

Abstract
Single particle tomography (SPT or subtomogram averaging) offers a powerful alternative to traditional 2-D single particle reconstruction for studying conformationally or compositionally heterogeneous macromolecules. It can also provide direct observation (without labeling or staining) of complexes inside cells at nanometer resolution. The development of computational methods and tools for SPT remains an area of active research. Here we present the EMAN2.1 SPT toolbox, which offers a full SPT processing pipeline, from particle picking to post-alignment analysis of subtomogram averages, automating most steps. Different algorithm combinations can be applied at each step, providing versatility and allowing for procedural cross-testing and specimen-specific strategies. Alignment methods include all-vs-all, binary tree, iterative single-model refinement, multiple-model refinement, and self-symmetry alignment. An efficient angular search, Graphic Processing Unit (GPU) acceleration and both threaded and distributed parallelism are provided to speed up processing. Finally, automated simulations, per particle reconstruction of subtiltseries, and per-particle Contrast Transfer Function (CTF) correction have been implemented. Processing examples using both real and simulated data are shown for several structures.
Keywords: Cryo-electron tomography, single particle tomography, subtiltseries, subtomogram, EMAN2
1. INTRODUCTION
Single particle cryo electron tomography, also known as cryoSPT or subtomogram averaging (Walz et al 1997), makes use of 3-D information from individual macromolecules, thereby helping to alleviate the ambiguity in traditional 2-D single particle analysis of distinguishing between particle orientation and particle conformation. Collecting more than one image for all particles, from different directions, has been proposed to be critical for validation of orientation determination accuracy (Henderson 2003), even for homogeneous particles that exist in a single conformation. Unsurprisingly, particle classification by conformation also often requires the use of additional images from tilted views (Brignole et al 2009); hence the emergence of the Random Conical Tilt (RCT) (Radermacher et al 1986) and Orthogonal Tilt Reconstruction (OTR) (Leschziner, & Nogales 2006) methods. Besides providing multiple images of isolated complexes to obtain 3-D structures, SPT also permits the study of macromolecules in crowded environments (e.g., inside cells) whose projections would overlap in 2-D. However, due to mechanical limitations, typical electron microscopes yield incomplete tomographic data with a “missing wedge” (Baumeister et al 1999) that produces anisotropic resolution in 3-D, which, along with other factors like radiation damage, limits overall resolution. These problems can be ameliorated by averaging multiple 3-D subtomograms after alignment and classification, to obtain structures with an improved signal to noise ratio (SNR) and a filled-in missing wedge, as previously demonstrated (see reviews (Briggs 2013; Schmid 2011)).
To date, cryoSPT is the only technique in structural biology than can produce nanometer resolution structures of compositionally and conformationally heterogeneous complexes in a frozen-hydrated state (Shahmoradian et al 2013), and in the crowded environment of the cell (Brandt et al 2010; Dai et al 2013; Schwartz et al 2012). Indeed, cryoSPT is gaining momentum because of its unique potential for structural biology in situ at nanometer resolution (Harapin et al 2013; Lučić et al 2013), prompting the emergence of several tools in the last few years (Castaño-Díez et al 2012).
Our software offers a full suite of tools for SPT processing, including novel algorithms and several overall strategies targeting different SPT problems. We also build on previously successful methods, such as cross-correlation map normalization to compensate for the missing wedge in the alignment step (Schmid & Booth 2008), efficient angular sampling and adaptive-filtering (Hrabe et al 2012), among others.
EMAN2 (Tang et al 2007) is one of the most widely used software packages for electron microscopy (EM) data processing. Our toolbox extends its capabilities to single particle tomography, building upon our previous developments in this area (Schmid et al 2006; Schmid, & Booth 2008). Earlier versions of several of the algorithms comprising this new suite have been used on a range of specimens (Chang et al 2010; Koyfman et al 2011; Miyazaki et al 2010; Shahmoradian et al 2013; Schmid et al 2012), including data using Zernike phase contrast (Murata et al 2010), and within cells (Dai et al 2013). EMAN2’s modular approach and its long tradition in SPR (Ludtke et al 1999) makes it well suited to provide tools for SPT, a rapidly developing field where the algorithms and approaches are still under active development. Within EMAN2.1’s modular infrastructure there are over 180 different image processing filters, including a range of normalization, masking, linear filtration and mathematical operations. This versatility is unmatched by any other SPT package, which can facilitate tackling a wide variety of challenging problems. We make use of HDF as the standard image format, which natively supports multiple volumes in a single image file, each with an independent extensible header, dramatically easing data organization in complex projects with many tomograms. Most other programs either rely on individual image files for each particle, or re-extract particle data from the full tomogram multiple times. In EMAN2 SPT, the extensible header of HDF stacks permits directly associating metadata generated during processing with the corresponding image data.
In addition to an intuitive graphical 3-D particle picker for subtomograms, we also provide the ability to extract the original tiltseries data on a per particle basis (i.e., “subtiltseries” associated with individual subtomograms). This task is complicated by the need to predict the position of every particle in every image of the tiltseries, which requires solving the accurate location of the tilt axis, often not in the center of the tomogram. Here we use an iterative approach that can extract subtiltseries successfully even when the alignment of the mother tiltseries is suboptimal and the tilt axis is assumed to go through the middle of the tomogram, parallel to the y-axis. Constituting the raw, 2-D particle data, subtiltseries can be preprocessed and subtomograms reconstructed with any of the multiple methods available in EMAN2. Of note, it has been suggested that direct Fourier inversion can produce more accurate 3-D reconstructions (Heymann et al 2008) than the more traditional WBP and SIRT (Radermacher 2006) methods used in the field. Furthermore, since gold fiducials are absent from tilt series in cases where they might interact with the specimen and, when present, they undergo beam-induced motion resulting in unavoidable errors in whole-frame alignment (Comolli, & Downing 2005), subtiltseries can allow for more accurate individual per particle alignment, refinement and reconstruction, provided that the data have enough contrast, as demonstrated for negatively stained data (Iwasaki et al 2005) and cryoET data (Zhang, & Ren 2012).
We also provide missing wedge analysis tools and several subtomogram alignment and classification schemes, including automated multiple-model refinement (MMR) (Klaholz et al 2004; Scheres et al 2005), shown to facilitate classification of heterogeneous particles in SPR (Chen et al 2013; Fischer et al 2010) and SPT (Schmitz et al 1996). Our default strategy for missing wedge compensation in particle-particle alignment is cross-correlation map normalization (Rath et al 2003), as extensively discussed in the context of SPT (Schmid & Booth 2008), and widely demonstrated through our previous subtomogram averaging publications (Chang et al 2010; Koyfman et al 2011; Miyazaki et al 2010; Shahmoradian et al 2013; Schmid et al 2012; Murata et al 2010; Dai et al 2013; Hong et al 2015).
We also introduce a novel self-symmetry alignment tool, which permits fast 3-D reference-free alignment of particles with symmetry. While eventually these particles will be iteratively refined with others, self-symmetry can provide unbiased initial averages as seeds for further refinement. An early version of this tool was critical in one previous study (Dai et al 2013).
Our toolbox includes post-averaging analysis tools useful for data interpretation, as well as fully automated SPT simulations. The latter can help to assess data collection and processing parameters exhaustively, and to test algorithms.
Several CTF correction approaches have been proposed for electron tomography (ET) (Eibauer et al 2012; Mindell, & Grigorieff 2003; Winkler, & Taylor 2003). The most successful of these (Schur et al 2013) achieved sub-nanometer resolution, but relied on near-perfect experimental data, with very accurate defocuses combined with excellent stage eucentricity, high particle symmetry and extensive data collection. Indeed, achieving better than 2 nm resolution remains extremely rare in this field. When attempted, CTF correction is normally applied to aligned tiltseries, before reconstructing the overall tomogram (Fernandez et al 2006; Xiong et al 2009). Here we have implemented a novel per particle CTF correction strategy, so far tested only using simulated data, for which all parameters are known, as a proof of concept. At present our available experimental datasets used for testing our core SPT algorithms lacked sufficient contrast for accurate defocus estimation and high-resolution contrast for useful CTF correction testing, but we expect that with new tomographic data emerging from direct detectors (Pfeffer et al 2015), CTF correction for SPT will become more routine over the next year.
2. MATERIALS AND METHODS
2.1. Software design
EMAN2 is a cross-platform open-source software suite for EM consisting of Python scripts built upon a diverse C++ image-processing library (Tang et al 2007), unlike several other SPT packages that depend on the commercial Matlab library (Castaño-Díez et al 2012; Hrabe et al 2012; Nicastro et al 2006). Binaries are available for Linux, Mac and Windows, with full source is available. Threaded and MPI parallelization, as well as GPU acceleration are provided. A workflow interface GUI is provided for SPT iterative refinement via the same graphical project manager (e2projectmanager.py) used for traditional single particle analysis. Many additional utilities beyond this GUI are available from the command-line.
2.2. CryoET data collection and reconstruction
To validate our methods and compare different available approaches, we used 15 previously reported cryoET tiltseries (Chang et al 2010) containing epsilon-15 (ε15) virus particles as a first benchmark specimen, as well as a previously reported ε15 cryoET tiltseries collected using a Zernike Phase Contrast (ZPC) phase plate (Murata et al 2010), permitting to obtain a higher resolution average. The sampling of both ε15 specimens was ~5.2 Å/pixel, and the subtomograms were extracted using a 256×256×256 box size. Since different types of specimen may have different processing requirements, we also processed GroEL and GroEL/GroES datasets previously reported (Förster et al 2008), featured in several other publications (Scheres et al 2009; Yu and Frangakis 2011; Heumann et al 2011) and considered a “quasi-standard” dataset in the field (Hrabe et al 2012). Given the extremely low sampling of the data (12 Å/pixel, box size of 32×32×32), we processed yet another GroEL dataset reported to yield a higher resolution subtomogram average (Bartesaghi et al. 2008), at 4.1 Å/pixel and with a box size of 128×128×128. We also collected our own tiltseries for TRiC chaperonin as follows: 2.5 µL of TRiC, purified as previously described (Shahmoradian et al 2013), at 1 mg/ml concentration in PBS buffer, were applied to 200 mesh holey-carbon copper Quantifoil grids and plunge-frozen in liquid ethane at liquid nitrogen temperature using a Vitrobot Mark III. The specimen was imaged using SerialEM (Mastronarde 2005) on a JEOL2100 microscope operated at 200KV. Eight tiltseries were recorded for TRiC at 3.79 Å/pixel sampling, over a +−60° tilt range with a tilt step of 5°, a cumulative dose of ~80 e/Å2 and a target ~3 µm defocus at the tilt axis. For the specimens for which we had access to tiltseries, the best were aligned after evaluation (6 for cryoET ε15, 1 for cryoET ZPC ε15, and 2 for TRiC) and reconstructed into tomograms using standard protocols in IMOD (Kremer et al 1996). Gold fiducials were manually selected and carefully centered in all tilt images. Finally, the particle count from our TRiC tiltseries was low; therefore, to expand our dataset, we also processed previously reported TRiC tiltseries (Shahmoradian et al 2013). In the end, the sampling of the combined subtomograms set was 4.4 Å/pixel while the box size was 128×128×128.
