

Abstract
Isolated dystonia is a disorder characterized by involuntary twisting postures arising from sustained muscle contractions. Although autosomal-dominant mutations in TOR1A, THAP1, and GNAL have been found in some cases, the molecular mechanisms underlying isolated dystonia are largely unknown. In addition, although emphasis has been placed on dominant isolated dystonia, the disorder is also transmitted as a recessive trait, for which no mutations have been defined. Using whole-exome sequencing in a recessive isolated dystonia-affected kindred, we identified disease-segregating compound heterozygous mutations in COL6A3, a collagen VI gene associated previously with muscular dystrophy. Genetic screening of a further 367 isolated dystonia subjects revealed two additional recessive pedigrees harboring compound heterozygous mutations in COL6A3. Strikingly, all affected individuals had at least one pathogenic allele in exon 41, including an exon-skipping mutation that induced an in-frame deletion. We tested the hypothesis that disruption of this exon is pathognomonic for isolated dystonia by inducing a series of in-frame deletions in zebrafish embryos. Consistent with our human genetics data, suppression of the exon 41 ortholog caused deficits in axonal outgrowth, whereas suppression of other exons phenocopied collagen deposition mutants. All recessive mutation carriers demonstrated early-onset segmental isolated dystonia without muscular disease. Finally, we show that Col6a3 is expressed in neurons, with relevant mRNA levels detectable throughout the adult mouse brain. Taken together, our data indicate that loss-of-function mutations affecting a specific region of COL6A3 cause recessive isolated dystonia with underlying neurodevelopmental deficits and highlight the brain extracellular matrix as a contributor to dystonia pathogenesis.
Introduction
Among the heterogeneous group of dystonias, isolated dystonia (formerly termed primary dystonia) represents a clinical subtype characterized by dystonia as the only clinical abnormality except for tremor.1,2 Monogenic disease transmission is particularly apparent in early-onset forms (<30 years of age), often in combination with a positive family history.3,4 Autosomal-dominantly inherited mutations in three genes, TOR1A (MIM: 605204), THAP1 (MIM: 609520), and GNAL (MIM: 139312) (responsible for DYT1 [MIM: 128100], DYT6 [MIM: 602629], and DYT25 [MIM 615073], respectively), have been implicated unequivocally in the pathogenesis of isolated dystonia, usually exhibiting incomplete penetrance.2–4 Whereas the typical DYT1 and DYT6 phenotypes are defined by early-onset generalized isolated dystonia, DYT25 is typically found among adult-onset cases with focal/segmental isolated dystonia.2,3 Autosomal-recessively inherited isolated dystonia has been described in some consanguineous pedigrees, with the assignment of two different entities (DYT2 [MIM: 224500] and DYT17 [MIM: 612406]),5–8 but no causative variants have been identified thus far. To identify additional genetic variants contributing to isolated dystonia, we employed exome sequencing in a German kindred segregating early-onset segmental isolated dystonia as an autosomal-recessive trait. We performed comprehensive mutational screening of the candidate gene in a large cohort of isolated dystonia subjects and controls and subsequent in vivo functional testing of the specific region mutated in all identified cases.
Subjects and Methods
Participants
Affected individuals were recruited at the Department of Neurology, Klinikum rechts der Isar, Technische Universität München, Munich, Germany, and were examined by neurologists specializing in movement disorders. The index family (F1, Figure 1) consisted of two siblings affected by a severe form of early-onset segmental isolated dystonia and a healthy sister, born to non-consanguineous healthy parents of German origin. Given the absence of a family history of neurological disease and the similarity in clinical presentation, we hypothesized that the affected siblings shared their disease phenotype in an autosomal-recessive manner. Mutations in known genes causing isolated dystonia (TOR1A, THAP1, ANO3 [MIM: 610110], GNAL) had been ruled out. The dystonia cohort screened for variants in the candidate gene COL6A3 (MIM: 120250) was composed of 367 unrelated German individuals with mainly focal/segmental isolated dystonia (15.5% early-onset cases [<30 years of age], average age of onset 46.9 ± 17.7 years, 67.0% female, positive family history in 13.1%; detailed demographics are shown in Table S4). All subjects had been tested for mutations in TOR1A (ΔGAG), THAP1, ANO3, and GNAL, and no mutations had been found. Ancestry-matched controls belong to a large general population cohort (KORA) based in the region around Augsburg in Southern Germany (average age at sampling 76.1 ± 6.3 years, 51.8% female).9 All samples were collected with the written informed consent of participants. The study was approved by the local ethics review board.
Figure 1.
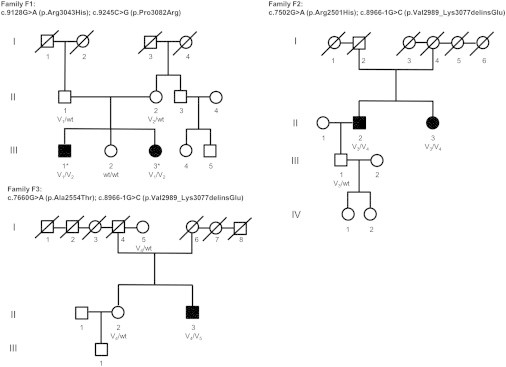
Family Pedigrees
Solid symbols denote individuals with isolated dystonia, open symbols are unaffected individuals, squares are males, circles are females, asterisks are individuals subjected to exome sequencing, and slashes are deceased individuals. COL6A3 genotypes are indicated by variant alleles: V1, c.9128G>A (p.Arg3043His); V2, c.9245C>G (p.Pro3082Arg); V3, c.7502G>A (p.Arg2501His); V4, c.8966−1G>C (p.Val2989_Lys3077delinsGlu); V5, c.7660G>A (p.Ala2554Thr); and wild-type alleles, WT.
Exome Sequencing and Variant Filtering
In the index family (F1, Figure 1), for two siblings (F1-III-1 and F1-III-3), blood-cell-derived genomic DNA libraries were captured with the SureSelect Human All Exon 50 Mb kit (Agilent Technologies), and DNA fragments were sequenced as 100 bp paired-end runs on an Illumina HiSeq2000 system to an average depth of coverage of at least 120×. Variants were identified by an analysis pipeline with Burrows-Wheeler Aligner (v.0.5.9) for read alignments against the human reference genome hg19 and Samtools (v.0.1.18) for variant calling. Custom Perl scripts were employed for variant annotation and classification. To eliminate common genetic alterations, variants with a minor allele frequency (MAF) ≥ 0.3% in 4,300 European American (EA) exomes of the NHLBI exome sequencing project (ESP), 3,640 in-house exomes, HapMap, and the 1000 Genomes project were discarded. Assuming a recessive disorder, only non-synonymous variants (NSVs) including missense, nonsense, stop-loss, and splice-site mutations, as well as small insertions and deletions present in the homozygous state or heterozygous but accompanied by a second rare (MAF < 0.3%) heterozygous NSV and shared by both exomes were retained as candidate alleles. All exome candidate variants were validated by Sanger sequencing and co-segregation of the variants with disease was tested in family F1. For whole-exome sequencing statistics, see Tables S1 and S2.
COL6A3 Mutational Screening
Primer pairs for amplification of all 43 coding exons and flanking sequences of COL6A3 (GenBank: NM_004369.3) were designed with the ExonPrimer software and sequences are shown in Table S8. PCR conditions are available upon request. COL6A3 exons 41 and 42 encoding the C4 domain of collagen VI α3 were analyzed in 367 German isolated dystonia cases and 376 KORA controls by direct sequencing. When a rare (MAF < 0.3%) NSV was identified, Sanger sequencing of the entire COL6A3 coding region ensued to detect additional rare NSVs. The remaining COL6A3 coding exons 2–40 and 43–44 were analyzed in 360 isolated dystonia cases and 373 KORA controls without rare exon 41/42 NSVs using Idaho’s LightScanner high-resolution melting (HRM) curve analysis according to standard protocols (Idaho Technology).10 In the case of altered melting patterns, Sanger sequencing was performed to detect the underlying sequence change.
