

Abstract
Next-generation sequencing provides the opportunity to practice predictive medicine based on identified variants. Putative loss-of-function (pLOF) variants are common in genomes and understanding their contribution to disease is critical for predictive medicine. To this end, we characterized the consequences of pLOF variants in an exome cohort by iterative phenotyping. Exome data were generated on 951 participants from the ClinSeq cohort and filtered for pLOF variants in genes likely to cause a phenotype in heterozygotes. 103 of 951 exomes had such a pLOF variant and 79 participants were evaluated. Of those 79, 34 had findings or family histories that could be attributed to the variant (28 variants in 18 genes), 2 had indeterminate findings (2 variants in 2 genes), and 43 had no findings or a negative family history for the trait (34 variants in 28 genes). The presence of a phenotype was correlated with two mutation attributes: prior report of pathogenicity for the variant (p = 0.0001) and prior report of other mutations in the same exon (p = 0.0001). We conclude that 1/30 unselected individuals harbor a pLOF mutation associated with a phenotype either in themselves or their family. This is more common than has been assumed and has implications for the setting of prior probabilities of affection status for predictive medicine.
Introduction
Putative loss-of-function (pLOF) variants including nonsense, frameshift, and splice site alterations are common in genomes.1 Genome sequence analysis can generate up to 800 pLOF mutations in a single genome, which is much higher than the estimate of true loss-of-function variants, which are estimated to be present at a level closer to 100 variants per person. The large number of pLOF alleles is attributable to sequencing and annotation errors, gene redundancy, hypomorphic alleles, true LOF alleles for carrier states, and LOF mutations in genes that are sensitive to haploinsufficiency.1,2 With the increasing use of next-generation sequencing technologies for predictive medicine, it is critical to be able to predict the consequences of pLOFs, especially in individuals without pre-existing clinical diagnoses. Although many bioinformatic approaches have been developed to classify missense alterations, there are few tools available to assess pLOF variants. General bioinformatics rules are starting to emerge but tools are not yet available that are capable of delivering accurate predictions of pathogenicity. Complicating the situation, genotype-phenotype correlation studies are typically performed on individuals selected for predefined phenotypes based on the researcher’s presupposition as to what the phenotype(s) comprise. The ability of this approach to discover novel phenotypic associations is inherently limited and can lead to an underestimate of causation for variants resulting in atypical phenotypes. To address this issue, we set out to characterize the phenotypic consequences of pLOF variants by using iterative phenotyping to understand the spectrum of these variants in the ClinSeq cohort,3 their consequences, and what, if any, variant attributes are correlated with disease phenotypes.
To enable the practical detection of a phenotype, we focused on pLOF variants in genes for which there was evidence that a heterozygous LOF variant could cause disease. We then performed a customized clinical evaluation of the participants with these variants to identify phenotypic characteristics in them or their close family members that could be attributable to the pLOF variant. We correlated these findings with a number of simple pathogenicity metrics of the variant, the haploinsufficiency score,4 and the combined annotation-dependent depletion (CADD) score.5
Subjects and Methods
Study Participants
The participants were 45 to 65 years of age at enrollment and were selected for a range of atherosclerosis phenotypes but not for personal or family histories of any other phenotype.3 An initial family history was collected on all participants via the U.S. Surgeon General’s family health history tool, My Family Health Portrait, which specifically asks users about their family history of heart disease, stroke, diabetes, and colon, breast, and ovarian cancers. A board-certified genetic counselor or geneticist spent approximately 30 min reviewing the family history with the participant in order to confirm the reported diagnoses, inquire broadly about other diseases in the family, and expand the pedigree to a minimum of three generations, as described.6 A clinical assessment was also conducted on all participants and included an echocardiogram and blood lipid panel, as described.3 The 951 participants included in this study include 483 males (51%) and 468 females (49%). Of these participants, 85% self-identified as being of Caucasian descent and non-Hispanic and 18% self-identified as being of Ashkenazi decent. The NHGRI institutional review board reviewed and approved this study and all subjects provided written informed consent.