2.3. Subtomogram and subtiltseries simulation
While testing on experimental data is the final test of any software, during development it can also be extremely valuable to test using simulated data, where the ground truth is known. This can also be valuable for end-users when encountering difficulties with specific specimens. Simulations can be used to mimic uncooperative datasets to help identify where potential difficulties may lie. We provide a set of completely automated tools for simulating subtiltseries and subtomograms in EMAN2 through e2spt_simulation.py, which randomizes the angular orientation (3 Euler angles) and center (X,Y,Z) of a given model and computes projections with a user-defined tilt range and tilt step. Simulated noise and CTF can optionally be applied to the particle projections in Fourier space. The applied CTF can take both the defocus gradient as well as the 3-D location of each particle in a tomogram into account to produce accurate perparticle variations in defocus in each tilt image, simulating a tomogram with perfect stage eucentricity. While this is an incomplete simulation of the full imaging process on the microscope, it is sufficient to validate the most important aspects of tomography. Experimental data for specimens with known structure remains the final test of the overall pipeline.
2.4 SPT pipeline
SPT processing with EMAN2 would typically start with a reconstructed tomogram. However, EMAN2 can optionally be used to reconstruct the aligned tilt series, to compare its direct Fourier inversion algorithms (several variants are available) to the real-space methods favored in IMOD.
A simplified version of the standard workflow most users would need to follow is presented schematically in Figure 1.
Figure 1. EMAN2 SPT workflow schematic.
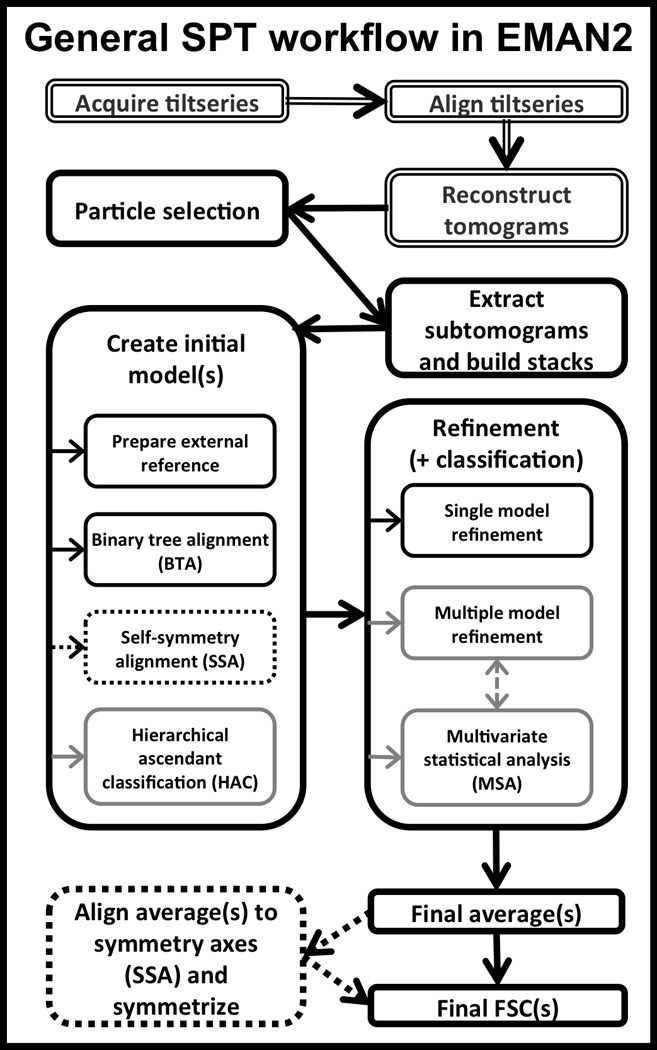
Double-line boxes indicate steps typically not performed with EMAN2. The bolded path in black is the quickest, default route for subtomogram alignment. If heterogeneity is suspected, alignment strategies outlined in grey might help. The dotted lines indicate steps that could be productive to carry out if the particles have symmetry.
2.4.1. Particle picking: subtomogram and subtiltseries extraction
The EMAN2.1 interactive SPT particle picker is e2spt_boxer.py (Figure 2). Selected particles are extracted on the fly, and tomograms can be dynamically filtered for better manual identification and centering. The Main Window (Figure 2A) displays 3 orthogonal X/Y/Z slices through the selected point in the tomogram, with optional multiplane averaging and lowpass filtration (Figure 2B), permitting precise centering of each particle in all 3 axes. The currently selected particle is also displayed as X/Y/Z projections and an isosurface of the subtomogram in the Single Particle View window, with an optional real-time lowpass filter applied (Figure 2D). Extracted particles can be saved in HDF5 or other formats supporting 3-D stacks or as multiple individual volume files. A stack of Z subtomogram reprojections (unaffected by the missing wedge) as shown in the Particle List window during particle picking (Figure 2C) is also saved for convenient later review of selected particles without reopening the much larger tomographic datasets.
Figure 2. ε15 virus visualized in EMAN2’s 3-D particle picking GUI for SPT.
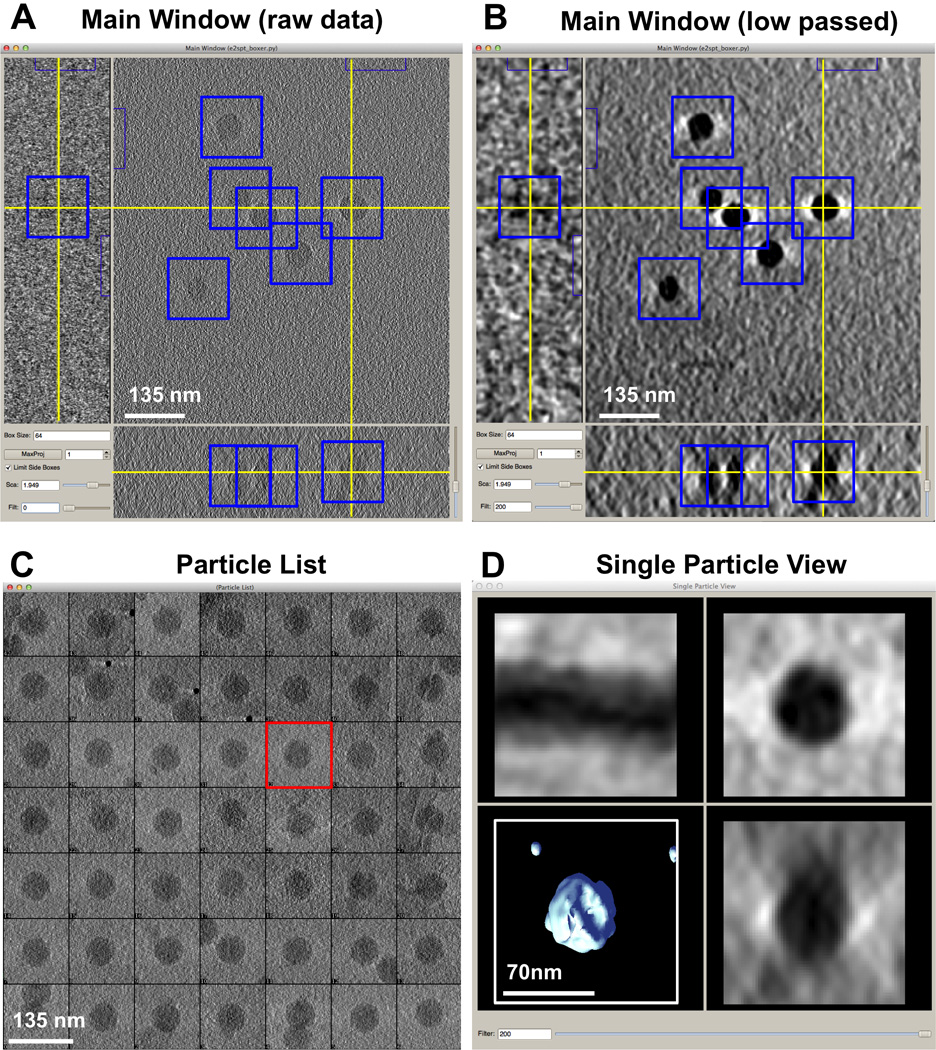
A) Main Window of the Graphical User Interface (GUI) provided by e2spt_boxer.py for subtomogram selection, showing three ~21Å thick orthogonal slices through a representative cryoET raw tomogram of ε15 in vitro. Blue boxes outline the subtomograms picked so far; the yellow cross denotes the center of the subtomogram currently selected. B) Same as A) but using the dynamic filter (bottom left of the Main Window) to lowpass the tomogram on the fly to 200Å. C) Particle List Window showing 2-D Z projections (X/Y plane) for all picked subtomograms (the subtomogram currently selected is outlined in red). D) Single Particle View Window showing three orthogonal projections and a 3-D interactive isosurface for the subtomogram currently selected, dynamically lowpass filtered to 200Å.
Subtomograms from different tomograms can be joined into a single 3-D stack using e2proc3d.py or e2buildstacks.py. The former also allows excluding undesirable particles by index. Subtomogram sets can be scrambled using e2spt_scramblestack.py to prevent per tomogram bias, which may impact some alignment approaches (e.g., if the combined stack is divided into n sub-stacks to generate n initial models, scrambling the stack will ensure that all sub-stacks contain particles from all the tomograms). Furthermore, the source tomogram and particle coordinates are stored in the header of extracted subtomograms, permitting each subtomogram to be tracked back to its original mother tomogram throughout the averaging process.