Splice Variant Analysis
The effect of the canonical COL6A3 exon 41 splice site mutation c.8966−1G>C on mRNA transcript was analyzed by cDNA sequencing. RNA was isolated from whole blood of three case subjects and a control individual via the PAXgene Blood miRNA kit from QIAGEN. Blood total RNA was reverse transcribed to cDNA with the SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen), and the relevant cDNA fragment was amplified with specific primers (Table S8). PCR amplicons were subsequently subjected to Sanger sequencing.
COL6A3 Exon Dosage Analysis and Copy-Number Variation Screening
To investigate the presence of compound heterozygous changes in an isolated dystonia subject positive for a single COL6A3 mutation (c.9128G>A [p.Arg3043His] in exon 41), dosage of COL6A3 exons 33–44 was analyzed with SYBR Green-based quantitative real-time PCR (qRT-PCR) on an ABI7900HT real-time PCR system as described.11 Primer sequences were designed with Primer3Plus (sequences available upon request). Additionally, screening for larger CNVs was done with the CytoScan HD Array from Affymetrix; data were analyzed with the Chromosome Analysis Suite software (Affymetrix).
Muscle Magnetic Resonance Imaging
Muscle MRI studies were performed at 3 Tesla (Siemens TRIO scanner). Sequences included axial T1-weighted images as well as axial and coronal short-tau inversion recovery (STIR) images of pelvis and thigh.
Fibroblast Culturing
Primary fibroblast cell lines were established from skin biopsy samples of isolated dystonia subjects, unaffected relatives, normal control probands, and individuals with Ullrich congenital muscular dystrophy (MIM: 254090) or Bethlem myopathy (MIM: 158810) obtained from the biobank of the MRC Centre for Neuromuscular Disease (Newcastle). Cells were grown in high-glucose Dulbecco’s Modified Eagle Medium (DMEM) supplemented by 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, and 1% amphotericin-B, in atmospheric oxygen and 5% CO2 at 37°C.
Collagen VI Immunolabeling in Human Fibroblasts
After 3 days of treatment with L-ascorbic acid (50 μg/ml/day), adherent human skin fibroblasts were fixed with 4% paraformaldehyde for 15 min, washed in phosphate-buffered saline (PBS), and blocked in 5% goat serum and 0.1% Triton X-100 (Sigma 21123) in PBS for 30 min at room temperature. Cells were incubated with primary antibody (monoclonal mouse anti-collagen VI, MAB1944, Millipore) at 1:2,000 concentration in blocking solution for 30 min. After PBS washes, cells were incubated with secondary antibody (AlexaFluor 555 goat anti-mouse, Life Technologies) at 1:1,000 concentration in blocking solution for 30 min. Cells were then treated with 4′,6-diamidino-2-phenylindole (DAPI, 100 ng/ml in PBS) for 3 min. After additional PBS washes, cells were visualized on a Life Technologies EVOS epifluorescent microscope.
Col6a3 Expression Analysis in Adult Mouse Brain
All animal experiments followed the NIH Guide for the Care and Use of Laboratory Animals under Stanford APLAC (Administrative Panel on Laboratory Animal Care) protocol #28777. qRT-PCR was carried out to assess the relative expression of Col6a3 in regions of the adult mouse brain (n = 3). Tissue from the following regions were collected: brainstem, dorsal midbrain (tectum), ventral midbrain (tegmentum), cerebellum, frontal cortex, striatum, and hippocampus. To extract total RNA, the RNeasy Mini kit from QIAGEN was used according to the manufacturer's protocol. Concentration and integrity of the RNA was assessed with the Agilent 2100 Bioanalyzer with RNA 6000 Nano chips. Reverse transcription was performed with the SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen) with 1,000 ng total RNA as template. qRT-PCR was performed on an ABI7900HT real-time PCR system employing commercially available TaqMan gene expression assays (Life Technologies) for Col6a3 and an endogenous reference gene (Polr2a). 75 ng cDNA were used per reaction. All samples were run in quadruplicate. Gene expression levels were determined via the ΔΔCT method.
In Situ Hybridization Analysis of Col6a3 in Adult Mouse Brain
Male C57BL6/J mice (age 6 months) were euthanized by cervical dislocation and brains were rapidly extracted and snap frozen in 2-methlybutane. Brains were sectioned on a cryostat and collected on Superfrost Plus slides (Fisher Scientific). Each subsequent step prior to prehybridization was followed by washes in diethylpyrocarbonate (DEPC)-treated PBS. Sections were fixed in 4% paraformaldehyde for 10 min and endogenous peroxidase was quenched in 0.3% H2O2 in DEPC-PBS for 30 min. Sections were then acetylated in 1.35% triethanolamine with 0.02 N HCl and 0.25% acetic anhydride. Next, sections were permeabilized in 1% Triton X-100 (Sigma 21123) in DEPC-PBS. Antigen retrieval was performed with 10 mM sodium citrate (pH 6) at 80°C for 10 min. Sections were prehybridized with hybridization buffer (50% formamide, 12.5% dextran sulfate, 750 mM NaCl, 125 mM Tris-HCl [pH 7.5], 1.25 μM EDTA [pH 8], 125 μg/ml salmon sperm DNA, 625 μg/ml total yeast RNA, 62.5 μg/ml yeast tRNA, 1.25× Denhardt’s solution, 0.1% sodium dodecyl sulfate [SDS], 0.1% sodium thiosulfate, 0.1 M dithiothreitol) for 4 hr. Digoxigenin-labeled riboprobes were in vitro transcribed with a labeling kit (Roche), diluted to 1 μg/ml in hybridization solution, and hybridized to sections overnight at 57°C. The riboprobes (sense and antisense) corresponded to nucleotides 4,667–5,569 of the mouse Col6a3 cDNA (GenBank: NM_001243008.1). After hybridization, sections were washed three times in 5× saline sodium citrate (SSC), soaked in 0.2× SSC at 57°C for 1 hr, and then rinsed in room temperature 0.2× SSC. For immunohistochemical detection of digoxigenin, slides were first blocked with TNB buffer (PerkinElmer) for 30 min, then incubated with peroxidase-conjugated anti-digoxigenin antibody (1:500, Roche) as well as antibody for cell type identification: mouse anti-NeuN (1:200, MAB377, Millipore) or mouse anti-glial fibrillary acidic protein (1:200, G3893, Sigma). After washes in TNT buffer (PerkinElmer), sections were incubated with Tyramide Signal Amplification Cy3 reagent for 45 min. After further washes in TNT buffer, sections were incubated with secondary antibody diluted 1:200 in TNB (AlexaFluor 488 goat anti-mouse, Life Technologies). Sections were then washed in PBS and coverslipped with FluoroGel (Electron Microscopy Sciences). Sections were visualized on a Zeiss Axio Imager.M2 epifluorescence microscope.
