basic fibroblast growth factor
- BSA
bovine serum albumin
- PFA
paraformaldehyde
- PBST
phosphate-buffered saline with 0.1% Triton-X 100
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors claim no conflict of interest.
References
- Akande MG, Aliu YO, Ambali SF, Ayo JO. Taurine mitigates cognitive impairment induced by chronic co-exposure of male Wistar rats to chlorpyrifos and lead acetate. Environ Toxicol Pharmacol. 2014;37:315–325. doi: 10.1016/j.etap.2013.11.023. [DOI] [PubMed] [Google Scholar]
- ATSDR . Toxicological profile for lead. U.S. Department of Health and Human Services, Public Health Serivce, Agency for Toxic Substances and Disease Registry; Atlanta, GA, US: 2007. [Google Scholar]
- Baranowska-Bosiacka I, Struzynska L, Gutowska I, Machalinska A, Kolasa A, Klos P, Czapski GA, Kurzawski M, Prokopowicz A, Marchlewicz M, Safranow K, Machalinski B, Wiszniewska B, Chlubek D. Perinatal exposure to lead induces morphological, ultrastructural and molecular alterations in the hippocampus. Toxicology. 2013;303:187–200. doi: 10.1016/j.tox.2012.10.027. [DOI] [PubMed] [Google Scholar]
- Barger SW, Smith-Swintosky VL, Rydel RE, Mattson MP. beta-Amyloid precursor protein mismetabolism and loss of calcium homeostasis in Alzheimer's disease. [Review] Ann N Y Acad Sci. 1993;695:158–164. doi: 10.1111/j.1749-6632.1993.tb23045.x. [DOI] [PubMed] [Google Scholar]
- Basha MR, Wei W, Bakheet SA, Benitez N, Siddiqi HK, Ge YW, Lahiri DK, Zawia NH. The fetal basis of amyloidogenesis: exposure to lead and latent overexpression of amyloid precursor protein and beta-amyloid in the aging brain. J Neurosci. 2005;25:823–829. doi: 10.1523/JNEUROSCI.4335-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer SA, Altman J, Russo RJ, Zhang X. Timetables of neurogenesis in the human brain based on experimentally determined patterns in the rat. Neurotoxicology. 1993;14:83–144. [PubMed] [Google Scholar]
- Bihaqi SW, Bahmani A, Subaiea GM, Zawia NH. Infantile exposure to lead and late-age cognitive decline: Relevance to AD. Alzheimer's & dementia : the journal of the Alzheimer's Association. 2013 doi: 10.1016/j.jalz.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi NY, Choi H, Park HH, Lee EH, Yu HJ, Lee KY, Joo Lee Y, Koh SH. Neuroprotective effects of amlodipine besylate and benidipine hydrochloride on oxidative stress-injured neural stem cells. Brain Res. 2014;1551:1–12. doi: 10.1016/j.brainres.2014.01.016. [DOI] [PubMed] [Google Scholar]
- Choi WS, Abel G, Klintworth H, Flavell RA, Xia Z. JNK3 mediates paraquat- and rotenone-induced dopaminergic neuron death. J Neuropathol Exp Neurol. 2010;69:511–520. doi: 10.1097/NEN.0b013e3181db8100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clelland CD, Choi M, Romberg C, Clemenson GD, Jr., Fragniere A, Tyers P, Jessberger S, Saksida LM, Barker RA, Gage FH, Bussey TJ. A functional role for adult hippocampal neurogenesis in spatial pattern separation. Science. 2009;325:210–213. doi: 10.1126/science.1173215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- Deng W, Saxe MD, Gallina IS, Gage FH. Adult-born hippocampal dentate granule cells undergoing maturation modulate learning and memory in the brain. J Neurosci. 2009;29:13532–13542. doi: 10.1523/JNEUROSCI.3362-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanasekaran DN, Reddy EP. JNK signaling in apoptosis. Oncogene. 2008;27:6245–6251. doi: 10.1038/onc.2008.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ercal N, Gurer-Orhan H, Aykin-Burns N. Toxic metals and oxidative stress part I: mechanisms involved in metal-induced oxidative damage. Current topics in medicinal chemistry. 2001;1:529–539. doi: 10.2174/1568026013394831. [DOI] [PubMed] [Google Scholar]
