

Abstract
An important component of plant evolution is the plethora of pathways producing more than 200,000 biochemically diverse specialized metabolites with pharmacological, nutritional and ecological significance. To unravel dynamics underlying metabolic diversification, it is critical to determine lineage-specific gene family expansion in a phylogenomics framework. However, robust functional annotation is often only available for core enzymes catalyzing committed reaction steps within few model systems. In a genome informatics approach, we extracted information from early-draft gene-space assemblies and non-redundant transcriptomes to identify protein families involved in isoprenoid biosynthesis. Isoprenoids comprise terpenoids with various roles in plant-environment interaction, such as pollinator attraction or pathogen defense. Combining lines of evidence provided by synteny, sequence homology and Hidden-Markov-Modelling, we screened 17 genomes including 12 major crops and found evidence for 1,904 proteins associated with terpenoid biosynthesis. Our terpenoid genes set contains evidence for 840 core terpene-synthases and 338 triterpene-specific synthases. We further identified 190 prenyltransferases, 39 isopentenyl-diphosphate isomerases as well as 278 and 219 proteins involved in mevalonate and methylerithrol pathways, respectively. Assessing the impact of gene and genome duplication to lineage-specific terpenoid pathway expansion, we illustrated key events underlying terpenoid metabolic diversification within 250 million years of flowering plant radiation. By quantifying Angiosperm-wide versatility and phylogenetic relationships of pleiotropic gene families in terpenoid modular pathways, our analysis offers significant insight into evolutionary dynamics underlying diversification of plant secondary metabolism. Furthermore, our data provide a blueprint for future efforts to identify and more rapidly clone terpenoid biosynthetic genes from any plant species.
Introduction
To elucidate the dynamics underlying metabolic diversification across multiple lineages, it is of paramount importance to identify and distinguish the complete set of orthologous and paralogous loci present within multiple genome annotations in a phylogenetic framework [1]. Two homologous genes are referred to as orthologs if they descend from one locus present in the common ancestor lineage and split due to speciation [2,3]. By definition, orthologous genes are embedded in chromosomal segments derived from the same ancestral genomic locus, thus sharing high inter-species synteny between closely related lineages [4]. In contrast, paralogous loci refer to homologs within one lineage and are due to, for example, tandem-, transposition- or whole genome duplications (WGDs) [5,6]. Large-scale synteny is not observed for paralogs derived from small-scale events like tandem- and transposition duplication. In contrast, paralogs derived from WGDs are located within intra-species syntenic genomic blocks, and can be referred to as ohnologs or syntelogs [7,8]. Supergene loci refer to clusters of genes in close genomic proximity, often causing linkage disequilibrium [9,10]. Tandem duplicates comprise arrays of paralog supergenes that are due to, for example, errors in meiosis like unequal crossing over and have been connected to metabolic diversification in plants [11–13].
Together with the continuous progress and use of next generation sequencing techniques, genome-wide analysis of syntelog distribution provided evidence for a history of ancient shared and/or lineage-specific polyploidy events for all flowering plant lineages [4]. For example, the lineage of the model plant Arabidopsis underwent at least five polyploidy events during evolution, two preceding and three following angiosperm evolution [14]. Among those, the most recent WGD event is commonly referred to as “At-α” and is shared by all other mustard family members, including the extant sister clade of the Aethionemeae [15,16]. The more ancient At-β WGD event is in turn shared by core species in the order Brassicales, and excepting early-branching lineages such as papaya [17,18] and therefore occurred after split of the Carica lineage. At-y refers to an older whole genome triplication (WGT) event with evidence in all Asterids (including tomato) and Rosids, grape (Vitales) and basal clades such as Pachysandra terminalis (Buxales) and Gunnera manicata (Gunnerales) [19,20]. Crops like Brassica rapa (Br-α WGT), Solanum lycopersicum and Solanum tuberosum (Sol-α WGD/WGT) also show evidence of ancient genome multiplications [21,22]. As a consequence, the level of “genome multiplicity” expected from the successive WGDs/WGTs in B. rapa (defined as “syntenic depth”) is 36x when compared to the 1x eudicot ancestor (3x due to At- y, 2x due to At-β, another 2x due to At-α as well as 3x due to Br- α, see above).
Evidence is now accumulating for significant impact of ancient and recent gene and genome duplication events to birth and diversification of key biological traits. Duplication was proposed to be a key factor in expansion of regulatory and enzymatic pathways involved in generation of >200,000 diverse biochemical secondary metabolites in the flowering plant lineage [23–25]. For example, a differential impact of various duplication modes has been revealed for plant resistance proteins [26]. Likewise, the last three polyploidy events of the Arabidopsis-lineage (see above) likely contributed to shaping the genetic versatility of the glucosinolate pathway, a class of plant secondary metabolites with beneficial effects to human health and nutrition [25]. Similarly, polyploidy has been brought in connection to the origin of C4-photosynthesis in Cleomaceae [27].
Little is known about the impact of genome duplication to diversification of isoprenoid pathways. Isoprenoids form a highly diverse class of metabolites commonly found in all kingdoms of life [28]. In angiosperms, for example, phytol side-chain substitutes of chlorophyll and carotenoid pigments as well as phytohormones like gibberellin or brassinosteroids are well-characterized isoprenoids involved in basic metabolic processes that are essential for plant growth and development [29–31]. To the knowledge of the authors, evidence for the connection of polyploidy to secondary metabolite pathway evolution is to date only available for glucosinolates biosynthesis in the mustard family [25]. Note that both glucosinolates and terpenoids are defined as specialized or secondary metabolites that play major roles in plant-insect interactions like, for example, attraction of beneficial organisms or defense against herbivores [32]. Boutanaev et al. investigated core terpene synthase (TPS) genes (which generate terpene scaffold diversity) and identified micro-syntenic clusters that have arisen within recent evolutionary history by gene duplication, acquisition of new function and genome reorganization [33]. Note that in concert with TPS genes, terpenoid biosynthesis depends on various independent pathways (referred to as modules hereafter). Here, we performed further extended comparative analysis of various independent terpenoid biosynthetic modules in context of gene- and genome duplication.
Briefly, a sequential combination of six distinct reaction modules acts in concert to convert primary metabolites to longer-chain compounds mediating designated biological function. Therefore, plant terpenoid biosynthesis displays “modular” organization, including (1) TPS genes, (2) IPP isomerases (IDI), (3) prenyltransferases (PTF), (4) genes from MVA and (5) MEP pathways as well as (6) triterpene-specific synthases (see Fig 1for a comprehensive overview). Notably, genes involved in the latter three modules share a common evolutionary origin (i.e. genes are homologous) as previously described based on analyses of Solanaceae species [34]. All terpenoids are synthesized from two universal C5-isoprenoid building blocks (a) isopentenyl diphosphate (IPP) and (b) its isomer dimethylallyl diphosphate (DMAPP). In plants, IPP is synthesized independently by the mevalonate (MVA, shown in black in Fig 1) and methylerythritol phosphate (MEP, Shown in purple in Fig 1) pathways. In contrast, DMAPP is synthesized by enzymes of the MEP pathway only [35]. Both DMAPP and IPP compounds can be isomerized by enzymes of the IPP isomerase type (IDI, shown in turquois in Fig 1) [36]. Due to the economic relevance of enzymes involved in MEP and MVA pathways as well as IPP isomerases, the underlying biochemistry has been thoroughly investigated. Note that both MVA and MEP pathways comprise sequential arrangements of consecutive reaction steps leading to formation of intermediate products [37]. Analysis of stoichiometry indicated dosage-dependent effects regarding both pathways in yeast [38]. Going beyond yeast, comparative network analysis of MVA and MEP pathways in prokaryotes and the model plant A. thaliana characterized dosage-dependent effects of enzymes in both pathways and elevations of corresponding metabolite concentrations in plants and humans. This indicates that enzymes involved in MVA and MEP pathways operate concentration-dependent across all kingdoms of life [37,39]. Similarly, genetic engineering of Escherichia coli in context of industrial terpenoid production revealed that enzymes of the IDI group function in a dosage-dependent manner [40]. This was confirmed by mechanistic investigations of IDI enzymes in Thermus thermophiles due to their relevance for a wide range of biotechnological applications [41]. Likewise, dosage-dependent effects have been revealed for plant-derived IDI enzymes. For example, the economic potential of in vitro production of caoutchouc led to cloning, heterologous expression and functional characterization (i.e. determination of biochemical function) of IDI loci from the rubber tree Hevea brasiliensis [42].
Fig 1. Overview of all plant specialized terpenoid biosynthetic modules.

Proteins involved in the mevalonic (MVA, shown in black) and methylerythritol phosphate (MEP, shown in purple) pathways synthesize the universal C5-isoprenoid building blocks isopentenyl diphosphate (IPP) and its isomer dimethylallyl diphosphate (DMAPP). Those compounds can be isomerized using enzymes of the IPP isomerase class (IDI, shown in turquois). Subsequently, the C5- blocks are transferred by enzymes of the prenyltransferase (PTF, shown in green) group to the isoprenoid intermediates with variable carbon backbone chain lengths (i.e. C10 for geranyl pyrophosphate; C15 for farnesyl pyrophosphate; C20 for geranylgeranyl phosphate and nerolidol diphosphate). Terpene synthase (core-TPS) gene products (shown in red) further catalyze biosynthesis of C10 (mono-), C15 (sesqui-) or C20 (di-) terpenes (end products, shown in yellow). C30 (shown in blue) refers to enzymes catalyzing biosynthesis of specific triterpenes (end products, shown in yellow). Likewise, prenyltransferases are involved in biosynthesis of longer-branched tetra- and polyterpenes (shown in green).
Enzymes of the prenyltransferase class (PTF, shown in green in Fig 1) subsequently catalyze formation of C10-prenyl diphosphate molecules. Moreover, they can mediate the (optional) elongation of the C10-backbone by the addition of further C5-isopentenyl diphosphate units necessary for formation of di- and sesquiterpenes including longer-chain (C25-C55) tetra- and polyterpenes [43–45]. Terpene synthases (encoded by TPS genes, shown in red in Fig 1) catalyze conversion of specific C10-, C15 or C20 isoprenoid precursors to specialized monoterpenes (C10), sesquiterpenes (C15) and diterpenes (C20), building a module further downstream within terpenoid biosynthesis, respectively [29,46]. Specialized triterpene synthases catalyze formation of pentacyclic triterpenes (such as lupane and squalene) (C30, shown in blue in Fig 1) [47–49]. Note that those compounds can be further modified in distant branches of plant secondary metabolism, for example to triterpene alcohols (such as lanosterol and cycloarthenol) with various bioactivities [50,51]. Entry to the aforementioned MEP pathway was previously proposed to be catalyzed by two divergent 1-deoxy-D-xylulose 5-phosphate synthase isoforms in S. lycopersicum (SlDXS1 / SlDXS2) and A. thaliana (AtDXS1 / AtDXS2) [28,52–54]. In tomato, DXS1 is ubiquitously expressed whereas DXS2 transcripts are abundant in a few tissue types including glandular trichomes. Trichomes are hair-like structures present in the aerial parts of many plant species. They exhibit tremendous diversity but are of general interest to plant breeders since they are often responsible for the production of plant secondary metabolites with various bioactivities, including terpenoids [55–57]. Interestingly, knock-down of DXS2 led to a differential distribution of mono- and sesquiterpenes within tomato glandular trichomes as well as to a significant increase of trichome density, giving rise to economic and ecological potential to this small gene family [52].
The core-TPS gene family has been most intensively studied in the model plant Arabidopsis thaliana. The ecotype Col-0, that contains 32 full-length functional and 8 pseudogenes of the terpene synthase type, of which about a third have been annotated to a designated biochemical function by functional characterization [29,58–64]. Most of the Col-0 core-TPS genes are constitutively expressed in roots, flowers or leaves for production of mono-, di- or sesquiterpenes whereas some are up-regulated under presence of specific stress-related stimuli [58,64,65]. Notably, 27 of the 32 Col-0 core-TPS genes comprise supergene clusters organized in 16 tandem arrays [11], whereas two of them constitute an ohnolog duplicate gene pair due to the most recent At-α ancient whole genome duplication event (see above) [8]. Beyond A. thaliana, efforts to identify core-TPS genes have been published for tomato (S. lycopersicum) [66], orange (C. sinensis) [67], eucalyptus (E. grandis) [68], grape (V. vinifera) [69], millet (S. bicolor), apple (M. domesticus) [70] and the basal Angiosperm Amborella (A. trichopoda) [71]. However, functional characterization (i.e. distinct biochemical function) of all TPS genes present within a species are currently available for tomato and the model plant Arabidopsis only. The complete set of biosynthetic elements involved in both MEP and MVA pathways as well as other terpenoid-associated prenyltransferases including triterpene-specific synthases has to-date only been described in Arabidopsis with a total of 34 genes [35]. Among those, 15 possess prenyltransferase activity, whereas nine and eight belong to the MVA and MEP pathway, respectively. Furthermore, the A. thaliana genome contains two genes encoding proteins of the IPP isomerase (IDI) type with similar bioactivities [35,36]. In total, the gene count of all modules within the complete terpenoid biosynthetic pathway therefore rises to 66 including 32 functional core-TPS genes in Arabidopsis, with a 64% (42 / 66) fraction of tandem duplicate supergenes and 21% (14 / 66) comprising ohnolog duplicate gene pairs dating back to the At-α, At-γ or At-β ancient whole genome multiplication events (see above) [8,11,72].
In this study, we employed a meta-method by combining evidence provided by sequence homology (BLAST), HMM Modelling (interpro scan) and genomic context (SynMap) for robust annotation of genes involved in all modules of terpenoid biosynthesis on a uniquely broad phylogenomics framework. First, we infer novel annotation for loci previously not brought in connection to terpenoid biosynthesis within 17 genome assembly including twelve major crops, thereby providing insights to diversification of plant secondary metabolism during 250 MA of flowering plant evolution. Second, we assessed and compared key factors contributing to copy number variation across all terpenoid biosynthetic modules, thereby providing evidence for the impact of gene- and genome duplication to metabolic diversification in plants. Third, we established a novel clade of duplicate genes with pleiotropic effects in control of trichome density and terpenoid biosynthesis, thereby providing data that support the concept of functional divergence following gene and genome duplication. In summary, our data offer significant insight into evolutionary dynamics underlying diversification of plant secondary metabolism. Furthermore, we provide a blueprint for future efforts to identify and more rapidly modify terpenoid biosynthetic genes across all modules in any flowering plant species.
Materials and Methods
Software prerequisites
All employed Perl and Python scripts required perl (strawberry v5.18) and Python (v2.7) libraries including Bioperl (v1.6.910) and Biopython (v1.63) modules, respectively. The iprscan_urllib.py-script for HMM-based domain annotation (see below) required SOAPy, NumPy and urllib Python modules. For BLAST screens, we employed the stand-alone command line version of NCBI BLAST 2.2.27+ (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/, last accessed on December 13th, 2014) [73]. Fisher’s exact test for count data was performed using the R package for statistical computing (www.r-project.org, last accessed on December 13th, 2014).
Genome annotations
In total, we analyzed 15 draft genomes as well as two gene-space assemblies represented by non-redundant transcriptomes. The complete sets of representative genes and proteins for 12 of these 17 datasets were downloaded using www.phytozome.net, last accessed on December 13th, 2014) [74] and the CoGe package for comparative genomics [4]. We included Amborella trichopoda EVM27 [71], Arabidopsis thaliana TAIR10 [75], Brassica rapa v1.1 [76], Carica papaya v0.5 [18], Citrus sinensis v1 [77], Eucalyptus grandis v1.1 [78], Glycine max Wm82.a2.v1 [79], Sorghum bicolor v1.4 [80], Solanum tuberosum v3.2.10 [81], Solanum lycopersicum v2.40 (Potato Genome Consortium 2012), Vitis vinifera Genoscope.12X [20] and Zea mays 5a.59 [82]. Tarenaya hasslerania v5 [83] (Weber, Schranz et al, unpublished data), Cleome gynandra v2 (Weber, Schranz et al. 2014, unpublished data) and Nicotiana benthamiana v0.42 [84] genome annotations were made available by the authors. Non-redundant transcriptomes of Cannabis sativa ChemDawg (marijuana) [85] and Lactuca sativa (Mitchelmoore et al, unpublished data) early-draft gene-space assembly of were extracted from Genbank [86].