Next-Generation Sequencing
DNA was isolated from whole blood via the salting-out method (QIAGEN), followed by phenol chloroform extraction. Solution hybridization exome capture was performed with either the SureSelect All Exon System (Agilent Technologies) or the Illumina TruSeq system (Illumina). Flow cell preparation and paired-end read sequencing were performed with either the GAIIx or HiSeq 2000 sequencer7 (Illumina). Image analyses and base calling were performed as described.7 Reads were aligned to hg19, NCBI 37, via novoalign (Novocraft Technologies). Samples were sequenced to sufficient coverage such that 85% of the targeted exome was called with high-quality variant detection (reported as genotype at every callable position). Genotypes were called using only those sequence bases with Phred base qualities of at least Q20 by Most Probable Genotype7 (MPG) and an MPG score of ≥10. Filters were applied with the VarSifter Next-Gen variant analysis software.8
Next-Generation Variant Analysis
Exome data were generated on 951 participants from the ClinSeq cohort. Variants were filtered for quality (MPG score ≥ 10). pLOF variants were defined as nonsense, frameshift, and canonical splice site (intronic +1, +2, −1, −2) variants, recognizing that not all of these represent true LOF. Variants were filtered (Figure 1) for genes in the Human Gene Mutation Database (HGMD, December 2012) and for pLOF in at least one reference sequence (RefSeq) transcript. We excluded variants with a minor allele frequency (MAF) ≥ 0.005 in ClinSeq or dbSNP. This filter was calculated based on number of calls at a position and was not adjusted for ancestry. Inheritance information from the Clinical Genomic Database (CGD), the Human Gene Mutation Database (HGMD, professional version), Online Mendelian Inheritance of Man (OMIM), and Genetics Home Reference were used to limit variants to those in genes with expected autosomal-dominant inheritance; additionally, pLOF variants previously reported not to cause disease in an autosomal-dominant pattern were removed. We excluded genes for which the only associated phenotype would be better considered to be a trait rather than a disease, such as hair color, recognizing that this distinction is subjective. No exclusion criteria for disease penetrance were used, even if the penetrance was expected to be low in the heterozygous state. In addition, classification of dominant inheritance of disease susceptibility due to pLOF variants required a minimum of two reported cases of pLOF variants in the gene causing disease according to HGMD. We removed variants with numerous low-quality calls, pharmacogenetic variants, variants in genes with unclear disease associations, and variants associated with transient childhood phenotypes or later-onset neurodegenerative disorders.
Figure 1.
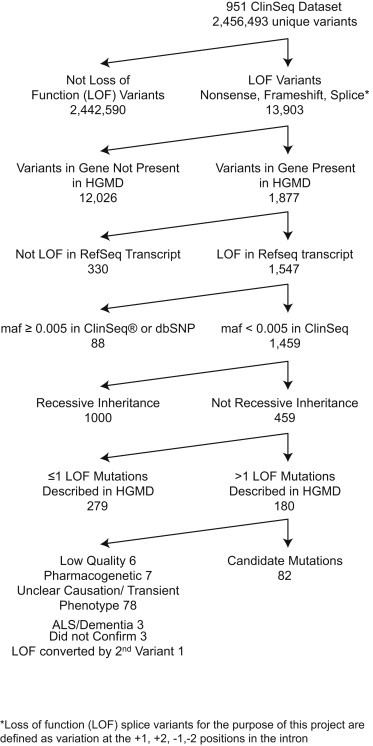
Filtering Tree Used to Identify Variants of Interest in Our Cohort of 951 Individuals
Sequence Confirmation
Primers were designed for all variants of interest and confirmation sequencing was performed with standard Sanger sequencing protocols (BigDye, LifeTechnologies).
Iterative Phenotyping
Re-contact was attempted for participants with a variant of interest to update their personal and family history, focusing on the phenotype normally associated with variants in the gene. Participants were invited to a follow-up clinic visit if a family history was insufficient to confirm or rule out a diagnosis. Follow-up visits focused on detecting phenotypic findings potentially related to the variant in question and included additional diagnostic procedures (e.g., cranial MRI or skin biopsies) that could confirm or refute the phenotype in some cases. When potentially informative relatives were available, saliva DNA was collected for segregation analysis (DNA Genotek) and, when appropriate, relatives were assessed in the clinic.
Variant Attribute Correlations
Variant analysis of pLOF variants was performed with correlates of pathogenicity (Table 1) including: if ≥5 pLOF mutations were present in that gene in HGMD, if there was a mutation in that exon in the public version of HGMD, if the identified mutation was logged in HGMD as a disease-causing (DM) mutation, if the pLOF was in the middle 90% of the gene (i.e., not in the first or last 5%), if MutationTaster9 predicted nonsense-mediated decay (NMD), if the variant was expected to result in a frameshift with >10 aberrant amino acids, and if the exon with the pLOF variant was in the dominant gene model as defined by presence in >75% of spliced expressed sequence tags (ESTs) for the gene. QuickCalcs was utilized to calculate statistical significance of associations via a two-tailed Fisher’s exact test. Analyses were run inclusive of all variants that could be defined as yes or no for each attribute. Specifically, splice site variants were not included in NMD or frame shift attributes because a clear outcome could not be predicted. All analyses were run a second time after removing variants identified in LDLR (MIM: 606945) because variants in this gene were over represented and the possibility existed that LDLR variants could in some way be different and skew results. Additionally, variants were analyzed via existing bioinformatics predictors of deleteriousness including combined annotation-dependent depletion (CADD v.1.0) scores5 and the haploinsufficiency index of the gene when available4 by the Mann-Whitney U test. For the haploinsufficiency index, each variant was assigned the haploinsufficiency index for the relevant gene and each gene was included one time in the calculation. The haploinsufficiency score was not a factor in deciding which genes should be included in the initial analysis.
