

Abstract
An expanded G4C2 repeat in C9orf72 represents the most common known genetic cause of amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD). However, the lower limit for pathological expansions is unknown (the suggested cutoff is 30 repeats). It has been proposed that the expansion might have occurred only once in human history and subsequently spread throughout the population. However, our present findings support a hypothesis of multiple origins for the expansion. We report a British-Canadian family in whom a ∼70-repeat allele from the father (unaffected by ALS or FTLD at age 89 years) expanded during parent-offspring transmission and started the first generation affected by ALS (four children carry an ∼1,750-repeat allele). Epigenetic and RNA-expression analyses further discriminated the offspring’s large expansions (which were methylated and associated with reduced C9orf72 expression) from the ∼70-repeat allele (which was unmethylated and associated with upregulation of C9orf72). Moreover, RNA foci were only detected in fibroblasts from offspring with large expansions, but not in the father, who has the ∼70-repeat allele. All family members with expansions were found to have an ancient known risk haplotype, although it was inherited on a unique 5-Mb genetic backbone. We conclude that small expansions (e.g., 70 repeats) might be considered “pre-mutations” to reflect their propensity to expand in the next generation. Follow-up studies might help explain the high frequency of ALS- or FTLD-affected individuals with an expansion but without a familial history (e.g., 21% among Finnish ALS subjects).
Main Text
It is now recognized that amyotrophic lateral sclerosis (ALS [MIM: 612069]) and frontotemporal lobar degeneration (FTLD [MIM: 600274]) have significant clinico-pathological overlap. ALS symptoms are caused by the degeneration of motor neurons in the cerebral cortex, brainstem, and spinal cord and lead to paralysis and death, most commonly as a result of respiratory failure;1 FTLD symptoms are caused by the degeneration of the frontal and temporal lobes of the cerebral cortex and lead to a behavioral and/or language disorder.2 Several overlapping genes have been associated with ALS and FTLD, providing strong evidence of common pathological mechanisms.3 An expanded G4C2 repeat in the non-coding region of C9orf72 (MIM: 614260) represents the most common known genetic cause of both ALS and FTLD in populations of European ancestry.4,5 It has been detected in up to 29% of FTLD and 50% of ALS subjects, as well as 88% of subjects with both FTLD and ALS symptoms, including familial and simplex individuals.4,6,7
The size of expansions is very variable, and whether these alleles have the same pathological significance is unknown. Even the lower limit for pathological expansions is not yet established, and the majority of studies use the originally suggested cutoff of 30 repeats.4,5 Because most of C9orf72 genotyping is done by repeat-primed PCR (rp-PCR), the exact size of the expansion cannot be determined beyond a certain length (i.e., when it is larger than 50–70 repeats). When measured by Southern blot, the estimated size of large expansions can vary from several hundred to thousands of repeats;8–10 however, some individuals show relatively small expansions (30–150 repeats).6,10,11 Whether these small expansions have pathogenic significance needs to be studied carefully. For example, we reported a Parkinson-disease-affected individual who carries 39 repeats that do not segregate with disease.6
The suggested C9orf72-related disease mechanisms are complex. One possibility is loss of function through haploinsufficiency, supported by the fact that individuals with expansions show ∼50% fewer C9orf72 transcripts,4,5 possibly as a result of epigenetic alterations. Indeed, we previously demonstrated that DNA hypermethylation of a CpG island adjacent to the 5′ end of the G4C2 repeat is expansion specific.12,13 Moreover, a higher degree of methylation was associated with familial ALS and shorter disease duration.12 Given that hypermethylation was only detected in a portion of individuals with expansions (36% in ALS), we recently tested whether the G4C2 expansion itself could be the main methylation site. Using a qualitative assay, we demonstrated that the G4C2 expansion is generally methylated in 100% of unrelated individuals with alleles of >90 repeats, whereas the small or intermediate alleles (2–90 repeats) are completely unmethylated.14
The search for the origin of the C9orf72 repeat expansion identified an ∼200-kb risk haplotype (known as the “Finnish” risk haplotype) in all individuals with expansions.15–19 It led to the common founder hypothesis proposing that the expansion occurred as a single event and subsequently spread throughout the population.18,20 In contrast, a multiple-origins hypothesis suggests that the risk haplotype formed a permissive allele associated with repeat instability and predisposed the G4C2 repeat to expand to a pathogenic allele on multiple occasions in human history.20,21 However, only a limited number of C9orf72 studies have used Southern blotting—in which a wild-type allele (2–30 repeats) gives a single band and large expansions appear as a smear—to size the expansion. Somatic instability can lead to very different repeat sizes within and among tissues of the same individual.9 This was recently confirmed in a report of an ALS subject with ∼3,000 repeats in CNS tissues but only ∼90 repeats in blood.21 Of note, this individual had the same risk haplotype as other subjects with C9orf72 repeat expansions, supporting the permissive nature of an ∼90-repeat allele to expand.
Observations of somatic instability of the G4C2 repeat are in favor of multiple origins of expansions, but direct evidence of this hypothesis could come from the investigation of unaffected parents of subjects with C9orf72 repeat expansions and affected by ALS or FTLD. In the current genetic and epigenetic study, we present a unique family in whom the repeat stretches from small to large expansions when passed to the offspring (the first generation affected by ALS).