Zebrafish Functional Assay
We used reciprocal BLAST against the Danio rerio genome and identified a sole zebrafish ortholog of col6a3 (ENSEMBL: ENSDART00000138754). To determine the effect of col6a3 suppression in zebrafish embryos, we designed and obtained three morpholinos from Gene Tools: col6a3 exon42 (MO1) (5′-ACTGTTTTCTGGACATAAGAACGTA-3′), col6a3 exon43 (MO2) (5′-TCCATTGTCACTTACTTGCTTCTGA-3′), and col6a3 exon46 (MO3) (5′-TTTGCATCACTTACTTAATTCTGGT-3′). To determine morpholino efficiency, total mRNA was extracted from control and morpholino-injected embryos and reverse transcribed to produce cDNA and PCR amplified at the site targeted by the MO. PCR amplicons were gel extracted and sequenced. Injected embryos were fixed at 2 dpf (days postfertilization) in Dent’s fixative (80% methanol, 20% DMSO) overnight at 4°C. After rehydration steps in decreasing series of methanol/PBS/Triton X-100, permeabilization, fixing, and blocking solution, embryos were incubated with anti-α acetylated tubulin (1:500; Sigma-Aldrich), overnight at 4°C. After washes in PBS/Triton X-100, embryos were incubated with AlexaFluor 488 goat anti-mouse (1:500, Life Technologies). Injected embryos were scored based on their motor neuron abnormalities, which included pathfinding errors, abnormal branching, and failure to extend. Abnormalities were separated in classes based on severity. All the experiments were repeated three times and a Pearson’s chi-square test (χ2) was used to determine the significant differences of the morphant phenotype.
Results
Exome Sequencing and Validation of Variants
Two siblings of the index family F1 presenting an almost identical phenotype of early-onset segmental isolated dystonia involving the face, neck, bulbar muscles, and upper limbs (Figure 1 and Table 1) were subjected to exome sequencing with the SureSelect Human All Exon 50 Mb kit from Agilent and the Illumina HiSeq2000 system. We generated ∼10 Gb of high-quality sequence per sample, with an average coverage of 127× (F1-III-1) and 125× (F1-III-3). At least 96% of the target regions were covered at 20× (Table S1). Variant filtering steps were applied under the postulation of an autosomal-recessive mode of disease inheritance. Focusing on rare (MAF < 0.3%) protein-altering genetic variation shared by both exomes, we found only a single gene, COL6A3, that harbored compound heterozygous non-synonymous substitutions (c.9128G>A [p.Arg3043His] and c.9245C>G [p.Pro3082Arg]) segregating with the disease (Tables S2 and S3). The exome data yielded two further candidate genes containing putatively compound heterozygous changes (LTBP1 [MIM: 150390] and SPNS3 [MIM: 611701]), yet Sanger sequencing analysis in family F1 revealed that all of these variants were derived from the healthy mother (Table S3). No shared homozygous mutations were observed. Sanger sequencing confirmed the presence of both COL6A3 variants in individuals F1-III-1 and F1-III-3, whereas the unaffected sister did not carry either mutation. Each parent was heterozygous for one of the variant alleles (Figure 1). The c.9128G>A (p.Arg3043His) variant was not found in 8,600 European American (EA) control chromosomes of the NHLBI exome sequencing project (NHLBI-ESP), whereas the c.9245C>G (p.Pro3082Arg) variant was present in 8 of 8,600 EA alleles (MAF = 0.0009). The very low frequency of both variants was confirmed by recently released exome data from the Exome Aggregation Consortium (ExAC), where c.9128G>A (p.Arg3043His) and c.9245C>G (p.Pro3082Arg) were observed in 22 of 66,716 (MAF = 0.0003) and in 95 of 66,660 (MAF = 0.001) European chromosomes, respectively. The substituted amino acid residues were completely conserved across mammalian species (Figure 2), and each variant was predicted as likely deleterious by bioinformatics prediction tools (SIFT, PolyPhen2, CADD; Table 2). Notably, both variants are located within the C-terminal fibronectin type-III (FN-III) motif (C4 domain) of the encoded collagen VI α3, corresponding to COL6A3 exons 41 and 42 (Figure 2).
Table 1.
Clinical Phenotypes of Five Isolated Dystonia Case Subjects with Biallelic COL6A3 Mutations in Three German Families
| Individual | Sex | Age at Sampling (years) | Age at Onset (years) | Site of Onset | Dystonia Distribution | Dystonic Features |
|---|---|---|---|---|---|---|
| Family 1 (F1): c.9128G>A (p.Arg3043His) + c.9245C>G (p.Pro3082Arg)a | ||||||
| F1-III-1 | M | 51 | 20 | neck | segmental | tremulous cervical dystonia; upper limb dystonia with dystonic action and postural tremor; writer’s cramp; oromandibular dystonia; laryngeal dystonia; trunk dystonia |
| F1-III-3 | F | 48 | 20 | neck | segmental | tremulous cervical dystonia; upper limb dystonia with dystonic action and postural tremor; writer’s cramp; oromandibular dystonia; laryngeal dystonia |
| Family 2 (F2): c.8966−1G>C (p.Val2989_Lys3077delinsGlu) + c.7502G>A (p.Arg2501His)a | ||||||
| F2-II-2 | M | 71 | 24 | neck | focal | cervical dystonia |
| F2-II-3 | F | 68 | 24 | hand | segmental | writer’s cramp; cervical dystonia |
| Family 3 (F3): c.8966−1G>C (p.Val2989_Lys3077delinsGlu) + c.7660G>A (p.Ala2554Thr)a | ||||||
| F3-II-3 | M | 62 | 6 | hand | segmental | writer’s cramp; cervical dystonia; oromandibular dystonia |
Numbering according to NCBI accessions GenBank: NM_004369.3 and NP_004360.2.
Figure 2.
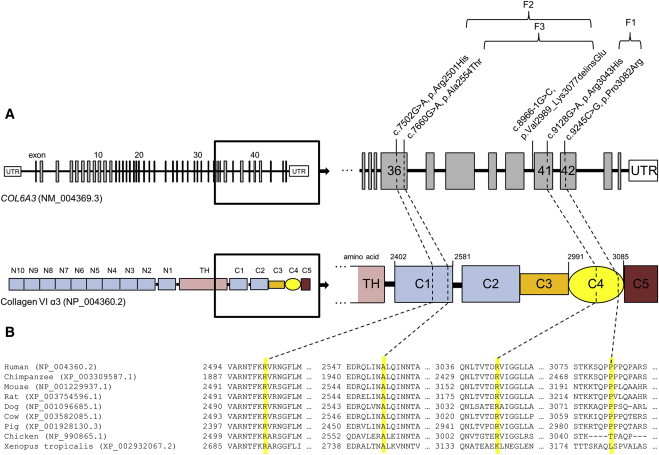
COL6A3 Mutations in Recessive Isolated Dystonia
(A) Schematic representation of the exon-intron structure of COL6A3 (GenBank: NM_004369.3) and domain organization of collagen VI α3 (GenBank: NP_004360.2) along with the localization of detected sequence variants. COL6A3 transcript GenBank: NM_004369.3 consists of 43 coding exons depicted as vertical bars. Collagen VI α3 is composed of a central triple helical domain (TH) flanked by large N- and C-terminal globular domains (N1–10 and C1–C5). Domains N1–N10 as well as C1–C2 contain amino acid motifs similar to type A domains of the von Willebrand factor whereas the C-terminal C3–C5 domains comprise additional protein motifs, a lysine-proline repeat structure (C3), a fibronectin type-III motif (C4), and a Kunitz protease inhibitor motif (C5). C-terminal globular domains and the corresponding coding exons are magnified to illustrate the position of identified compound heterozygous variants in exon 36/domain C1 and exon 41–42/domain C4 in three German isolated dystonia-affected families (F1, F2, F3).
(B) Multiple sequence alignment of collagen VI α3 in vertebrate species using the RefSeq database and ClustalW2. The affected amino acid residues are highlighted in yellow.
Table 2.