The advantages of processing extracted per-particle subtiltseries rather than extracting subtomograms directly after reconstruction have been previously demonstrated (Iwasaki et al 2005; Zhang & Ren 2012; Bartesaghi et al 2012). In EMAN2, subtiltseries extraction from aligned tiltseries is automated through e2spt_subtilt.py, using the 3-D coordinates of subtomograms to geometrically estimate their position in each tilt image. A value for ice thickness can be entered by the user or automatically estimated from the difference between the largest and the smallest Z coordinate from the set of extracted subtomograms. Alternatively, 2-D coordinates from the zero-tilt image can also be provided. If only 2-D coordinates are provided, or the ice thickness value is largely inaccurate, or the alignment of the tilt series is suboptimal (i.e., the direction of the tilt axis and the value for the tilt angles are inaccurate), the geometrical prediction of the position of the particles in all 2-D images across the tiltseries can be significantly off. To compensate, each particle can be progressively tracked, centered and extracted, using the previous tilt image as a reference. For example, in a tilt series collected from −60° to +60° with a 2° tilt step, the zero tilt image of each particle would serve as a reference to adjust the center of its corresponding particle views in the −2° and +2° tilt images; in turn, the +4° tilt image of a particle would be centered against the +2° image, while the +6° degree image would be centered against the already centered image from the +4° view; so on and so forth. Optional preprocessing options to facilitate alignment (lowpass and highpass filtering, thresholding, shrinking, masking, etc.) are incorporated into the algorithm. Extracted subtiltseries could be subsequently reconstructed into subtomograms with e2make3d.py using any of several available reconstruction methods (the default in EMAN2 is Fourier inversion), refined individually (Zhang, & Ren 2012), or used for novel SPT-SPA hybrid approaches (Bartesaghi et al 2012). Our subtiltseries extraction tool also allows keeping different subsets or ranges of particles. Finally, we also provide a tool for stacking full micrographs into tiltseries automatically or to reorder/manipulate the images of an existing tiltseries (e2spt_tiltstacker.py), which can be useful when tiltseries are acquired manually using data collection schemes that might vary, such as collecting bidirectional tiltseries starting from 0 tilt, either collecting the positive range of angles first and the negative last, or vice versa.
2.4.2 Per particle CTF correction
Per particle CTF correction, using subtiltseries, is performed with e2spt_ctf.py, provided that the defocus at the tilt axis can be accurately measured (Fernandez et al 2006; Mindell, & Grigorieff 2003), derived (Eibauer et al 2012) or assumed (Schur et al 2013; Zanetti et al 2009). Of note, instead of computing a flat defocus gradient along X as existing “strip-based tiling” approaches (Winkler, & Taylor 2003; Fernandez et al 2006) do, we implement a novel strategy for CTF correction using 3-D information to optimize the defocus on a per particle basis, since the distance Δx of a particle to the tilt axis and Δz to the middle Z-plane of the tomogram will induce different defocus shifts in its projections, depending on tilt angle, and many microscope stages have poor eucentricity. We demonstrate below (section 3.3) that the defocus shift can be significant for thick specimens (e.g., complexes inside cells or their vicinity) and can, in principle, be corrected on a per-particle basis, taking into account the z-position of each particles in the tomogram.
2.4.3 Subtomogram preprocessing
Preprocessing can improve the accuracy and speed of subtomogram alignment, even if it is still largely an ad-hoc process. EMAN2.1 provides optional automated preprocessing, including contrast reversal, normalization, masking (up to three different masks applied simultaneously), user-selected filters applied sequentially, thresholding, shrinking and box-size clipping to accelerate alignment while retaining oversampling benefits from using the full-size box for averaging. Subtomograms can also be lowpass filtered dynamically based on the even/odd FSC of the reconstruction from a previous iteration, similar to the approach used in 2-D single particle analysis. Any two volumes being aligned can be optionally filtered to have their radial power spectra match. This is particularly useful when using external references or when aligning particles from different tomograms to each other (since they might have been imaged under significantly different conditions).
2.4.4 Initial model generation
There are multiple strategies for generating initial models. In traditional single particle reconstruction, a single iterative procedure has been found to work well for most specimens (Tang et al 2007). Unfortunately, extensive testing with real data has shown that iterative sequential averaging without a reference does not always work reliably with subtomograms where missing-wedge bias may be a concern. Rather, different strategies can be helpful, or in some cases necessary for processing specific datasets. Conceptually, the ideal approach would be to perform exhaustive all-vs-all comparisons between every pair of particles; however, computing an n2 similarity matrix with any significant number of subtomograms rapidly becomes prohibitively computationally intensive. As such, all-vs-all alignment is normally applied to small sets or subsets of particles, to full sets using greatly constrained angular parameters, or as a method of last resort. Although other algorithms are much more efficient, they can be more prone to failure for certain types of structures. It is currently impossible to accurately predict the best approach for any arbitrary project (Castaño-Díez et al 2012); hence the importance of simulation tools and of automating data processing as much as possible, to efficiently experiment with multiple combinations of processing parameters and algorithms.
In the EMAN2 SPT toolbox, programs to generate initial averages include a binary tree alignment (BTA) algorithm (e2spt_binarytree.py), hierarchical ascendant classification (HAC, e2spt_hac.py) and a novel self-symmetry alignment strategy (SSA, e2symsearch3d.py). All alignment programs take a subset parameter that allows running quick tests on a fraction of the data for parameter optimization and timing estimates.
BTA averages the largest power-of-2 subset of particles from a given set, in a pairwise fashion, iteratively, until the tree converges.
Our implementation of HAC (Ward Jr 1963) or “all-vs-all” only averages unique best pairs and can be run supervised or unsupervised (to convergence). That is, unlike standard implementations of HAC or “all-vs-all”, we impose more stringent similarity requirements on particle mergers, thus increasing the probability of obtaining a good initial average by following a “unique best pairs” averaging policy. For example, if subtomograms A, B, C and D are available for averaging, and A–B turns out to be the best pair, these two particles will be averaged. If the second best pair is A–C, the average cannot occur because A has already been taken up to average with B. However, if the third best pair is C–D, traditional, unconstrained HAC would merge these two subtomograms since they are both still available (they haven’t been averaged with anything else). Our conservative approach, on the other hand, does not permit this merger, since C correlated better with A than it did with D. Rather, it is best for C to remain free to potentially join the average of A and B in a subsequent iteration. Criteria to terminate the algorithm automatically if similarity scores degrade or to preclude certain particle mergers are also implemented (e.g, cross-correlation thresholds and class size limits).
SSA aligns individual subtomograms to the symmetry axes, using a self-symmetry search. It generates a copy of the particle, applies symmetry assuming correct alignment, and then calculates the normalized cross-correlation between the symmetrized and unsymmetrized maps. A multidimensional simplex minimizer (http://www.gnu.org/software/gsl/) varies the translational center (X,Y,Z) and angular orientation (three Euler angles) of the raw particle, symmetry is applied to it in the new orientation, and a new score is computed. Since the simplex minimizer does not guarantee finding the global minimum, the process is repeated, starting with n random orientations. For high symmetries, particularly at low resolution, the energy surface tends to be fairly smooth, so this process generally converges to the correct global minimum with relatively small n. However, the algorithm can be applied to particles with any degree of symmetry.
Our three initial model generation methods scale differently with number of particles; i.e., the slower, more conservative methods can achieve better results at the cost of computational efficiency, and yet the faster methods are useful since conservative alignment is not always necessary to obtain a good subtomogram average.
Binary tree alignment (BTA) aligns fewer particles than our other initial model generation methods since it takes a subset equal to the largest power of two as the initial dataset. On the other hand, self-symmetry alignment bypasses fine-alignment preprocessing since orientations are sampled randomly instead of divided into coarse and fine alignment steps. Furthermore, SSA significantly reduces the angular search space since orientations need to be scanned only within a single asymmetric unit. This isn’t true when two particles are being aligned respect to one another and neither is aligned to the symmetry axes. In the latter case, two of the three Euler angles need to sample the full range of angular space. Finally, when applying symmetry, averages derived using either BTA or hierarchical ascendant classification (HAC) would require an additional step to align them to the symmetry axes before imposing symmetry. In terms of runtime, HAC scales as O(N^2), where N is the number of particles in the dataset. BTA scales as O(log N) and SSA scales as O(N). After initial model generation, iterative refinement against a single model also scales as O(N) and refining against multiple-models (MMR) scales as O(N*M), where M is the number of reference models.
Clearly, absolute alignment time will depend on how finely angular space is scanned, the box size and sampling of the data, and the power of the computational resources used. However, we provide tools to benchmark the alignment time of two subtomograms of any given size (e2spt_timetest.py). If running alignments on a Linux system with GPUs and building EMAN2 from source, the alignment time of a GPU and the speedup versus a CPU can also be benchmarked. Relative speedup will depend on the specific model of the CPU and the GPU, as well as on the specific box size used. We find, unsurprisingly, that even numbered box sizes with small prime factors usually perform the best (http://blake.bcm.edu/emanwiki/EMAN2/BoxSize). Sometimes, scaling up to a larger box size actually results in faster alignments. For example, subtomograms with a box size of 3983 will take over 3 times longer to align than particles with the slightly larger size of 4003. In fact, boxes 4003 run faster than all smaller sizes down to 3653, with a box of 3643 running only marginally faster.
Contrary to previous assertions (Castaño-Díez 2012), boxes > 1283 voxels are routinely used in SPT on a wide range of specimens such as carboxysomes (Schmid 2011), viruses (Schmid et al 2012) and axonemes (Koyfman 2011). We predict that as SPT strives to achieve higher resolution, finer image sampling will easily result in subtomograms much larger than 1283, reaching 5123 or more for large macromolecular assemblies, such as centrioles (Li 2011) and the nuclear pore complex (Maimon et al 2012).
2.4.5 Subtomogram alignment, classification and averaging
Iterative refinement programs include e2spt_classaverage.py and e2spt_multirefine.py, which can be seeded with preexisting references, or automatically use other SPT programs for de novo initial average computation (2.4.4). If using a preexisting reference, sampling, box size and other factors can be automatically adjusted with e2match.py. This program handles format conversion as necessary (by calling e2pdb2mrc.py and/or e2proc3d.py) and applies the scaling, clipping or padding necessary to produce a sensible reference for the data. All SPT alignment programs permit splitting datasets into two independent subsets, mimicking the single particle analysis “gold standard” (Henderson et al 2012; Scheres 2012). If external references are used for refinement, they are automatically phase-randomized to prevent model bias (Chen et al 2013a).
Subtomogram alignment is broken down into coarse and fine alignment cycles for efficiency (except for SSA, because it samples orientations randomly), scanning equidistant points in angular space, which in EMAN2 can be trivially restricted to any given asymmetric unit or subregion, accommodating for project-specific needs. Coarse alignment finds multiple best-matching, but approximate, peaks; then, fine alignment scans the vicinity of each coarse peak with a fine angular sampling. The angular ranges and steps can be specified manually, or the programs can automatically compute optimal values based on resolution and accuracy targets. Automatic calculation of alignment parameters can reduce computation times by limiting angular oversampling and relieve the user from manually calculating optimal values. These values depend on the imaging sampling size (Å/pixel), taking into account any shrinking factors specified for preprocessing, as well as on the hydrodynamic radius of the particle and the target resolution. Symmetry plays an important role in identifying angular search ranges. In many situations where it might seem that symmetry could be used to restrict the search space, very little restriction is possible. The only situation in which significant search-space reductions are possible is when a symmetrized reference is used, and that reference has been aligned to the appropriate symmetry axis. In this situation one of the Euler angles can be limited to the maximum angular extent of the asymmetric triangle of the reference.