In accordance with the ethical review board, informed consent was obtained from all study participants. PED25 is a Canadian family of British origin (Figure 2). The parents, 9685 (II-1) and 9686 (II-2), had five children, two of whom, 9548 (III-2) and 8665 (III-5), were diagnosed with ALS at Sunnybrook Health Sciences Centre according to the revised El Escorial Criteria.1 Both parents (90 and 89 years old) are alive and, according to neurological assessments, do not show symptoms of ALS or FTLD. The detailed clinical findings can be found in the Supplemental Data (“Clinical Descriptions”).
Figure 2.
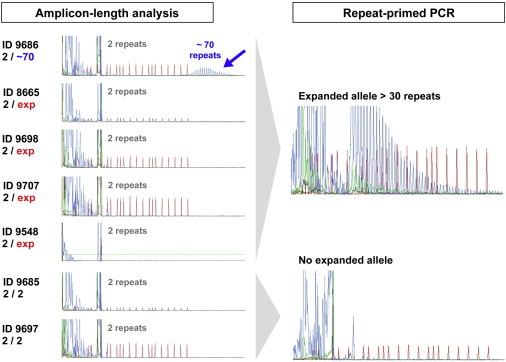
Results of C9orf72 Genotyping
PCR products of amplicon-length analysis and rp-PCR were separated on an ABI3100 DNA Analyzer and visualized by GeneScan software. Although five family members were identified to have expansions in the rp-PCR, only individual 9686 had a small expansion of ∼70 repeats.
Samples of blood DNA were collected from seven family members in two generations (Figure 1). The C9orf72 G4C2 repeat was genotyped by a two-step strategy as described previously6 with minor modifications. In the first step, undiluted PCR products were resolved on a 3100 DNA analyzer (amplicon-length analysis). In the second step, rp-PCR was used for determining the presence of large expansions.4,5
Figure 1.
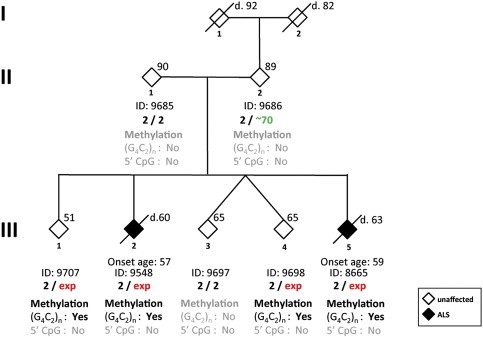
Pedigree of Family PED25
Individual ID and C9orf72 genotype are shown beneath the corresponding diamond. Arabic numbers indicate the repeat number, and “exp” represents the expansion allele. The age at time of examination is shown in the upper right corner. The age of death is indicated by a prefix of “d.” The age of onset is indicated for individuals with disease above the ID number. For protecting confidentiality, the gender of family members is masked. The results of methylation analyses for the 5′ CpG island and for (G4C2)n methylation are shown beneath the C9orf72 genotype. “Yes” represents the presence of methylation, and “no” represents the absence of methylation.
The rp-PCR revealed repeat expansions in five family members, including the unaffected 89-year-old father (9686 [II-2]) and his four children, two of whom are affected by ALS (Figures 1 and 2). Notably, the amplicon-length analysis revealed that the father has a small expansion of ∼70 repeats, whereas his children have larger expansions beyond a detectable range (Figure 2). Issues of paternity were excluded by genotyping for eight randomly selected autosomal short tandem repeat (STR) markers (D2S2166, D5S406, D7S493, D10S1651, D11S908, D15S978, D18S452, and D21S266; data not shown). Sanger sequencing of other ALS- and FTLD-associated genes (SOD1, FUS, and GRN) in the ALS-affected proband (9548 [III-2]) was performed as reported previously22 and did not reveal additional pathogenic mutations. So far, no clinical sign of ALS or dementia was found for two of four children with a large expansion: 9698 (III-4; age 65 years) and 9707 (III-1; age 51 years). This is most likely due to the high clinical heterogeneity, including a variable age of onset, in C9orf72-related diseases. For instance, we previously reported a pair of monozygotic twins who both have large expansions and have been discordant for ALS for at least 6 years.23 However, the penetrance of the pathological expansion in C9orf72 reaches ∼100% by age 80 years.15,24
To estimate the size of the G4C2 repeat, we conducted Southern blotting as previously described.9 In brief, a total of 7–10 μg of DNA was digested with XbaI (Promega) and electrophoresed on 0.8% agarose gel. Subsequently, DNA was transferred to a positively charged nylon membrane (Roche), cross-linked, and then hybridized with a digoxigenin (DIG)-labeled probe. Expansions were visualized with anti-DIG antibody (Roche) and CDP-star substrate (Roche) on X-ray film. DNA sufficient for Southern blotting of six PED25 family members (not 8665) was obtained. For one deceased family member (9548), DNA was also obtained from multiple autopsy tissues (cerebellar vermis, orbitofrontal cortex, primary motor cortex, superior temporal gyrus, hippocampus, and medulla dissected from the fresh right hemisphere, as well as the spinal cord, heart, kidney, liver, and skin). In addition, Southern blotting was done for fibroblasts derived from skin biopsy of three family members with large expansions (9698, 9707, and 9548). The fibroblasts were generated from 3-mm dermal explants and cultured in 6-well plates pre-coated with 0.1% gelatin. Cells were maintained in DMEM with high glucose (11995, Invitrogen), supplemented with 20% (v/v) fetal bovine serum (GIBCO), 100 U/ml penicillin, and 100 μg/ml streptomycin (GIBCO). Cells were then maintained at 37°C and 5% CO2 in a humidified incubator.