Mutations in COL6A3 in Individuals with Isolated Dystonia
| Exona | Genomic Position (hg19) | Variation Nucleotidea | Variation Amino Acida | Mutation Type | dbSNP142 | Frequency NHLBI-ESP (EA) | Frequency ExAC (European) | SIFT Prediction | PolyPhen-2 Prediction | CADD Prediction12 | Affected Individual |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 36 | chr2: 238,253,159 | c.7502G>A | p.Arg2501His | missense | rs541928674 | not found | 0.00001 | damaging | probably damaging | 22.8 | F2-II-2, F2-II-3 |
| 36 | chr2: 238,253,001 | c.7660G>A | p.Ala2554Thr | missense | NA | not found | not found | tolerated | probably damaging | 23.4 | F3-II-3 |
| 41 | chr2: 238,243,533 | c.8966−1G>C | p.Val2989_Lys3077delinsGlu | canonical splice | NA | not found | 0.00003 | NA | NA | 23.5 | F2-II-2, F2-II-3, F3-II-3 |
| 41 | chr2: 238,243,370 | c.9128G>A | p.Arg3043His | missense | rs552651651 | not found | 0.0003 | damaging | benign | 13.5 | F1-III-1, F1-III-3 |
| 42 | chr2: 238,242,176 | c.9245C>G | p.Pro3082Arg | missense | rs182976977 | C = 8/G = 8,592 | 0.001 | tolerated | possibly damaging | 15.7 | F1-III-1, F1-III-3 |
Overview of compound heterozygous COL6A3 mutations identified in isolated dystonia cases. In silico predictions of the deleterious potential of the mutations assessed by SIFT, PolyPhen-2, and CADD are shown. A CADD score ≥ 10 indicates that the variant is predicted to be among the 10% most deleterious substitutions that can affect the human genome, a score ≥ 20 indicates that the variant is among the 1% most deleterious.12 Abbreviations are as follows: NHLBI-ESP, National Heart, Lung, and Blood Institute-exome sequencing project; EA, European American; ExAC, Exome Aggregation Consortium (encompassing ∼33,000 European exomes); NA, not available.
Numbering according to NCBI accessions GenBank: NM_004369.3 and NP_004360.2.
COL6A3 Mutational Screening
We hypothesized that COL6A3 mutations might be implicated in recessively inherited isolated dystonia and that the collagen VI α3 C4 domain might constitute a mutational hotspot for this condition. Therefore, we Sanger sequenced exons 41 and 42 of COL6A3 in 367 German cases with various forms of isolated dystonia, including 15.5% early-onset cases and 13.1% with a positive family history (Table S4), as well as in 376 ancestry-matched controls. This led to identification of six different rare (MAF < 0.3%) sequence changes (Table S5), one of them (c.8966−1G>C) affecting the invariant splice recognition site of exon 41 in two independent early-onset isolated dystonia samples (F2-II-2, F3-II-3, Figure 1). The c.8966−1G>C variant was absent in the NHLBI-ESP cohort (EA) as well as in 752 control alleles tested by us, and was found in only 2 of 66,458 European alleles at ExAC (MAF = 0.00003). Analysis of whole-blood mRNA transcripts showed that the variant produces an in-frame 89-amino-acid deletion from the C4 domain (residues 2,989–3,077) while inserting a single codon for glutamate (p.Val2989_Lys3077delinsGlu), corresponding to skipping of exon 41 (Figure S1). Subsequent sequence analysis of the entire COL6A3 coding region uncovered a second rare nucleotide substitution in each of the two splicing mutation carriers, c.7502G>A (p.Arg2501His) in individual F2-II-2 and c.7660G>A (p.Ala2554Thr) in individual F3-II-3, respectively (Figure 1, Table 2). These variants affected highly conserved codons in exon 36 of COL6A3 (encoding the C1 domain) and were classified as deleterious by PolyPhen2 and CADD; neither variant was seen in the NHLBI-ESP dataset or in our control sample. At ExAC, c.7502G>A (p.Arg2501His) occurred in one of 66,732 European chromosomes (MAF = 0.00001), and c.7660G>A (p.Ala2554Thr) was absent in this population. Sequencing of DNA obtained from all available family members demonstrated that the identified mutations co-segregated with an early-onset isolated dystonia phenotype in each pedigree. In the family of individual F2-II-2 (family F2, Figure 1), the affected sister (F2-II-3) also carried both mutant alleles, c.7502G>A (p.Arg2501His) and c.8966−1G>C, whereas the unaffected son (F2-III-1) was heterozygous for only the c.7502G>A (p.Arg2501His) substitution. In family F3 positive for c.7660G>A (p.Ala2554Thr) and c.8966−1G>C, the sister (F3-II-2) of individual F3-II-3 was healthy and solely harbored the c.8966−1G>C splicing mutation. This change was also present in an unaffected sister of the deceased father (F3-I-5), indicating that it was inherited from the paternal side (Figure 1).
In a further five case subjects and three control subjects with rare exon 41/42 variants, no additional coding mutations were discovered by exonic sequencing of COL6A3 (Table S5). Of note, the exon 41 c.9128G>A (p.Arg3043His) alteration was found again in an early-onset isolated dystonia subject with a family history suggestive of autosomal-recessive disease inheritance. However, further studies, including dosage analysis of exons 33–44 (encoding the C1–C5 domains) as well as screening for larger CNVs, failed to identify a second COL6A3 aberration. Finally, a HRM-based screen to investigate the entire COL6A3 coding regions in case subjects and control subjects without exon 41/42 variants (n = 360 and n = 373, respectively) did not identify any further recessive mutation carriers (Table S6).
Phenotypic Profile Associated with Recessive COL6A3 Mutations
As shown in Table 1, the clinical picture of COL6A3-associated isolated dystonia was characterized by an early symptom onset before 25 years of age. Three individuals reported onset in the neck, and two had onset in the hand, and in all but a single focal case dystonia gradually progressed to other body regions within a few years, resulting in segmental distribution at final exam. Cranio-cervical involvement was observed in all five cases, and four out of five individuals exhibited additional writer’s cramp. Also, the siblings of the index family F1 presented laryngeal dystonia as well as upper limb dystonic action and postural tremor. Inter-familial phenotypic heterogeneity was evident, but between affected relatives of the same family clinical manifestations were strikingly similar. Notably, COL6A3 has previously been linked to neurological disease, with dominantly as well as recessively acting mutations causing a clinical spectrum of skeletal muscle diseases such as Ullrich congenital muscular dystrophy (UCMD [MIM: 254090]) and Bethlem myopathy (BM [MIM: 158810]).13,14 In our cases, however, clinical investigations combined with muscle MRI did not yield signs of muscular involvement (for details see Table S7). Furthermore, we could not detect abnormalities characteristic of COL6A3-related muscular disease or collagen VI organization defects in fibroblast cultures derived from isolated dystonia individuals. In both dystonia proband and control cells, immunocytochemistry displayed a regular distribution of extra- and intracellular collagen VI, in contrast to dermal fibroblasts from UCMD- and BM-affected individuals (Figure S2).
Expression of Col6a3 in Adult Mouse Brain
COL6A3 located on chromosome 2q37 encodes the α3-subunit of the heterotrimeric type VI collagen, an extracellular matrix (ECM) protein capable of generating microfibrillar networks.13,14 Collagen VI has been shown to be ubiquitously expressed, but previous research had concentrated on determining its significance in peripheral tissues.13,14 Using qRT-PCR, we found Col6a3 to be widely expressed throughout the adult mouse brain including striatum and cerebellum, with highest mRNA levels in brainstem and midbrain (Figure S3). In situ hybridization analysis confirmed particularly robust Col6a3 mRNA expression in the pontine gray and the septal nuclei. Moreover, in situ hybridization combined with cell-type-specific antibody staining revealed neurons, but not astroglia, as the cellular source of Col6a3 (Figure S4).