Table 1.
pLOF Variants Identified in Our Cohort of 951 Individuals Followed up with Iterative Phenotyping
| Gene Name | cDNA Nomenclature | Protein Nomenclature | Disease | Phenotype Classification | ≥5 pLOF in HGMD | HGMD DM | HGMD Public Mutations in Exon | Dominant Transcript | fs∗ > 10 Amino Acids | Middle 90% of CDS | NMD Predicted by MutationTaster | Haploinsufficiency Score | CADD Score | Genomic Position (hg19) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BRCA1 | NM_007294.3; c.68_69del | NP_009225.1; p.Glu23Valfs∗17 | familial breast-ovarian cancer 1 | positive | yes | yes | yes | yes | yes | no | yes | 0.999 | 14.15 | chr17: g.41276045_41276046del |
| BRCA1 | NM_007294.3; c.547+2T>A | NP_009225.1; splice | familial breast-ovarian cancer 1 | positive | yes | yes | yes | yes | splice | yes | splice | 0.999 | 12.29 | chr17: g.41251790A>T |
| BRCA2 | NM_000059.3; c.5946del | NP_000050.2; p.Ser1982Argfs∗22 | familial breast-ovarian cancer 2 | positive | yes | yes | yes | yes | yes | yes | yes | 0.972 | 45.00 | chr13: g.32914438del |
| BRCA2 | NM_000059.3; c.8297del | NP_000050.2; p.Thr2766Asnfs∗11 | familial breast-ovarian cancer 2 | positive | yes | yes | yes | yes | no | yes | yes | 0.972 | 47.00 | chr13: g.32937636del |
| F11 | NM_000128.3; c.403G>T | NP_000119.1; p.Glu135∗ | factor XI deficiency | positive | yes | yes | yes | yes | no | yes | yes | 0.104 | 17.26 | chr4: g.187195347G>T |
| FLCN | NM_144606.5; c.918G>A | NP_653207.1; p.Trp306∗ | Birt-Hogg-Dubé | positive | yes | no | yes | no | no | yes | no | 0.312 | 24.80 | chr17: g.17124804C>T |
| FLCN | NM_144997.5; c.1285dup | NP_659434.2; p.His429Profs∗27 | Birt-Hogg-Dubé | positive | yes | yes | yes | no | yes | yes | yes | 0.312 | 31.00 | chr17: g.17119709 dup |
| HOXD13 | NM_000523.3; c.820C>T | NP_000514.2; p.Arg274∗ | brachydactyly-syndactyly syndrome | positive | yes | yes | yes | yes | no | yes | no | 0.826 | 28.40 | chr2: g.176959246C>T |
| KCNQ4 | NM_004700.3; c.1725del | NP_004691.2; p.Ile576Serfs∗40 | deafness, autosomal dominant 2A | positive | no | no | no | yes | yes | yes | no | 0.218 | 37.00 | chr1: g.41300749del |
| KRT16 | NM_005557.3; c.614+1G>A | NP_005548.2; splice | pachyonychia congenita | positive | no | no | no | yes | splice | yes | splice | 0.266 | 21.30 | chr17: g.39767890C>T |
| LDLR | NM_000527.3; c.2061dup | NP_000518.1; p.Asn688Glnfs∗29 | hypercholesterolemia | positive | yes | yes | yes | yes | yes | yes | yes | 0.469 | 15.65 | chr19: g.11231119dup |
| LDLR | NM_000527.3; c.653del | NP_000518.1; p.Gly218Valfs∗47 | hypercholesterolemia | positive | yes | yes | yes | yes | yes | yes | yes | 0.469 | 15.88 | chr19: g.11216234del |
| LDLR | NM_000527.3; c.261G>A | NP_000518.1; p.Trp87∗ | hypercholesterolemia | positive | yes | yes | yes | yes | no | yes | yes | 0.469 | 26.40 | chr19: g.11213410G>A |
| LDLR | NM_000527.3; c.564C>G | NP_000518.1; p.Tyr188∗ | hypercholesterolemia | positive | yes | yes | yes | yes | no | yes | yes | 0.469 | 19.01 | chr19: g.11216146C>G |
| LDLR | NM_000527.3; c.2478del | NP_000518.1; p.Val827Serfs∗102 | hypercholesterolemia | positive | yes | yes | yes | yes | yes | no | no | 0.469 | 25.00 | chr19: g.11240274del |
| LDLR | NM_000527.3; c.313+1G>A | NP_000518.1; splice | hypercholesterolemia | positive | yes | yes | yes | yes | splice | yes | splice | 0.469 | 15.97 | chr19: g.11213463G>A |
| LDLR | NM_000527.3; c.694+2T>C | NP_000518.1; splice | hypercholesterolemia | positive | yes | yes | yes | yes | splice | yes | splice | 0.469 | 16.15 | chr19: g.11216278T>C |
| MYH7 | NM_000257.2; c.732+1G>A | NP_000248.2; splice | cardiomyopathy | positive | yes | yes | yes | yes | splice | yes | splice | NA | 18.63 | chr14: g.23900793C>T |
| PKD1 | NM_000296.3; c.5968_5969del | NP_000287.3; p.Arg1990Glufs∗59 | polycystic kidney disease 1 | positive | yes | yes | yes | yes | yes | yes | yes | NA | 8.26 | chr16: g.2159202_2159203del |
| PMS2 | NM_000535.5; c.943C>T | NP_000526.1; p.Arg315∗ | mismatch repair cancer syndrome | positive | yes | yes | yes | yes | no | yes | yes | NA | 27.90 | chr7: g.6031649G>A |