De novo protein annotation of early-draft gene-space assemblies
Non-redundant transcriptome data of Cannabis sativa ChemDawg (marijuana) and Lactuca sativa derive from unpublished early-draft gene-space assembly (see above) and therefore contain significant parts of non-coding sequences as well as putative sequencing errors. We therefore subjected both datasets to the translatedna.py script v1.75 (https://github.com/jenhantao/HiSeq/blob/master/translatedna.py, last accessed on December 13th, 2014). First, single mRNA sequences were translated in all six frames. Second, the peptide fragment encoded by the largest open reading frame was printed. All other parts were discarded. The output comprises an approximation on the non-redundant set of proteins for both species.
Confirmation and expansion of multi-gene families associated with the terpenoid biosynthetic module in Arabidopsis thaliana (“run 1”)
Functional annotation of target genes across all organisms was an interlaced approach consisting of 3 independent BLAST screens (run 1–3). For run 1 in A. thaliana, we obtained 32 core-TPS genes from [29] as well as 34 genes acting in modules further up- and downstream in terpenoid biosynthesis [35]. We queried all 66 sequences against the TAIR10 A. thaliana genome annotation in a BLAST screen without e-value threshold (forward run). We extracted all target sequences and queried them back against the A. thaliana TAIR10 genome annotation with an applied target sequence maximum threshold of 2 (under consideration of self-hits produced by Col-0 genes within this pool) (reverse run). After removal of self-hits, we scored loci as associated with the A. thaliana terpenoid specialized metabolism if they were part of the target sequence pool in the forward run, and aligned to a terpenoid biosynthetic gene as defined by [29,35] in the reverse run. We thereby created an extended set of A. thaliana TP-associated loci (85 genes).
Species-wise determination of putative homologous gene anchors (“run 2”)
In the next step for large-scale specialized terpenoid biosynthetic gene identification, run 2 determined unidirectional best BLAST hits for both (a) protein and (b) coding DNA sequences between A. thaliana Col-0 and all other 14 genome annotations in a screen without e-value thresholds (for early-draft gene-space assemblies, only protein data were used). Since terpenoid biosynthetic loci can comprise multiple domain types connected by partially conserved linkers, the BLAST approach can result in false positives due to short but highly conserved highest-scoring sequence pairs (HSPs) in functionally non-relevant (i.e. structural) parts of the protein. Therefore, we developed a python script to discard target sequences with a query/target sequence length ratio below 0.5 and above 2.0 as previously described to avoid false positive BLAST results due to short but highly conserved highest-scoring sequence pairs (HSPs) in functionally non-relevant (i.e. structural) parts of the protein [26]. We determined (c) additional, length-filtered HSP pairs (based on both CDS and proteins) for these loci within the aforementioned length ratio scope to form a 2nd line of evidence for homolog gene detection as previously described [26].
Syntelog / ohnolog determination
Calculation of pairwise syntenic blocks within and between genomes is based on integer programming [87] but implemented to an easy-to-use web interface termed CoGe package for comparative genomics (www.genomevolution.org, last accessed on December 13th, 2014) [4,88]. Within all genome assemblies, we determined genes sharing the same genomic context to counterparts in the A. thaliana Col-0 genome annotation (defined as syntelogs) using the DAGchainer [89] and Quota-Align [87] algorithms implemented to the “SynMap” function within CoGe. To mask noise generated by successive duplication(s) of ohnolog blocks including segmental duplications, we applied Quota-Align ratios for the “coverage depth”-parameter that are consistent with the syntenic depth (defined as the level of “genome multiplicity” expected from the multiplication of successive WGDs/WGTs) calculated for each genome annotation. For merging of adjacent syntenic blocks, we applied a threshold of n = 350 gene spacers. For within-species ohnolog counterparts of target genes, we applied the “Synfind” function within the CoGe package (https://genomeevolution.org/CoGe/SynFind.pl, last accessed on December 13th, 2014). To decrease false-positive scoring of recent segmental duplications, we set maximum threshold values of 1.5 for the Ks-value averages between duplicate gene copies. This facilitates selective scoring of ohnolog duplicate pairs within genomic blocks that are due to polyploidy as previously described [4]. Please note that we appended URLs to regenerate genome-wide ohnolog identification for 13 out of 17 genomes subjected to this analysis (see Results section).
Determination of tandem duplicate gene copies
Following a widely-used method for tandem duplicate identification, we queried the complete set of proteins encoded in the whole genome assembly against itself in a BLAST screen without any e-value threshold (this ensures the identification of most homologs including highly diverged ones) and filtered our final set of target sequences from above outside a window of n = 10 allowed gene spacers in both directions from the query sequences (this ensures the identification of adjacent duplicates organized in arrays among all homologs scored above) as previously described for the identification of tandem duplicates [11]. We acknowledged that determination of genome-wide tandem duplicate frequencies following this approach decreases in accuracy with increased degrees of assembly fragmentation (i.e. total number of scaffolds/contigs). This means that false-negatives singletons are more likely scored in genomes with many short scaffolds (“gene-space assemblies”) compared to annotations with few scaffolds in the size-range of chromosome pseudo-molecules which is due to the lack of information on the relative order of scaffolds. Similarly, it is not possible to score tandem duplicates based on non-redundant transcriptomes because those represent collections of single transcripts without information of the genomic context. As a result, our analysis of tandem duplicate fractions was restricted to 13 genome assemblies.
Scoring of putative gene transposition duplicate pairs among Arabidopsis DXS-like genes
Scoring of gene transposition duplicate pairs among DXS genes involved three steps. First, we obtained all tandem- and ohnolog duplicates present within the gene family as described above. Second, we queried CDS sequences of non-tandem/non-ohnolog duplicate target genes against the Arabidopsis genome in a BLAST screen without e-value threshold. Third, we generated (B)LastZ two-way alignments of the genomic regions that harbor (a) query as well as (b) highest-scoring non-self target sequence within a 40 kb window (20 kb on each side). This was accomplished using the GEvo function from the CoGe comparative genomics package (http://genomevolution.org/CoGe/GEvo.pl, last accessed on December 13th, 2014) [4]. Graphical highlights of transposon-like sequences have been customized by choosing “show other features” in the “results visualization” tab. We scored DXS-like gene pairs as gene transposition duplicates if they comprise highest-scoring sequence pairs embedded in otherwise non-syntenic regions, while both loci showing evidence for adjacent fragments of transposable elements as previously described [25].
Determination of anchor paralogs and generation of extended multi-gene family pools across all analyzed species (“run 3”)
Since ortholog detection based on unidirectional or reciprocal best BLAST hits can miss many “real” orthologs in duplicate-rich species like animals or plants [90], a separate run was necessary to increase accuracy. For run 3, we defined the initial homologous genes set as the merged set consisting of five HSP partner groups (first group: based on length-filtered protein pairs; second group: based on non-length-filtered protein pairs; third group: based on non-length-filtered CDS pairs; fourth group: based on length-filtered CDS pairs; fifth group: based on syntelogs, see above for length filter criteria). We thereby created a set of putative homologous loci anchoring all A. thaliana gene families in all other analyzed genome annotations (“anchor pool”). In a next step, we performed a BLAST search without e-value thresholds to query all homologous anchor genes against all 17 genomes in a species-wise manner to determine putative paralogs of the anchor gene set (“run 3 forward”). We extracted all target sequences and queried them against the A. thaliana Col-0 TAIR10 genome annotation with a target sequence maximum threshold of 2 (“run 3 reverse”). After removal of self-hits, we scored loci as associated with terpenoid biosynthesis within their species if they align to any member of the extended terpenoid biosynthetic loci in A. thaliana (see above). We defined all members of this pool as homologous to the anchor pool if they were not present within the set of homologous anchor genes (see above).
Hidden Markov Modeling and prediction of protein domains
Since we included highest-scoring sequence partners based on BLAST as well as syntelogs, the above-mentioned extended multi-gene family pool of terpenoid biosynthetic genes is based on both sequence homology and genomic location of its members. However, we observed an erosion of synteny across lineages relative to their phylogenetic distance. Furthermore, DNA sequence homology decreases with phylogenetic distance due to wobble rules for the 3rd codon position. Likewise, the protein sequence homology between distant multi-gene family members can decrease due to synonymous substitutions of amino acids belonging to the same chemical class (i.e. aliphatic, aromatic, basic, cyclic). Therefore, we applied a final filtering step to remove false-positive loci from the extended terpenoid biosynthetic genes pool across all genomes (including the extended terpenoid biosynthetic genes pool in Arabidopsis, see above). Using the iprscan_urllib.py script provided by the European Molecular Biology Laboratory (EMBL, Heidelberg, Germany) (https://www.ebi.ac.uk/Tools/webservices/download_clients/python/urllib/iprscan_urllib2.py, last accessed on December 13th, 2014), we queried every member of the terpenoid biosynthetic genes pool (including the extended set determined for A. thaliana, see above) to 14 algorithms that apply Hidden Markov Models for (protein domain) signature recognition (BlastProDom, FPrintScan, HMMPIR, HMMPfam, HMMSmart, HMMTigr, ProfileScan, HAMAP, PatternScan, SuperFamily, SignalPHMM, TMHMM, HMMPanther and Gene3D) [91]. We overcame the one-sequence-at-a-time limitation of the EMBL server by writing batch wrappers for 25x-fold parallelization. As a result, we mapped all protein domains present in the putative multi-gene family pool onto their genes in less than a day, and discarded all false positive genes from the whole set (i.e. genes not encoding at least one domain common to at least one reaction module). Referencing of all identified genes to distinct terpenoid biosynthetic modules was based on presence of module-specific protein domains.
Multiple protein alignments
To generate multiple alignments of protein sequences, the stand-alone 64-bit version of MAFFT v7 was employed (http://mafft.cbrc.jp/alignment/software/, last accessed on December 13th, 2014) [92]. First, all terpenoid biosynthetic proteins were aligned species-wise using the command line {mafft.bat—anysymbol—thread 4—threadit 0—reorder—auto input > output} Mesquite v2.75 (http://mesquiteproject.org, last accessed on December 13th, 2014) was used with multi-core preferences to trim MAFFT multiple alignments down to gap-free sites. Trimmed blocks were re-aligned using MAFFT with the command line {mafft.bat—anysymbol—thread 4—threadit 0—reorder—maxiterate 1000—retree 1 –localpair input > output}.
Microarray-based gene expression analysis in Arabidopsis
To test differential and trichome-specific expression of DXS-like genes in Arabidopsis, we have used a Col-0 wild type trichome-specific transcriptome dataset (available at the TrichOME database, http://www.planttrichome.org/, last accessed on December 13th, 2014) [93]. Normalized values of three independent experiments performed with the ATH1 microarray were generated and averaged as described [94]. For calculation of relative gene expression, we referenced the bHLH-motif containing house-keeping gene AT4G34720 [95].
Quantitative PCR-based gene expression analysis in tomato (S. lycopersicum)
Leaves, stems and roots were collected in triplicate from 4-week-old Solanum lycopersicum cultivar Moneymaker plants. Part of the stems were left intact and part were used for trichome isolation by shaking the stems in liquid nitrogen. Frozen isolated trichomes, stems that remained after trichome removal, intact stems, leaves and roots were ground to a fine powder and subjected to RNA isolation with Tri Reagent (Sigma) and DNase treatment (TURBO DNase, Ambion) according to the manufacturer’s instructions. cDNA was synthesized from 1 μg of total RNA using the RevertAid kit (Fermentas). RT-qPCR was used to study the expression of 1-deoxy-d-xylulose 5-phosphate synthase isoforms 1, 2 and 3 (DXS1, DXS2, and DXS3) in cDNA derived from different tissues. Gene specific primers were designed using Primer3Plus (DXS1-F: 5’-ATTGGGATATGGCTCAGCAG-3’; DXS1-R: 5’-CAGTGGTTTGCAGAAACGTG-3’; DXS2-F: 5’-TTTACCGACCGCAACCTTAG-3’; DXS2-R: 5’-GTGCTTGAGGTCCAATTTGC-3’; DXS3-F: 5’- AATGGAGCCTTCACTTCACC-3’; DXS3-R: 5’-ACCCAGCTGCAAATGTTACC-3’). Tomato RUB1 conjugating enzyme-encoding (RCE1) gene (Gen-Bank accession no. AY004247) (RCE-F: 5’- GATTCTCTCTCATCAATCAATTCG-3’ and RCE-R ‘5-GAACGTAAATGTGCCACCCATA-3’) was used for normalization. PCR reactions were prepared in duplicate by mixing cDNA equivalents of 10 ng RNA with the SYBR Green Real-Time PCR master mix (Invitrogen) and 300 nM of each primer. Quantification of the transcript level was performed in an ABI 7500 Real-Time PCR System (Applied Biosystems) with the following cycling program: 2 min, 50°C, 15 min 95°C, 45 cycles of 15 sec at 95°C and 1 min at 60°C followed by a melting curve analysis. At the end of each run, amplified products were sequenced to verify their identity. Relative expression values were calculated using the efficiency δCt method as previously described [96]. All wet-lab expression analysis were performed in four independent biological and three technical replicates.
Phylogenetic and similarity/identity analysis
We performed Bayesian Markov chain Monte Carlo (MCMC) analysis using MrBayes version 3.2.2 (http://mrbayes.sourceforge.net/, last accessed on December 13th, 2014) [97] with the following parameters: Dirichlet model; uniform gamma shape parameter variation 0.00–200.00; 50 million generations; 2 independent runs, 4 chains each; temperature heating 0.2; sample taking every 5000 generations; burn-in time at 12500000 samples. Bayesian inference trees were constructed using the CIPRES package (http://www.phylo.org/sub_sections/portal/, last accessed on December 13th, 2014) [98]. Model convergence was checked in Tracer version 1.5 (http://tree.bio.ed.ac.uk/software/tracer/, last accessed on December 13th, 2014) [99]. FigTree v1.3.1 was used to generate and edit phylogenetic trees (http://tree.bio.ed.ac.uk/software/figtree/, last accessed on December 13th, 2014) [100]. Results were scored reliable once the effective sampling size of all parameters was above 100. Tree branches supported with posterior probabilities (PP) below 0.7 were considered weak and above 0.9 as strong. Protein sequence similarity analysis were performed using the Needle program from the EMBOSS software package (http://emboss.sourceforge.net/, last accessed on December 13th, 2014) [101].
Ethics Statement
The authors hereby state that no specific permissions were required for any activities and/or locations that are connected to this research. Likewise, the authors hereby confirm that the research summarized in this article did not involve endangered or protected species. In addition, the authors hereby state clearly that all sampling procedures and/or experimental manipulations were reviewed/specifically approved and no field permit was required. Wageningen University & Research Center and all other institutes affiliated with this work comprise legal entities that do not act on any basis that is prohibited by local, state or federal law.
Results
(Re)annotation of the Arabidopsis terpenoid biosynthetic inventory and expansion of genes associated with all reaction modules
As an initial step for identification of genes involved in all terpenoid biosynthetic modules, we reviewed current literature to pool all published Arabidopsis core-TPS genes with biosynthetic elements acting further up- and downstream in the pathway (Fig 1) [29,35,61]. As a result, we generated a list of 66 biosynthetic elements previously identified in the model plant (Table 1). This compilation represents a patchwork of information containing both genes found by functional studies as well as genes with computationally inferred association to terpenoid metabolism. Hence, uniform standards of gene identification have not been applied for curation of this initial list.