In Vivo Functional Testing of Specific col6a3 Exon Suppression via Zebrafish Embryos
All COL6A3-related isolated dystonia cases in our study harbored at least one mutant substitution affecting exon 41. This finding raised the possibility that disruption of the function of this very specific region of the gene might account for the specificity of the phenotype and thus differentiate between neurological pathologies causing dystonia and collagen organization defects found in muscular disease. To test this hypothesis directly, we performed in vivo studies with zebrafish as a model organism. Reciprocal BLAST of the zebrafish genome with human COL6A3 identified a sole ortholog (63% similarity, 44% identity). First, we tested for the expression of this locus during early development. We have previously generated RNA-seq data from zebrafish embryo heads at 5 days post fertilization for the purpose of testing the expression of other genes relevant to neurodevelopmental disorders (NCBI Gene Expression Omnibus, GEO: GSE63191).15 We therefore measured the abundance of col6a3, as well as four dominant dystonia genes (tor1a, thap1, gnal, ano3) relative to gapdh; we found robust expression of col6a3 (Table S9). We thus proceeded with the design of a morpholino (MO1) that inhibits the splicing of zebrafish exon 42, corresponding to human exon 41 (Figure S5). Injection of MO1 caused dose-dependent motor neuron pathfinding, branching, and extension errors at 48 hr post fertilization (Figure 3); we observed no overt phenotypes pathognomonic of collagen defects, such as the stiffening and profound body curvature defects seen previously in zebrafish collagen mutants and morphants.16 These observations were in sharp contrast to the phenotypes seen upon suppression of the splicing of two other col6a3 exons: exon 43 (MO2), an exon with no human ortholog, or exon 46 (MO3), corresponding to exon 42 in humans (Figure S5). In both instances, we saw no defects in the elaboration and branching of motor neurons. However, embryos injected with either MO showed characteristic, dose-dependent body curvature defects (Figure 3).
Figure 3.
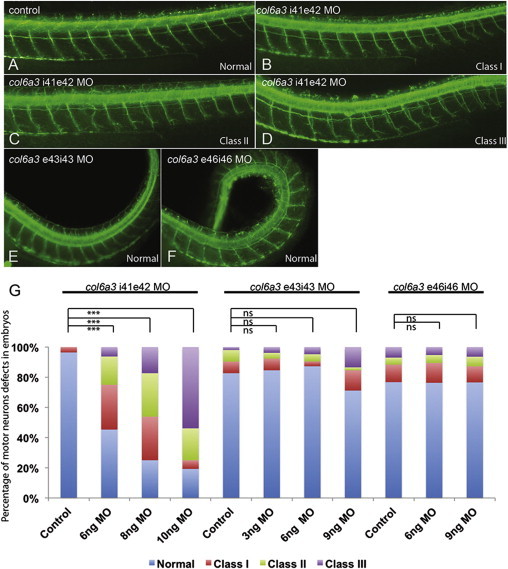
Knockdown of the Human COL6A3 Exon 41 Ortholog in Zebrafish Embryos Causes Motor Neuron Pathfinding Errors
(A–F) Representative lateral views of 2 days post fertilization zebrafish embryos stained with acetylated tubulin. In control embryos (A), motor neurons have completed migration toward the myotome. In MO1-injected embryos (B–D), secondary axons exit the periphery but fail to migrate along the common path. This phenotype is concentration dependent: class I corresponding to one motor neuron abnormality (B); class II to two or three (C); and class III to four or more motor neuron abnormalities (D). Embryos injected with MO2 (E) and MO3 (F) do not present pathfinding errors but develop a body curvature defect reminiscent of collagen deposition mutants previously described.16
(G) Percentage of affected embryos under the conditions being evaluated above. ∗∗∗p < 0.0001; ns = nonsignificant.
Discussion
As with other neurological disorders,17 isolated dystonia is thought to encompass a heterogeneous collection of rare monogenic conditions.3,4 Our work has identified, in three families with early-onset segmental isolated dystonia, compound heterozygous mutations in the α3-subunit gene of collagen VI, COL6A3, highlighting biallelic mutations in this gene as an autosomal-recessive cause of isolated dystonia. Employing an unbiased whole-exome approach in a German kindred affected by recessive isolated dystonia, we detected two compound heterozygous missense mutations, both mapping to the FN-III motif (C4 domain) encoding exons 41 and 42 of COL6A3 (c.9128G>A [p.Arg3043His] and c.9245C>G [p.Pro3082Arg]), which segregated with the disease status according to our strict filtering criteria. The causative nature of these changes was supported by the discovery of a COL6A3 exon 41 skipping mutation (c.8966−1G>C) co-segregating with different exon 36 missense mutations (c.7502G>A [p.Arg2501His], c.7660G>A [p.Ala2554Thr]) in two further pedigrees presenting a similar clinical phenotype as the index case subjects. Consistent with disease-causing mutations, the observed alterations were either extremely rare or absent from an extensive number of control alleles, and the missense substitutions disrupted amino acid residues highly conserved across evolution. Notably, in the ExAC dataset, the c.9128G>A (p.Arg3043His) variant and the c.9245C>G (p.Pro3082Arg) variant were each reported in two South Asian exomes in the homozygous state. Nonetheless, this finding does not argue against their pathogenicity, because only individuals with severe pediatric disease phenotypes have been removed from this dataset. Thus, it seems possible that either these homozygous alleles display reduced penetrance (a phenomenon documented previously in isolated dystonia)4 or that the two homozygote subjects have or will develop dystonia, which could manifest in late adolescence or young adulthood and might be of only moderate severity. In contrast to the other mutations identified in our study, c.9245C>G (p.Pro3082Arg) does not appear to be ultra-rare, with a frequency of 0.001 in the European ExAC population. However, it can be anticipated that there are several carriers for recessive disorders in the general population. Frequencies as high as 0.001 at ExAC can also be observed for COL6A3 variants causing collagen VI-related myopathies in the compound heterozygous status.18 In a fourth potentially recessive isolated dystonia-affected family positive for the exon 41 (c.9128G>A [p.Arg3043His]) change, the absence of a second COL6A3 coding region alteration raised the possibility of intronic or intergenic regulatory mutations contributing to COL6A3-associated isolated dystonia, a mechanism demonstrated recently in collagen-related diseases.19,20 Likewise, disruption of the second allele could have been missed in the remaining dystonia subjects that tested positive for only one COL6A3 mutation in our targeted exonic screen. Additional exploration of COL6A3 non-coding regions and/or CNV analysis might be helpful to establish the diagnosis in such cases.