| PPARG | NM_015869.4; c.1495del | NP_056953.2; p.Glu499Argfs∗12 | lipodystrophy, familial partial 3 | positive | yes | no | yes | yes | yes | no | no | 0.514 | 37.00 | chr3: g.12475621del |
| PROS1 | NM_000313.3; c.601+1G>A | NP_000304.2; splice | protein S deficiency | positive | yes | no | yes | yes | splice | yes | splice | NA | 13.58 | chr3: g.93624627C>T |
| SFTPC | NM_003018.3; c.322C>T | NP_003009.2; p.Gln108∗ | surfactant metabolism dysfunction 2 | positive | yes | no | yes | yes | no | yes | yes | 0.343 | 20.90 | chr8: g.22020713C>T |
| SGCE | NM_003919.2; c.21G>A | NP_003910.1; p.Trp7∗ | dystonia 11, myoclonic | positive | yes | no | yes | yes | no | no | yes | 0.475 | 28.10 | chr7: g.94285390C>T |
| SLC4A1 | NM_000342.3; c.253_257dup | NP_000333.1; p.Asn87Argfs∗24 | spherocytosis 4 | positive | yes | no | yes | yes | yes | yes | yes | 0.199 | 20.40 | chr17: g.42338095_42338099dup |
| SLC4A1 | NM_000342.3; c.2354del | NP_000333.1; p.Leu785Argfs∗44 | spherocytosis 4 | positive | yes | no | yes | yes | yes | yes | yes | 0.199 | 38.00 | chr17: g.42328914del |
| TGIF1 | NM_170695.2; c.22del | NP_733796.2; p.Val8Cysfs∗126 | holoprosencephaly 4 | positive | yes | no | no | no | yes | no | yes | NA | 14.89 | chr18: g.3451999del |
| TNFRSF13B | NM_012452.2; c.204dup | NP_036584.1; p.Leu69Thrfs∗12 | immunoglobulin A deficiency 2 | positive | yes | yes | yes | yes | yes | yes | yes | NA | 10.72 | chr17: g.16852293dup |
| ARID1B | NM_020732.3; c.1762G>T | NP_065783.3; p.Glu588∗ | Coffin-Siris syndrome | negative | yes | no | no | no | no | yes | yes | NA | 23.60 | chr6: g.157192772G>T |
| ATP2C1 | NM_014065.2; c.1656del | NP_054784.2; p.Met553Cysfs∗16 | Hailey-Hailey disease | negative | yes | no | no | no | yes | yes | no | 0.875 | 32.00 | chr3: g.130735044del |
| BRCA2 | NM_000059.3; c.5482_5486del | NP_000050.2; p.Lys1828Valfs∗4 | familial breast-ovarian cancer 2 | negative | yes | no | yes | yes | no | yes | yes | 0.972 | 26.90 | chr13: g.32913974_32913978del |
| BRCA2 | NM_000059.3; c.10094_10095insGAATTATATC | NP_000050.2; p.Ser3366Asnfs∗5 | familial breast-ovarian cancer 2 | negative | yes | no | yes | yes | no | no | yes | 0.972 | NA | chr13: g.32972744_32972745ins GAATTATATC |
| COL10A1 | NM_000493.3; c.1300_1322del | NP_000484.2; p.Gly434Lysfs∗10 | metaphyseal chondrodysplasia, Schmid type | negative | yes | no | yes | no | no | yes | no | 0.133 | 17.70 | chr6: g.116441961_116441983del |
| CREB3L3 | NM_032607.1; c.732dup | NP_115996.1; p.Lys245Glufs∗130 | hypertriglyceridemia | negative | no | no | no | yes | yes | yes | no | 0.156 | 22.70 | chr19: g.4168365dup |
| DMD | NM_004006.2; c.10247G>A | NP_003997.1; p.Trp3416∗ | Duchene muscular dystrophy | negative | yes | no | yes | yes | no | yes | yes | 0.493 | 53.00 | chrX: g.31196064C>T |
| DSG1 | NM_001942.2; c.3106C>T | NP_001933.2; p.Arg1036∗ | keratosis palmoplantaris striata I | negative | yes | no | no | yes | no | no | no | 0.736 | 39.00 | chr18: g.28935265C>T |
| EPHA2 | NM_004431.3; c.1420del | NP_004422.2; p.Arg474Alafs∗19 | cataract 6 | negative | no | no | no | yes | yes | yes | yes | 0.635 | 37.00 | chr1: g.16462158del |
| GLI2 | NM_005270.4; c.149−1G>A | NP_005261.2; splice | holoprosencephaly 9 | negative | yes | no | no | yes | splice | no | no | NA | 13.83 | chr2: g.121684936G>A |
| GLI3 | NM_000168.5; c.76C>T | NP_000159.3; p.Arg26∗ | Greig cephalopolysyndactyly syndrome | negative | yes | no | no | yes | no | no | yes | 0.996 | 36.00 | chr7: g.42262777G>A |
| GNAS | NM_080425.2; c.758del | NP_536350.2; p.Ser253Thrfs∗437 | pseudohypoparathyroidism | negative | yes | no | no | no | yes | yes | yes | 0.533 | 9.36 | chr20: g.57429078del |
| HSPB1 | NM_001540.3; c.438dup | NP_001531.1; p.Gly147Argfs∗14 | Charcot-Marie-Tooth disease 2 | negative | no | no | yes | yes | yes | yes | no | 0.578 | 28.70 | chr7: g.75933310dup |
| MSH6 | NM_000179.2; c.730C>T | NP_000170.1; p.Gln244∗ | mismatch repair cancer syndrome | negative | yes | yes | yes | yes | no | yes | yes | 0.971 | 15.91 | chr2: g.48025852C>T |