Southern blot analysis of blood DNA confirmed the substantial difference in expansion size between the two generations. The father showed two distinct bands corresponding to a wild-type allele (two repeats) and a small expansion (∼70 repeats). In contrast, four of his children demonstrated a large smear similar in length, and the most abundant expansion sizes were ∼650 and ∼1,750 repeats (Figure 3). Large expansions were also revealed in all investigated autopsy tissues of ALS-affected individual 9548, in whom repeat numbers over 3,500 were detected in CNS tissues; in contrast, the cerebellum showed smaller expansions (∼1,750 repeats; Figure 4), a general phenomenon observed in subjects with C9orf72 repeat expansions.9 Fibroblasts showed a relatively small expansion as a well-defined band (∼750 repeats; Figure 4). In the liver, we observed an expansion band corresponding to ∼3,000 repeats; in other non-CNS tissues, we detected a higher level of somatic instability (not shown) comparable to that seen in blood (Figure 4).
Figure 3.
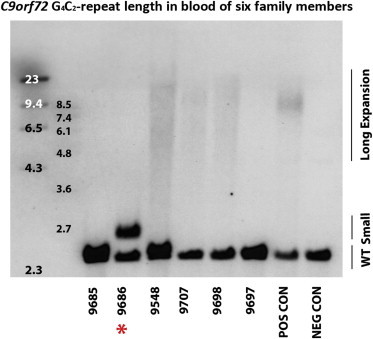
Southern Blot Results from Members of Family PED25
Southern blot analysis using blood DNA confirmed the presence of a small expansion in 9686 (highlighted by the red star), the presence of large expansions in 9698, 9707, and 9548, and the absence of an expansion in 9685 and 9697. The molecular-weight marker is shown on the left side of the Southern blot. “POS CON” and “NEG CON” refer to positive and negative control individuals, respectively.
Figure 4.
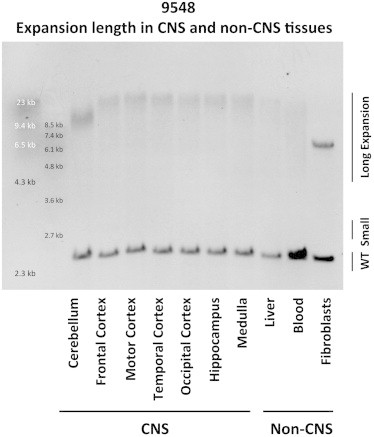
Southern Blot Results from Autopsy Tissues of Individual 9548
Southern blot analysis using genomic DNA extracted from autopsy tissues confirmed the presence of large expansions in CNS and non-CNS tissues from individual 9548.
To study the epigenetic profile of C9orf72 in family PED25, we used an aliquot from the same DNA preparation as for Southern blot. In addition to generating fibroblasts from 9698, 9707, and 9548, we generated fibroblasts for two more family members (9697 and 9686) and two unrelated healthy control individuals.
The methylation level of the CpG island 5′ to the G4C2 repeat was estimated via direct bisulfite sequencing as described previously.12 In brief, each DNA sample was sequenced after bisulfite conversion, after which all unmethylated C nucleotides were converted to T nucleotides, and methylated C remain unchanged. A total of 26 CpG sites were studied at the CpG island 5′ to the G4C2 repeat and were classified as either unmethylated (T peak) or methylated (T-C double peaks). None of the family members demonstrated increased DNA methylation at the CpG island 5′ to the repeat in blood (Figure 1).
Methylation of the G4C2 repeat itself was studied with our recently reported qualitative (G4C2)n-methylation assay.14 In brief, each sample was amplified by rp-PCR with primers specific to methylated or unmethylated DNA after bisulfite conversion. The primers were labeled by FAM for amplification of methylated DNA (blue channel) and HEX for amplification of unmethylated DNA (green channel). Data were visualized by Genotyper Software (version 3.6, Applied Biosystems). If the G4C2 repeat was methylated in all DNA copies, only the blue channel was expected to have products, whereas if the G4C2 repeat was unmethylated in all DNA copies, only the green channel was expected to have products. If the G4C2 repeat was methylated in some DNA copies, both channels would have products. We used a yes/no test to evaluate the methylation of ∼30 repeats at the 5′ end of the expansion (this test was sensitive enough to detect methylation in a mixture of DNA standards containing only 5% highly methylated DNA).14
The (G4C2)n-methylation assay demonstrated that all four children with large expansions had CpG methylation of the G4C2 repeat itself (Figures 1 and 5). The autopsy tissues of 9548 were also shown to have methylation of the expanded G4C2 repeat (Figure S1). In contrast, (G4C2)n methylation was not detected in the blood DNA of the two family members without an expansion (9685 and 9697) or of the father (9686), who has the ∼70-repeat allele (Figure 5). Fibroblasts generated from the skin of the father did not show (G4C2)n methylation either (Figure S2). Surprisingly, fibroblasts from all three family members with large expansions (9698, 9707, and 9548) also did not have (G4C2)n methylation (Figure S2). Importantly, for the deceased individual (9548), we obtained enough skin DNA, which clearly demonstrated (G4C2)n methylation (Figure S1B). Our results suggest that C9orf72-related phenotypes in fibroblast-based functional studies should be interpreted with caution because the epigenetic profile has been reset in such cell lines.