Table 1. The published terpenoid biosynthetic module in Arabidopsis.
| Gene ID | Annotation | Tandem duplicate | Bowers pair B | Reference C |
|---|---|---|---|---|
| Isopentenyl diphosphate (IPP) isomerases | ||||
| AT3G02780 | IDI1 | - | A12N076 | Campbell et al., 1998 |
| AT5G16440 | IDI2 | - | A12N076 | Campbell et al., 1998 |
| Mevalonic acid (MVA) pathway | ||||
| AT1G31910 | HMG1 | Yes | - | Benveniste et al., 2002 |
| AT1G76490 | PMK | Yes | C2N120 | Caelles et al., 1989 |
| AT2G17370 | HMG2 | - | C2N120 | Caelles et al., 1989 |
| AT2G38700 | MVD1 | - | A11N067 | Cordier et al., 1999 |
| AT3G54250 | MVD2 | - | A11N067 | Benveniste et al., 2002 |
| AT4G11820 | HMGS | - | - | Montamat et al., 1995 |
| AT5G27450 | MK | - | - | Riou et al., 1994 |
| AT5G47720 | ACT1 | Yes | - | Ahumada et al., 2008 |
| AT5G48230 | ACT2 | Yes | - | Ahumada et al., 2008 |
| Methylerythritol phosphate (MEP) pathway | ||||
| AT1G63970 | MDS | - | - | Hsieh and Goodman, 2006 |
| AT2G02500 | MCT | - | - | Rohdich et al., 2000 |
| AT2G26930 | CMK | - | - | Hsieh et al., 2008 |
| AT4G15560 | DXS1 | - | A15N013 | Lange et al., 2003 |
| AT4G34350 | HDR | - | - | Hsieh and Goodman, 2005 |
| AT5G11380 D | DXS3 | - | - | Lange et al., 2003 |
| AT5G60600 | HDS | - | - | Rodríguez-Concepción et al., 2002 |
| AT5G62790 | DXR | - | - | Schwender et al., 1999 |
| Prenyltransferases (PTF) | ||||
| AT1G49530 | GGPS1 | Yes | - | Zhu et al., 1997a |
| AT2G18620 | GGPS2 | Yes | A10N118 | Wang and Dixon, 2009 |
| AT2G18640 | GGPS3 | Yes | - | Okada et al., 2000 |
| AT2G23800 | GGPS4 | Yes | A10N309 | Zhu et al., 1997b |
| AT2G34630 | GPS1 | - | - | Bouvier et al., 2000 |
| AT3G14510 | GGPS5 | Yes | - | Finkelstein et al., 2002 |
| AT3G14530 | GGPS6 | Yes | - | Wang and Dixon, 2009 |
| AT3G14550 | GGPS7 | Yes | - | Okada et al., 2000 |
| AT3G20160 | GGPS8 | - | - | Zhu et al., 1997a |
| AT3G29430 | GGPS9 | Yes | - | Finkelstein et al., 2002 |
| AT3G32040 | GGPS10 | Yes | - | Finkelstein et al., 2002 |
| AT4G17190 | FPS1 | Yes | A21N001 | Cunillera et al., 2000 |
| AT4G36810 | GGPS11 | - | A10N118 | Okada et al., 2000 |
| AT4G38460 | GGR | - | - | Oh et al., 2002 |
| AT5G47770 | FPS2 | Yes | A21N001 | Delourme et al., 1994 |
| Core terpene synthases | ||||
| AT1G31950 | TPS29 | Yes | - | Lange et al., 2003 |
| AT1G33750 | TPS22 | - | - | Lange et al., 2003 |
| AT1G48800 | TPS28 | Yes | - | Lange et al., 2003 |
| AT1G61120 | TPS4 | Yes | - | Herde et al., 2008 |
| AT1G61680 | TPS14 | Yes | - | Chen et al., 2003 |
| AT1G66020 | TPS26 | Yes | - | Lange et al., 2003 |
| AT1G70080 | TPS6 | Yes | - | Lange et al., 2003 |
| AT1G79460 | TPS32 | Yes | - | Yamaguchi et al., 1998 |
| AT2G23230 | TPS05 | Yes | - | Dal Bosco et al., 2003 |
| AT2G24210 | TPS10 | - | - | Bohlmann et al., 2000 |
| AT3G14490 | TPS17 | Yes | - | Dal Bosco et al., 2003 |
| AT3G14520 | TPS18 | Yes | - | Lange et al., 2003 |
| AT3G14540 | TPS19 | Yes | - | Lange et al., 2003 |
| AT3G25810 | TPS24 | Yes | - | Chen et al., 2003 |
| AT3G25820 | TPS27 | Yes | - | Chen et al., 2004 |
| AT3G25830 | TPS23 | Yes | - | Chen et al., 2004 |
| AT3G29110 | TPS16 | Yes | - | Lange et al., 2003 |
| AT3G29190 | TPS15 | Yes | - | Lange et al., 2003 |
| AT3G29410 | TPS25 | Yes | - | Dal Bosco et al., 2003 |
| AT3G32030 | TPS30 | Yes | - | Lange et al., 2003 |
| AT4G02780 | TPS31 | - | - | Mann et al., 2010 |
| AT4G13280 | TPS12 | Yes | - | Ro et al., 2006 |
| AT4G13300 | TPS13 | Yes | - | Ro et al., 2006 |
| AT4G15870 | TPS1 | Yes | - | Aubourg et al., 1997 |
| AT4G16730 | TPS2 | Yes | - | Huang et al., 2010 |
| AT4G16740 | TPS3 | Yes | - | Fäldt et al., 2003 |
| AT4G20200 | TPS7 | Yes | - | Lange et al., 2003 |
| AT4G20210 | TPS8 | Yes | A21N124 | Tholl and Lee, 2011 |
| AT4G20230 | TPS9 | Yes | - | Dal Bosco et al., 2003 |
| AT5G23960 | TPS21 | - | - | Chen et al., 2003 |
| AT5G44630 | TPS11 | - | A21N124 | Tholl et al., 2005 |
| AT5G48110 | TPS20 | Yes | - | Dal Bosco et al., 2003 |
Gene abbreviations are adapted from the Arabidopsis Information Resource A .
A TAIR10, www.arabidopsis.org, last accessed on December 13th, 2014.
B Ohnolog pair according to Bowers et al., 2003 [8].
C for a comprehensive review, see Tholl and Lee, 2011 and Phillips et al., 2008.
D assosciation of AtDXS3 to MEP pathway is subject of scientific debate (see Phillips et al., 2008).
In a next step, we therefore screened for additional members of all involved gene families within the Col-0 genome that may have been missed in previous studies in an interlaced bioinformatics approach (see Materials & Methods section). Briefly, we combined layers of information based on sequence similarity, gene synteny and Hidden Markov Modelling. First, we queried all 66 genes against the Col-0 genome in a sensitive BLAST screen (no e-value cutoff). After removing self hits, we included every target sequence for further analysis if it formed a highest-scoring sequence pair with a distant member of the initial list. This included identification of ohnologs as well as annotation and mapping of protein domains. Interestingly, this led to evidence for 19 additional genes that produce highest-scoring sequence pairs (HSPs) with genes mentioned in Table 1. In essence, these genes comprise tandem-, transposition- as well as ohnolog duplicate copies of genes previously known to be associated with terpenoid metabolism (Table 2). Note that segmental duplications are excluded from this analysis due to technical reasons (see below). Interestingly, 18 of those 19 genes are annotated as triterpene-specific synthases. Thirteen of those have been functionally characterized in previous efforts [47–49,102]. Five further ones lack functional data but have been assigned to three-letter code nomenclature in the Arabidopsis information resource based on computational inferences (i.e. sequence homology). Interestingly, all those loci include an encoded oxysqualene synthase domain (S1 Table). In contrast, one is a DXS-like gene with its closest homolog involved in the MEP pathway (Table 2). Note that the initial set of query genes applied to our first, above-mentioned BLAST analysis did not contain triterpene-specific synthases (Table 1). Therefore, our results indicate sequence homology of triterpene-specific synthases to genes of the core-TPS as well as the prenyltransferase class. Based on these findings, we hypothesize that all three groups go back to one common ancestral gene family with subsequent rounds of duplication going hand in hand with functional diversification as previously described in many cases including proline-rich proteins [103].
Table 2. The extended terpenoid phenotypic module in Arabidopsis, including triterpene- specific (C30) synthases.
| Gene ID | Annotation | Description A | Tandem duplicate | Bowers pair B | Reference C |
|---|---|---|---|---|---|
| Methylerythritol phosphate (MEP) pathway | |||||
| AT4G15560 | DXPS2 | Desoxy-xylulosephosphatesynthase 2 | Yes | A15N013 | Lange et al., 2003 |
| Triterpene-specific synthases | |||||
| AT1G62730 | - | N/A; Squalene/phytoene synthase | No | - | Wang et al., 2008 |
| AT1G66960 | LUP5 | Lupeol synthase 5 | Yes | - | Herrera et al., 1998 |
| AT1G78480 | - | N/A; Prenyltransferase/squalene oxidase | Yes | - | Hanada et al., 2010 |
| AT1G78500 | PEN6 | Pentacyclic triterpene synthase 6 | Yes | - | Husselstein-Muller et al., 2001 |
| AT1G78950 | LUP4 | Lupeol synthase 4 | Yes | - | Benveniste et al., 2002 |
| AT1G78955 | CAMS1 | Camelliol synthase 1 | Yes | - | Kushiro et al., 1998 |
| AT1G78960 | LUP2 | Lupeol synthase 2 | Yes | - | Herrera et al., 1998 |
| AT1G78970 | LUP1 | Lupeol synthase 1 | Yes | - | Herrera et al., 1998 |
| AT3G29255 | - | N/A; Squalene cyclase (InterPro:IPR018333) | Yes | - | this manuscript |
| AT2G07050 | CAS1 | Cycloartenol synthase 1 | - | - | Lange et al., 2003 |
| AT3G45130 | LAS1 | Lanosterol synthase 1 | - | - | Benveniste et al., 2002 |
| AT4G15340 | PEN1 | Pentacyclic triterpene synthase 1 | Yes | - | Husselstein-Muller et al., 2001 |
| AT4G15370 | PEN2 | Pentacyclic triterpene synthase 2 | Yes | - | Husselstein-Muller et al., 2001 |
| AT5G36150 | PEN3 | Pentacyclic triterpene synthase 3 | - | - | Husselstein-Muller et al., 2001 |
| AT5G42600 | MRN1 | Marernal Synthase 1 | - | - | Benveniste et al., 2002 |
| AT5G48010 | THAS1 | Thalianol Synthase 1 | Yes | - | Benveniste et al., 2002 |
| Function not clear | |||||
| AT1G48820 | - | N/A; tandem duplicate of TPS28 | Yes | - | Lange et al., 2003 |
| AT2G37140 | - | N/A; best BLAST hit is TPS1 | - | - | Lange et al., 2003 |
Three letter gene abbreviations are adapted from the Arabidopsis Information Resource A .
A TAIR10, www.arabidopsis.org, last accessed on December 13th, 2014.
B Ohnolog pair according to Bowers et al., 2003.
C For a comprehensive review, see Tholl and Lee, 2011.
To account for the whole range of sequence diversity found among Arabidopsis terpenoid biosynthetic genes, we merged Table 1A and 1B and obtained pool of 85 genes putatively involved in all Col-0 terpenoid biosynthetic modules (“extended set”) (sum of all Col-0 gene entries in S2 Table). Initially, we dissected all members into three groups based on putative affiliation with a certain module (Fig 1): (a) prenyltransferases and triterpene-specific synthases, (b) core terpene synthases and (c) genes involved in MEP and MVA pathways including IPP isomerases, respectively. Visual comparison of duplicate fractions revealed striking differences between the subsets but also when comparing to the fraction of duplicates among all protein-coding genes (Fig 2). For the whole set of 85 genes, we found a 68%-fraction of tandem duplicate supergene clusters and a 15%-fraction of ohnolog duplicate pairs (Fig 2A, Table 3 and Table 4). For subgroup (a), we report a 70%-fraction of tandem duplicate supergenes and a 13%-fraction of ohnolog duplicate copies (Fig 2B). In contrast, 94% of subgroup (b) comprise members of tandem arrays, while the ohnologs fraction drops to 6% (Fig 2C, Table 3 and Table 4). Interestingly, subgroup (c) contains only 16% of tandem duplicate genes but a 27% fraction of genes retained after ancient polyploidy events (Fig 2D).
Fig 2. Circos ideogram showing 5 Arabidopsis chromosomes with the extended set of genes associated with major terpenoid biosynthetic modules.

A. Gene inventory of the complete terpenoid biosynthetic pathway after initial expansion of published modules. Tandem duplicate supergenes are marked in red. Singletons are marked in green. Ohnolog duplicate gene pairs are marked in blue. Central pie chart shows a 68% tandem duplicate supergenes fraction. B. Subset of prenyltransferases and specific triterpene synthases marked in yellow. Central pie chart shows a 70% tandem duplicate supergenes fraction. C. Subset of core terpene synthase (TPS) genes marked in bright green. Central pie chart shows an 84% tandem duplicate supergenes fraction. D. Subset of genes associated with MEP and MVA pathways, including IPP isomerases, marked in blue. Central pie chart shows a 16% tandem duplicate supergenes fraction.
Table 3. Tandem Duplicates fractions among terpenoid specialized biosynthetic module in 13 A genomes.
| Species | Genome-wide | core-TPS genes | MEP-pathway | MVA-pathway | IPP-isomerases | Prenyl-transferases | Triterpene synthases | Average B |
|---|---|---|---|---|---|---|---|---|
| A. thaliana | 15% | 94% * | - | 33% | - | 73% * | 68% * | 68% * |
| B. rapa | 20% | 51% * | - | 7% | - | 42% | 40% | 33% * |
| T. hasslerania | 17% | 62% * | 18% | 7% | - | 53% * | 20% | 37% * |
| C. papaya | 17% | 52% * | - | - | - | - | 54% * | 32% * |
| C. sinensis | 44% | 37% | 50% | 56% | 100% | 25% | 37% | 39% |
| E. grandis | 33% | 73% * | 18% | 19% | - | 60% | 75% * | 63% * |
| G. max | 73% | 42% * | 12% * | - | 50% | 7% * | 33% * | 20% |
| V. vinifera | 32% | 91% * | 21% | 35% | - | - | 96% * | 78% * |
| S. lycopersicum | 28% | 80% * | - | 13% | 50% | 55% | 55% | 56% * |
| S. tuberosum | 51% | 51% | - | 21% * | - | - | 25% | 36% * |
| S. bicolor | 35% | 68% * | 20% | - | - | 14% | 76% * | 52% * |
| Z. mays | 44% | 42% | 31% | 40% | 33% | 10% | 58% | 40% |
| A. trichopoda | 24% | 57% * | 18% | - | - | - | 67% | 34% |
| Average B | 32% | 59% | 13% | 17% | 17% | 25% | 53% | 46% * |
Minus indicates absence of tandem duplicates. Asterisks indicate significant enrichment compared to genome-wide tandem duplicate fraction based on fisher's exact test on count data (p-value threshold: 0.01). For absolute gene numbers and p-values, see S5 Table.
A C. Sativa, L. sativa and N. benthamiana and C. gynandraare excluded from this analysis due to technical reasons (see Materials & Methods section).
B Averages based on numbers of tandem and singleton genes, not on percentage values since gene counts in subsets are not equal.
Table 4. Ohnolog duplicates fractions among the terpenoid specialized biosynthetic module in 13 A genomes.
| Species | Genome-wide | CoGe-link C | core-TPS genes | MEP-pathway | MVA-pathway | IPP-isomerases | Prenyl-transferases | Triterpene synthases | Average B |
|---|---|---|---|---|---|---|---|---|---|
| A. thaliana | 22% | bit.ly/1t7DH7A | 6% | 13% | 22% | 100% * | 33% * | 5% | 15% |
| B. rapa | 53% | bit.ly/1uVTlHt | 26% | 54% * | 73% * | 100% * | 74% * | 33% | 49% |
| T. hasslerania | 48% | bit.ly/1r0khkj | 26% | 27% | 60% * | 100% * | 47% | 45% | 40% |
| C. papaya | 7% | bit.ly/1yt11Ap | - | - | - | - | 17% * | - | 2% |
| C. sinensis | 6% | bit.ly/1xRKTJh | 3% | - | 22% * | - | - | - | 3% |
| E. grandis | 18% | bit.ly/1p2oGrm | 10% | - | 50% * | - | - | - | 11% |
| G. max | 62% | bit.ly/1yt2QNw | 48% | 56% | 85% * | 100% * | 53% | 27% | 57% |
| V. vinifera | 22% | bit.ly/1uefADr | 2% | - | 15% | - | 40% * | - | 4% |
| S. lycopersicum | 19% | bit.ly/1xsTXoT | 14% | - | 33% * | - | - | - | 12% |
| S. tuberosum | 11% | bit.ly/1yWDKGZ | 4% | - | 17% * | - | - | - | 6% |
| S. bicolor | 23% | bit.ly/1xRLLxE | - | - | 27% * | - | 29% * | 20% | 11% |
| Z. mays | 27% | bit.ly/11xv3rs | 8% | 23% | 55% * | 66% * | 60% * | 16% | 28%* |
| A. trichopoda | 7% | bit.ly/1x3SpxM | - | 1% | 17% * | - | 33% * | - | 8% |
| Average B | 28% | 10% | 19% | 42% | 48% | 33% | 10% | 18% |
Minus indicates absence of ohnolog duplicates. Asterisks indicate above-average fraction of ohnolog duplicates compared to the genome-wide background. For absolute values, see S5 Table.