| MSH6 | NM_000179.2; c.4068_4071dup | NP_000170.1; p.Lys1358Aspfs∗2 | mismatch repair cancer syndrome | negative | yes | no | yes | yes | no | no | no | 0.971 | 40.00 | chr2: g.48033984_48033987dup |
| MYH6 | NM_002471.3; c.1716dup | NP_002462.2; p.Lys573Glufs∗63 | congenital heart defects | negative | no | no | no | no | yes | yes | yes | 0.855 | 26.80 | chr14: g.23868115dup |
| MYOC | NM_000261.1; c.814C>T | NP_000252.1; p.Arg272∗ | glaucoma 1A, primary open angle | negative | yes | yes | yes | yes | no | yes | no | 0.635 | 28.30 | chr1: g.171605766G>A |
| MYOC | NM_000261.1; c.1102C>T | NP_000252.1; p.Gln368∗ | glaucoma 1A, primary open angle | negative | yes | yes | yes | yes | no | yes | no | 0.635 | 18.23 | chr1: g.171605478G>A |
| OFD1 | NM_003611.2; c.936−2A>G | NP_003602.1; splice | oral-facial-digital syndrome 1 | negative | yes | no | no | no | splice | yes | splice | 0.217 | 13.69 | chrX: g.13769366A>G |
| PAX3 | NM_000438.5; c.630G>A | NP_000429.2; p.Trp210∗ | Waardenburg syndrome I | negative | yes | no | no | no | no | no | no | 0.993 | 14.14 | chr2: g.223158842C>T |
| PRKCSH | NM_002743.2; c.−77−2A>C | NP_002734.2; splice | polycystic liver disease | negative | yes | no | no | yes | splice | no | splice | 0.263 | 8.53 | chr19: g.11546860A>C |
| RAD51D | NM_002878.3; c.904−2A>T | NP_002869.3; splice | breast and ovarian cancer 4 | negative | yes | no | no | yes | splice | yes | splice | NA | 11.05 | chr17: g.33428057T>A |
| RP1 | NM_006269.1; c.6304C>T | NP_006260.1; p.Gln2102∗ | retinitis pigmentosa 1 | negative | yes | no | yes | no | no | no | no | 0.430 | 45.00 | chr8: g.55542746C>T |
| RUNX1 | NM_001754.4; c.−59−1G>A | NP_001745.2; splice | platelet disorder, familial, with associated myeloid malignancy | negative | yes | no | no | yes | splice | no | splice | 0.812 | 17.35 | chr21: g.36421256C>T |
| SDHC | NM_003001.3; c.43C>T | NP_002992.1; p.Arg15∗ | paragangliomas 3 | negative | yes | yes | yes | yes | no | yes | yes | NA | 29.60 | chr1: g.161293426C>T |
| SMAD3 | NM_001145103.1; c.72del | NP_001138575.1; p.Arg25Glyfs∗47 | Loeys-Dietz syndrome, type 3 | negative | yes | no | no | no | yes | yes | yes | 1.000 | 10.52 | chr15: g.67430436del |
| TGIF1 | NM_173207.1; c.38C>G | NP_775299.1; p.Ser13∗ | holoprosencephaly 4 | negative | yes | no | no | no | no | no | yes | NA | 22.90 | chr18: g.3447775C>G |
| THRA | NM_199334.3; c.54−1G>A | NP_955366.1; splice | hypothyroidism, nongoitrous 6 | negative | no | no | no | yes | splice | no | splice | 0.558 | 16.64 | chr17: g.38233123G>A |
| TRPC6 | NM_004621.5; c.1649dup | NP_004612.2; p.His550Glnfs∗10 | focal segmental glomerulosclerosis 2 | negative | no | no | no | yes | no | yes | yes | NA | 24.50 | chr11: g.101347127dup |
| TTN | NM_133378.4; c.29362+1G>A | NP_596869.4; splice | cardiomyopathy | negative | yes | no | no | no | splice | yes | splice | NA | 15.95 | chr2: g.179547423C>T |
| TTN | NM_133379.3; c.16321C>T | NP_596870.2; p.Arg5441∗ | cardiomyopathy | negative | yes | no | no | no | no | no | no | NA | 55.00 | chr2: g.179610806G>A |
| TTN | NM_133379.3; c.14844_14845del | NP_596870.2; p.Tyr4949∗ | cardiomyopathy | negative | yes | no | no | no | no | yes | no | NA | 47.00 | chr2: g.179612285_179612286del |
| TTN | NM_133437.3; c.10670dup | NP_597681.3; p.Leu3558Thrfs∗9 | cardiomyopathy | negative | yes | no | no | yes | no | yes | yes | NA | 46.00 | chr2: g.179621021dup |
| XRCC2 | NM_005431.1; c.643C>T | NP_005422.1; p.Arg215∗ | breast cancer | negative | no | yes | yes | yes | no | yes | no | 0.366 | 13.00 | chr7: g.152345927G>A |
| DSPP | NM_014208.3; c.1289C>G | NP_055023.2; p.Ser430∗ | dentinogenesis imperfecta, Shields type II | indeterminate | yes | no | yes | no EST data | no | yes | no | 0.802 | 18.94 | chr4: g.88535103C>G |
| SALL4 | NM_020436.3; c.2788_2789delinsC | NP_065169.1; p.Gly930Glnfs∗13 | Duane-radial ray syndrome | indeterminate | yes | no | no | yes | yes | yes | no | 0.632 | 37.00 | chr20: g.50401177_50401178delinsG |