Figure 5.
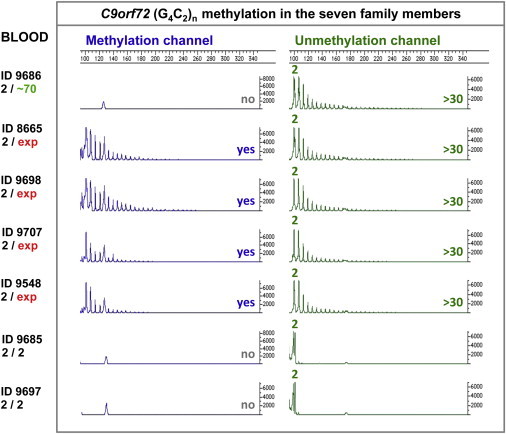
Chromatograms of the (G4C2)n-Methylation Assay in Blood Samples from Family PED25
Methylated and unmethylated G4C2-repeat alleles were detected by a methylation channel and a non-methylation channel, respectively. Detectable repeat size was marked along with the corresponding peak.
For assessing C9orf72 expression, total RNA was extracted from blood with the RNeasy Mini Kit (QIAGEN) and reverse transcribed to cDNA with oligo dT primers and the AffinityScript Multiple Temperature cDNA Synthesis Kit (Agilent Technologies). RNA integrity was checked on an Agilent 2100 Bioanalyzer, and only samples with an RNA integrity number > 7 were used. Real-time PCR was performed on an ABI Prism 7500 System (Life Technologies). We used inventoried TaqMan expression assays for C9orf72 (Hs00376619_m1) and five housekeeping genes including HPRT1 (MIM: 308000; Hs99999909_m1), UBC (MIM: 191340; Hs00824723_m1), B2M (MIM: 109700; Hs99999907_m1), GAPDH (MIM: 138400; Hs00266705_g1) and TBP (MIM: 600075; Hs00427620_m1) (Applied Biosystems). Samples were run in triplicate. All tested housekeeping genes were evaluated for expression stability via the geNorm algorithm incorporated in the software qbase+ (Biogazelle),25 which determined HPRT1, UBC, and B2M to be the suitable reference genes for the blood samples and GAPDH and UBC to be suitable reference genes for the fibroblasts samples. Relative quantification was calculated with the ddCt method after normalization to the corresponding reference genes.
The inventoried TaqMan gene-expression assay detected all three known C9orf72 transcripts in blood from five family members, four of whom (in addition to two normal control individuals) also had fibroblasts available to be tested (Figure 6). Expression of C9orf72 was correlated with the status of (G4C2)n methylation. Blood RNA from family members with methylated large G4C2-repeat expansions (n = 3) showed less gene expression than did blood RNA from family members with normal alleles (n = 2). In contrast, fibroblast DNA from the same three individuals with large expansions was not methylated at the G4C2 repeat and expressed C9orf72 at a level similar to that in fibroblast DNA from individuals with normal alleles (Figure 6). Surprisingly, C9orf72 mRNA levels were higher in the blood and fibroblasts of the father (who has the unmethylated 70-repeat allele) than in those of the control individuals (Figure 6). A follow-up test confirmed that his C9orf72 expression in blood cells was higher than that in three additional unrelated normal control individuals (9446, 9593, and 9600; Figure S4).
Figure 6.
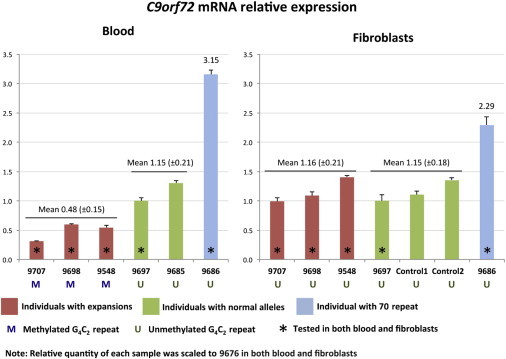
Relative Quantification of C9orf72 mRNA in Blood and Fibroblast Samples from Family PED25
Both blood and fibroblasts of individuals with expansions (9548, 9698, and 9707) were tested. We also tested blood of individuals with normal alleles (9685 and 9697), as well as fibroblasts of 9697 and two normal control individuals. The relative quantity of each sample was scaled to that of individual 9676. Error bars represent the SE of the triplicate reactions.
To assess RNA foci in individuals with wild-type alleles and G4C2 expansions, we carried out RNA fluorescence in situ hybridization on fibroblasts by using a locked nucleic acid probe recognizing the sense strand of the repeat expansion in C9orf72 according to previously published protocols26,27 (Figure S5). Fibroblasts from an unrelated healthy control individual were examined alongside those of the PED25 family members, including 9697 (with normal alleles), 9686 (with the 70-repeat allele), and 9698, 9707, and 9548 (with large expansions). RNA foci were detected in the fibroblasts from all three individuals with large expansions: 9698 (26.0% of cells, n = 3 experiments), 9707 (20.4% of cells, n = 3 experiments), and 9548 (27.5% of cells, n = 3 experiments) (Figure S5). These values are comparable to those in a published report, which found that 15%–45% of fibroblasts from subjects with expansions were positive for sense-strand RNA foci.26 In contrast, no or negligible levels of RNA-foci-positive cells were detected in fibroblasts from 9686, who has the 70-repeat allele (0.006% of cells, n = 2 experiments); similar results were observed for the control individual (0% of cells, n = 3 experiments) and 9697 (0% of cells, n = 3 experiments) (Figure S5). These results further support that the 70-repeat allele is not likely to be pathogenic.