A C. sativa and L. sativa, N. benthamiana and C. gynandra are excluded from this analysis due to technical restrictions.
B Averages based on numbers of tandem and singleton genes, not on percentage values since gene count in subsets is not equal.
C Link to the CoGe platform for comparative genomics for online-regeneration of the analysis for ohnolog identification.
In summary, we found a connection to biosynthesis of mono-, di- and sesquiterpenes for 19 additional genes in Arabidopsis that are homologous to but absent from the published set of terpenoid biosynthetic genes. Likewise, we showed an asymmetric distribution of duplicates among genes involved in different modules of terpenoid biosynthesis in Arabidopsis.
Protein domain annotation of extended genes set associated with all terpenoid biosynthetic modules in Arabidopsis
Increasing phylogenetic distance of plant species can lead to increased sequence diversity in homologs while the broad class of biological function remains unchanged [1,104]. For example, amino acid substitutions within the same chemical group (i.e. aliphatic, aromatic) may have little or no effects on protein function, but may result in decreased accuracy in orthologous and paralogous gene detection by sequence homology (such as BLAST) [90,105,106]. We therefore performed Hidden Markov Modelling (HMM)-driven protein motif searches and annotation among all subsets of genes involved in the extended set of Col-0 terpenoid biosynthetic genes in order to screen for additional homologs (see Materials & Methods section). Briefly, we submitted all 85 target sequences to the “Interpro5” algorithm that performs parallelized prediction of protein domains (see Materials & Methods section) [91,107–109]. This is based on machine learning for pattern recognition rather than direct sequence comparisons. As the “training” dataset for domain modelling for the submitted protein sequences, Interpro5 uses the HMM-generated profiles of all protein motif entries and associated sequences present within the pfam and various other databases [110]. Notably, benchmarking of profile HMMs and the BLAST algorithm previously revealed a higher sensitivity of HMM-based methods that is mirrored by an increased alignment quality [111].
As a result of HMM-driven protein domain annotation, we obtained a collection of all motifs encoded by all genes present in the initial set (Table 5). First, we pooled all terpenoid pathway-associated genes from the extended set into (a) core-TPS proteins, (b) IPP isomerases, (c) genes involved in the MEP pathway, (d) MVA pathway-associated proteins, (e) prenyltransferases as well as (f) triterpene-specific synthases and subjected all six sets to the Interpro5 algorithm [91], thereby querying a total of 14 protein motif databases. Five among those recognized motifs shared by every single member of at least 2 pools and were selected for further analysis: Interpro, Pfam, Panther, Gene3D as well as Superfamily [107,110,112–114] (Table 5). In a next step, we screened for protein motif entries within these 5 databases that are specific for any of the 6 aforementioned subsets of genes associated with all Col-0 terpenoid biosynthetic modules (S1 Table). Interestingly, our approach identified 38 domains associated with more than one subgroup due to accurate modelling of protein domain signatures (Table 5). Those were found either for both core-TPS proteins and prenyltransferases, or for both core-TPS genes and triterpene-specific synthases. Together with the sequence homology determined in the initial BLAST screen that formed the extended set of Col-0 target genes, this illustrates that those gene families are similar in terms of both sequence and domain structure as described above and hence might share a common evolutionary origin and function (S1 Table). As a result, it is not possible to affiliate one distinct homologous genes to one of these three subsets based on domain composition in every case without functional data at hand, also when utilizing specific combinations of two or more domains. In summary, we performed in-depth investigation of protein domains among all enzymes involved in every Arabidopsis terpenoid biosynthetic module, thereby curating a set of all detectable domains involved in the terpenoid biosynthetic pathway within the Arabidopsis model plant.
Table 5. Overview of protein domain annotation for the extended set of Arabidopsis terpenoid biosynthetic genes A .
| Database | Predicted domains | Predicted domains specific for functional module | Genes with predicted domains | Genes with module-specific domains |
|---|---|---|---|---|
| Interpro | 64 | 49 | 85 | 48 |
| Panther | 20 | 18 | 85 | 59 |
| Pfam | 25 | 17 | 85 | 43 |
| Superfamily | 16 | 10 | 84 | 11 |
| Gene3D | 16 | 9 | 83 | 10 |
| Total | 141 (100%) | 103 of 141 (73%) | 85 of 85 (100%) | 59 of 85 (69%) |
A 85 target genes in the extended set of Arabidopsis terpenoid biosynthetic genes.
Annotation of genes in all terpenoid biosynthetic modules across 17 target species based on both sequence homology and protein domain composition
We obtained a list of 1,904 protein-coding genes with putative annotation to a terpenoid biosynthetic module. To cross-reference every member to one of the six designated functional modules (Fig 1), we mapped all aforementioned protein motifs onto all target genes. For genes with ambiguous domain composition (i.e. presence of 38 domains without clear referencing to one functional module within the terpenoid biosynthesis, see above), we used the annotation of its highest-scoring target sequence alignment in Arabidopsis. Depending on sequence homology as well as on presence/absence of the aforementioned module-specific protein domains (S1 Table), we describe a total of 840 core terpene synthase genes (shown red in Fig 1), 190 prenyltransferases (shown in green in Fig 1), 338 triterpene-specific synthases (shown in blue in Fig 1) as well as 219 and 278 genes associated with the MEP (shown in purple in Fig 1) and MVA pathways (shown in black in Fig 1), respectively. Likewise, we found a total of 39 IPP isomerases (shown in turquois in Fig 1), summing up to 1,904 target genes in total (Fig 3). Please note that all sequence identifiers are appended in S2 Table.
Fig 3. Illustration showing the complete set of genes associated with all terpenoid biosynthetic modules identified in this study across 17 genome assemblies, based on the HMM-generated profiles of Table 5.

For core-TPS genes, numbers of previously published full-length target genes is included if available. Asterisks indicate number of previously identified full-length TPS open reading frames and hence putative number of functional terpene synthase enzymes. Incomplete protein fragments are not included.
Compared to the total number of protein-coding genes present in the genome, V. vinifera (grapevine) possesses the most expanded inventory of terpenoid biosynthetic genes including all modules, but also for individual modules like core terpene synthases, triterpene-specific synthases and both MEP and MVA pathways. In contrast, the highest number of prenyltransferases relative to the total number of protein-coding genes is encoded by the C4-species C. gynandra. The small gene family of IPP isomerases is most abundant (i.e. target gene count compared to number of all genes per genome) in B. rapa. Note that the B. rapa genome possesses the highest syntenic depth level among all species analyzed in this study (S3 Table).
In contrast, the basal Angiosperm A. trichopoda possesses the leanest inventory relative to the number of all protein-coding genes when looking at all terpenoid biosynthetic modules. Same counts for triterpene-specific synthases and for genes associated with the MVA pathway. For core-TPS genes, the G. max (soybean) genome encodes the smaller relative number of proteins. For prenyltransferases, we found that the L. sativa genome encodes the smallest relative number. In contrast, the MEP pathway in S. tuberosum (potato) recruits the lowest number of genes compared to all of its protein-coding genes. Finally, we found the lowest relative number of IPP isomerases within the C. papaya genome (S3 Table). In summary, we provide evidence for annotation of 1,904 genes to every major module of terpenoid biosynthesis within 17 target genomes, many of which have not been connected to this trait so far. Similar to functional annotation of the Arabidopsis genome, computational inferences of gene function comprise an important step for the future collection of functional data in wet-lab experiments [75].
General and subset-specific cross-referencing of supergene clusters and ohnolog duplicates to terpenoid biosynthetic elements among all species
After curating a set of 1,904 target genes across 17 species, we first scored supergenes organized in tandem arrays as well as ohnolog duplicates due to polyploidy events. Second, we compared the obtained duplicate fractions between all six modules of terpenoid biosynthesis. For detection of potential enrichment or depletion of duplicate frequencies within these subsets, a species-wise comparison to the genome-wide average of tandem/ohnolog duplicates fraction was necessary. Due to technical reasons, these genome-wide fractions can’t be accurately determined for C. gynandra, N. benthamiana, C. sativa and L. sativa (for Cannabis and Lactuca, non-redundant RNAseq data are available only whereas the C. gynandra and N. benthamiana assemblies are highly fragmented, leading to a highly error-prone determination of genome-wide duplicates fractions, see Materials & Methods section). Therefore, our genome-wide analysis of duplicates fractions was restricted to 13 genome assemblies.
On average, 46% of all curated genes associated with terpenoid biosynthesis comprise supergenes with duplicates organized in tandem arrays. Compared to the 32% average observed for the genome-wide tandem duplicate fraction determined across the 13 genome assemblies subjected to this part of our analysis, our results highlight a significant enrichment of supergene clusters for terpenoid biosynthetic genes according to statistical analysis based on Fisher’s exact test on count data (Table 3, S4 Table). Next, we investigated the species-wise fractions of tandem duplicates among all identified terpenoid biosynthetic genes for comparison to the respective genome-wide background. Similar to our findings for genome-wide tandem duplicate fractions across all analyzed genomes, the significant enrichment for supergene clusters holds up for all organisms except C. sinensis, G. max, Z. mays and A. trichopoda (Table 3). However, comparison of duplicate frequencies within different functional modules of terpenoid biosynthesis across the 13 genomes subjected to tandem duplicate analysis did reveal certain subsets that are enriched for duplicates (five genomes were not applicable to this analysis due to technical reasons, see above). For example, triterpene-specific synthases are significantly enriched for tandem arrayed supergenes compared to the genome-wide background in G. max. Similarly, core-TPS genes are enriched for tandem duplicates compared to genome-wide average in the basal angiosperm A. trichopoda (Table 3). We have found that only Citrus and maize lack significant enrichment for tandem duplicates among all subsets of genes involved in terpenoid biosynthesis. Based on the enriched fraction of tandem duplicates specific for certain terpenoid biosynthetic modules, we deducted a general pattern. Both core-TPS genes and triterpene-specific synthases were found to be significantly more enriched for tandem duplicates across most of the analyzed species, whereas MEP and MVA pathways as well as IPP isomerase functions retained few or no supergene clusters within most analyzed species (Table 3, S4 Table).
In a next step, we determined the cumulative fraction of duplicate genes retained after ancient polyploidy events (ohnologs). Similar to the analysis of tandem duplicates, ohnolog identification relies on gene contextual information and is hence not applicable to highly fragmented gene-space assemblies or translated transcriptome datasets (see above) [4,89]. Please note that we appended URLs for online-regeneration of ohnolog identification in 13 genomes out of 17 genomes (Table 4). We again measured genome-wide averages wherever possible and compared them to the fractions among all subsets as described above for tandem duplicate supergenes (Table 4). On average, 18% of all genes associated with all modules of terpenoid biosynthesis comprise ohnolog duplicate gene copies. Compared to the 28% fraction of genome-wide ohnolog merged across all analyzed species, Fisher’s exact test on count data indicates absence of significant ohnolog enrichment for this set (S5 Table). In contrast, species-wise analysis revealed a significant enrichment of ohnologs among all terpenoid biosynthetic genes identified in Z. mays (Table 4). Moreover, analysis of species-specific ohnolog distributions among different terpenoid biosynthetic modules highlighted differential trends. In essence, we revealed patterns of above-average ohnolog retention opposite to those described for tandem duplicates. For example, dosage-independent modules like core-TPS synthases and triterpene-specific synthases contain below-average ohnolog fractions in all analyzed species (Table 4), while recruiting highest fractions of supergene clusters as shown above (Table 3).Strikingly, genes associated with dosage-dependent modules like the MVA pathway and IPP isomerases show the highest fractions of ohnolog duplicates merged across all genomes (Table 4). In contrast, both subsets include low fractions of tandem duplicates compared to other subsets (Table 3).
However, ohnolog fractions of dosage-dependent modules vary greatly between different species in many cases. The small gene family of IPP isomerases, for example, consists of 100% ohnolog duplicates within Arabidopsis, Brassica, Tarenaya as well as Glycine. In contrast, we did not detect retained ohnologs within this gene families within Carica, Citrus, Eucalyptus, Vitis, all analyzed Solanaceae as well as Sorghum based on the applied preferences. This is likely due to technical reasons (see Materials & Methods section). Briefly, the scoring method of SynMap depends on presence of long colinear regions and hence the N50 value indicating the “fragmentation” of the assembly. This means that false-negatives are more likely scored in genomes with many short scaffolds compared to few in the size-range of chromosome pseudo-molecules, due to the lack of information on the relative order of scaffolds.
In summary, we showed above-average fractions of ohnologs combined with below-average fractions of supergene clusters recruited by two dosage-dependent terpenoid biosynthetic modules (IPP isomerases and genes involved in the MVA pathway) (Table 3, Table 4). In addition, we revealed a below-average rate of ohnolog retention combined with a significantly increased rate of tandem duplicates for stoichiometrically insensitive genes (i.e. genes that are not acting in a dosage-dependent way) like core-terpene synthases as well as triterpene-specific synthases.
Identification and phylogenetic analysis of key genes controlling isoprenoid profiles and trichome density
The aforementioned biosynthetic inventory of all plant terpenoid biosynthetic modules is necessary and sufficient for production of related compounds with designated biochemical function. However, some terpenoids are autotoxic and can only be produced in high amounts in specialized hair-like aerial structures termed glandular trichomes [56,115] where they are stored or secreted to the surface in order to facilitate ecological interactions (i.e. repelling herbivores or attracting beneficial organisms). Biogenesis and distribution of trichomes is controlled by various biosynthetic and regulatory processes, often mediated by pleiotropic genes [116,117]. In this context, it has recently become evident that trichome density on the leaf surface is amongst other factors influenced by a class of pleiotropic genes that also catalyzes the entry step to the MEP pathway [35]. In tomato, two deoxy-xylulosephosphate synthase genes (DXS) have previously been identified. Interestingly, differential and tissue-specific expression was observed: While DXS1 is ubiquitously expressed, DXS2 was found to be abundant in only a few tissues including trichomes. Reduction of DXS2 expression in cultivated tomato led to an increase in glandular trichome density [52]. To identify additional DXS-like homologs, we screened our curated genes set and found evidence for 79 encoded proteins within all genomes subjected to our analysis (Fig 4, S5 Table). In addition, we included four DXS-like genes that were previously identified in the moss Physcomitrella patens to reconstruct the evolutionary history of all 83 target genes during Angiosperm radiation [53,118].
Fig 4. Phylogenetic relationships among 83 DXS-like proteins.

Brassicaceae and Cleomaceae are marked in blue. Solanaceae are marked in red. Proteins encoded in the basal Angiosperm Amborella are marked in green. The moss Physcomitrella comprises the outgroup and is marked in yellow. DXS-like genes group in three distinct clades since the origin of Angiosperms. Notably, all analyzed Brassicaceae have lost DXS2-like genes. However, the model plant Arabidopsis contains one highly diverged member of clade two that groups closer to clade one than to any other clade two homologs (marked by black arrow).