Automated subtomogram classification can be accomplished through 1) unsupervised hierarchical ascendant classification with constraints (e2spt_hac.py), 2) multiple model refinement (e2spt_refinemulti.py) or 3) by using correlation thresholds to discard low correlating particles during iterative refinement (e2stp_classaverage.py). 3-D multivariate statistical analysis with k-means classification is also possible by sequentially running a series of programs: e2spt_classaverage.py (to align all the subtomograms to a common reference), followed by e2msa.py, e2basis.py and e2classifykmeans.py, and e2spt_classaverage.py again (to refine the alignment within each class). Finally, averages of classified particles are averaged in Fourier space, thresholding out low pixels, to better account for and smooth out the effects of missing data (including both the missing wedge and gaps between tilts in Fourier space).
We also provide a tool to extract sub-subtomograms from distal symmetry-related positions of 3D volumes, e2melonball.py (e.g., the density surrounding the 12 5-fold locations on the surface of a virus capsid, or any symmetry-related densities from particles with any given symmetry), similar to methodology previously demonstrated (Briggs et al 2005; Huiskonen et al 2007). Then, these regions can be treated as subtomogram stacks subjected to further alignment and classification.
We provide many miscellaneous tools for post-averaging analyses as well as a missing wedge visualization tool (e2spt_wedge.py), which can help assess qualitatively whether the missing wedge has been properly filled, particularly for small particle sets or when a structure shows suspicious features. Orthogonal projections (easily obtainable with e2orthoproject.py) of a volume displaying a particle’s Fourier-amplitudes, and orthogonal slices through it (e2slicer.py), can also quickly reveal whether the missing wedge has been filled in or not. Orthogonal projections and slices in both real and Fourier space can also aid in analyzing structural features of a subtomogram average and should always complement isosurfaces, since choosing surface thresholds is often somewhat arbitrary. Finally, rotational self-correlation plots (computable for any 3-D structure) as well as plots for rotational correlation between different maps or images (either varying azimuth, altitude, or both simultaneously) can be generated with e2rotationalplot.py. Such analyses can substantiate the success of subtomogram averaging in instances when the specimen possess known symmetry or pseudo-symmetry, but was not used while processing the data.
3. RESULTS
3.1. Building initial averages
To evaluate the performance of the various available strategies, we began by testing ε15 (a ~70 nm icosahedral virus), using a representative 20-particle set, as described above (2.4.1) (Figure 2). We compared the performance of the three initial model generation methods described (2.4.4), BTA, HAC and SSA (Figure 3). We aligned the same 20 subtomograms in each case and computed the FSC of each average against the known high-resolution ε15 structure (EMDB 1176) (Jiang et al 2006). The resolution of both the BTA (Figure 3A) and the HAC (Figure 3C) averages is ~65Å according to their FSC with the ε15 model. On the other hand, SAA yielded a much better initial average (Figure 3B) at ~48Å resolution. In terms of relative efficiency, BTA converged within minutes (15 iterations on 20 processors) and was ~10× faster than HAC run to convergence (190 iterations, taking less than 2 hrs on the same processors)(see section 2.4.4 for details on the scalability and efficiency of different alignment methods). Lastly, SSA was ~3× faster than HAC and ~3× slower than BTA given an arbitrary number of 20 starting orientations. Of note, GPU acceleration can be used to provide an additional speedup of 5–50×, depending on specific conditions and hardware. Best practices for initial model generation are addressed in the Discussion.
Figure 3. Initial models and iterative refinement of cryoET ε15 subtomograms using different alignment approaches.
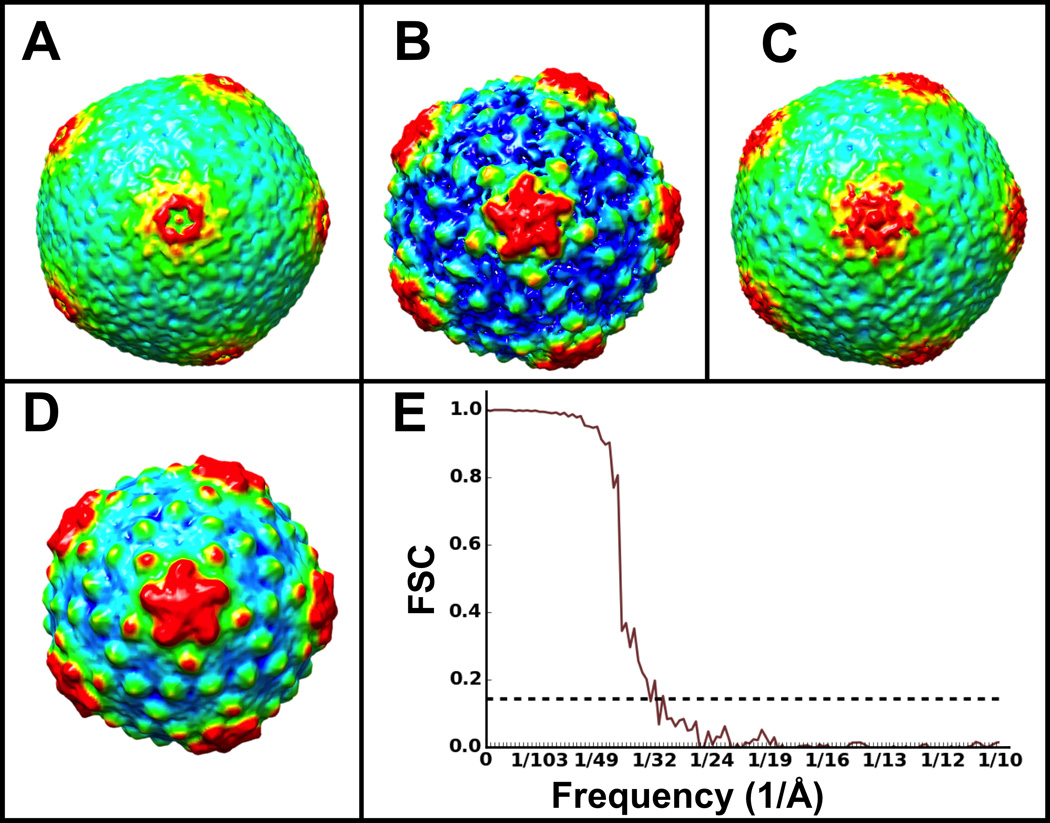
Initial icosahedral averages of 20 subtomograms using A) binary tree (BTA), B) self-symmetry (SSA) and C) “all-vs-all” (HAC) alignments. D) Average of a full set of 220 subtomograms using B) as an initial model for gold-standard iterative refinement. E) Gold-standard FSC for the structure in D).
3.2 Iterative refinement of subtomograms
Satisfied that our best reference-free ε15 initial model (produced by SSA) had a good distribution of Fourier amplitudes and exhibited the virus’s known structural features, we used it to seed iterative refinement of the same 20 ε15 subtomograms and achieved a map which improved from 48Å to 43Å resolution. To test whether the resolution was particle-count limited, we extracted a total of 220 subtomograms and seeded gold-standard iterative refinement with our 43Å map phase-randomized to 60Å (i.e., two separately phase-randomized copies of the map were used to refine two independent 110 particle subsets), ultimately excluding the worst 10% of the particles from averaging based on correlation. The resolution of our final map (Figure 3D) improved to ~35 Å, according to the FSC=0.143 threshold (Figure 3E). In light of previous analyses of resolution vs. subtomogram number for comparable ε15 data (Murata et al 2010), we have approached the maximum resolution achievable with this specific dataset with its 6–9 µm defocus range, and indeed it is slightly improved compared to the 4 nm resolution previously reported (Chang et al 2010).
To benchmark our pipeline on data containing higher-resolution information, we also processed an ε15 cryoET Zernike Phase Contrast (ZPC) dataset, also previously reported (Murata et al 2010). We divided a 133 subtomogram set randomly into seven subsets and computed an initial model from each using HAC. We then symmetrized each of these models using SSA and selected the best one (based on the resemblance of its features to the known structure of ε15) as a reference to seed iterative gold-standard icosahedral refinement with the entire set of particles. It is important to note that for phase plate data, applying a highpass filter can sometimes be essential to accomplish accurate alignment since it ameliorates the cut-on frequency artifact that characterizes ZPC images (Danev & Nagayama 2008). The data converged to a final ~24 Å resolution (Figure 4B,D,E), in excellent agreement with the ~25 Å resolution value previously reported for this dataset (Muarata et al 2010). We then spherically masked the vertex containing the tail from the known ε15 model and compared all 12 vertices of the particles in our dataset to it, to bring all the tails to the same vertex, thereby breaking the symmetry. After averaging, we achieved a symmetry-free map (Figure 4A,C) at ~37 Å resolution (Figure 4E), slightly lower than the resolution reported previously without enforcing symmetry on the average of this dataset. However, the method by Murata and colleagues involved careful manual orientation of the viruses tails, as well as extensive visual supervision; on the other hand, our structure was accomplished without manual or visual intervention, by using our aligner that automatically scans symmetry-related positions through e2spt_classaverage.py. Our final cryoET-ZPC averages were sharpened by application of a structure factor, a highpass filter, and by wiener filtering using the gold-standard FSC curve for each map, to prevent the strong ripples from the phase plate from obscuring fine features.
Figure 4. Reference-free iterative refinement of cryoET-ZPC ε15 subtomograms with and without symmetry.