To examine whether PED25 members share a haplotype, we studied six STR markers (D9S171, D9S1679, D9S259, D9S2154, D9S161, and D9S319) spanning a 5-Mb region flanking C9orf72. STR markers were amplified by PCR with one FAM-labeled primer, diluted, and analyzed on an ABI 3730 DNA Analyzer (Applied Biosystems). Alleles were scored with GeneMapper Software version 5 (Applied Biosystems) and normalized to CEPH standard genotypes. STR genotypes were obtained for six members of family PED25 and two expansion-affected individuals from the published family VSM-20.4 In addition, we genotyped three unrelated persons with small expansions in C9orf72 and 11 subjects with repeats in the long wild-type range (21–30 repeats). The details of all STR assays are available upon request. TaqMan SNP-genotyping assays were performed for rs4879515 (C_31009935_20) and rs3849942 (C_27515934_20; Thermo Fisher Scientific) on a 7900HT Fast Real-Time PCR System; genotype calls were made with SDS 2.4 software (Applied Biosystems).
Haplotype analysis showed that the father (9686, who has the small expansion) and his children with large expansions (9548, 9707, and 9698), but not family members without expansions (9685 and 9697), share a 5-Mb haplotype flanking C9orf72 (Table 1). Interestingly, STR alleles in PED25 family members with expansions were different from those observed in family VSM-204 (Table 1) or in any of the investigated subjects with small expansions or with repeats in the long wild-type range (Table S1 and Figure S3). Assessment of SNPs mapping within 100 kb of the expansion and tagging the reported “Finnish” C9orf72 risk haplotype4 revealed that the father and his children with large expansions do share a much smaller haplotype with family VSM-20. We also detected the same risk haplotype in subjects with small expansions and subjects with repeats in the long wild-type range, including control individuals (Table S1).
Table 1.
STR Markers and SNPs for Members of Family PED25
| Marker | Mb |
Family PED25 |
VSM-20 (a) |
VSM-20 (b) |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
9685 |
9686 |
9548 |
9707 |
9698 |
9697 |
||||||||||||
| a1 | a2 | a1 | a2 | a1 | a2 | a1 | a2 | a1 | a2 | a1 | a2 | a1 | a2 | a1 | a2 | ||
| D9S171 | 24.53 | 167 | 185 | 167 | 167 | 167 | 185 | 167 | 167 | 167 | 185 | 167 | 185 | 185 | 183 | 185 | 185 |
| D9S1679 | 24.78 | 135 | 135 | 131 | 137 | 131 | 135 | 131 | 135 | 131 | 135 | 137 | 135 | 135 | 135 | 135 | 131 |
| D9S259 | 26.02 | 288 | 292 | 288 | 280 | 288 | 292 | 288 | 288 | 288 | 292 | 280 | 292 | 288 | 280 | 288 | 288 |
| D9S2154 | 26.17 | 138 | 150 | 142 | 148 | 142 | 150 | 142 | 138 | 142 | 150 | 146 | 150 | 146 | 150 | 146 | 134 |
| rs4879515∗ | 27.48 | C | C | T | C | T | C | T | C | T | C | C | C | T | T | T | C |
| rs3849942∗ | 27.54 | G | G | A | G | A | G | A | G | A | G | G | G | A | A | A | G |
| C9orf72∗ | 27.57 | 2 | 2 | EXP | 2 | EXP | 2 | EXP | 2 | EXP | 2 | 2 | 2 | EXP | 2 | EXP | 5 |
| D9S161 | 27.63 | 119 | 127 | 119 | 127 | 119 | 119 | 119 | 127 | 119 | 119 | 127 | 119 | 117 | 117 | 117 | 129 |
| D9S319 | 29.56 | 159 | 159 | 163 | 171 | 163 | 159 | 163 | 159 | 163 | 159 | 171 | 159 | 167 | 167 | 167 | 163 |
Segregation of the repeat expansion in C9orf72 and genetic markers flanking the area of the repeat expansion was observed in family PED25. The “C9orf72” row displays the number of repeats on allele 1 (a1) and allele 2 (a2). Repeat expansions are indicated with “EXP.” Asterisks denote the shared C9orf72 risk haplotype.
In summary, we report a British-Canadian family in whom a paternal ∼70-repeat allele in C9orf72 expanded considerably in the next generation and passed large G4C2 expansions (∼1,750 repeats) on to four children. Notably, it is possible that an individual can carry a small expansion in blood but a large expansion in the CNS.9 Because the father is alive, we can only study DNA from his blood and fibroblasts; however, it is unlikely that a large expansion would occur in his CNS tissues, given that he remains unaffected by ALS or FTLD even at 89 years of age and that penetrance of pathological G4C2 expansions was found to be almost 100% by 80 years of age.15,24 Thus, the possibility that he will develop ALS or FTLD in the coming years is low. In contrast, two of his children with large expansions developed ALS in their 50s. Importantly, one of them (the deceased individual) showed large expansions in all studied CNS and non-CNS tissues. Our observation that ALS segregates with large but not small expansions demands a better-defined cutoff for pathogenic repeat number. Small expansions could be considered pre-mutations because of their potential instability, which could lead to larger expansions in the next generation.