| Positive variants | 26/28 | 18/28 | 25/28 | 25/28 | 13/22 | 23/28 | 17/22 | NA | NA | |||||
| Negative variants | 27/34 | 5/34 | 21/34 | 12/34 | 7/27 | 22/34 | 14/28 | NA | NA | |||||
| p Value | 0.1662 | 0.0001 | 0.0193 | 0.0001 | 0.0234 | 0.1589 | 0.0780 | 0.1514 | 0.5409 |
Variants are listed as positive, negative, or indeterminate based on participant phenotypic findings. Variants were considered positive if at least one participant with that variant was classified as positive for the phenotype. Seven attributes considered predictors of pathogenicity are shown along with haploinsufficiency score for the gene and CADD score for the variant.
We attempted to catalog the penetrance of the gene-phenotype pairs (see Table S2) in an effort to correlate known penetrance with our finding of a positive or negative phenotype. We sourced this information from GeneReviews first, and if not described there, searched in OMIM, and then the primary literature.
Results
Variant Filtering and Classification
Variants were filtered for quality (MPG score ≥ 10), yielding 2,456,493 variants (average of 100,664 variants per individual). pLOF variants were defined as nonsense, frameshift, and canonical splice site variants knowing that not all of these are true LOF (yielding 13,903 variants, average of 484 per individual). Variants were further filtered (Figure 1) so as to produce a list of pLOF variants in genes where pLOF variants were considered highly likely to cause a phenotype in the heterozygous state. The filtered list contained 82 variants in 103 participants (Table S1).
Iterative Phenotyping
An attempt was made to re-contact all 103 participants with a pLOF variant of interest. Fifteen participants were lost to follow-up, five individuals declined follow-up phenotyping, and four individuals had family members who were reported as potentially positive by family history but declined to participate, leaving 79 participants (Table S2) with 64 variants (Table 1). Of these 79 participants, 34 had findings or family histories that could be attributed to the variant (28 variants in 18 genes), 2 had indeterminate findings (2 variants, 1 each in 2 genes), and 43 had negative findings or a negative family history for associated traits (34 variants in 28 genes).
In 15 individuals, a positive personal/family history for disease was verified by initial family history alone (spherocytosis [MIM: 612653], polycystic kidney disease [MIM: 173900], cancer, and hypercholesterolemia [MIM: 143890]). One individual with a mutation in SLC4A1 (MIM: 109270) had a personal and family history of spherocytosis with an affected mother, two affected siblings, two affected children, and additional affected relatives in the pedigree. Another individual with a mutation in PKD1 (MIM: 601313) had a clear personal and family history of polycystic kidney disease with an affected father, two affected siblings, and two affected children. Six individuals had a positive personal or family history for cancer. Five had a positive personal (n = 1) and/or family (n = 5) history for breast and ovarian cancer (MIM: 604370) with mutations in BRCA1 (MIM: 113705) (n = 2) and BRCA2 (MIM: 600185) (n = 3). To determine positive family history for breast and ovarian cancer, one case of premenopausal breast or ovarian cancer was required to have occurred and at least one additional case of breast, ovarian, or prostate cancer was required. In one predominantly male pedigree, the family history was determined to be positive based on a single case of breast cancer at the age of 50 in the male participant’s mother. One individual had a positive personal and family history for a mismatch repair cancer syndrome (MIM: 276300) with a mutation in PMS2 (MIM: 600259). The participant had colon cancer and the participant’s mother was deceased (from liver cancer thought to be due to metastatic disease originating in the colon). Seven individuals with mutations in LDLR had a personal and family history of hypercholesterolemia and were under treatment for their disease.