On the clinical level, the symptomatology of COL6A3-associated isolated dystonia differed from recessive DYT2 dystonia, a disorder typically characterized by lower-limb-onset dystonia followed by rapid generalization.5–7 In contrast, our case subjects developed initial symptoms in the neck or hand and the distribution remained segmental with cranio-cervical predominance. There was some phenotypic overlap with recessively transmitted DYT17 dystonia mapping to chromosome 20, but this type of isolated dystonia has been reported in only a single family and refinement of its phenotype has never been accomplished.8
COL6A3 mutations have been implicated previously in a continuum of skeletal muscle phenotypes that range from severe UCMD to mild BM, with both recessive and dominant transmission patterns.13,14 Allelic conditions have been recognized increasingly in dystonia21,22 and have also been reported in collagen VI-associated disorders.23 Moreover, it is notable that many forms of congenital muscular dystrophies and myopathies show an increased incidence of brain abnormalities, intimating a molecular link between skeletal muscle pathology and central nervous system (CNS) disease.24,25 The variable phenotypic expression associated with COL6A3 mutations might be due to their domain-specific localization. The ECM molecule collagen VI is composed of three α chains, α1(VI), α2(VI), and α3(VI) encoded by COL6A1 (MIM: 120220), COL6A2 (MIM: 120240), and COL6A3, respectively, each with N-terminal and C-terminal non-collagenous domains connected by a central triple helix.13,14 All isolated dystonia-related COL6A3 mutations occurred in the C-terminal tail of the α3 (VI) chain with at least one exon 41 mutation in each family, whereas myopathy-causing mutations cluster in the N-terminal and triple helical segments.18,26–29 Essentially for proper muscle function, the collagen VI α chains assemble intracellularly as monomers, and subsequently form dimers and tetramers that are secreted into the extracellular space.13,14 Notably, given our mutational distribution, the α3(VI) C2-C5 domains are not required for intracellular collagen VI assembly and the α3(VI) C3-C4 domains are not critical for extracellular microfibril formation.30 Neither recessive nor dominant UCMD- and BM-affected case subjects have been attributed to α3(VI) C4 domain mutations.18,26–29 Furthermore, several lines of evidence indicate that large parts of the α3(VI) C terminus are proteolytically cleaved, suggesting microfibril-independent functions for these domains.31–33 Consistent with this, the C termini of various collagens have been implicated in fundamental biological processes, such as proliferation and differentiation.34,35
The ECM represents an important functional component of the CNS, participating in various aspects of neurodevelopment and neurodegeneration.36–38 The molecular roles of collagens in the CNS are not limited to structural adhesion activities but comprise a wide range of cellular processes including neural circuit formation and maintenance.39,40 For instance, C1 domains of type IV collagen coordinate synaptogenesis and the stability of synaptic networks.41 Involvement of type VI collagen in CNS physiology is poorly understood, yet emerging data suggest its neuroprotective potential.42,43 Now, we show that the α3-subunit gene of collagen VI is widely expressed in adult mouse brain, including motor regions thought to have a central role in the pathogenesis of dystonia, and determine neurons as its cellular source. Moreover, by using a functional zebrafish assay, we demonstrated that selective knockdown of the human COL6A3 exon 41 ortholog negatively impacts axonal targeting mechanisms. A similar knockdown phenotype in zebrafish has been described for collagen XVIII,44 mutations that have been linked to ataxia-epilepsy phenotypes45 and Knobloch syndrome (MIM: 267750), a rare neurodevelopmental disorder.46 We hypothesize that the effect seen in our zebrafish experiments could hamper both the establishment of neural circuitry during early development as well as synaptic remodeling processes in brain maturation and in the adult brain. In light of recent studies supporting the pivotal role of ECM molecules in synaptic plasticity,47 it seems conceivable that the exon 41-encoded part of the collagen VI α3 C4 domain (FN-III motif) might be implicated in the organization of structural plasticity. The FN-III motif of other ECM proteins has been shown previously to be involved in axonal outgrowth of neurons, thereby modulating long-term potentiation in the hippocampus.48 Although the precise mechanisms by which COL6A3 mutations cause isolated dystonia remain to be elucidated, it is intriguing to consider that abnormal synaptic plasticity is viewed as a pathophysiological hallmark of isolated dystonia.49–52 Defects in synaptic function might account for maladaptive organization of sensorimotor loops, resulting in consolidation of erratic motor programs.51,52 The microstructural defects underlying aberrant plasticity in isolated dystonia might be established during embryonic and early postnatal development, characterizing isolated dystonia as a neurodevelopmental circuit disorder.53,54 Based on our genetic and functional evidence, we propose that (1) mutant COL6A3 might represent one of the molecular factors contributing to irregular sensorimotor circuit formation, and (2) the resulting plasticity changes might reflect a pathophysiological substrate for the development of dystonic movements. Because the large size of COL6A3 renders high-quality in vitro transcription of mRNA essentially impossible, it was technically not feasible to perform rescue experiments with dystonia-related missense alleles in our zebrafish model. Accordingly, future functional studies are warranted to determine how these missense substitutions mediate the phenotypic effect. Moreover, further genetic screening is required to clarify the prevalence of COL6A3-related isolated dystonia and to delineate the associated mutational and clinical spectrum.
In conclusion, we have identified COL6A3 mutations as an autosomal-recessive cause of isolated dystonia. Strikingly, all three families in our study have at least one mutation in exon 41, suppression of which induces specific neuronal pathologies in vivo. This suggests that perturbation of this specific region might be necessary to develop isolated dystonia. The involvement of a collagen gene in isolated dystonia highlights the ECM as a functional compartment in dystonia and substantially expands the scope for further research and drug discovery for this debilitating disorder.
Acknowledgments
We thank all individuals with dystonia and their family members who participated in this study. We are gratefully indebted to Jelena Golic, Susanne Lindhof, Sybille Frischholz, and Regina Feldmann (Institut für Humangenetik, Helmholtz Zentrum München, Munich, Germany) and Melanie Plastini (Center for Sleep Sciences and Medicine, Stanford University, Palo Alto, USA) for expert technical assistance, and our study nurses Silke Zwirner and Antje Lüsebrink (Neurologische Klinik und Poliklinik, Klinikum rechts der Isar, Technische Universität München, Munich, Germany). We thank Dr. Kornelia Kreiser, Abteilung für Diagnostische und Interventionelle Neuroradiologie, Klinikum rechts der Isar, Technische Universität München (Munich, Germany) for the performance of muscle MRI. We thank Erik Tilch for help with CADD. This study was funded by in-house institutional funding from Technische Universität München and Helmholtz Zentrum München, Munich, Germany, by seed funding from the Center for Human Disease Modeling, Duke University, and by P50 MH094268 to N.K. Recruitment of case and control cohorts was supported by institutional funding from Helmholtz Zentrum München, Munich, Germany, and government funding from the German Bundesministerium für Bildung und Forschung (BMBF, 03.2007-02.2011 FKZ 01ET0713). Fibroblasts were obtained from the Medical Research Council (MRC) Centre for Neuromuscular Diseases Biobank (Newcastle), which is part of EuroBioBank. The EU funded projects Neuromics (No. 305121) and RD-Connect (No. 305444). N.K. is a Distinguished Brumley Professor. M.Z. received intramural funding from the Langmatz-Stiftung.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://browser.1000genomes.org