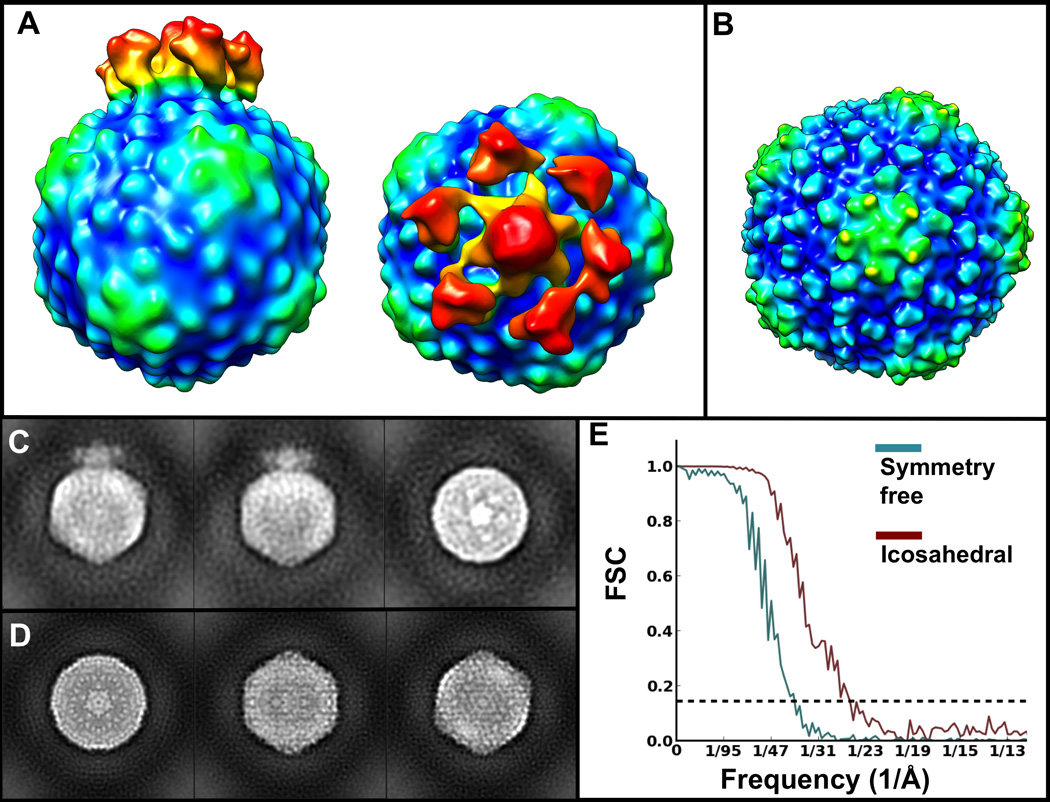
A) Symmetry-free average of 86 ε15 subtomograms. B) Average of 66 ε15 subtomograms refined with icosahedral symmetry enforced. C) Orthogonal projections of the structure in A). D) Orthogonal projections of the structure in B). E) Gold-standard FSC curves for the maps in A) (teal) and B) (brown), indicating ~37 Å and ~24 Å resolution at the 0.143 cutoff.
As a proof of concept and to benchmark our routines on a smaller and lower-symmetry specimen, we iteratively refined GroEL and GroEL/ES subtomograms previously reported (Förster et al 2008). These subtomogram have been repeatedly processed such that they’ve been claimed to constitute a “quasi-standard” SPT dataset (Hrabe et al 2012). We refined these particles without using any preexisting references and without imposing symmetry at any step, keeping the best 80% of particles for averaging and discarded the rest as possible false positives. We achieved maps at ~38 Å (GroEL) (Figure 5A,C) and ~39 Å gold-standard resolution (GroEL/GroES) (Figure 5B,C). Both maps show strong 7-fold symmetry (Figure 5D), even though they were refined and averaged without taking symmetry into account.
Figure 5. GroEL and GroEL/GroES reference-free and symmetry-free subtomogram averages.
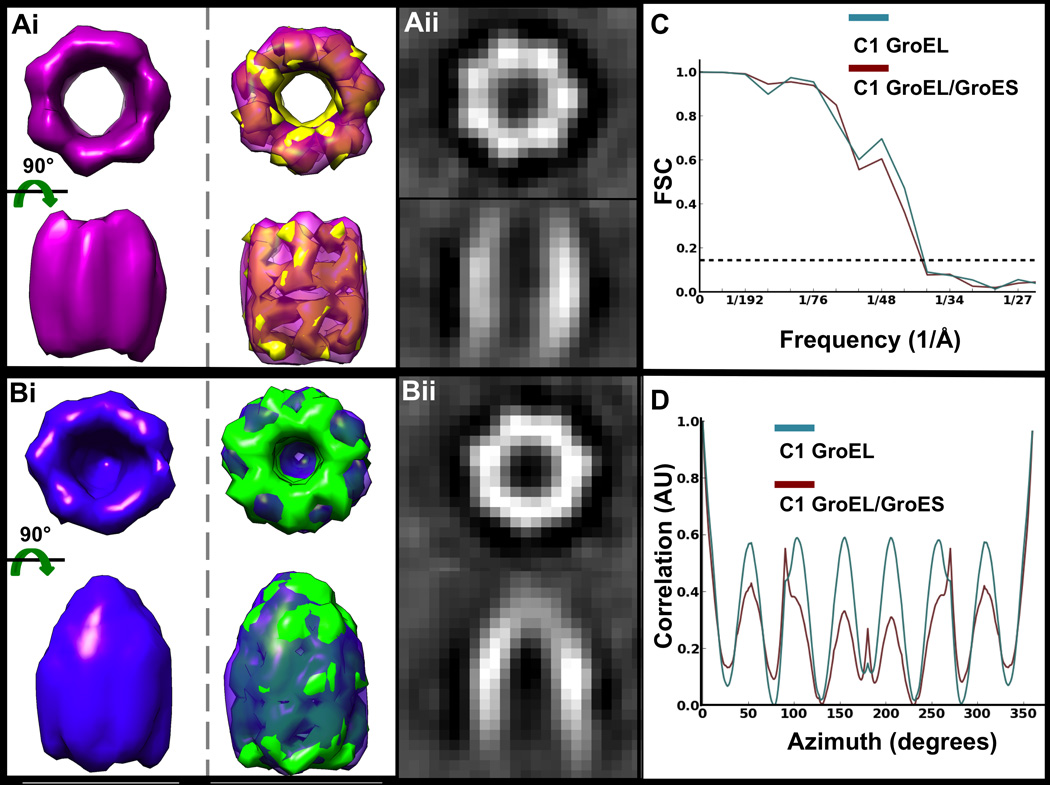
Reference-free and symmetry-free averages of Ai) 170 GroEL and Bi) 456 GroEL/GroES subtomograms shown as isosurfaces (top row), superimposed with their respective known crystal structures converted to EM density and filtered to the same resolution as the maps (PDB ID 3CAU for GroEL and PDB ID 1PCQ for GroEL/GroES). Aii, Bii) Corresponding orthogonal slices through the middle of the raw volumes shown in Ai) and Bi). C) Gold-standard FSC curves for the maps in Ai) (teal) and Bi) (maroon), indicating ~38 Å and ~39 Å resolution, respectively. D) Rotational self-correlation plots for the maps in Ai) (teal) and Bi) (maroon), demonstrating strong 7-fold symmetry in both structures, even though symmetry was not enforced or used during refinement. for a symmetry-free structure from this dataset using roughly the same number of particles (~66% of the entire dataset).
We also processed a 345 GroEL dataset reported to yield a higher-resolution subtomogram average (Bartesaghi et al 2008). Again, we didn’t use preexisting references; rather, our refinement program successfully built initial models from the data itself (one from each of the even and odd halves of the dataset), using the default binary tree alignment method. However, for this target we did refine the data imposing D7 symmetry to obtain the best possible average (Figure 6). We iteratively discarded the worst 33% of particles by correlation, attaining a final map with a gold-standard resolution of ~24 Å (Figure 6C), in good agreement with the reported ~27 Å resolution for a symmetry-free structure from this dataset using roughly the same number of particles (~66% of the entire dataset).
Figure 6. Reference-free D7 GroEL subtomogram average.
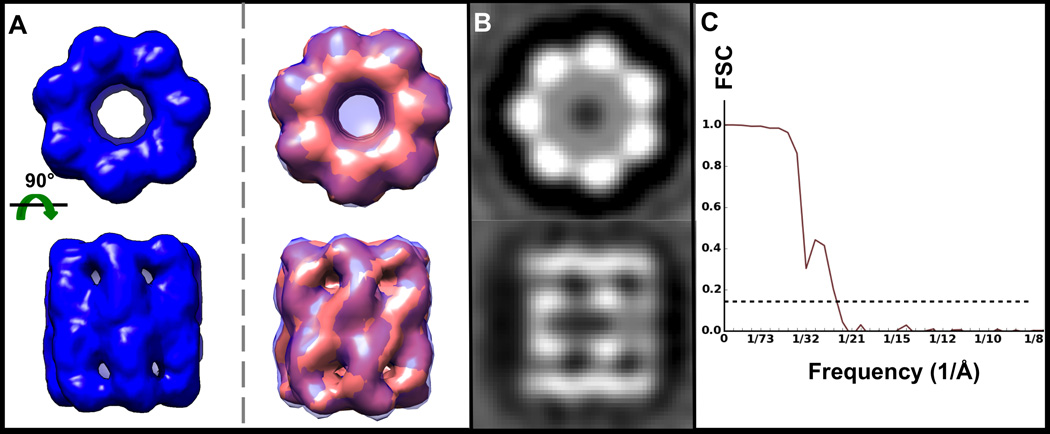
A) Average of 226 GroEL subtomograms iteratively refined imposing D7 symmetry, shown as an isosurface (left, blue) and super imposed with the known crystal structure (right, pink; PDB ID 3CAU) converted to EM density and filtered to the same resolution. B) Corresponding orthogonal projections of the map in A). C) Gold standard FSC curve for the map in A) indicating ~24 Å resolution.
Finally, we processed our own TRiC chaperonin dataset and complemented it with particles from previously reported tiltseries (Shahmoradian et al 2013), which we reconstructed into tomograms, to yield a final set of 550 subtomograms. Our gold-standard symmetry-free and reference-free average (Figure 7), keeping the top 66% of particles, converged to ~18 Å resolution (Figure 7D), and exhibits all of TRiC’s asymmetric features (Cong et al 2012), which deviate from its known pseudo-8 fold symmetry; namely, the one subunit that “sticks out” (Muñoz et al 2011), as well as the “notches” that separate the subunits into pseudo-dimers at the intermediate domains, conferring pseudo-4 fold symmetry to the chaperonin. This structure is significantly improved in terms of both resolution and features, compared to those previously reported for TRiC (Shahmoradian et al 2013). Of note, TRiC shows no symmetry or pseudo-symmetry at all in its flexible apical domains, (Figure 7C, slice 1), in contrast to other chaperonins such as the thermosome and MmCPN, and thus constitutes a much more challenging specimen.
Figure 7. Symmetry-free and reference-free TRiC subtomogram average (open state).