In 15 individuals, a negative individual/family history for disease was verified by family history alone: cancer (BRCA1, n = 2; BRCA2, n = 3; MSH6 [MIM: 600678], n = 3; RAD51D [MIM: 602954], n = 1; XRCC2 [MIM: 600375], n = 1), intellectual disability (ARID1B [MIM: 614556], n = 1), Duchenne muscular dystrophy (MIM: 310200) (DMD [MIM 300377], n = 1), and metaphyseal chondrodysplasia (MIM: 156500) (COL10A1 [MIM: 120110], n = 3). Ten participants with variants in genes where LOF variants are known to contribute to cancer susceptibility were determined to be negative for a personal or family history of relevant cancers. One individual with a BRCA1 mutation had a small family with a predominance of males and no occurrences of breast cancer in the few women. Two individuals with mutations in BRCA2 and one individual with a mutation in RAD51D had multiple cases of breast, ovarian, and/or prostate cancer in their families but no cases of premenopausal breast or ovarian cancer and were therefore considered to be negative. One individual with a mutation in MSH6 reported six cases of cancer on the maternal side of the pedigree including lung cancer, cervical cancer, and breast cancer. We judged this as unaffected, because these are not cancers typically associated with MSH6. A single individual with a mutation in CREB3L3 (MIM: 611998) related to hypertriglyceridemia (MIM: 145750) was considered negative based on phenotyping (lipid panel) at the time of enrollment. Six individuals had variants predicted to cause disorders that should be detectable with echocardiography: congenital heart defects (MIM: 614089) (MYH6 [MIM: 160710], n = 1), aneurysms-osteoarthritis syndrome (MIM: 613795) (SMAD3 [MIM: 603109], n = 1), or cardiomyopathy (MIM: 604145, 613765) (TTN [MIM: 188840], n = 4). All had normal echocardiography studies at the time of enrollment.
Forty-one individuals underwent follow-up phenotyping at the NIH, and one at an outside medical facility. Nineteen of these individuals were positive for associated phenotypic features on examination (Table S2). Eight individuals had positive biochemical findings with an otherwise normal exam. Five individuals had decreased factor XI (F11 [MIM: 264900]) and one individual had decreased protein S (PROS1 [MIM: 176880]). Although both of these findings can affect coagulation, heterozygotes are typically asymptomatic. Two individuals had abnormally low IgA levels (TNFRSF13B [MIM: 604907]), which can contribute to common variable immune deficiency (MIM: 240500) with low expressivity. Positive findings on evaluation of participants without a contributing family history included left ventricular non-compaction (MIM: 613426) on echocardiography and MRI (MYH7 [MIM: 160760], n = 1), presence of spherocytes on the peripheral blood smear with mild anemia, elevated reticulocyte count and total bilirubin (SLC4A1, n = 1), Birt-Hogg Dube syndrome (BHDS [MIM: 135150]) confirmed by findings of lesions consistent with fibrofolliculomas on skin biopsy in one individual and suspected BHDS with a history of skin papules and detection of lung cysts on chest CT in the second participant (FLCN [MIM: 607273], n = 2), and hearing loss as measured by a formal hearing test (KCNQ4 [MIM: 603537], n = 1). Two participants had findings suggestive of disease related to the identified variant. One individual had micro signs of holoprosencephaly (MIM: 142946) including a high-arched palate and an underdeveloped upper frenulum (TGIF1 [MIM: 602630]). One individual had abnormal lung diffusion capacity with dyspnea on climbing stairs with an SFTPC (MIM: 178620) mutation; mutations in SFTPC have been shown to cause adult lung disease due to surfactant metabolism dysfunction10 (MIM: 610913). In four individuals, the phenotype of relatives contributed to our assessment of the phenotype. One individual with a mutation in SGCE (MIM: 604149) had findings of dystonia (MIM: 159900) on exam and her son, who was positive for the mutation, had severe writer’s cramp reported on an iterative family history, a reported manifestation of SGCE mutations.11 Another individual with a PPARG (MIM: 601487) mutation had a family history of lipodystrophy incorrectly diagnosed as Dunnigan type lipodystrophy (MIM: 151660). On exam the participant had lipodystrophy and laboratory abnormalities including mild liver function elevations, mild hypertriglyceridemia with low HDL, and intermittently elevated fasting glucose concentrations with mild hyperinsulinemia. One individual with a mutation in KRT16 (MIM: 148067), which is known to cause pachyonychia congenita (MIM: 167200), had abnormal sweating and blistering of the feet and reported having three similarly affected sons on iterative family history, who were also positive for the identified mutation. Lastly, one individual with a mutation in HOXD13 (MIM: 142989) reported to cause brachydactyly-syndactyly syndrome12 (MIM: 160713) had apparently short digits and reported a niece who had toes that were short enough to restrict her ability to wear sandals. The niece was not available for segregation analysis.