ClustalW2, http://www.ebi.ac.uk/Tools/msa/clustalw2/
ExAC Browser, http://exac.broadinstitute.org/
ExonPrimer, http://ihg.gsf.de/ihg/ExonPrimer.html
Gene Expression Omnibus (GEO), http://www.ncbi.nlm.nih.gov/geo/
International HapMap Project, http://hapmap.ncbi.nlm.nih.gov/
NHLBI Exome Sequencing Project (ESP) Exome Variant Server, http://evs.gs.washington.edu/EVS/
OMIM, http://www.omim.org/
PolyPhen-2, http://www.genetics.bwh.harvard.edu/pph2/
Primer3Plus, http://primer3plus.com/cgi-bin/dev/primer3plus.cgi
References
- 1.Albanese A., Bhatia K., Bressman S.B., Delong M.R., Fahn S., Fung V.S., Hallett M., Jankovic J., Jinnah H.A., Klein C. Phenomenology and classification of dystonia: a consensus update. Mov. Disord. 2013;28:863–873. doi: 10.1002/mds.25475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balint B., Bhatia K.P. Dystonia: an update on phenomenology, classification, pathogenesis and treatment. Curr. Opin. Neurol. 2014;27:468–476. doi: 10.1097/WCO.0000000000000114. [DOI] [PubMed] [Google Scholar]
- 3.Charlesworth G., Bhatia K.P., Wood N.W. The genetics of dystonia: new twists in an old tale. Brain. 2013;136:2017–2037. doi: 10.1093/brain/awt138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ozelius L.J., Bressman S.B. Genetic and clinical features of primary torsion dystonia. Neurobiol. Dis. 2011;42:127–135. doi: 10.1016/j.nbd.2010.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Giménez-Roldán S., Delgado G., Marín M., Villanueva J.A., Mateo D. Hereditary torsion dystonia in gypsies. Adv. Neurol. 1988;50:73–81. [PubMed] [Google Scholar]
- 6.Khan N.L., Wood N.W., Bhatia K.P. Autosomal recessive, DYT2-like primary torsion dystonia: a new family. Neurology. 2003;61:1801–1803. doi: 10.1212/01.wnl.0000099076.17187.9a. [DOI] [PubMed] [Google Scholar]
- 7.Moretti P., Hedera P., Wald J., Fink J. Autosomal recessive primary generalized dystonia in two siblings from a consanguineous family. Mov. Disord. 2005;20:245–247. doi: 10.1002/mds.20228. [DOI] [PubMed] [Google Scholar]
- 8.Chouery E., Kfoury J., Delague V., Jalkh N., Bejjani P., Serre J.L., Mégarbané A. A novel locus for autosomal recessive primary torsion dystonia (DYT17) maps to 20p11.22-q13.12. Neurogenetics. 2008;9:287–293. doi: 10.1007/s10048-008-0142-4. [DOI] [PubMed] [Google Scholar]
- 9.Wichmann H.E., Gieger C., Illig T., MONICA/KORA Study Group KORA-gen—resource for population genetics, controls and a broad spectrum of disease phenotypes. Gesundheitswesen. 2005;67(1):S26–S30. doi: 10.1055/s-2005-858226. [DOI] [PubMed] [Google Scholar]
- 10.van der Stoep N., van Paridon C.D., Janssens T., Krenkova P., Stambergova A., Macek M., Matthijs G., Bakker E. Diagnostic guidelines for high-resolution melting curve (HRM) analysis: an interlaboratory validation of BRCA1 mutation scanning using the 96-well LightScanner. Hum. Mutat. 2009;30:899–909. doi: 10.1002/humu.21004. [DOI] [PubMed] [Google Scholar]
- 11.Schormair B., Kemlink D., Roeske D., Eckstein G., Xiong L., Lichtner P., Ripke S., Trenkwalder C., Zimprich A., Stiasny-Kolster K. PTPRD (protein tyrosine phosphatase receptor type delta) is associated with restless legs syndrome. Nat. Genet. 2008;40:946–948. doi: 10.1038/ng.190. [DOI] [PubMed] [Google Scholar]
- 12.Kircher M., Witten D.M., Jain P., O’Roak B.J., Cooper G.M., Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bönnemann C.G. The collagen VI-related myopathies: muscle meets its matrix. Nat. Rev. Neurol. 2011;7:379–390. doi: 10.1038/nrneurol.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Allamand V., Briñas L., Richard P., Stojkovic T., Quijano-Roy S., Bonne G. ColVI myopathies: where do we stand, where do we go? Skelet. Muscle. 2011;1:30. doi: 10.1186/2044-5040-1-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borck G., Hög F., Dentici M.L., Tan P.L., Sowada N., Medeira A., Gueneau L., Thiele H., Kousi M., Lepri F. BRF1 mutations alter RNA polymerase III-dependent transcription and cause neurodevelopmental anomalies. Genome Res. 2015;25:155–166. doi: 10.1101/gr.176925.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mangos S., Lam P.Y., Zhao A., Liu Y., Mudumana S., Vasilyev A., Liu A., Drummond I.A. The ADPKD genes pkd1a/b and pkd2 regulate extracellular matrix formation. Dis. Model. Mech. 2010;3:354–365. doi: 10.1242/dmm.003194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McClellan J., King M.C. Genetic heterogeneity in human disease. Cell. 2010;141:210–217. doi: 10.1016/j.cell.2010.03.032. [DOI] [PubMed] [Google Scholar]
- 18.Briñas L., Richard P., Quijano-Roy S., Gartioux C., Ledeuil C., Lacène E., Makri S., Ferreiro A., Maugenre S., Topaloglu H. Early onset collagen VI myopathies: Genetic and clinical correlations. Ann. Neurol. 2010;68:511–520. doi: 10.1002/ana.22087. [DOI] [PubMed] [Google Scholar]
- 19.Bovolenta M., Neri M., Martoni E., Urciuolo A., Sabatelli P., Fabris M., Grumati P., Mercuri E., Bertini E., Merlini L. Identification of a deep intronic mutation in the COL6A2 gene by a novel custom oligonucleotide CGH array designed to explore allelic and genetic heterogeneity in collagen VI-related myopathies. BMC Med. Genet. 2010;11:44. doi: 10.1186/1471-2350-11-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richards A.J., McNinch A., Whittaker J., Treacy B., Oakhill K., Poulson A., Snead M.P. Splicing analysis of unclassified variants in COL2A1 and COL11A1 identifies deep intronic pathogenic mutations. Eur. J. Hum. Genet. 2012;20:552–558. doi: 10.1038/ejhg.2011.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heinzen E.L., Arzimanoglou A., Brashear A., Clapcote S.J., Gurrieri F., Goldstein D.B., Jóhannesson S.H., Mikati M.A., Neville B., Nicole S., ATP1A3 Working Group Distinct neurological disorders with ATP1A3 mutations. Lancet Neurol. 2014;13:503–514. doi: 10.1016/S1474-4422(14)70011-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lohmann K., Klein C. The many faces of TUBB4A mutations. Neurogenetics. 2014;15:81–82. doi: 10.1007/s10048-014-0399-8. [DOI] [PubMed] [Google Scholar]
- 23.Karkheiran S., Krebs C.E., Makarov V., Nilipour Y., Hubert B., Darvish H., Frucht S., Shahidi G.A., Buxbaum J.D., Paisán-Ruiz C. Identification of COL6A2 mutations in progressive myoclonus epilepsy syndrome. Hum. Genet. 2013;132:275–283. doi: 10.1007/s00439-012-1248-1. [DOI] [PubMed] [Google Scholar]
- 24.Waite A., Tinsley C.L., Locke M., Blake D.J. The neurobiology of the dystrophin-associated glycoprotein complex. Ann. Med. 2009;41:344–359. doi: 10.1080/07853890802668522. [DOI] [PubMed] [Google Scholar]
- 25.Waite A., Brown S.C., Blake D.J. The dystrophin-glycoprotein complex in brain development and disease. Trends Neurosci. 2012;35:487–496. doi: 10.1016/j.tins.2012.04.004. [DOI] [PubMed] [Google Scholar]