As previously reported, the Physcomitrella DXS-like genes form a monophyletic clade that groups distant to all other analyzed Angiosperm target genes. Strikingly, we have identified multiple gene family members that remained un-characterized within all other analyzed annotations except maize. Within the Angiosperm clades, we found that DXS-like genes always group in three distinct clades that form monophyletic groups rooted by basal members present in the Amborella genome (Fig 4), which is commonly placed at or near the base of the flowering plant lineage [71,119]. Within those clades, we observed grouping of closely related species consistent with the evident phylogenetic relationships of these species as a whole (Fig 4). However, our analysis also revealed clade-specific differences. First, we did not detect any proteins grouping to clade three within the non-redundant Cannabis transcriptome (S5 Table). Second, whereas most organisms encode at least one member of every clade, Brassicaceae and Cleomaceae have lost DXS-like genes belonging to clade two (Fig 4, S5 Table). Interestingly, the model plant Arabidopsis forms the only exception, because it possesses one DXS2 locus (AtDXS2/AtDXL1 or AT3G21500) that is highly diverged from any other members present in that clade (marked by black arrow in Fig 4). Our analysis revealed that AtDXS2 forms a basal sister to all other clade two members and groups closer to its clade one ortholog present in the basal Angiosperm Amborella compared to any other clade two members. Note that first evidence supports functional specialization at both the expression and biochemical level within the plant DXS family in Arabidopsis (see introduction section) [53]. In this context, the authors reveal the occurrence and putative relevance of lineage-specific gene duplications. Therefore, the plant DXS family emerges as an interesting model to examine the molecular evolutionary basis of plant secondary metabolism diversification, giving rise to further investigation of this gene family in a broader phylogenomics framework, as we presented in this study.
Next, we assessed the contribution of gene and genome duplication to DXS-like gene family composition among four further genome annotations with most accurate determination of ohnolog blocks (Table 6). To our knowledge, the contribution of genome duplication to DXS-like family evolution has previously not been assessed to that extend. For A. thaliana, B. rapa, T. hasslerania and G. max, we found 24 DXS-like genes in total, organized in eight duplicate groups (defined as set of genes comprising descendants from one distinct ancestral singleton due to one or more rounds of duplication) and distributed across all three DXS-like clades (Fig 4, S5 Table). Strikingly, 100% of those are due to ancient polyploidy events, either directly when forming pairs (WGD) or triplets (WGT) of ohnolog copies or indirectly when forming tandem- or transposition duplicates (GTD) of ohnolog group members (Table 6). In A. thaliana, for example, DXS1 (AT4G15560) and DXS2 (AT3G21500) form the ohnolog duplicate gene pair A15N013, dating back to the At-α WGD event [8,72] (Table 1A and 1B). The encoded proteins share 78.8% of protein sequence similarity (Table 6). Likewise, the corresponding genes are differentially expressed and pleiotropic (see introduction section; i.e. involved in terpenoid biosynthesis, plastid development and trichome formation [35,53,54,120,121]). Further analysis of DXS3 (AT5G11380) indicated its putative origin due to gene transposition duplication of DXS1. First, both genes form a highest-scoring sequence pair based on our BLAST analysis after removal of self-hits in Arabidopsis (see Materials & Methods section). Second, both genes are embedded in a non-syntenic genomic regions that contain remnants of transposon-like sequences (Fig 5). Considering the increased phylogenetic distance between this pair of genes and its reduced degree of protein sequence similarity (Table 6) compared to the pair of DXS1/DXS2 (Fig 4), this illustrates that genetic versatility within the Arabidopsis DXS family was further leveraged by a gene transposition duplication (GTD). Taken together, these results give rise to the onset of functional diversification of the A15N013 ohnolog pair following the At-α WGD event in Brassicaceae (see Discussion section). Similarly, short sequence duplication may have contributed to functional diversification of DXS-like genes. Based on those results, we further assessed the impact of various duplication modes to all other identified DXS-like genes in all analyzed genome assemblies including analysis of expression and sequence diversity.
Table 6. Overview of gene and genome duplication responsible for DXS-like cluster extension; shown are all target genes for four genomes A .
| Species | Gene Identifier | Clade | Origin of Duplication | Duplicate Group | Similarity B to Duplicate copy | Identity B to Duplicate copy |
|---|---|---|---|---|---|---|
| A. thaliana | AT3G21500 | 1 | At-α WGD (A15N013) | 1 | 78.8% | 72.5% |
| A. thaliana | AT4G15560 | 1 | At-α WGD (A15N013) | 1 | ||
| A. thaliana | AT5G11380 | 3 | GTD (AT4G15560) C | 1 | 68.6% | 53.3% |
| B. rapa | Bra001832 | 1 | Br-α WGT D | 2 | 77.9%- 81.8% | 73.4%- 77.3% |
| B. rapa | Bra012779 | 1 | Br-α WGT | 2 | 92.0% | 93.7% |
| B. rapa | Bra033495 | 1 | Br-α WGT | 2 | ||
| B. rapa | Bra008967 | 3 | GTD (Bra033495) C | 2 | 67.0% | 52.6% |
| T. hasslerania | Th2v17645 | 3 | Tandem (Th2v17646) E | 3 | 4.3% | 6.5% |
| T. hasslerania | Th2v17646 | 3 | Tandem (Th2v17645) E | 3 | ||
| T. hasslerania | Th2v18234 | 1 | Th-α WGT D | 4 | 92.4%- 93.3% | 88.8%- 89.5% |
| T. hasslerania | Th2v26234 | 1 | Th-α WGT | 4 | 87.6% | 83.9% |
| T. hasslerania | Th2v25487 | 1 | Th-α WGT | 4 | ||
| G. max | Glyma07g38260 | 1 | Glycine WGD (I) | 5 | 94.4% | 91.7% |
| G. max | Glyma17g02480 | 1 | Glycine WGD (I) | 5 | ||
| G. max | Glyma15g10610 | 1 | Glycine WGD (I) | 5 | 51.7% | 48.9% |
| G. max | Glyma13g28470 | 1 | Glycine WGD (I) | 5 | ||
| G. max | Glyma04g07400 | 3 | Glycine WGD (II) | 6 | 97.0% | 94.3% |
| G. max | Glyma06g07491 | 3 | Glycine WGD (II) | 6 | ||
| G. max | Glyma17g07400 | 2 | Glycine WGD (III) | 7 | 45.4% | 44.8% |
| G. max | Glyma13g01280 | 2 | Glycine WGD (III) | 7 | ||
| G. max | Glyma18g28830 | 2 | Glycine WGD (IV) | 8 | 96.8% | 94.1% |
| G. max | Glyma08g37670 | 2 | Glycine WGD (IV) | 8 | ||
| G. max | Glyma08g37680 | 2 | Tandem (Glyma08g37670) | 8 | 97.6% | 96.1% |
| G. max | Glyma09g33320 | 2 | Segmental (Glyma08g37670) F | 8 | 92.2% | 86.5% |
| - | - | - | - | average | 77.6% | 73.4% |
A Analysis restricted to Genomes with most accurate identification of ohnologs due to technical limitation.
B Based on encoded protein sequence.
C Origin of GTD Duplicate based on lowest blastp e-value for alignment to other family members.
D Embedded in most fractionated subgenome; similarity and identity scores shown relative to ohnologs in both other subgenomes.
E Note significant length difference of both genes in this array; low similarity and identity scores indicate annotation error dividing one ORF into two neighboring genes. Both values are excluded for calculation of average.
F Gene scored as Segmental Duplicate due to high synteny score of harbouring region while other members of duplicate group are sufficient to cover the synthenic depth of this genome (i.e. no WGT evident).
Fig 5. (B)LastZ two-way multiple alignment of 40kb-windows harboring the putative Arabidopsis gene transposition duplicate gene pair DXS3 (AT5G11380) (upper lane, marked in purple) and DXS1 (AT4G15560) (lower lane, marked in purple).
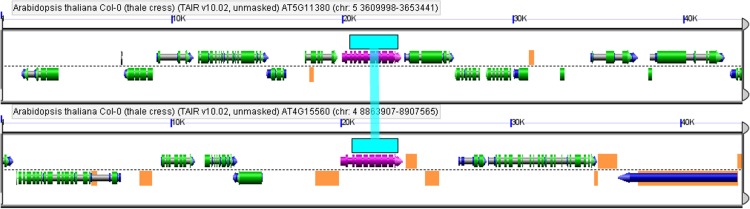
Non-syntenic coding sequences are marked in green. Both duplicate copies form a highest-scoring sequence pair (marked in turquoise). Transposon-like sequences are marked in orange. Pseudogenes are marked in blue. Analysis can be regenerated online following the CoGe link https://genomevolution.org/r/eooq (last accessed on December 13th, 2014).
Initially, we assessed divergence levels among both pairs of DXS-like protein sequences and compared those following various modes of duplication by testing for differential and tissue-specific expression of all three DXS-like genes in Arabidopsis. Please note that glandular trichomes are absent in the model plant [122]. Notably, DXS1 is the only member of its gene family that is annotated to “trichome specific up-regulation” in the plant ontology database (PO:0000282) [123–125]. However, we confirmed expression of all three loci in Arabidopsis non-glandular trichomes (and various other tissue types) based on publically available microarray data [94]. Furthermore, we uncovered consistent patterns of differential expression across several tissue types. Compared to housekeeping gene expression, DXS1 transcript are most abundant in all analyzed tissues. The ohnolog duplicate DXS2 shows lowest expression levels, whereas the transposed duplicate DXS3 forms an intermediate across all analyzed tissues (Fig 6).
Fig 6. Comparative tissue-specific expression of all Arabidopsis DXS-like genes relative to the bHLH housekeeping gene.

Values comprise averages of four independent ATH1 microarray experiments (Experiment ID: E-MEXP-2008, see Materials & Methods section). Notably, DXS1 is the only member with annotation to “trichome” plant ontology (PO:0000282). The error bars represent the standard error.
To assess and compare DXS-like gene family divergence in further species, we have performed two separate approaches. First, we performed DXS-like gene expression analysis. Second, we assessed and compared the protein sequence identities of DXS-like duplicate groups due to different duplication events.
Gene duplication can result in transposition of the novel duplicate copy to a distant genomic location, leading to the presence of other cis-acting elements including promotors or enhancers that influence gene expression [126,127]. This results in sub-functionalization of segregants on the expression level. To extend the aforementioned findings concerning sub-functionalization of DXS genes in A. thaliana, we have tested expression of S. lycopersicum target genes in every clade. In addition to increased expression of DXS2 in trichomes and global expression of DXS1 that was previously made evident [52] (Fig 7), we have uncovered that transcript levels of DXS3 are almost 2-fold higher in trichomes compared to any other analyzed tissue type (Fig 8).
Fig 7. Transcript levels of S. lycopersicum DXS-like genes in different parts of the plant (leaves, roots, stems, stems without (W/O) trichomes, and isolated stem trichomes) relative to those of the reference gene RCE1 (Solyc10g039370.1.1).
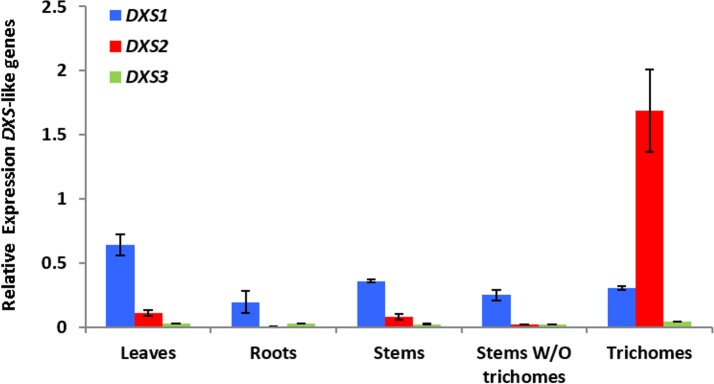
Transcript levels were determined by real-time qPCR with four biological and three technical replicates for each biological sample. The error bars represent the standard error.
Fig 8. Transcript levels of the S. lycopersicum DXS3 gene in different parts of the plant (leaves, roots, stems, stems without (W/O) trichomes, and isolated stem trichomes) relative to those of the reference gene RCE1 (Solyc10g039370.1.1).
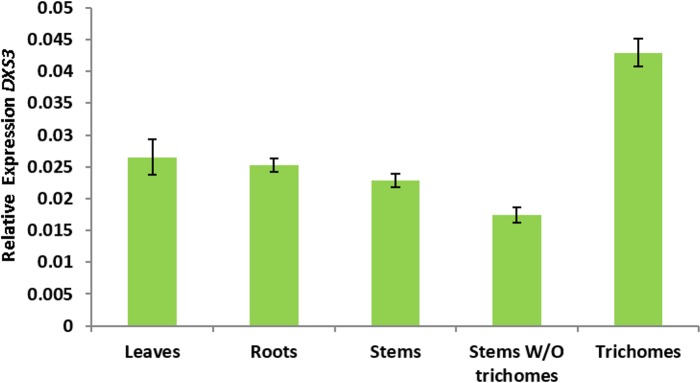
Transcript levels were determined by real-time qPCR with four biological replicates and three technical replicates for each biological sample. The error bars represent the standard error.
In addition to frequent changes in gene expression, recent analysis revealed an accelerated rate of amino acid changes when comparing ohnolog duplicates to their paralogs [128]. High rates of amino acid substitutions lead to decreased levels of protein sequence identities when comparing gene copies due to different duplication modes. For example, polyploidy facilitated rapid diversification of protein sequences and sub-functionalization on a biochemical level in several cases, including glucosinolate biosynthesis, resistance proteins of the NB-LRR type as well as L-type lectin receptor-like kinases [25,26,129]. In all three cases, functional diversification among certain duplicate pairs correlates with differentially decreased protein sequence identities when comparing “novel” gene copies due to certain duplication events. Therefore, we assessed protein sequence similarity/identity among all other seven DXS-like duplicate groups (i.e. sets of genes due to duplication of one distinct ancestral singleton), thereby screening for indications of putative sub- or neo-functionalization (Table 6). Values for protein sequence similarity (identity) range from 45.4% (44.8%) (G. max, duplicate group 7) to 96.8% (94.1%) (G. max, duplicate group 8). In summary, DXS-like proteins share an average of 77.6% (73.4%) for sequence similarity (identity) among all groups, thereby reaching a cumulative divergence level similar to that observed in A. thaliana, for which data on differential target gene expression following gene and genome duplication are available (see above).
In summary, we have analyzed three clades of DXS-like genes present in every analyzed genome annotation. We have assessed differential and tissue-specific expression for two distant lineages, thereby collecting indications for putative sub-functionalization following gene and genome duplication within this group of target genes. To further support this hypothesis, more sequence and expression data are necessary from basal angiosperms in order to facilitate comparison of the observed profiles in a more ancestral state.
Discussion
A combination of synteny, sequence similarity and protein domain modelling facilitates large-scale gene identification and novel annotations in all modules of terpenoid biosynthesis
In a genome informatics approach, we combined a novel and easy-to-follow meta-method for gene and supergene cluster identification with a custom pipeline for de novo protein annotation for large-scale identification of biosynthetic elements associated with plant secondary metabolism. The method provided in this article is novel because it integrates information provided by the genomic location of target genes to information on sequence homology and to the information on encoded protein domain composition. Our method can be applied to a collection of final stage genome annotations, early-stage gene-space assemblies as well as non-redundant transcriptomes, thereby facilitating uniform standards for gene identification. In this context, we analyzed various datasets of different quality for a flowering-plant wide comparative survey of genes building up a major pathway of plant specialized metabolism. In summary, we curated a set of genes associated with all modules of terpenoid biosynthesis and determined key factors shaping metabolic diversification in an Angiosperm-wide scale.
First, we investigated 17 species including twelve major crops. During this initial part of our analysis, we discovered previously uncharacterized genes of the (a) TERPENE SYNTHASE- as well as the (b) DXS-like types in all species except Arabidopsis and Eucalyptus. These gene families have often been subjected to species-specific analysis in the past because they are involved in (a) generating a diverse set of terpenoid compounds and (b) in control of trichome density on the leaf surface, thereby providing significant economic and ecologic potential. The provided data on novel annotations of target genes in most species elucidated the power of our approach in a proof-of-concept and may act as a blueprint for future efforts to more rapidly find and clone functional core-TPS and DXS-like genes from any flowering plant in context of plant breeding and biotechnology.