Although we do not have access to a sperm sample from the elderly father, the expansion event most likely happened in his germ cells rather than occurring four times separately in each child and reaching a similar size (Figure 3). Notably, many diseases caused by repeat expansions are predominantly inherited through paternal transmissions, most likely as a result of germ-cell-specific mutations.28 Our study is in favor of the multiple-origins hypothesis of C9orf72 expansions.21 Indeed, it does not seem plausible that the expansion occurred just once in human history, given that such events are still ongoing (i.e., in family PED25).
It was shown that wild-type C9orf72 alleles (<20 repeats) are stable between generations.6 In contrast, intermediate alleles (20–30 repeats) and small expansions (30–150 repeats) were reported to be susceptible to unfaithful inheritance8,10 or somatic instability.9,29,30 This might suggest a mechanism for the enlargement of repeat number, implying that the critical repeat length makes it difficult for the DNA machinery to maintain the precise number of repeats. Another mechanism could involve the C9orf72 risk haplotype as a prerequisite for the repeat expansion to occur. Both mechanisms support the multiple-origins hypothesis of C9orf72 expansions. In family PED25, we observed an ancient C9orf72 risk haplotype inherited on a unique genetic backbone. The father and offspring with expansions each carry a distinct 5-Mb haplotype that is not observed in family VSM-20 or other investigated subjects, but a smaller, ∼100-kb region that is part of a known risk haplotype was present in family PED25 and in VSM-20 family members with large expansions or alleles with 21–30 repeats. Notably, SNPs tagging the risk haplotype are common in white individuals; for instance, rs3849942, a surrogate marker for the repeat expansion, has a minor allele frequency of 23% in control individuals.4 Sequencing of the entire risk haplotype in subjects with C9orf72 repeat expansions and normal control individuals with the minor rs3849942 allele might reveal the variant(s) that predispose individuals to G4C2-repeat instability.
Results of the epigenetic, expression, and RNA-foci analyses further discriminate large expansions from the 70-repeat allele, arguing against its pathogenic nature. In contrast to the large expansions in the offspring, the 70-repeat allele is not methylated (Figure 5). Intriguingly, detecting methylation in CNS tissues required more than twice the amount of input DNA than in non-CNS tissues, which might indicate a lower level of methylation in the CNS. Therefore, it is tempting to speculate that methylation of the repeat expansion is a protective mechanism to minimize the production of toxic RNA and dipeptides, which lead to collateral damage (haploinsufficiency). Indeed, we demonstrated that the presence of (G4C2)n methylation is linked to decreased expression of C9orf72. In blood, methylation of large G4C2 expansions was associated with reduced C9orf72 mRNA levels, whereas in fibroblasts derived from the same subjects, (G4C2)n methylation was not detected, and C9orf72 was expressed at normal levels (Figure 6). Remarkably, C9orf72 mRNA levels were increased in both blood and fibroblasts from the PED25 father (who has the unmethylated 70-repeat allele), reminiscent of what was seen for FMR1 (MIM: 309550), a gene in which mutations cause fragile X mental retardation syndrome (MIM: 300624) as a result of a CGG-repeat expansion. Methylation of a fully expanded pathogenic FMR1 allele (>200 CGG repeats) led to transcriptional silencing of FMR1,31 whereas the level of FMR1 mRNA was elevated by 2- to 5-fold in unaffected individuals with pre-mutations (55–200 CGG repeats).32 For C9orf72, the 70-repeat allele might also be considered a “pre-mutation” to reflect its propensity for substantial expansion in the next generation.
In conclusion, our data suggest that the repeat expansion in C9orf72 might have occurred on multiple occasions in human history and encourage the investigation of families similar to PED25. Specifically, it would be important to investigate the unaffected parents of individuals with disease to establish a better cutoff for pathological repeat number, which is critical in the clinical utility of genetic screening, as well as for functional studies of C9orf72. Such investigations might help explain the high frequency of simplex cases of C9orf72 repeat expansions, each of which could be the first generation of a familial form of ALS and/or FTLD. Indeed, the incidence of C9orf72 repeat expansions in subjects without a family history of ALS or FTLD is unusually high for a monogenic disorder (e.g., 21% among Finnish individuals with ALS).5
Acknowledgments
This work was supported by the W. Garfield Weston Foundation (E.R., Z.X., J.R., J.K., and L.Z.), the James Hunter Family ALS Initiative (L.Z. and J.R.), an ALS Canada Bernice Ramsey Discovery Grant (Y.Y. and L.Z.), and NIH grant R01 NS080882. J.R. is a Tier 2 Canada Research Chair in ALS; P.M.M. holds an Alzheimer Society of Canada Doctoral Award; P.M. holds an American ALS Association Milton Safenowitz Postdoctoral Fellowship; J.R.M. is a James Hunter Senior Research Fellow; and M.v.B. is supported by the Milton Safenowitz Post-Doctoral Fellowship for ALS Research from the ALS Association.