- 26.Demir E., Sabatelli P., Allamand V., Ferreiro A., Moghadaszadeh B., Makrelouf M., Topaloglu H., Echenne B., Merlini L., Guicheney P. Mutations in COL6A3 cause severe and mild phenotypes of Ullrich congenital muscular dystrophy. Am. J. Hum. Genet. 2002;70:1446–1458. doi: 10.1086/340608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baker N.L., Mörgelin M., Peat R., Goemans N., North K.N., Bateman J.F., Lamandé S.R. Dominant collagen VI mutations are a common cause of Ullrich congenital muscular dystrophy. Hum. Mol. Genet. 2005;14:279–293. doi: 10.1093/hmg/ddi025. [DOI] [PubMed] [Google Scholar]
- 28.Baker N.L., Mörgelin M., Pace R.A., Peat R.A., Adams N.E., Gardner R.J., Rowland L.P., Miller G., De Jonghe P., Ceulemans B. Molecular consequences of dominant Bethlem myopathy collagen VI mutations. Ann. Neurol. 2007;62:390–405. doi: 10.1002/ana.21213. [DOI] [PubMed] [Google Scholar]
- 29.Lampe A.K., Zou Y., Sudano D., O’Brien K.K., Hicks D., Laval S.H., Charlton R., Jimenez-Mallebrera C., Zhang R.Z., Finkel R.S. Exon skipping mutations in collagen VI are common and are predictive for severity and inheritance. Hum. Mutat. 2008;29:809–822. doi: 10.1002/humu.20704. [DOI] [PubMed] [Google Scholar]
- 30.Lamandé S.R., Mörgelin M., Adams N.E., Selan C., Allen J.M. The C5 domain of the collagen VI alpha3(VI) chain is critical for extracellular microfibril formation and is present in the extracellular matrix of cultured cells. J. Biol. Chem. 2006;281:16607–16614. doi: 10.1074/jbc.M510192200. [DOI] [PubMed] [Google Scholar]
- 31.Trüeb B., Winterhalter K.H. Type VI collagen is composed of a 200 kd subunit and two 140 kd subunits. EMBO J. 1986;5:2815–2819. doi: 10.1002/j.1460-2075.1986.tb04573.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chu M.L., Zhang R.Z., Pan T.C., Stokes D., Conway D., Kuo H.J., Glanville R., Mayer U., Mann K., Deutzmann R. Mosaic structure of globular domains in the human type VI collagen alpha 3 chain: similarity to von Willebrand factor, fibronectin, actin, salivary proteins and aprotinin type protease inhibitors. EMBO J. 1990;9:385–393. doi: 10.1002/j.1460-2075.1990.tb08122.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aigner T., Hambach L., Söder S., Schlötzer-Schrehardt U., Pöschl E. The C5 domain of Col6A3 is cleaved off from the Col6 fibrils immediately after secretion. Biochem. Biophys. Res. Commun. 2002;290:743–748. doi: 10.1006/bbrc.2001.6227. [DOI] [PubMed] [Google Scholar]
- 34.O’Reilly M.S., Boehm T., Shing Y., Fukai N., Vasios G., Lane W.S., Flynn E., Birkhead J.R., Olsen B.R., Folkman J. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 1997;88:277–285. doi: 10.1016/s0092-8674(00)81848-6. [DOI] [PubMed] [Google Scholar]
- 35.Ortega N., Werb Z. New functional roles for non-collagenous domains of basement membrane collagens. J. Cell Sci. 2002;115:4201–4214. doi: 10.1242/jcs.00106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dityatev A., Seidenbecher C.I., Schachner M. Compartmentalization from the outside: the extracellular matrix and functional microdomains in the brain. Trends Neurosci. 2010;33:503–512. doi: 10.1016/j.tins.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 37.Novak U., Kaye A.H. Extracellular matrix and the brain: components and function. J. Clin. Neurosci. 2000;7:280–290. doi: 10.1054/jocn.1999.0212. [DOI] [PubMed] [Google Scholar]
- 38.Bonneh-Barkay D., Wiley C.A. Brain extracellular matrix in neurodegeneration. Brain Pathol. 2009;19:573–585. doi: 10.1111/j.1750-3639.2008.00195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hubert T., Grimal S., Carroll P., Fichard-Carroll A. Collagens in the developing and diseased nervous system. Cell. Mol. Life Sci. 2009;66:1223–1238. doi: 10.1007/s00018-008-8561-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fox M.A. Novel roles for collagens in wiring the vertebrate nervous system. Curr. Opin. Cell Biol. 2008;20:508–513. doi: 10.1016/j.ceb.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 41.Fox M.A., Sanes J.R., Borza D.B., Eswarakumar V.P., Fässler R., Hudson B.G., John S.W., Ninomiya Y., Pedchenko V., Pfaff S.L. Distinct target-derived signals organize formation, maturation, and maintenance of motor nerve terminals. Cell. 2007;129:179–193. doi: 10.1016/j.cell.2007.02.035. [DOI] [PubMed] [Google Scholar]
- 42.Cheng J.S., Dubal D.B., Kim D.H., Legleiter J., Cheng I.H., Yu G.Q., Tesseur I., Wyss-Coray T., Bonaldo P., Mucke L. Collagen VI protects neurons against Abeta toxicity. Nat. Neurosci. 2009;12:119–121. doi: 10.1038/nn.2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheng I.H., Lin Y.C., Hwang E., Huang H.T., Chang W.H., Liu Y.L., Chao C.Y. Collagen VI protects against neuronal apoptosis elicited by ultraviolet irradiation via an Akt/phosphatidylinositol 3-kinase signaling pathway. Neuroscience. 2011;183:178–188. doi: 10.1016/j.neuroscience.2011.03.057. [DOI] [PubMed] [Google Scholar]
- 44.Schneider V.A., Granato M. The myotomal diwanka (lh3) glycosyltransferase and type XVIII collagen are critical for motor growth cone migration. Neuron. 2006;50:683–695. doi: 10.1016/j.neuron.2006.04.024. [DOI] [PubMed] [Google Scholar]
- 45.Paisán-Ruiz C., Scopes G., Lee P., Houlden H. Homozygosity mapping through whole genome analysis identifies a COL18A1 mutation in an Indian family presenting with an autosomal recessive neurological disorder. Am. J. Med. Genet. B. Neuropsychiatr. Genet. 2009;150B:993–997. doi: 10.1002/ajmg.b.30929. [DOI] [PubMed] [Google Scholar]
- 46.Sertié A.L., Sossi V., Camargo A.A., Zatz M., Brahe C., Passos-Bueno M.R. Collagen XVIII, containing an endogenous inhibitor of angiogenesis and tumor growth, plays a critical role in the maintenance of retinal structure and in neural tube closure (Knobloch syndrome) Hum. Mol. Genet. 2000;9:2051–2058. doi: 10.1093/hmg/9.13.2051. [DOI] [PubMed] [Google Scholar]
- 47.Dityatev A., Schachner M., Sonderegger P. The dual role of the extracellular matrix in synaptic plasticity and homeostasis. Nat. Rev. Neurosci. 2010;11:735–746. doi: 10.1038/nrn2898. [DOI] [PubMed] [Google Scholar]
- 48.Strekalova T., Sun M., Sibbe M., Evers M., Dityatev A., Gass P., Schachner M. Fibronectin domains of extracellular matrix molecule tenascin-C modulate hippocampal learning and synaptic plasticity. Mol. Cell. Neurosci. 2002;21:173–187. doi: 10.1006/mcne.2002.1172. [DOI] [PubMed] [Google Scholar]
- 49.Breakefield X.O., Blood A.J., Li Y., Hallett M., Hanson P.I., Standaert D.G. The pathophysiological basis of dystonias. Nat. Rev. Neurosci. 2008;9:222–234. doi: 10.1038/nrn2337. [DOI] [PubMed] [Google Scholar]
- 50.Neychev V.K., Gross R.E., Lehéricy S., Hess E.J., Jinnah H.A. The functional neuroanatomy of dystonia. Neurobiol. Dis. 2011;42:185–201. doi: 10.1016/j.nbd.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Quartarone A., Pisani A. Abnormal plasticity in dystonia: Disruption of synaptic homeostasis. Neurobiol. Dis. 2011;42:162–170. doi: 10.1016/j.nbd.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 52.Quartarone A., Hallett M. Emerging concepts in the physiological basis of dystonia. Mov. Disord. 2013;28:958–967. doi: 10.1002/mds.25532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ledoux M.S., Dauer W.T., Warner T.T. Emerging common molecular pathways for primary dystonia. Mov. Disord. 2013;28:968–981. doi: 10.1002/mds.25547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Niethammer M., Carbon M., Argyelan M., Eidelberg D. Hereditary dystonia as a neurodevelopmental circuit disorder: Evidence from neuroimaging. Neurobiol. Dis. 2011;42:202–209. doi: 10.1016/j.nbd.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.