- Garthe A, Behr J, Kempermann G. Adult-generated hippocampal neurons allow the flexible use of spatially precise learning strategies. PloS one. 2009;4:e5464. doi: 10.1371/journal.pone.0005464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano G, Klintworth HM, Kavanagh TJ, Costa LG. Apoptosis induced by domoic acid in mouse cerebellar granule neurons involves activation of p38 and JNK MAP kinases. Neurochem Int. 2007;52:1100–1105. doi: 10.1016/j.neuint.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman E. Time after time: environmental influences on the aging brain. Environmental health perspectives. 2014;122:A238–243. doi: 10.1289/ehp/122-A238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Patzlaff NE, Jobe EM, Zhao X. Isolation of multipotent neural stem or progenitor cells from both the dentate gyrus and subventricular zone of a single adult mouse. Nat Protoc. 2012;7:2005–2012. doi: 10.1038/nprot.2012.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaako-Movits K, Zharkovsky T, Romantchik O, Jurgenson M, Merisalu E, Heidmets LT, Zharkovsky A. Developmental lead exposure impairs contextual fear conditioning and reduces adult hippocampal neurogenesis in the rat brain. Int J Dev Neurosci. 2005;23:627–635. doi: 10.1016/j.ijdevneu.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Kim AH, Khursigara G, Sun X, Franke TF, Chao MV. Akt Phosphorylates and Negatively Regulates Apoptosis Signal-Regulating Kinase 1. Molecular and cellular biology. 2001;21:893–901. doi: 10.1128/MCB.21.3.893-901.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klintworth H, Newhouse K, Li T, Choi WS, Faigle R, Xia Z. Activation of c-Jun N-Terminal Protein Kinase Is a Common Mechanism Underlying Paraquat- and Rotenone-Induced Dopaminergic Cell Apoptosis. Toxicological sciences : an official journal of the Society of Toxicology. 2007;97:149–162. doi: 10.1093/toxsci/kfm029. [DOI] [PubMed] [Google Scholar]
- Kyriakis JM, Avruch J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiological reviews. 2012;92:689–737. doi: 10.1152/physrev.00028.2011. [DOI] [PubMed] [Google Scholar]
- Leal RB, Cordova FM, Herd L, Bobrovskaya L, Dunkley PR. Lead-stimulated p38MAPK-dependent Hsp27 phosphorylation. Toxicology and applied pharmacology. 2002;178:44–51. doi: 10.1006/taap.2001.9320. [DOI] [PubMed] [Google Scholar]
- Liu CM, Zheng GH, Ming QL, Sun JM, Cheng C. Protective effect of puerarin on lead-induced mouse cognitive impairment via altering activities of acetyl cholinesterase, monoamine oxidase and nitric oxide synthase. Environmental toxicology and pharmacology. 2013a;35:502–510. doi: 10.1016/j.etap.2013.02.009. [DOI] [PubMed] [Google Scholar]
- Liu CM, Zheng GH, Ming QL, Sun JM, Cheng C. Protective effect of quercetin on lead-induced oxidative stress and endoplasmic reticulum stress in rat liver via the IRE1/JNK and PI3K/Akt pathway. Free radical research. 2013b;47:192–201. doi: 10.3109/10715762.2012.760198. [DOI] [PubMed] [Google Scholar]
- Ming GL, Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 2011;70:687–702. doi: 10.1016/j.neuron.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newhouse K, Hsuan SL, Chang SH, Cai B, Wang Y, Xia Z. Rotenone-Induced Apoptosis Is Mediated by p38 and JNK MAP Kinases in Human Dopaminergic SH-SY5Y Cells. Toxicol Sci. 2004;79:137–146. doi: 10.1093/toxsci/kfh089. [DOI] [PubMed] [Google Scholar]
- Pan YW, Wang W, Xia Z. Assessment of adult neurogenesis in mice. Curr Protoc Toxicol. 2013 doi: 10.1002/0471140856.tx1220s56. Chapter 12:Unit12 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan YW, Kuo CT, Storm DR, Xia Z. Inducible and Targeted Deletion of the ERK5 MAP Kinase in Adult Neurogenic Regions Impairs Adult Neurogenesis in the Olfactory Bulb and Several Forms of Olfactory Behavior. PloS one. 2012a;7:e49622. doi: 10.1371/journal.pone.0049622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan YW, Zou J, Wang W, Sakagami H, Garelick MG, Abel G, Kuo CT, Storm DR, Xia Z. Inducible and conditional deletion of extracellular signal-regulated kinase 5 disrupts adult hippocampal neurogenesis. J Biol Chem. 2012b;287:23306–23317. doi: 10.1074/jbc.M112.344762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payton M, Riggs KM, Spiro A, 3rd, Weiss ST, Hu H. Relations of bone and blood lead to cognitive function: the VA Normative Aging Study. Neurotoxicology and teratology. 1998;20:19–27. doi: 10.1016/s0892-0362(97)00075-5. [DOI] [PubMed] [Google Scholar]
- Qian Y, Tiffany-Castiglioni E. Lead-induced endoplasmic reticulum (ER) stress responses in the nervous system. Neurochemical research. 2003;28:153–162. doi: 10.1023/a:1021664632393. [DOI] [PubMed] [Google Scholar]
- Qian Y, Falahatpisheh MH, Zheng Y, Ramos KS, Tiffany-Castiglioni E. Induction of 78 kD glucose-regulated protein (GRP78) expression and redox-regulated transcription factor activity by lead and mercury in C6 rat glioma cells. Neurotoxicity research. 2001;3:581–589. doi: 10.1007/BF03033212. [DOI] [PubMed] [Google Scholar]