Second, we identified various genes that have previously not been associated with a distinct function and established computational inferences to encoded prenyltransferases and triterpene-specific synthases across all lineages. These enzymes are commonly associated with the biosynthesis of di-, sesqui-, tri-, tetra- and polyterpenes. Assessing similarities to core-TPS genes in both coding sequence on the DNA level and protein domain composition, we provided indications for the common evolutionary origin shared among all three gene families. Furthermore, we monitored the underlying variation of gene copy number in a phylogenomics framework and thereby described a framework that increased genetic versatility to create the necessary basis for metabolic diversification within a timeframe of 250 MA corresponding to flowering plant radiation.
Third, our approach identified homologs of all genes currently annotated to MVA and MEP pathways including DXS-like genes in Arabidopsis across all analyzed genomes. Large-scale annotation of genes employed by those pathways has to date not yet been made available for every analyzed species except Arabidopsis, tomato and potato. In this context, our study provides and important prerequisite for future efforts aiming at metabolic engineering within any of the analyzed crop lineages.
Both gene- and genome duplication mediated a dramatic increase of genetic versatility underlying modular terpenoid biosynthesis in all species
In the next part of our analysis, we screened for gene and genome duplication events that affected copy number of all loci involved in distant modules of terpenoid biosynthesis across all investigated species. In this context, genetic versatility is defined as the number of homologs within one gene family. Including novel annotations of previously un-identified genes to all six modules (see above), we described a 376%-increase of terpenoid biosynthetic gene copy number (“genetic versatility”) ranging from the leanest state found in the basal Angiosperm Amborella (50 genes) up to most versatile genotypes found for Vitis that has been subjected to extensive human domestication (188 genes). Merging the genetic inventory associated with all six modules, we revealed that this increase is driven by a combination of gene and genome duplication across 250 MA corresponding with the radiation time of flowering plants. However, individual differences apply when considering single terpenoid biosynthetic modules separately. To our knowledge, this is the to-date most intensive and systematic study of plant gene family expansion that influenced metabolic diversification in a phylogenomics framework.
Please note that segmental duplications are excluded from our analysis. In this context, we acknowledged an error rate due to false-positive scoring as ohnolog duplicates affecting ancient segmental duplications of large genomic regions. Briefly: It is currently not possible to accurately distinguish large segmental duplications from fractionated blocks due to genome multiplications in all cases. Likewise, very short segmental duplications with high degree of fractionation may be accidentally scored as a series of distant gene transposition duplication. This is mainly due to technical reasons, because the SynMap algorithm controls scoring of synteny merely based on a function of collinear genes in a certain density as previously described [4,87,88]. However, most segmental duplications that did not emerge roughly at the same time than any of the investigated genome duplications will display significantly different averages for ka/ks values, and are therefore excluded from synteny analysis due to the cut-offs thresholds applied in the SynMap preferences (see Materials & Methods section).
Enzymes catalyzing the committed step of end product biosynthesis are more often encoded by supergene clusters due to tandem duplication
We highlighted a consistently asymmetric distribution of supergene clusters across all terpenoid biosynthetic modules. Generally, core terpene synthases as well as triterpene-specific synthases comprise enzymes catalyzing the committed step for biosynthesis of designated end products (mono-, di-, sesqui-, and triterpenes). We revealed that those are most enriched for tandem duplicate copies across all analyzed genomes. Please note that in alignment with this these findings, the role of syntenic core-TPS supergene clusters that include adjacent loci involved in different modules was recently made evident for diversification of terpenoid pathway assembly during radiation of various Angiosperm clades (see below) [33]. Moreover, it has become evident that single-featured polymorphisms affecting those genes are sufficient to alter, amongst others, herbivore behavior in otherwise isogenic lines [130–132]. In the opinion of the authors, such processes may have correlated with human efforts of plant domestication and crop breeding in multiple cases. It seems possible that sub-functionalization following tandem duplication of target genes influenced key traits (i.e. scent, taste), making the plant more suitable for further selection. This hypothesis is supported by the high target gene count for highly domesticated species with high content of terpenoids (like Vitis, Cannabis and Lactuca). Although Eucalyptus possesses the highest terpenoid biosynthetic gene count among all species analyzed in this study, it did not undergo major processes of human domestication [78]. However, several herbivores are known to respond differently to Eucalyptus inter- and intraspecific variation of secondary metabolite profiles with potential effects on target gene evolution [133]. Please also note that intensive domestication may also lead to a low TPS gene count in some cases, for example as a result of selection towards different key traits negatively influenced by genes in linkage disequilibrium to TPS genes [134].
Dosage-dependent enzymes in modules mediating intermediate reaction steps are more often encoded by ohnolog duplicates–Introducing a two-step model for rapid plant pathway diversification
Compared to the above-mentioned asymmetric distribution of tandem duplicate copies across all subsets of genes involved in terpenoid biosynthesis, we reported opposite tendencies for retained ohnologs. We made evident that multi-gene family members involved in the MVA pathway as well as IPP isomerases more often tend to originate from whole genome multiplication events. For the MVA pathway, ohnolog fractions greatly outreach genome-wide averages for all genome annotations except papaya. IPP isomerases comprise 100% of retained ohnologs in Brassicaceae, Cleomaceae as well as Glycine. These groups of gene copies are due to duplication of a distinct ancestral singleton (“duplicate groups”) but encode enzymes involved in different terpenoid functional modules, working together by catalyzing neighboring reactions and isomerization of intermediate products (IPP or MVA/MEP modules). According to the gene balance hypothesis, duplicate loci are preferentially retained when functioning together in a dosage-dependent way [6,135]. In this context, we showed an asymmetric ohnologs distribution among the modules acting up- and downstream of core terpene scaffold synthesis.
Based on those findings, we hypothesize a two-step mechanism for the rapid plant pathway- and trait diversification observed in nature. This proposed mechanism depends on both gene- and genome duplication and affects different groups of genes at different times. In a first step, ancient polyploidy plays a paramount role by mediating the described expansion of certain genetic networks involved in plant primary metabolism (like MEP/MVA and IDI loci, see Fig 1), thereby creating a certain degree of “pathway redundancy”. Due to stoichiometric effects, the following post-polyploidy rate of plant survival depends on parallel retention of most (if not all) duplicated genes present in affected metabolic modules. Both functional diversification of ohnolog duplicates and/or incomplete module retention may lead to detrimental effects due to altered fractions of primary metabolite concentrations, as previously hypothesized and backed up by gene network analysis in context of mustard family evolution [135,136]. In a second step, more recent, short sequence duplications (including tandem and gene transposition duplication) creates an extended pool of trans-acting elements (like, for example, additional core-TPS or DXS genes). Since increased copy number of those genes does not lead to detrimental effects due to stoichiometry as described above, functional diversification may create extended capabilities to catalyze biosynthesis of extended product ranges (novel functions). The aforementioned polyploidy-induced primary module duplication created a superabundance of primary metabolites, thereby providing a “playground” for the evolution of novel functions catalyzed by novel gene copies due to short sequence duplicates.
In a nutshell, our results provided evidence for a partial polyploidy-driven expansion of plant secondary metabolism and strongly supported the gene-balance hypothesis for the dosage-dependent subset of involved key genes. Such trends have often been suggested for plants [14,23,137], but solid evidence on a genetic level was to-date only available for glucosinolates and plant resistance proteins of the NB-LRR type [25,26].
Duplicate gene copies of ancestral singletons diversified in metabolic function following gene and genome duplication: the case of DXS-like genes
Recent analysis strongly support the concept of functional specialization following gene duplication as the evolutionary fate explaining retention of the duplicated gene pair DXS1/DXS2 in Arabidopsis [53]. Based on this approach, we performed follow-up analysis of DXS-like gene family evolution on a broader phylogenomics scale. In summary, we showed that certain sets of duplicate gene copies that descend from duplication of one ancestral singleton (i.e. duplicate groups) contain genes encoding different enzymes for the same pathway in Arabidopsis and tomato. Some of those convey pleiotropic effects due to published annotation to different traits (i.e. control of trichome density and terpenoid biosynthesis). Additionally, we identified common protein motifs present (a) within and (b) across different modules of terpenoid biosynthesis. We conclude an expansion of isoprenoid pathways by gene family diversification following gene and genome duplication, thereby resulting in the complex, modular architecture of terpenoid biosynthesis and the plethora of produced compounds observed across the Angiosperm clade. Because supergene clusters tend to be younger than genes preferentially retained after ancient polyploidy events [11,138], ohnologs are likely prone to acquire additional roles over time as previously described (sub- and neo-functionalization) [135,139,140].
Moreover, we have found evidence for the preferential (i.e. above-average) retention of IPP genes following various independent, successive polyploidy events for the Brassicaceae-Cleomaceae sister group system [83]. Similar to DXS-like proteins, IPP isomerases convey pleiotropic functions because they are relevant for the biosynthesis of other isoprenoid compounds beyond plant terpenoid biosynthesis. They also have been brought in connection with plant development in Arabidopsis, thereby mediating a check-point for primary metabolism (e.g. hormones) and different branches of specialized metabolism [141–143]. The observed trend of IDI over-retention is consistent for species-specific WGT events (Th-α for Tarenaya and Br-α for Brassica) as well as for the more ancient At-α WGD event shared by all Brassicaceae [8,17,72,76,144]. Similarly, we observed a rising IDI gene counts following soybean polyploidy. We concluded a preferential retention of this gene family following polyploidy that might be due to reported dosage-sensitivity (see Introduction section) and is likely visible especially in the aforementioned genomes due to their high levels of syntenic depth (i.e. high levels of genome multiplicity due to more successive WGDs/WGTs compared to other genomes). However, the case of Arabidopsis provides an exception which might be due to its reductive genome state that has been previously reported for the genus of the model plant [145].
Furthermore, our results further support the concept of sub-functionalization among DXS-like genes on a broader phylogenomics scale than previously reported [53]. In addition, we assessed and compared the differential impact of various duplication modes (i.e. WGD and short sequence duplication) to functional diversification of DXS-like genes, thereby uncovering novel aspects shaping target gene family evolution. Similar to IDI loci, DXS-like genes have been associated with more than one trait. Two among three DXS-like genes in Arabidopsis comprise the retained ohnolog pair A15N013, dating back to the At-α that is shared by all Brassicaceae. While both DXS1 (AT4G15560) and DXS2 (AT3G21500) are annotated to the MEP pathway, DXS1 is also involved in plastid development [8,52,72,120,121]. In addition to the reported control of isoprenoid profiles, functional evidence for control of trichome density on the leaf surface has been made evident [52]. Initially, we discovered a whole new clade of DXS-like genes with members in Solanaceae and Brassicaceae including Arabidopsis. Next, we scored the contribution of ohnolog retention to the set of target loci identified the Brassicaceae-Cleomaceae sister group system as well as the legume G. max. We showed that all target genes within the aforementioned four genome annotations date back to ancient polyploidy events, either directly by comprising ohnolog duplicate groups or indirectly by comprising tandem- or transposition copies of ohnologs. Furthermore, we unraveled phylogenetic relationships within the target gene family that groups to three clades of encoded DXS-like proteins. We brought those clades in connection with a expression polymorphisms following gene- and genome duplication in tomato and the model plant Arabidopsis, thereby elucidating another case of putative sub-functionalization following duplication.
Modified terpenes: Future work or going beyond the plant terpenoid biosynthetic module
Recently, Boutanaev et al. published a very conclusive investigation of core-TPS gene diversification across an evolutionary timeframe similar to the scope of our study (see introduction) [33]. The authors defined an (incomplete) “terpenome” that merely consists of (some, but not all present) core-TPS genes and supergene clusters that consist of both core-TPS and CYP-like genes. CYP-like genes encode cytochrome P450 enzymes that catalyze downstream modifications of various secondary metabolite core structures including alkaloids, glucosinolates and terpene post-modification reactions [146–148]. The authors infer an important role of (micro)synteny and TPS/CYP-locus linkage disequilibrium for terpenoid pathway assembly in plants, and suggest a differential mechanism of trait diversification in monocots and dicots [33]. However, terpenoid biosynthesis in plants is modular because it consists of more than just the core-TPS gene family (Fig 1). Likewise, CYP-like genes are not the only family mediating terpene post-modification reactions [35]. Due to our large-scale annotation of terpenoid biosynthetic genes across all pathway modules within 17 representative genomes, our results provide a valuable basis for future efforts to further investigate the role of synteny and genetic linkage disequilibrium in context of a more complete “terpenome”. This includes the possibility to better elucidate the effects of genetic co-segregation with many other gene families that convey terpene downstream modifications, similar to the aforementioned case study published by Boutanaev et al. [33]. Such gene families may include, for example, UDP glucuronosyltransferases and many other pleiotropic genes involved in biosynthesis of terpenoids and, beyond that, various other plant secondary metabolites [149]. Ultimately, the data provided in our study will facilitate a better understanding of plant secondary metabolite pathway assembly in Angiosperms with various implications for plant breeding and metabolic engineering in context of medicine, flavor, fragrance and pigment production.
Supporting Information
(XLS)
(XLS)
Numbers are quotients of the module-wise gene count of terpenoid biosynthetic pathways and the number of all protein-coding genes within the whole genome. Species with highest and lowest relative pathway size among all analyzed species are color-coded as indicated in the legend.
(XLS)
(XLS)
Red indicates absence of tandem/ohnolog duplicates. Green indicates significant enrichment among terpenoid biosynthetic genes compared to background with threshold of 0.01.
(XLS)
Acknowledgments
We would like to thank Mariam Neckzei for her help with all illustrations.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was funded by a Netherlands Organization for Scientific Research (NWO) Ecogenomics grant (M.E.S.).