Contributor Information
Lorne Zinman, Email: lorne.zinman@sunnybrook.ca.
Ekaterina Rogaeva, Email: ekaterina.rogaeva@utoronto.ca.
Supplemental Data
References
- 1.Brooks B.R., Miller R.G., Swash M., Munsat T.L., World Federation of Neurology Research Group on Motor Neuron Diseases El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. Other Motor Neuron Disord. 2000;1:293–299. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- 2.Neary D., Snowden J.S., Gustafson L., Passant U., Stuss D., Black S., Freedman M., Kertesz A., Robert P.H., Albert M. Frontotemporal lobar degeneration: a consensus on clinical diagnostic criteria. Neurology. 1998;51:1546–1554. doi: 10.1212/wnl.51.6.1546. [DOI] [PubMed] [Google Scholar]
- 3.Hardy J., Rogaeva E. Motor neuron disease and frontotemporal dementia: sometimes related, sometimes not. Exp. Neurol. 2014;262(Pt B):75–83. doi: 10.1016/j.expneurol.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 4.DeJesus-Hernandez M., Mackenzie I.R., Boeve B.F., Boxer A.L., Baker M., Rutherford N.J., Nicholson A.M., Finch N.A., Flynn H., Adamson J. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Renton A.E., Majounie E., Waite A., Simón-Sánchez J., Rollinson S., Gibbs J.R., Schymick J.C., Laaksovirta H., van Swieten J.C., Myllykangas L., ITALSGEN Consortium A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xi Z., Zinman L., Grinberg Y., Moreno D., Sato C., Bilbao J.M., Ghani M., Hernández I., Ruiz A., Boada M. Investigation of c9orf72 in 4 neurodegenerative disorders. Arch. Neurol. 2012;69:1583–1590. doi: 10.1001/archneurol.2012.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cruts M., Gijselinck I., Van Langenhove T., van der Zee J., Van Broeckhoven C. Current insights into the C9orf72 repeat expansion diseases of the FTLD/ALS spectrum. Trends Neurosci. 2013;36:450–459. doi: 10.1016/j.tins.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 8.Beck J., Poulter M., Hensman D., Rohrer J.D., Mahoney C.J., Adamson G., Campbell T., Uphill J., Borg A., Fratta P. Large C9orf72 hexanucleotide repeat expansions are seen in multiple neurodegenerative syndromes and are more frequent than expected in the UK population. Am. J. Hum. Genet. 2013;92:345–353. doi: 10.1016/j.ajhg.2013.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Blitterswijk M., DeJesus-Hernandez M., Niemantsverdriet E., Murray M.E., Heckman M.G., Diehl N.N., Brown P.H., Baker M.C., Finch N.A., Bauer P.O. Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. Lancet Neurol. 2013;12:978–988. doi: 10.1016/S1474-4422(13)70210-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dols-Icardo O., García-Redondo A., Rojas-García R., Sánchez-Valle R., Noguera A., Gómez-Tortosa E., Pastor P., Hernández I., Esteban-Pérez J., Suárez-Calvet M. Characterization of the repeat expansion size in C9orf72 in amyotrophic lateral sclerosis and frontotemporal dementia. Hum. Mol. Genet. 2014;23:749–754. doi: 10.1093/hmg/ddt460. [DOI] [PubMed] [Google Scholar]
- 11.Dobson-Stone C., Hallupp M., Loy C.T., Thompson E.M., Haan E., Sue C.M., Panegyres P.K., Razquin C., Seijo-Martínez M., Rene R. C9ORF72 repeat expansion in Australian and Spanish frontotemporal dementia patients. PLoS ONE. 2013;8:e56899. doi: 10.1371/journal.pone.0056899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xi Z., Zinman L., Moreno D., Schymick J., Liang Y., Sato C., Zheng Y., Ghani M., Dib S., Keith J. Hypermethylation of the CpG island near the G4C2 repeat in ALS with a C9orf72 expansion. Am. J. Hum. Genet. 2013;92:981–989. doi: 10.1016/j.ajhg.2013.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xi Z., Rainero I., Rubino E., Pinessi L., Bruni A.C., Maletta R.G., Nacmias B., Sorbi S., Galimberti D., Surace E.I. Hypermethylation of the CpG-island near the C9orf72 G4C2-repeat expansion in FTLD patients. Hum. Mol. Genet. 2014;23:5630–5637. doi: 10.1093/hmg/ddu279. [DOI] [PubMed] [Google Scholar]
- 14.Xi Z., Zhang M., Bruni A.C., Maletta R.G., Colao R., Fratta P., Polke J.M., Sweeney M.G., Mudanohwo E., Nacmias B. The C9orf72 repeat expansion itself is methylated in ALS and FTLD patients. Acta Neuropathol. 2015;129:715–727. doi: 10.1007/s00401-015-1401-8. [DOI] [PubMed] [Google Scholar]
- 15.Majounie E., Renton A.E., Mok K., Dopper E.G., Waite A., Rollinson S., Chiò A., Restagno G., Nicolaou N., Simon-Sanchez J., Chromosome 9-ALS/FTD Consortium. French research network on FTLD/FTLD/ALS. ITALSGEN Consortium Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012;11:323–330. doi: 10.1016/S1474-4422(12)70043-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ishiura H., Takahashi Y., Mitsui J., Yoshida S., Kihira T., Kokubo Y., Kuzuhara S., Ranum L.P., Tamaoki T., Ichikawa Y. C9ORF72 repeat expansion in amyotrophic lateral sclerosis in the Kii peninsula of Japan. Arch. Neurol. 2012;69:1154–1158. doi: 10.1001/archneurol.2012.1219. [DOI] [PubMed] [Google Scholar]