- Sava V, Velasquez A, Song S, Sanchez-Ramos J. Adult hippocampal neural stem/progenitor cells in vitro are vulnerable to the mycotoxin ochratoxin-A. Toxicological sciences : an official journal of the Society of Toxicology. 2007;98:187–197. doi: 10.1093/toxsci/kfm093. [DOI] [PubMed] [Google Scholar]
- Schneider JS, Anderson DW, Wade TV, Smith MG, Leibrandt P, Zuck L, Lidsky TI. Inhibition of progenitor cell proliferation in the dentate gyrus of rats following post-weaning lead exposure. Neurotoxicology. 2005;26:141–145. doi: 10.1016/j.neuro.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. Journal of cellular and molecular medicine. 2005;9:59–71. doi: 10.1111/j.1582-4934.2005.tb00337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart WF, Schwartz BS, Simon D, Kelsey K, Todd AC. ApoE genotype, past adult lead exposure, and neurobehavioral function. Environ Health Perspect. 2002;110:501–505. doi: 10.1289/ehp.02110501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart WF, Schwartz BS, Davatzikos C, Shen D, Liu D, Wu X, Todd AC, Shi W, Bassett S, Youssem D. Past adult lead exposure is linked to neurodegeneration measured by brain MRI. Neurology. 2006;66:1476–1484. doi: 10.1212/01.wnl.0000216138.69777.15. [DOI] [PubMed] [Google Scholar]
- Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, Takeda K, Minowa O, Miyazono K, Noda T, Ichijo H. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO reports. 2001;2:222–228. doi: 10.1093/embo-reports/kve046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toscano CD, Guilarte TR. Lead neurotoxicity: from exposure to molecular effects. Brain research Brain research reviews. 2005a;49:529–554. doi: 10.1016/j.brainresrev.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Toscano CD, Guilarte TR. Lead neurotoxicity: from exposure to molecular effects. Brain Res Rev. 2005b;49:529–554. doi: 10.1016/j.brainresrev.2005.02.004. [DOI] [PubMed] [Google Scholar]
- van Wijngaarden E, Campbell JR, Cory-Slechta DA. Bone lead levels are associated with measures of memory impairment in older adults. Neurotoxicology. 2009;30:572–580. doi: 10.1016/j.neuro.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verina T, Rohde CA, Guilarte TR. Environmental lead exposure during early life alters granule cell neurogenesis and morphology in the hippocampus of young adult rats. Neuroscience. 2007;145:1037–1047. doi: 10.1016/j.neuroscience.2006.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Pan YW, Zou J, Li T, Abel GM, Palmiter RD, Storm DR, Xia Z. Genetic Activation of ERK5 MAP Kinase Enhances Adult Neurogenesis and Extends Hippocampus-Dependent Long-Term Memory. J Neurosci. 2014;34:2130–2147. doi: 10.1523/JNEUROSCI.3324-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisskopf MG, Wright RO, Schwartz J, Spiro A, 3rd, Sparrow D, Aro A, Hu H. Cumulative lead exposure and prospective change in cognition among elderly men: the VA Normative Aging Study. Am J Epidemiol. 2004;160:1184–1193. doi: 10.1093/aje/kwh333. [DOI] [PubMed] [Google Scholar]
- White LD, Cory-Slechta DA, Gilbert ME, Tiffany-Castiglioni E, Zawia NH, Virgolini M, Rossi-George A, Lasley SM, Qian YC, Basha MR. New and evolving concepts in the neurotoxicology of lead. Toxicology and applied pharmacology. 2007;225:1–27. doi: 10.1016/j.taap.2007.08.001. [DOI] [PubMed] [Google Scholar]
- Wu J, Basha MR, Brock B, Cox DP, Cardozo-Pelaez F, McPherson CA, Harry J, Rice DC, Maloney B, Chen D, Lahiri DK, Zawia NH. Alzheimer's disease (AD)-like pathology in aged monkeys after infantile exposure to environmental metal lead (Pb): evidence for a developmental origin and environmental link for AD. J Neurosci. 2008;28:3–9. doi: 10.1523/JNEUROSCI.4405-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing Effects of ERK and JNK-p38 MAP Kinases on Apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- Yoon SO, Kim MM, Park SJ, Kim D, Chung J, Chung AS. Selenite suppresses hydrogen peroxide-induced cell apoptosis through inhibition of ASK1/JNK and activation of PI3-K/Akt pathways. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2002;16:111–113. doi: 10.1096/fj.01-0398fje. [DOI] [PubMed] [Google Scholar]