References
- 1. Lyons E, Freeling M (2008) How to usefully compare homologous plant genes and chromosomes as DNA sequences. The Plant Journal 53: 661–673. 10.1111/j.1365-313X.2007.03326.x [DOI] [PubMed] [Google Scholar]
- 2. Koonin EV (2005) Orthologs, paralogs, and evolutionary genomics 1. Annu Rev Genet 39: 309–338. [DOI] [PubMed] [Google Scholar]
- 3. Fitch WM (1970) Distinguishing homologous from analogous proteins. Syst Zool 19: 99–113. [PubMed] [Google Scholar]
- 4. Lyons E, Pedersen B, Kane J, Alam M, Ming R, Tang H, et al. (2008) Finding and comparing syntenic regions among Arabidopsis and the outgroups papaya, poplar, and grape: CoGe with rosids. Plant Physiol 148: 1772–1781. 10.1104/pp.108.124867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ohno S (1970) Evolution by gene duplication New York: Springer Publishing Group. 160 p. [Google Scholar]
- 6. Freeling M (2009) Bias in plant gene content following different sorts of duplication: tandem, whole-genome, segmental, or by transposition. Annu Rev Plant Biol 60: 433–453. 10.1146/annurev.arplant.043008.092122 [DOI] [PubMed] [Google Scholar]
- 7. Wolfe K (2000) Robustness—it's not where you think it is. Nat Genet 25: 3–4. [DOI] [PubMed] [Google Scholar]
- 8. Bowers JE, Chapman BA, Rong J, Paterson AH (2003) Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 422: 433–438. [DOI] [PubMed] [Google Scholar]
- 9. Joron M, Papa R, Beltran M, Chamberlain N, Mavarez J, Baxter S, et al. (2006) A conserved supergene locus controls colour pattern diversity in Heliconius butterflies. PLoS Biol 4: e303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Smith JM (1989) Evolutionary genetics: Oxford University Press. [Google Scholar]
- 11. Rizzon C, Ponger L, Gaut BS (2006) Striking similarities in the genomic distribution of tandemly arrayed genes in Arabidopsis and rice. PLoS Comput Biol 2: e115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kliebenstein DJ, Lambrix VM, Reichelt M, Gershenzon J, Mitchell-Olds T (2001) Gene duplication in the diversification of secondary metabolism: tandem 2-oxoglutarate-dependent dioxygenases control glucosinolate biosynthesis in Arabidopsis. Plant Cell 13: 681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Leister D (2004) Tandem and segmental gene duplication and recombination in the evolution of plant disease resistance gene. Trends Genet 20: 116–122. [DOI] [PubMed] [Google Scholar]
- 14. Jiao Y, Wickett NJ, Ayyampalayam S, Chanderbali AS, Landherr L, Ralph PE, et al. (2011) Ancestral polyploidy in seed plants and angiosperms. Nature 473: 97–100. 10.1038/nature09916 [DOI] [PubMed] [Google Scholar]
- 15. Schranz ME, Mohammadin S, Edger PP (2012) Ancient whole genome duplications, novelty and diversification: the WGD Radiation Lag-Time Model. Curr Opin Plant Biol 15: 147–153. 10.1016/j.pbi.2012.03.011 [DOI] [PubMed] [Google Scholar]
- 16. Haudry A, Platts AE, Vello E, Hoen DR, Leclercq M, Williamson RJ, et al. (2013) An atlas of over 90,000 conserved noncoding sequences provides insight into crucifer regulatory regions. Nat Genet 45: 891–898. 10.1038/ng.2684 [DOI] [PubMed] [Google Scholar]
- 17. Barker MS, Vogel H, Schranz ME (2009) Paleopolyploidy in the Brassicales: analyses of the Cleome transcriptome elucidate the history of genome duplications in Arabidopsis and other Brassicales. Genome Biol Evol 1: 391–399. 10.1093/gbe/evp040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ming R, Hou S, Feng Y, Yu Q, Dionne-Laporte A, Saw JH, et al. (2008) The draft genome of the transgenic tropical fruit tree papaya (Carica papaya Linnaeus). Nature 452: 991–996. 10.1038/nature06856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Vekemans D, Proost S, Vanneste K, Coenen H, Viaene T, Ruelens P, et al. (2012) Gamma Paleohexaploidy in the Stem Lineage of Core Eudicots: Significance for MADS-Box Gene and Species Diversification. Mol Biol Evol 29: 3793–3806. 10.1093/molbev/mss183 [DOI] [PubMed] [Google Scholar]
- 20. Jaillon O, Aury JM, Noel B, Policriti A, Clepet C, Casagrande A, et al. (2007) The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature 449: 463–467. [DOI] [PubMed] [Google Scholar]
- 21. Tang H, Lyons E (2012) Unleashing the genome of brassica rapa. Front Plant Sci 3: 172 10.3389/fpls.2012.00172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Consortium TTG (2012) The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485: 635–641. 10.1038/nature11119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Freeling M, Thomas BC (2006) Gene-balanced duplications, like tetraploidy, provide predictable drive to increase morphological complexity. Genome Res 16: 805–814. [DOI] [PubMed] [Google Scholar]
- 24. Hartmann T (2007) From waste products to ecochemicals: fifty years research of plant secondary metabolism. Phytochemistry 68: 2831–2846. [DOI] [PubMed] [Google Scholar]
- 25. Hofberger JA, Lyons E, Edger PP, Pires JC, Schranz ME (2013) Whole genome and tandem duplicate retention facilitated glucosinolate pathway diversification in the mustard family. Genome Biol Evol 5: 2155–2173. 10.1093/gbe/evt162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hofberger JA, Zhou B, Tang H, Jones JD, Schranz ME (2014) A novel approach for multi-domain and multi-gene family identification provides insights into evolutionary dynamics of disease resistance genes in core eudicot plants. BMC Genomics 15: 966 10.1186/1471-2164-15-966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. van den Bergh E, Külahoglu C, Bräutigam A, Hibberd JM, Weber AP, Zhu X-G, et al. (2014) Gene and genome duplications and the origin of C 4 photosynthesis: Birth of a trait in the Cleomaceae. Current Plant Biology 1: 2–9. [Google Scholar]
- 28. Phillips MA, Leon P, Boronat A, Rodriguez-Concepcion M (2008) The plastidial MEP pathway: unified nomenclature and resources. Trends Plant Sci 13: 619–623. 10.1016/j.tplants.2008.09.003 [DOI] [PubMed] [Google Scholar]
- 29. Aubourg S, Lecharny A, Bohlmann J (2002) Genomic analysis of the terpenoid synthase (AtTPS) gene family of Arabidopsis thaliana. Mol Genet Genomics 267: 730–745. [DOI] [PubMed] [Google Scholar]
- 30. Benveniste P (2002) Sterol metabolism. Arabidopsis Book 1: e0004 10.1199/tab.0004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cheng A-X, Lou Y-G, Mao Y-B, Lu S, Wang L-J, Chen X-Y (2007) Plant Terpenoids: Biosynthesis and Ecological Functions. Journal of Integrative Plant Biology 49: 179–186. [Google Scholar]
- 32. Gershenzon J, Dudareva N (2007) The function of terpene natural products in the natural world. Nat Chem Biol 3: 408–414. [DOI] [PubMed] [Google Scholar]
- 33. Boutanaev AM, Moses T, Zi J, Nelson DR, Mugford ST, Peters RJ, et al. (2015) Investigation of terpene diversification across multiple sequenced plant genomes. Proc Natl Acad Sci U S A 112: E81–88. 10.1073/pnas.1419547112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Matsuba Y, Nguyen TT, Wiegert K, Falara V, Gonzales-Vigil E, Leong B, et al. (2013) Evolution of a complex locus for terpene biosynthesis in solanum. Plant Cell 25: 2022–2036. 10.1105/tpc.113.111013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Tholl D, Lee S (2011) Terpene Specialized Metabolism in Arabidopsis thaliana. Arabidopsis Book 9: e0143 10.1199/tab.0143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Phillips MA, D'Auria JC, Gershenzon J, Pichersky E (2008) The Arabidopsis thaliana type I isopentenyl diphosphate isomerases are targeted to multiple subcellular compartments and have overlapping functions in isoprenoid biosynthesis. The Plant Cell Online 20: 677–696. 10.1105/tpc.107.053926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vranová E, Coman D, Gruissem W (2013) Network analysis of the MVA and MEP pathways for isoprenoid synthesis. Annual review of plant biology 64: 665–700. 10.1146/annurev-arplant-050312-120116 [DOI] [PubMed] [Google Scholar]
- 38. Gruchattka E, Hädicke O, Klamt S, Schütz V, Kayser O (2013) In silico profiling of Escherichia coli and Saccharomyces cerevisiae as terpenoid factories. Microbial cell factories 12: 84 10.1186/1475-2859-12-84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shack S, Gorospe M, Fawcett TW, Hudgins WR, Holbrook NJ (1999) Activation of the cholesterol pathway and Ras maturation in response to stress. Oncogene 18: 6021–6028. [DOI] [PubMed] [Google Scholar]
- 40. Campos N, Rodriguez-Concepcion M, Sauret-Gueto S, Gallego F, Lois L, Boronat A (2001) Escherichia coli engineered to synthesize isopentenyl diphosphate and dimethylallyl diphosphate from mevalonate: a novel system for the genetic analysis of the 2-C-methyl-D-erythritol 4-phosphate pathway for isoprenoid biosynthesis. Biochem J 353: 59–67. [PMC free article] [PubMed] [Google Scholar]
- 41. Johnston JB (2007) Mechanistic Investigations of Types I and II Isopentenyl Diphosphate Isomerase: ProQuest. [Google Scholar]
- 42.Han K-H, Kang H-S, Oh S-K, Shin D-h, Yang J-M (2001) Isopentenyl diphosphate isomerase from Hevea brasiliensis and rubber producing method using the same. Google Patents.
- 43. Kang JH, Gonzales-Vigil E, Matsuba Y, Pichersky E, Barry CS (2014) Determination of residues responsible for substrate and product specificity of Solanum habrochaites short-chain cis-prenyltransferases. Plant Physiol 164: 80–91. 10.1104/pp.113.230466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Akhtar TA, Matsuba Y, Schauvinhold I, Yu G, Lees HA, Klein SE, et al. (2013) The tomato cis-prenyltransferase gene family. Plant J 73: 640–652. 10.1111/tpj.12063 [DOI] [PubMed] [Google Scholar]
- 45. Oldfield E, Lin FY (2012) Terpene biosynthesis: modularity rules. Angew Chem Int Ed Engl 51: 1124–1137. 10.1002/anie.201103110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bohlmann J, Meyer-Gauen G, Croteau R (1998) Plant terpenoid synthases: molecular biology and phylogenetic analysis. Proc Natl Acad Sci U S A 95: 4126–4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kolesnikova MD, Wilson WK, Lynch DA, Obermeyer AC, Matsuda SPT (2007) Arabidopsis camelliol C synthase evolved from enzymes that make pentacycles. Organic letters 9: 5223–5226. [DOI] [PubMed] [Google Scholar]
- 48. Shibuya M, Xiang T, Katsube Y, Otsuka M, Zhang H, Ebizuka Y (2007) Origin of structural diversity in natural triterpenes: direct synthesis of seco-triterpene skeletons by oxidosqualene cyclase. J Am Chem Soc 129: 1450–1455. [DOI] [PubMed] [Google Scholar]
- 49. Lodeiro S, Xiong Q, Wilson WK, Kolesnikova MD, Onak CS, Matsuda SP (2007) An oxidosqualene cyclase makes numerous products by diverse mechanisms: a challenge to prevailing concepts of triterpene biosynthesis. J Am Chem Soc 129: 11213–11222. [DOI] [PubMed] [Google Scholar]
- 50. Moses T, Pollier J, Thevelein JM, Goossens A (2013) Bioengineering of plant (tri)terpenoids: from metabolic engineering of plants to synthetic biology in vivo and in vitro. New Phytol 200: 27–43. 10.1111/nph.12325 [DOI] [PubMed] [Google Scholar]
- 51. Laszczyk MN (2009) Pentacyclic triterpenes of the lupane, oleanane and ursane group as tools in cancer therapy. Planta Med 75: 1549–1560. 10.1055/s-0029-1186102 [DOI] [PubMed] [Google Scholar]
- 52. Paetzold H, Garms S, Bartram S, Wieczorek J, Uros-Gracia EM, Rodriguez-Concepcion M, et al. (2010) The isogene 1-deoxy-D-xylulose 5-phosphate synthase 2 controls isoprenoid profiles, precursor pathway allocation, and density of tomato trichomes. Mol Plant 3: 904–916. 10.1093/mp/ssq032 [DOI] [PubMed] [Google Scholar]
- 53. Carretero-Paulet L, Cairo A, Talavera D, Saura A, Imperial S, Rodriguez-Concepcion M, et al. (2013) Functional and evolutionary analysis of DXL1, a non-essential gene encoding a 1-deoxy-D-xylulose 5-phosphate synthase like protein in Arabidopsis thaliana. Gene 524: 40–53. 10.1016/j.gene.2012.10.071 [DOI] [PubMed] [Google Scholar]
- 54. Heyndrickx KS, Vandepoele K (2012) Systematic Identification of Functional Plant Modules through the Integration of Complementary Data Sources. Plant Physiology 159: 884–901. 10.1104/pp.112.196725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tissier A (2012) Glandular trichomes: what comes after expressed sequence tags? The Plant Journal 70: 51–68. 10.1111/j.1365-313X.2012.04913.x [DOI] [PubMed] [Google Scholar]
- 56. Markus Lange B, Turner GW (2013) Terpenoid biosynthesis in trichomes—current status and future opportunities. Plant biotechnology journal 11: 2–22. 10.1111/j.1467-7652.2012.00737.x [DOI] [PubMed] [Google Scholar]
- 57. Wagner GJ, Wang E, Shepherd RW (2004) New approaches for studying and exploiting an old protuberance, the plant trichome. Ann Bot 93: 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tholl D, Chen F, Petri J, Gershenzon J, Pichersky E (2005) Two sesquiterpene synthases are responsible for the complex mixture of sesquiterpenes emitted from Arabidopsis flowers. Plant J 42: 757–771. [DOI] [PubMed] [Google Scholar]
- 59. Yamaguchi S, Sun T, Kawaide H, Kamiya Y (1998) The GA2 locus of Arabidopsis thaliana encodes ent-kaurene synthase of gibberellin biosynthesis. Plant Physiol 116: 1271–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bohlmann J, Martin D, Oldham NJ, Gershenzon J (2000) Terpenoid Secondary Metabolism in Arabidopsis thaliana: cDNA Cloning, Characterization, and Functional Expression of a Myrcene-β-Ocimene Synthase. Archives of Biochemistry and Biophysics 375: 261–269. [DOI] [PubMed] [Google Scholar]
- 61. Chen F, Tholl D, D'Auria JC, Farooq A, Pichersky E, Gershenzon J (2003) Biosynthesis and emission of terpenoid volatiles from Arabidopsis flowers. Plant Cell 15: 481–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chen F, Ro DK, Petri J, Gershenzon J, Bohlmann J, Pichersky E, et al. (2004) Characterization of a root-specific Arabidopsis terpene synthase responsible for the formation of the volatile monoterpene 1,8-cineole. Plant Physiol 135: 1956–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Fäldt J, Arimura G-i, Gershenzon J, Takabayashi J, Bohlmann J (2003) Functional identification of AtTPS03 as (E)-β-ocimene synthase: a monoterpene synthase catalyzing jasmonate-and wound-induced volatile formation in Arabidopsis thaliana. Planta 216: 745–751. [DOI] [PubMed] [Google Scholar]
- 64. Herde M, Gartner K, Kollner TG, Fode B, Boland W, Gershenzon J, et al. (2008) Identification and regulation of TPS04/GES, an Arabidopsis geranyllinalool synthase catalyzing the first step in the formation of the insect-induced volatile C16-homoterpene TMTT. Plant Cell 20: 1152–1168. 10.1105/tpc.106.049478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Huang M, Abel C, Sohrabi R, Petri J, Haupt I, Cosimano J, et al. (2010) Variation of herbivore-induced volatile terpenes among Arabidopsis ecotypes depends on allelic differences and subcellular targeting of two terpene synthases, TPS02 and TPS03. Plant Physiol 153: 1293–1310. 10.1104/pp.110.154864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Falara V, Akhtar TA, Nguyen TT, Spyropoulou EA, Bleeker PM, Schauvinhold I, et al. (2011) The tomato terpene synthase gene family. Plant Physiol 157: 770–789. 10.1104/pp.111.179648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dornelas MC, Mazzafera P (2007) A genomic approach to characterization of the Citrus terpene synthase gene family. Genetics and Molecular Biology 30: 832–840. [Google Scholar]
- 68. Külheim C, Yeoh SH, Maintz J, Foley WJ, Moran GF (2009) Comparative SNP diversity among four Eucalyptus species for genes from secondary metabolite biosynthetic pathways. BMC Genomics 10: 452 10.1186/1471-2164-10-452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Martin DM, Aubourg S, Schouwey MB, Daviet L, Schalk M, Toub O, et al. (2010) Functional annotation, genome organization and phylogeny of the grapevine (Vitis vinifera) terpene synthase gene family based on genome assembly, FLcDNA cloning, and enzyme assays. BMC Plant Biol 10: 226 10.1186/1471-2229-10-226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Nieuwenhuizen NJ, Green SA, Chen XY, Bailleul EJD, Matich AJ, Wang MY, et al. (2013) Functional Genomics Reveals That a Compact Terpene Synthase Gene Family Can Account for Terpene Volatile Production in Apple. Plant Physiol 161: 787–804. 10.1104/pp.112.208249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Amborella Genome P (2013) The Amborella genome and the evolution of flowering plants. Science 342: 1241089 10.1126/science.1241089 [DOI] [PubMed] [Google Scholar]
- 72. Thomas BC, Pedersen B, Freeling M (2006) Following tetraploidy in an Arabidopsis ancestor, genes were removed preferentially from one homeolog leaving clusters enriched in dose-sensitive genes. Genome Res 16: 934–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215: 403–410. [DOI] [PubMed] [Google Scholar]
- 74. Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, et al. (2012) Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res 40: D1178–D1186. 10.1093/nar/gkr944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Swarbreck D, Wilks C, Lamesch P, Berardini TZ, Garcia-Hernandez M, Foerster H, et al. (2008) The Arabidopsis Information Resource (TAIR): gene structure and function annotation. Nucleic Acids Res 36: D1009–D1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang X, Wang H, Wang J, Sun R, Wu J, Liu S, et al. (2011) The genome of the mesopolyploid crop species Brassica rapa. Nat Genet 43: 1035–1039. 10.1038/ng.919 [DOI] [PubMed] [Google Scholar]
- 77. Xu Q, Chen LL, Ruan X, Chen D, Zhu A, Chen C, et al. (2013) The draft genome of sweet orange (Citrus sinensis). Nat Genet 45: 59–66. 10.1038/ng.2472 [DOI] [PubMed] [Google Scholar]
- 78. Myburg AA, Grattapaglia D, Tuskan GA, Hellsten U, Hayes RD, Grimwood J, et al. (2014) The genome of Eucalyptus grandis. Nature 510: 356–362. 10.1038/nature13308 [DOI] [PubMed] [Google Scholar]
- 79. Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, et al. (2010) Genome sequence of the palaeopolyploid soybean. Nature 463: 178–183. 10.1038/nature08670 [DOI] [PubMed] [Google Scholar]
- 80. Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, et al. (2009) The Sorghum bicolor genome and the diversification of grasses. Nature 457: 551–556. 10.1038/nature07723 [DOI] [PubMed] [Google Scholar]
- 81. Xu X, Pan S, Cheng S, Zhang B, Mu D, Ni P, et al. (2011) Genome sequence and analysis of the tuber crop potato. Nature 475: 189–195. 10.1038/nature10158 [DOI] [PubMed] [Google Scholar]
- 82. Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, et al. (2009) The B73 maize genome: complexity, diversity, and dynamics. Science 326: 1112–1115. 10.1126/science.1178534 [DOI] [PubMed] [Google Scholar]
- 83. Cheng S, van den Bergh E, Zeng P, Zhong X, Xu J, Liu X, et al. (2013) The Tarenaya hassleriana genome provides insight into reproductive trait and genome evolution of crucifers. Plant Cell 25: 2813–2830. 10.1105/tpc.113.113480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Bombarely A, Rosli HG, Vrebalov J, Moffett P, Mueller LA, Martin GB (2012) A draft genome sequence of Nicotiana benthamiana to enhance molecular plant-microbe biology research. Mol Plant Microbe Interact 25: 1523–1530. 10.1094/MPMI-06-12-0148-TA [DOI] [PubMed] [Google Scholar]
- 85. van Bakel H, Stout JM, Cote AG, Tallon CM, Sharpe AG, Hughes TR, et al. (2011) The draft genome and transcriptome of Cannabis sativa. Genome Biol 12: R102 10.1186/gb-2011-12-10-r102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Wheeler DL (2008) GenBank. Nucleic Acids Res 36: D25–D30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Tang H, Lyons E, Pedersen B, Schnable JC, Paterson AH, Freeling M (2011) Screening synteny blocks in pairwise genome comparisons through integer programming. BMC Bioinformatics 12: 102 10.1186/1471-2105-12-102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Woodhouse MR, Tang H, Freeling M (2011) Different gene families in Arabidopsis thaliana transposed in different epochs and at different frequencies throughout the rosids. Plant Cell 23: 4241–4253. 10.1105/tpc.111.093567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Haas BJ, Delcher AL, Wortman JR, Salzberg SL (2004) DAGchainer: a tool for mining segmental genome duplications and synteny. Bioinformatics 20: 3643–3646. [DOI] [PubMed] [Google Scholar]
- 90. Dalquen DA, Dessimoz C (2013) Bidirectional best hits miss many orthologs in duplication-rich clades such as plants and animals. Genome Biol Evol 5: 1800–1806. 10.1093/gbe/evt132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Zdobnov EM, Apweiler R (2001) InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17: 847–848. [DOI] [PubMed] [Google Scholar]
- 92. Katoh K, Misawa K, Kuma K, Miyata T (2002) MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res 30: 3059–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Dai X, Wang G, Yang DS, Tang Y, Broun P, Marks MD, et al. (2010) TrichOME: a comparative omics database for plant trichomes. Plant Physiol 152: 44–54. 10.1104/pp.109.145813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Marks MD, Wenger JP, Gilding E, Jilk R, Dixon RA (2009) Transcriptome analysis of Arabidopsis wild-type and gl3-sst sim trichomes identifies four additional genes required for trichome development. Mol Plant 2: 803–822. 10.1093/mp/ssp037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Czechowski T, Stitt M, Altmann T, Udvardi MK, Scheible WR (2005) Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol 139: 5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Livak KJ, Schmittgen TD (2001) Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2− ΔΔCT Method. methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- 97. Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, et al. (2012) MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol 61: 539–542. 10.1093/sysbio/sys029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Miller M, Holder M, Vos R, Midford P, Liebowitz T, Chan L, et al. (2009) The CIPRES Portals. CIPRES; Available: http://wwwphyloorg/sub_sections/portal. [Google Scholar]
- 99. Drummond AJ, Rambaut A (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7: 214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Rambaut A (2009) FigTree version 1.3. 1. Computer program distributed by the author. Available: http://treebioedacuk/software/figtree/.