- 17.Ratti A., Corrado L., Castellotti B., Del Bo R., Fogh I., Cereda C., Tiloca C., D’Ascenzo C., Bagarotti A., Pensato V., SLAGEN Consortium C9ORF72 repeat expansion in a large Italian ALS cohort: evidence of a founder effect. Neurobiol. Aging. 2012;33:e7–e14. doi: 10.1016/j.neurobiolaging.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 18.Smith B.N., Newhouse S., Shatunov A., Vance C., Topp S., Johnson L., Miller J., Lee Y., Troakes C., Scott K.M. The C9ORF72 expansion mutation is a common cause of ALS+/-FTD in Europe and has a single founder. Eur. J. Hum. Genet. 2013;21:102–108. doi: 10.1038/ejhg.2012.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van der Zee J., Gijselinck I., Dillen L., Van Langenhove T., Theuns J., Engelborghs S., Philtjens S., Vandenbulcke M., Sleegers K., Sieben A., European Early-Onset Dementia Consortium A pan-European study of the C9orf72 repeat associated with FTLD: geographic prevalence, genomic instability, and intermediate repeats. Hum. Mutat. 2013;34:363–373. doi: 10.1002/humu.22244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pliner H.A., Mann D.M., Traynor B.J. Searching for Grendel: origin and global spread of the C9ORF72 repeat expansion. Acta Neuropathol. 2014;127:391–396. doi: 10.1007/s00401-014-1250-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fratta P., Polke J.M., Newcombe J., Mizielinska S., Lashley T., Poulter M., Beck J., Preza E., Devoy A., Sidle K. Screening a UK amyotrophic lateral sclerosis cohort provides evidence of multiple origins of the C9orf72 expansion. Neurobiol. Aging. 2015;36:e1–e7. doi: 10.1016/j.neurobiolaging.2014.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rubino E., Rainero I., Chiò A., Rogaeva E., Galimberti D., Fenoglio P., Grinberg Y., Isaia G., Calvo A., Gentile S., TODEM Study Group SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology. 2012;79:1556–1562. doi: 10.1212/WNL.0b013e31826e25df. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xi Z., Yunusova Y., van Blitterswijk M., Dib S., Ghani M., Moreno D., Sato C., Liang Y., Singleton A., Robertson J. Identical twins with the C9orf72 repeat expansion are discordant for ALS. Neurology. 2014;83:1476–1478. doi: 10.1212/WNL.0000000000000886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Williams K.L., Fifita J.A., Vucic S., Durnall J.C., Kiernan M.C., Blair I.P., Nicholson G.A. Pathophysiological insights into ALS with C9ORF72 expansions. J. Neurol. Neurosurg. Psychiatry. 2013;84:931–935. doi: 10.1136/jnnp-2012-304529. [DOI] [PubMed] [Google Scholar]
- 25.Hellemans J., Mortier G., De Paepe A., Speleman F., Vandesompele J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol. 2007;8:R19. doi: 10.1186/gb-2007-8-2-r19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lagier-Tourenne C., Baughn M., Rigo F., Sun S., Liu P., Li H.R., Jiang J., Watt A.T., Chun S., Katz M. Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. Proc. Natl. Acad. Sci. USA. 2013;110:E4530–E4539. doi: 10.1073/pnas.1318835110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mizielinska S., Grönke S., Niccoli T., Ridler C.E., Clayton E.L., Devoy A., Moens T., Norona F.E., Woollacott I.O., Pietrzyk J. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science. 2014;345:1192–1194. doi: 10.1126/science.1256800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pearson C.E. Slipping while sleeping? Trinucleotide repeat expansions in germ cells. Trends Mol. Med. 2003;9:490–495. doi: 10.1016/j.molmed.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 29.Waite A.J., Bäumer D., East S., Neal J., Morris H.R., Ansorge O., Blake D.J. Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol. Aging. 2014;35 doi: 10.1016/j.neurobiolaging.2014.01.016. 1779.e5–1779.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buchman V.L., Cooper-Knock J., Connor-Robson N., Higginbottom A., Kirby J., Razinskaya O.D., Ninkina N., Shaw P.J. Simultaneous and independent detection of C9ORF72 alleles with low and high number of GGGGCC repeats using an optimised protocol of Southern blot hybridisation. Mol. Neurodegener. 2013;8:12. doi: 10.1186/1750-1326-8-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pieretti M., Zhang F.P., Fu Y.H., Warren S.T., Oostra B.A., Caskey C.T., Nelson D.L. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell. 1991;66:817–822. doi: 10.1016/0092-8674(91)90125-i. [DOI] [PubMed] [Google Scholar]
- 32.Tassone F., Hagerman R.J., Taylor A.K., Gane L.W., Godfrey T.E., Hagerman P.J. Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in the fragile-X syndrome. Am. J. Hum. Genet. 2000;66:6–15. doi: 10.1086/302720. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.