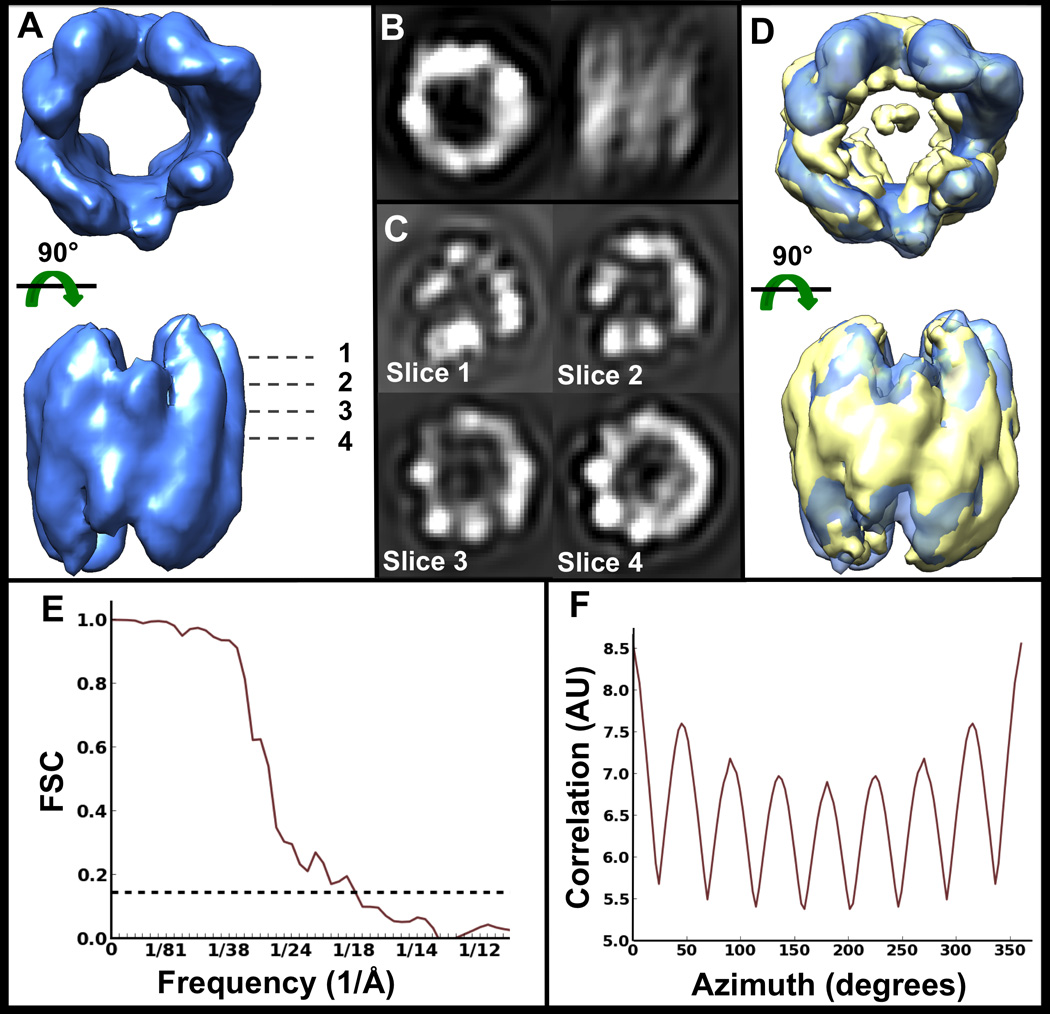
A) Isosurface of average of 362 TRiC subtomograms, iteratively refined without symmetry. B) Orthogonal projections of the map in A). C) Slices along z at different heights through the volume in A). Slice 4 is right through the middle of the volume. D) Map in A) superimposed with the known structure of TRiC resolved by cryoEM (EMDB1960), filtered to the same resolution. E) Gold-standard FSC curve for the map in A) indicating ~18 Å resolution. F) Rotational self-correlation plot for the map in A), demonstrating strong pseudo-8 fold symmetry, even though symmetry was not enforced or used in any way during refinement.
3.3 Per particle CTF correction
To demonstrate our methodology and implemented algorithms for CTF correction, we used simulated data for which the ground-truth for all parameters is known. We simulated subtiltseries (Methods 2.3) of randomly oriented GroEL particles (PDB ID 3CAU) (Ludtke et al 2008) by projecting them from −60° to +60° with a 4° tilt step and added CTF considering a non-flat defocus gradient (i.e., the particles were not co-planar) based on a target defocus of 3µ at the tilt axis, with particles distributed through a 400 nm thick slab in z (Figure 8A). It has been claimed that the defocus must be estimated within < ~100 nm error to achieve SPT at subnanometer resolution (Schur et al 2013). Therefore, the ice thickness limit for which flat defocus-gradient assumptions (co-planar particles) could be expected to guarantee productive CTF-correction at subnanometer resolution for all particles would be < ~200 nm. However, our CTF correction analyses (Figure 8) suggest that subnanometer resolution can still be achieved even if many particles in a dataset do not satisfy these requirements, as long as a substantial fraction are within this distance of the central plane. Our entire simulated subtomogram set (240 particles; 3,360 asymmetric units), applying full CTF, was subjected to our SPT processing pipeline. As expected, the FSC of the average without CTF correction (Figure 8B) against the known GroEL structure clearly fails to even approach subnanometer resolution (Figure 8E). On the other hand, applying the standard strip-based tiling CTF correction method (Figure 8C) (i.e., assuming the particles in the simulated specimen are all coplanar in a thin layer of ice) does achieve subnanometer resolution (Figure 8E), even though roughly half of the particles had defocus errors larger than 100 nm (Figure 8A). Finally, applying our depth-aware defocus gradient correction improves the resolution of the final average compared to applying the standard correction (Figure 8D,E).
Figure 8. Non-flat, depth-aware defocus gradient model improves CTF correction for SPT in 3-D, compared to standard strip-based tiling approach.
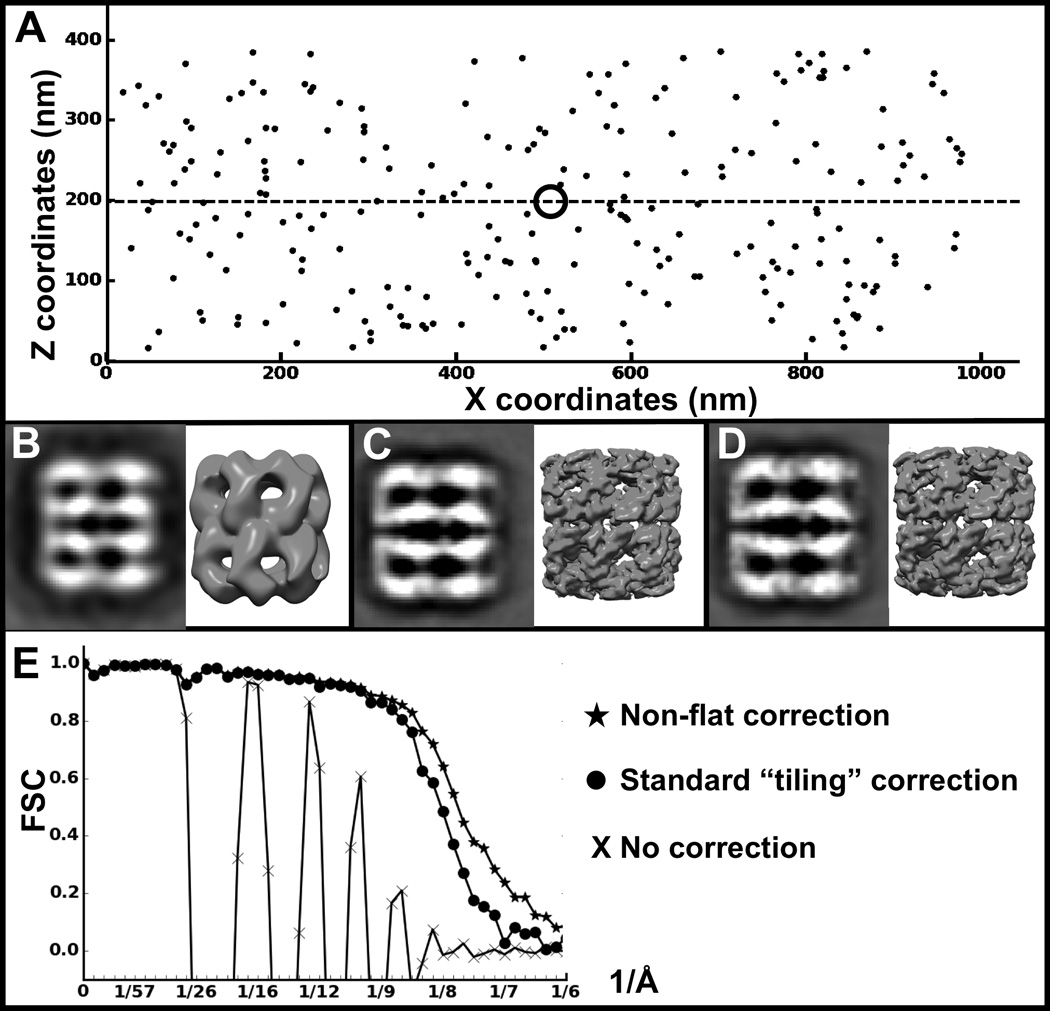
A) Spatial distribution of simulated subtomograms. B) Projection and isosurface of a subtomogram average using 240 simulated subtomograms without CTF correction, C) with CTF correction assuming coplanar particles with a simple tilt defocus gradient, and D) using our proposed particle depth-adjusted defocus CTF correction. The dashed line indicates the central z-plane, and the ring indicates the tilt axis. E) FSC curves between the known GroEL structure (PDB ID 3CAU) and the structures in B–D.
4. DISCUSSION
EMAN2 provides efficient processing capabilities with many unique features for the entire single particle tomography pipeline. It integrates most of the tools and methods available in the community in a single place, and introduces an interactive 3-D particle-picking tool that permits rapid and accurate subtomogram extraction. In addition to extensively automating well-established routines for alignment and classification, it automates multiple-model refinement and a novel self-symmetry alignment (SSA) approach. Our BTA strategy to generate initial models works well when the particles are relatively homogeneous and is much faster than the other alternatives. However, for flexible or pseudo-symmetric molecules like TRiC, this method can be prone to getting stuck in local minima, since random pairs might represent particles in substantially different conformations or that align in spurious, pseudo-symmetry-related orientations. Under such scenarios, HAC is more likely to yield accurate alignments (albeit more slowly), since it exhaustively performs “all-vs-all” comparisons, averaging unique-best-pairs, and can be constrained for classification (Förster et al 2008). SSA works well and relatively quickly for large-mass, high-contrast, high-symmetry particles as demonstrated here with ε15 and previously with other viruses (Dai et al 2013). SSA will also work on particles of lower symmetry and small size, provided that the per-particle contrast and SNR are sufficient. We have, in testing, successfully aligned averages of a few particles of GroEL (D7) and TRiC (D1) to their symmetry axes.