- 101. Rice P, Longden I, Bleasby A (2000) EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16: 276–277. [DOI] [PubMed] [Google Scholar]
- 102. Xiang T, Shibuya M, Katsube Y, Tsutsumi T, Otsuka M, Zhang H, et al. (2006) A New Triterpene Synthase from Arabidopsis t haliana Produces a Tricyclic Triterpene with Two Hydroxyl Groups. Organic letters 8: 2835–2838. [DOI] [PubMed] [Google Scholar]
- 103. Dvorakova L, Cvrckova F, Fischer L (2007) Analysis of the hybrid proline-rich protein families from seven plant species suggests rapid diversification of their sequences and expression patterns. BMC Genomics 8: 412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Abel S, Savchenko T, Levy M (2005) Genome-wide comparative analysis of the IQD gene families in Arabidopsis thaliana and Oryza sativa. BMC Evol Biol 5: 72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Wall DP, Fraser HB, Hirsh AE (2003) Detecting putative orthologs. Bioinformatics 19: 1710–1711. [DOI] [PubMed] [Google Scholar]
- 106. Dessimoz C, Gabaldon T, Roos DS, Sonnhammer EL, Herrero J, Quest for Orthologs Consortium (2012) Toward community standards in the quest for orthologs. Bioinformatics 28: 900–904. 10.1093/bioinformatics/bts050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Apweiler R, Attwood TK, Bairoch A, Bateman A, Birney E, Biswas M, et al. (2001) The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Res 29: 37–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Hunter S, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, et al. (2009) InterPro: the integrative protein signature database. Nucleic Acids Res 37: D211–215. 10.1093/nar/gkn785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, et al. (2014) InterProScan 5: genome-scale protein function classification. Bioinformatics 30: 1236–1240. 10.1093/bioinformatics/btu031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, et al. (2004) The Pfam protein families database. Nucleic Acids Res 32: D138–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Soding J (2005) Protein homology detection by HMM-HMM comparison. Bioinformatics 21: 951–960. [DOI] [PubMed] [Google Scholar]
- 112. Yeats C, Maibaum M, Marsden R, Dibley M, Lee D, Addou S, et al. (2006) Gene3D: modelling protein structure, function and evolution. Nucleic Acids Res 34: D281–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, et al. (2003) PANTHER: a library of protein families and subfamilies indexed by function. Genome Res 13: 2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Gough J, Karplus K, Hughey R, Chothia C (2001) Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. Journal of Molecular Biology 313: 903–919. [DOI] [PubMed] [Google Scholar]
- 115. Lipka V, Panstruga R (2005) Dynamic cellular responses in plant-microbe interactions. Curr Opin Plant Biol 8: 625–631. [DOI] [PubMed] [Google Scholar]
- 116. Hauser MT (2014) Molecular basis of natural variation and environmental control of trichome patterning. Front Plant Sci 5: 320 10.3389/fpls.2014.00320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Pattanaik S, Patra B, Singh SK, Yuan L (2014) An overview of the gene regulatory network controlling trichome development in the model plant, Arabidopsis. Front Plant Sci 5: 259 10.3389/fpls.2014.00259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Rensing SA, Lang D, Zimmer AD, Terry A, Salamov A, Shapiro H, et al. (2008) The Physcomitrella genome reveals evolutionary insights into the conquest of land by plants. Science 319: 64–69. [DOI] [PubMed] [Google Scholar]
- 119. Soltis PS, Soltis DE (2013) Angiosperm phylogeny: A framework for studies of genome evolution: Springer. [Google Scholar]
- 120. Estévez JM, Cantero A, Romero C, Kawaide H, Jiménez LF, Kuzuyama T, et al. (2000) Analysis of the expression of CLA1, a gene that encodes the 1-deoxyxylulose 5-phosphate synthase of the 2-C-methyl-D-erythritol-4-phosphate pathway in Arabidopsis. Plant Physiol 124: 95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Mandel MA, Feldmann KA, Herrera‐Estrella L, Rocha‐Sosa M, León P (1996) CLA1, a novel gene required for chloroplast development, is highly conserved in evolution. The Plant Journal 9: 649–658. [DOI] [PubMed] [Google Scholar]
- 122. Guimil S, Dunand C (2007) Cell growth and differentiation in Arabidopsis epidermal cells. J Exp Bot 58: 3829–3840. [DOI] [PubMed] [Google Scholar]
- 123. Plant Ontology Consortium (2002) The Plant Ontology consortium and plant ontologies. International Journal of Genomics 3: 137–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Avraham S, Tung CW, Ilic K, Jaiswal P, Kellogg EA, McCouch S, et al. (2008) The Plant Ontology Database: a community resource for plant structure and developmental stages controlled vocabulary and annotations. Nucleic Acids Res 36: D449–454. 10.1093/nar/gkm908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Jaiswal P, Avraham S, Ilic K, Kellogg EA, McCouch S, Pujar A, et al. (2005) Plant Ontology (PO): a Controlled Vocabulary of Plant Structures and Growth Stages. Comp Funct Genomics 6: 388–397. 10.1002/cfg.496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Wang X, Weigel D, Smith LM (2013) Transposon variants and their effects on gene expression in Arabidopsis. PLoS Genet 9: e1003255 10.1371/journal.pgen.1003255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Adams KL (2007) Evolution of duplicate gene expression in polyploid and hybrid plants. J Hered 98: 136–141. [DOI] [PubMed] [Google Scholar]
- 128. Qiu Y, Liu S-L, Adams KL (2014) Frequent changes in expression profile and accelerated sequence evolution of duplicated imprinted genes in Arabidopsis. Genome biology and evolution 6: 1830–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Hofberger JA, Nsibo DL, Govers F, Bouwmeester K, Schranz ME (2015) A complex interplay of tandem- and whole-genome duplication drives expansion of the L-type lectin receptor kinase gene family in the brassicaceae. Genome Biol Evol 7: 720–734. 10.1093/gbe/evv020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Kamolsukyunyong W, Sukhaket W, Ruanjaichon V, Toojinda T, Vanavichit A (2013) Single-feature polymorphism mapping of isogenic rice lines identifies the influence of terpene synthase on brown planthopper feeding preferences. Rice (N Y) 6: 18 10.1186/1939-8433-6-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Sallaud C, Giacalone C, Topfer R, Goepfert S, Bakaher N, Rosti S, et al. (2012) Characterization of two genes for the biosynthesis of the labdane diterpene Z-abienol in tobacco (Nicotiana tabacum) glandular trichomes. Plant J 72: 1–17. 10.1111/j.1365-313X.2012.05068.x [DOI] [PubMed] [Google Scholar]
- 132.Mendes MD, Barroso JG, Oliveira MM, Trindade H (2014) Identification and characterization of a second isogene encoding γ-terpinene synthase in Thymus caespititius. J Plant Physiol. [DOI] [PubMed]
- 133. Lawler I, Foley W, Eschler B, Pass D, Handasyde K (1998) Intraspecific variation in Eucalyptus secondary metabolites determines food intake by folivorous marsupials. Oecologia 116: 160–169. [DOI] [PubMed] [Google Scholar]
- 134. Trontin C, Tisné S, Bach L, Loudet O (2011) What does Arabidopsis natural variation teach us (and does not teach us) about adaptation in plants? Curr Opin Plant Biol 14: 225–231. 10.1016/j.pbi.2011.03.024 [DOI] [PubMed] [Google Scholar]
- 135. Edger PP, Pires JC (2009) Gene and genome duplications: the impact of dosage-sensitivity on the fate of nuclear genes. Chromosome Res 17: 699–717. 10.1007/s10577-009-9055-9 [DOI] [PubMed] [Google Scholar]
- 136. Bekaert M, Edger PP, Pires JC, Conant GC (2011) Two-phase resolution of polyploidy in the Arabidopsis metabolic network gives rise to relative and absolute dosage constraints. Plant Cell 23: 1719–1728. 10.1105/tpc.110.081281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Fawcett JA, Maere S, Van de Peer Y (2009) Plants with double genomes might have had a better chance to survive the Cretaceous-Tertiary extinction event. Proc Natl Acad Sci U S A 106: 5737–5742. 10.1073/pnas.0900906106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Fang L, Cheng F, Wu J, Wang X (2012) The Impact of Genome Triplication on Tandem Gene Evolution in Brassica rapa. Front Plant Sci 3: 261 10.3389/fpls.2012.00261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Roulin A, Auer PL, Libault M, Schlueter J, Farmer A, May G, et al. (2013) The fate of duplicated genes in a polyploid plant genome. The Plant Journal 73: 143–153. 10.1111/tpj.12026 [DOI] [PubMed] [Google Scholar]
- 140. Mühlhausen S, Kollmar M (2013) Whole genome duplication events in plant evolution reconstructed and predicted using myosin motor proteins. BMC evolutionary biology 13: 202 10.1186/1471-2148-13-202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Chen R, Harada Y, Bamba T, Nakazawa Y, Gyokusen K (2012) Overexpression of an isopentenyl diphosphate isomerase gene to enhance trans-polyisoprene production in Eucommia ulmoides Oliver. BMC Biotechnol 12: 78 10.1186/1472-6750-12-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Berthelot K, Estevez Y, Deffieux A, Peruch F (2012) Isopentenyl diphosphate isomerase: A checkpoint to isoprenoid biosynthesis. Biochimie 94: 1621–1634. 10.1016/j.biochi.2012.03.021 [DOI] [PubMed] [Google Scholar]
- 143. Okada K, Kasahara H, Yamaguchi S, Kawaide H, Kamiya Y, Nojiri H, et al. (2008) Genetic evidence for the role of isopentenyl diphosphate isomerases in the mevalonate pathway and plant development in Arabidopsis. Plant Cell Physiol 49: 604–616. 10.1093/pcp/pcn032 [DOI] [PubMed] [Google Scholar]
- 144. Schranz ME, Mitchell-Olds T (2006) Independent ancient polyploidy events in the sister families Brassicaceae and Cleomaceae. Plant Cell 18: 1152–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Hu TT, Pattyn P, Bakker EG, Cao J, Cheng JF, Clark RM, et al. (2011) The Arabidopsis lyrata genome sequence and the basis of rapid genome size change. Nat Genet 43: 476–481. 10.1038/ng.807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Zhao YJ, Cheng QQ, Su P, Chen X, Wang XJ, Gao W, et al. (2014) Research progress relating to the role of cytochrome P450 in the biosynthesis of terpenoids in medicinal plants. Appl Microbiol Biotechnol 98: 2371–2383. 10.1007/s00253-013-5496-3 [DOI] [PubMed] [Google Scholar]
- 147. Naur P, Petersen BL, Mikkelsen MD, Bak S, Rasmussen H, Olsen CE, et al. (2003) CYP83A1 and CYP83B1, two nonredundant cytochrome P450 enzymes metabolizing oximes in the biosynthesis of glucosinolates in Arabidopsis. Plant Physiol 133: 63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Glenn WS, Runguphan W, O'Connor SE (2013) Recent progress in the metabolic engineering of alkaloids in plant systems. Curr Opin Biotechnol 24: 354–365. 10.1016/j.copbio.2012.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Carretero-Paulet L, Fares MA (2012) Evolutionary dynamics and functional specialization of plant paralogs formed by whole and small-scale genome duplications. Mol Biol Evol 29: 3541–3551. 10.1093/molbev/mss162 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLS)
(XLS)
Numbers are quotients of the module-wise gene count of terpenoid biosynthetic pathways and the number of all protein-coding genes within the whole genome. Species with highest and lowest relative pathway size among all analyzed species are color-coded as indicated in the legend.
(XLS)
(XLS)
Red indicates absence of tandem/ohnolog duplicates. Green indicates significant enrichment among terpenoid biosynthetic genes compared to background with threshold of 0.01.
(XLS)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.