Different algorithms can also be combined in novel ways. For example, one can compute an accurate initial average with HAC using a subset of particles, then use SSA on this initial average to improve it, and finally seed symmetry-enforced iterative refinement. Interestingly, for both in vitro and in silico benchmark specimens, subtomograms that failed to align to the symmetry axis by SSA, as visualized individually after symmetrization, largely overlapped with those yielding the lowest correlation against known models. This suggests that, for complexes with symmetry, SSA could be automated to discard subtomograms of lesser quality.
Validation remains a central issue for SPT as it is for 2-D single particle analysis. We propose that gold standard methods, already routinely applied in single particle analysis, should also be adopted in SPT, at least in cases where the number of particles exceeds some minimum value (50–100). This can help provide robust resolution numbers, even if it does not entirely solve the issues of noise/model bias. Clearly, reconstructions performed without any external references will be free of model bias issues, but it may not always be possible to achieve good SPT averages without a reference. As additional validation, the same dataset could be processed using two or more completely different strategies and the corresponding averages compared. Based on past experience, the results would be similar, but certainly not identical, offering some information about the reliability of specific regions of the structure. Reporting FSC curves explicitly, as well as generating orthogonal projections, their FFTs and slices through structures should be standard practices in the field, as these can help reveal common problems that may occur. In EMAN2, most of these validation proposals are either automated or possible with minimal human effort.
The availability of a wide range of tools, diverse preprocessing options and use of a modern 3-D stack-friendly file format empowers users to tackle structural studies of challenging specimens in a variety of contexts. By making multiple strategies available within one environment users can readily devise hybrid processing pipelines to tackle otherwise intractable problems. Wider adoption of direct detection devices in the community is now producing experimental data with unprecedented resolution and contrast. The development of experimental new methods such as particle depth-aware CTF correction and subtiltseries extraction will permit users to experiment with emerging methodologies.
Acknowledgements
Research supported by NIH grants R01GM080139 and P41GM103832. We thank Dr. Judith Frydman for the TRiC specimen.
Abbreviations
- cryoSPT
Single particle tomography cryo electron microscopy
- SPT
single particle tomography
- BTA
binary tree alignment
- HAC
hierarchical ascendant classification
- SSA
self-symmetry alignment
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bammes BE, Jakana J, Schmid MF, Chiu W. Radiation damage effects at four specimen temperatures from 4 to 100K. Journal of structural biology. 2010;169(3):331–341. doi: 10.1016/j.jsb.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bammes BE, Rochat RH, Jakana J, Chen D-H, Chiu W. Direct electron detection yields cryo-EM reconstructions at resolutions beyond 3/4 Nyquist frequency. Journal of structural biology. 2012;177(3):589–601. doi: 10.1016/j.jsb.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartesaghi A, Sprechmann P, Liu J, Randall G, Sapiro G, Subramaniam S. Classification and 3D averaging with missing wedge correction in biological electron tomography. Journal of structural biology. 2008;162(3):436–450. doi: 10.1016/j.jsb.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartesaghi A, Lecumberry F, Sapiro G, Subramaniam S. Protein Secondary Structure Determination by Constrained Single-Particle Cryo-Electron Tomography. Structure. 2012;20(12):2003–2013. doi: 10.1016/j.str.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumeister W, Grimm R, Walz J. Electron tomography of molecules and cells. Trends in cell biology. 1999;9(2):81–85. doi: 10.1016/s0962-8924(98)01423-8. [DOI] [PubMed] [Google Scholar]
- Bernard Heymann J, Cardone G, Winkler DC, Steven AC. Computational resources for cryo-electron tomography in Bsoft. Journal of structural biology. 2008;161(3):232–242. doi: 10.1016/j.jsb.2007.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt F, Carlson L-A, Hartl FU, Baumeister W, Grünewald K. The three-dimensional organization of polyribosomes in intact human cells. Molecular cell. 2010;39(4):560–569. doi: 10.1016/j.molcel.2010.08.003. [DOI] [PubMed] [Google Scholar]
- Briggs JA. Structural biology in situ the potential of subtomogram averaging. Current opinion in structural biology. 2013;23(2):261–267. doi: 10.1016/j.sbi.2013.02.003. [DOI] [PubMed] [Google Scholar]
- Briggs JA, Huiskonen JT, Fernando KV, Gilbert RJ, Scotti P, Butcher SJ, Fuller SD. Classification and three-dimensional reconstruction of unevenly distributed or symmetry mismatched features of icosahedral particles. Journal of structural biology. 2005;150(3):332–339. doi: 10.1016/j.jsb.2005.03.009. [DOI] [PubMed] [Google Scholar]
- Brignole EJ, Smith S, Asturias FJ. Conformational flexibility of metazoan fatty acid synthase enables catalysis. Nature structural & molecular biology. 2009;16(2):190–197. doi: 10.1038/nsmb.1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardone G, Grünewald K, Steven AC. A resolution criterion for electron tomography based on cross-validation. Journal of structural biology. 2005;151(2):117–129. doi: 10.1016/j.jsb.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Castaño-Díez D, Kudryashev M, Arheit M, Stahlberg H. Dynamo: A flexible, user-friendly development tool for subtomogram averaging of cryo-EM data in high-performance computing environments. Journal of structural biology. 2012;178(2):139–151. doi: 10.1016/j.jsb.2011.12.017. [DOI] [PubMed] [Google Scholar]
- Chang JT, Schmid MF, Haase-Pettingell C, Weigele PR, King JA, Chiu W. Visualizing the Structural Changes of Bacteriophage Epsilon15 and Its Salmonella Host during Infection. Journal of molecular biology. 2010;402(4):731–740. doi: 10.1016/j.jmb.2010.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D-H, Madan D, Weaver J, Lin Z, Schröder GF, Chiu W, Rye HS. Visualizing GroEL/ES in the Act of Encapsulating a Folding Protein. Cell. 2013;153(6):1354–1365. doi: 10.1016/j.cell.2013.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, McMullan G, Faruqi AR, Murshudov GN, Short JM, Scheres SH, Henderson R. High-resolution noise substitution to measure overfitting and validate resolution in 3D structure determination by single particle electron cryomicroscopy. Ultramicroscopy. 2013a;135:24–35. doi: 10.1016/j.ultramic.2013.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comolli LR, Downing KH. Dose tolerance at helium and nitrogen temperatures for whole cell electron tomography. Journal of structural biology. 2005;152(3):149–156. doi: 10.1016/j.jsb.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Cong Y, Schröder GF, Meyer AS, Jakana J, Ma B, Dougherty MT, Schmid MF, Reissmann S, Levitt M, Ludtke SL. Symmetry-free cryo-EM structures of the chaperonin TRiC along its ATPase-driven conformational cycle. The EMBO journal. 2012;31(3):720–730. doi: 10.1038/emboj.2011.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai W, Fu C, Raytcheva D, Flanagan J, Khant HA, Liu X, Rochat RH, Haase-Pettingell C, Piret J, Ludtke SJ, Nagayama K, Schmid MF, King JA, Chiu W. Visualizing virus assembly intermediates inside marine cyanobacteria. Nature. 2013;502(7473):707–710. doi: 10.1038/nature12604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danev R, Nagayama K. Single particle analysis based on Zernike phase contrast transmission electron microscopy. Journal of structural biology. 2008;161(2):211–218. doi: 10.1016/j.jsb.2007.10.015. [DOI] [PubMed] [Google Scholar]
- Eibauer M, Hoffmann C, Plitzko JM, Baumeister W, Nickell S, Engelhardt H. Unraveling the structure of membrane proteins in situ by transfer function corrected cryo-electron tomography. Journal of Structural Biology. 2012;180(3):488–496. doi: 10.1016/j.jsb.2012.09.008. [DOI] [PubMed] [Google Scholar]
- Fernandez JJ, Li S, Crowther RA. CTF determination and correction in electron cryotomography. Ultramicroscopy. 2006;106(7):587–596. doi: 10.1016/j.ultramic.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Fischer N, Konevega AL, Wintermeyer W, Rodnina MV, Stark H. Ribosome dynamics and tRNA movement by time-resolved electron cryomicroscopy. Nature. 2010;466(7304):329–333. doi: 10.1038/nature09206. [DOI] [PubMed] [Google Scholar]
- Förster F, Pruggnaller S, Seybert A, Frangakis AS. Classification of cryo-electron sub-tomograms using constrained correlation. Journal of structural biology. 2008;161(3):276–286. doi: 10.1016/j.jsb.2007.07.006. [DOI] [PubMed] [Google Scholar]
- Harapin J, Eibauer M, Medalia O. Structural Analysis of Supramolecular Assemblies by Cryo-Electron Tomography. Structure. 2013;21(9):1522–1530. doi: 10.1016/j.str.2013.08.003. [DOI] [PubMed] [Google Scholar]
- Henderson R, Sali A, Baker ML, Carragher B, Devkota B, Downing KH, Egelman EH, Feng Z, Frank J, Grigorieff N. Outcome of the first electron microscopy validation task force meeting. Structure. 2012;20(2):205–214. doi: 10.1016/j.str.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heumann JM, Hoenger A, Mastronarde DN. Clustering and variance maps for cryo-electron tomography using wedge-masked differences. Journal of structural biology. 2011;175(3):288–299. doi: 10.1016/j.jsb.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong C, Pietilä MK, Fu CJ, Schmid MF, Bamford DH, Chiu W. Lemon-shaped halo archaeal virus His1 with uniform tail but variable capsid structure. Proceedings of the National Academy of Sciences. 2015;112(8):2449–2454. doi: 10.1073/pnas.1425008112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrabe T, Chen Y, Pfeffer S, Kuhn Cuellar L, Mangold A-V, Förster F. PyTom: A python-based toolbox for localization of macromolecules in cryo-electron tomograms and subtomogram analysis. Journal of structural biology. 2012;178(2):177–188. doi: 10.1016/j.jsb.2011.12.003. [DOI] [PubMed] [Google Scholar]
- Huiskonen JT, Jäälinoja HT, Briggs JA, Fuller SD, Butcher SJ. Structure of a hexameric RNA packaging motor in a viral polymerase complex. Journal of structural biology. 2007;158(2):156–164. doi: 10.1016/j.jsb.2006.08.021. [DOI] [PubMed] [Google Scholar]
- Iwasaki K, Mitsuoka K, Fujiyoshi Y, Fujisawa Y, Kikuchi M, Sekiguchi K, Yamada T. Electron tomography reveals diverse conformations of integrin αIIbβ3 in the active state. Journal of structural biology. 2005;150(3):259–267. doi: 10.1016/j.jsb.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Jiang W, Chang J, Jakana J, Weigele P, King J, Chiu W. Structure of epsilon15 bacteriophage reveals genome organization and DNA packaging/injection apparatus. Nature. 2006;439(7076):612–616. doi: 10.1038/nature04487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaholz BP, Myasnikov AG, Van Heel M. Visualization of release factor 3 on the ribosome during termination of protein synthesis. Nature. 2004;427(6977):862–865. doi: 10.1038/nature02332. [DOI] [PubMed] [Google Scholar]
- Koyfman AY, Schmid MF, Gheiratmand L, Fu CJ, Khant HA, Huang D, He CY, Chiu W. Structure of Trypanosoma brucei flagellum accounts for its bihelical motion. Proceedings of the National Academy of Sciences. 2011;108(27):11105–11108. doi: 10.1073/pnas.1103634108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. Journal of structural biology. 1996;116(1):71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Leschziner AE, Nogales E. The orthogonal tilt reconstruction method: an approach to generating single-class volumes with no missing cone for ab initio reconstruction of asymmetric particles. Journal of structural biology. 2006;153(3):284–299. doi: 10.1016/j.jsb.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Li S, Fernandez J-J, Marshall WF, Agard DA. Three-dimensional structure of basal body triplet revealed by electron cryo-tomography. The EMBO Journal. 2011;31(3):552–562. doi: 10.1038/emboj.2011.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lučić V, Rigort A, Baumeister W. Cryo-electron tomography: the challenge of doing structural biology in situ. The Journal of cell biology. 2013;202(3):407–419. doi: 10.1083/jcb.201304193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludtke SJ, Baker ML, Chen D-H, Song J-L, Chuang DT, Chiu W. De novo backbone trace of GroEL from single particle electron cryomicroscopy. Structure. 2008;16(3):441–448. doi: 10.1016/j.str.2008.02.007. [DOI] [PubMed] [Google Scholar]
- Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. Journal of structural biology. 1999;128(1):82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- Maimon T, Elad N, Dahan I, Medalia O. The human nuclear pore complex as revealed by cryo-electron tomography. Structure. 2012;20(6):998–1006. doi: 10.1016/j.str.2012.03.025. [DOI] [PubMed] [Google Scholar]
- Maiorca M, Hanssen E, Kazmierczak E, Maco B, Kudryashev M, Hall R, Quiney H, Tilley L. Improving the quality of electron tomography image volumes using pre-reconstruction filtering. Journal of structural biology. 2012;180(1):132–142. doi: 10.1016/j.jsb.2012.05.019. [DOI] [PubMed] [Google Scholar]
- Maiorca M, Millet C, Hanssen E, Abbey B, Kazmierczak E, Tilley L. Local regularization of tilt projections reduces artifacts in electron tomography. Journal of structural biology. 2014;186(1):28–37. doi: 10.1016/j.jsb.2014.03.009. [DOI] [PubMed] [Google Scholar]
- Mastronarde DN. Automated electron microscope tomography using robust prediction of specimen movements. Journal of structural biology. 2005;152(1):36–51. doi: 10.1016/j.jsb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Mindell JA, Grigorieff N. Accurate determination of local defocus and specimen tilt in electron microscopy. Journal of structural biology. 2003;142(3):334–347. doi: 10.1016/s1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- Miyazaki Y, Irobalieva RN, Tolbert BS, Smalls-Mantey A, Iyalla K, Loeliger K, DSouza V, Khant H, Schmid MF, Garcia EL. Structure of a conserved retroviral RNA packaging element by NMR spectroscopy and cryo-electron tomography. Journal of molecular biology. 2010;404(5):751–772. doi: 10.1016/j.jmb.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz IG, Yébenes H, Zhou M, Mesa P, Serna M, Park AY, Montoya G. Crystal structure of the open conformation of the mammalian chaperonin CCT in complex with tubulin. Nature structural & molecular biology. 2011;18(1):14–19. doi: 10.1038/nsmb.1971. [DOI] [PubMed] [Google Scholar]
- Murata K, Liu X, Danev R, Jakana J, Schmid MF, King J, Nagayama K, Chiu W. Zernike phase contrast cryo-electron microscopy and tomography for structure determination at nanometer and subnanometer resolutions. Structure. 2010;18(8):903–912. doi: 10.1016/j.str.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicastro D, Schwartz C, Pierson J, Gaudette R, Porter ME, McIntosh JR. The molecular architecture of axonemes revealed by cryoelectron tomography. Science. 2006;313(5789):944–948. doi: 10.1126/science.1128618. [DOI] [PubMed] [Google Scholar]
- Pfeffer S, Woellhaf MW, Herrmann JM, Förster F. Organization of the mitochondrial translation machinery studied in situ by cryoelectron tomography. Nature Communications. 2015;6 doi: 10.1038/ncomms7019. [DOI] [PubMed] [Google Scholar]
- Rath BK, Hegerl R, Leith A, Shaikh TR, Wagenknecht T, Frank J. Fast 3D motif search of EM density maps using a locally normalized cross-correlation function. Journal of structural biology. 2003;144(1):95–103. doi: 10.1016/j.jsb.2003.09.029. [DOI] [PubMed] [Google Scholar]
- Radermacher M, Wagenknecht T, Verschoor A, Frank J. A new 3-D reconstruction scheme applied to the 50S ribosomal subunit of E. coli. Journal of microscopy. 1986;141(1):RP1–RP2. doi: 10.1111/j.1365-2818.1986.tb02693.x. [DOI] [PubMed] [Google Scholar]
- Radermacher M. Electron tomography. New York: Springer; 2006. Weighted back-projection methods; pp. 245–273. [Google Scholar]
- Scheres SH, Valle M, Nuñez R, Sorzano CO, Marabini R, Herman GT, Carazo J-M. Maximum-likelihood multi-reference refinement for electron microscopy images. Journal of molecular biology. 2005;348(1):139–149. doi: 10.1016/j.jmb.2005.02.031. [DOI] [PubMed] [Google Scholar]
- Scheres SH, Melero R, Valle M, Carazo JM. Averaging of electron subtomograms and random conical tilt reconstructions through likelihood optimization. Structure. 2009;17(12):1563–1572. doi: 10.1016/j.str.2009.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH, Chen S. Prevention of overfitting in cryo-EM structure determination. Nature methods. 2012;9(9):853–854. doi: 10.1038/nmeth.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid MF. Single-particle electron cryotomography (cryoET) Adv Protein Chem Struct Biol. 2011;82:37–65. doi: 10.1016/B978-0-12-386507-6.00002-6. [DOI] [PubMed] [Google Scholar]
- Schmid MF, Booth CR. Methods for aligning and for averaging 3D volumes with missing data. Journal of structural biology. 2008;161(3):243–248. doi: 10.1016/j.jsb.2007.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid MF, Hecksel CW, Rochat RH, Bhella D, Chiu W, Rixon FJ. A tail-like assembly at the portal vertex in intact herpes simplex type-1 virions. PLoS pathogens. 2012;8(10):e1002961. doi: 10.1371/journal.ppat.1002961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid MF, Paredes AM, Khant HA, Soyer F, Aldrich HC, Chiu W, Shively JM. Structure of Halothiobacillus neapolitanus Carboxysomes by Cryo-electron Tomography. Journal of molecular biology. 2006;364(3):526–535. doi: 10.1016/j.jmb.2006.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz H, Reedy MC, Reedy MK, Tregear RT, Winkler H, Taylor KA. Electron tomography of insect flight muscle in rigor and AMPPNP at 23 C. Journal of molecular biology. 1996;264(2):279–301. doi: 10.1006/jmbi.1996.0641. [DOI] [PubMed] [Google Scholar]
- Schur FK, Hagen W, de Marco A, Briggs JA. Determination of protein structure at 8.5 Å resolution using cryo-electron tomography and subtomogram averaging. Journal of structural biology. 2013 doi: 10.1016/j.jsb.2013.10.015. [DOI] [PubMed] [Google Scholar]
- Schwartz CL, Heumann JM, Dawson SC, Hoenger A. A detailed, hierarchical study of Giardia lamblia's ventral disc reveals novel microtubule-associated protein complexes. PloS one. 2012;7(9):e43783. doi: 10.1371/journal.pone.0043783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahmoradian SH, Galaz-Montoya JG, Schmid MF, Cong Y, Ma B, Spiess C, Frydman J, Ludtke SJ, Chiu W. TRiCs tricks inhibit huntingtin aggregation. eLife. 2013;2 doi: 10.7554/eLife.00710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. EMAN2: an extensible image processing suite for electron microscopy. Journal of structural biology. 2007;157(1):38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Walz J, Typke D, Nitsch M, Koster AJ, Hegerl R, Baumeister W. Electron tomography of single ice-embedded macromolecules: three-dimensional alignment and classification. Journal of structural biology. 1997;120(3):387–395. doi: 10.1006/jsbi.1997.3934. [DOI] [PubMed] [Google Scholar]
- Ward JH., Jr Hierarchical grouping to optimize an objective function. Journal of the American statistical association. 1963;58(301):236–244. [Google Scholar]
- Winkler H, Taylor KA. Focus gradient correction applied to tilt series image data used in electron tomography. Journal of structural biology. 2003;143(1):24–32. doi: 10.1016/s1047-8477(03)00120-5. [DOI] [PubMed] [Google Scholar]
- Xiong Q, Morphew MK, Schwartz CL, Hoenger AH, Mastronarde DN. CTF determination and correction for low dose tomographic tilt series. Journal of structural biology. 2009;168(3):378–387. doi: 10.1016/j.jsb.2009.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Frangakis AS. Classification of electron sub-tomograms with neural networks and its application to template-matching. Journal of structural biology. 2011;174(3):494–504. doi: 10.1016/j.jsb.2011.02.009. [DOI] [PubMed] [Google Scholar]
- Zanetti G, Riches JD, Fuller SD, Briggs JA. Contrast transfer function correction applied to cryo-electron tomography and sub-tomogram averaging. Journal of structural biology. 2009;168(2):305–312. doi: 10.1016/j.jsb.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Ren G. IPET and FETR: experimental approach for studying molecular structure dynamics by cryo-electron tomography of a single-molecule structure. PloS one. 2012;7(1):e30249. doi: 10.1371/journal.pone.0030249. [DOI] [PMC free article] [PubMed] [Google Scholar]