Twenty-one individuals with variants in 18 different genes underwent follow-up phenotyping at the NIH and were negative on examination. Four individuals had variants in MYOC (MIM: 601652) that were predicted to cause glaucoma (MIM: 137750) but had normal eye examinations and intraocular pressures. Two other individuals had normal eye examinations in the presence of variants in EPHA2 (MIM: 176946) (n = 1) associated with cataract (MIM: 116600) formation or RP1 (MIM: 603937) (n = 1) with retinitis pigmentosa (MIM: 180100). The remaining variants predicted a variety of disease conditions and were assessed with the appropriate tests. In addition to a general physical exam, testing included audiology evaluations (PAX3 [MIM: 606597], n = 1), skin examinations (APT2C1 [MIM: 604384], n = 1; DSG1 [MIM: 125670], n = 1), cranial MRI (TGIF1, n = 1; GLI3 [MIM: 165240], n = 1), renal ultrasound (TRPC6 [MIM: 603652], n = 1; OFD1 [MIM: 300170], n = 1), liver ultrasound (PRKCSH [MIM: 177060], n = 2), hormonal testing (GNAS [MIM: 139320], n = 1; THRA [MIM: 190120], n = 1; GLI2 [MIM: 165230], n = 1), platelet function studies (RUNX1 [MIM: 151385], n = 1), neck, chest, pelvic, and abdominal CT (SDHC [MIM: 602413], n = 1), and nerve conduction velocity (NCV) and electromyography (EMG) (HSPB1 [MIM: 602195], n = 1), all of which were normal. In addition to normal examinations, a targeted family history was reassessed for these individuals and was determined to be noncontributory.
Two individuals had indeterminate findings on examination. One individual with a variant in DSPP (MIM: 125485) was examined for evidence of dentinogenesis imperfecta (MIM: 125490, 125500) with dental X-rays. Another individual with a variant in SALL4 (MIM: 607343) was examined for abnormalities associated with Duane-radial ray syndrome (MIM: 607323).
All X-linked variants (DMD, OFD1, PLP1 [MIM: 300401], ZIC3 [MIM: 300265]) were identified in females. OFD1 mutations cause an X-linked dominant condition whereas all others are X-linked recessive. Male relatives were available only for the DMD variant; two male relatives were positive for the variant and negative for muscle symptoms.
Bioinformatics
Many variant prediction algorithms (SIFT, PolyPhen) do not pertain to pLOF variants. Furthermore, it is not clear how individual attributes of a pLOF variant (e.g., predicted NMD, position of variant in gene, or presence of variant in an obligate exon) are correlated to pathogenicity in individuals. Although prior clinical reports are often used to support pathogenicity for specific variants, it is clear that not all reported pathogenic mutations cause disease13 and that the absence of prior clinical reports of pathogenicity is not sufficient to refute causation. For the group of individuals that could be assessed for findings of disease, variants were classified as either positive or negative. A variant was determined to be positive if at least one individual with the variant had a positive phenotype. Attributes were correlated with positive or negative status of the variants (Table 1). Attributes that were positively correlated with a positive status included a prior report of pathogenicity for the variant (p = 0.0001; 18 of 28 positive variants, 5 of 34 negative variants), prior report of other mutations in the same exon in the public version of HGMD as displayed on the UCSC Genome Browser (p = 0.0001; 25 of 28 positive variants, 12 of 34 negative variants), an alteration predicting the addition of more than ten aberrant amino acids (0.0234; 13 of 22 positive variants, 7 of 27 negative variants; splice site variants were not included in this analysis), and the presence of the mutation in more than 75% of overlapping ESTs (p = 0.0193; 25 of 28 positive variants, 21 of 34 negative variants). Attributes that were not correlated included ≥5 LOF variants reported in the gene (26 of 28 positive variants, 27 of 34 negative variants), presence of the mutation in the middle 90% of the gene (23 of 28 positive variants, 22 of 34 negative variants), and NMD according to MutationTaster (17 of 22 positive variants, 14 of 28 negative variants, prediction of NMD was not available for all variants). Neither the CADD score,5 which combines many variant attributes and is a predictor of deleteriousness, nor the haploinsufficiency value4 of the gene showed a correlation with affection status. Correcting for multiple testing via the Bonferroni method, only a prior report of pathogenicity for the variant and a prior report of other mutations in the same exon in the public version of HGMD as displayed on the UCSC Genome Browser were significant (p ≤ 0.006). We identified no correlation of the attribute of phenotype being present or absent with the known penetrance of the gene-disease dyad (data not shown).
Discussion
Among 951 participants, we identified 103 (11.1%, 95% CI 9.0%–13.0%) with a rare, heterozygous pLOF variant in a gene that can cause disease via loss-of-function alleles. Of the 79 who were phenotypically assessed, the overall yield of positive phenotypes was 34/79 (43.0%, 95% CI 32.1%–53.9%), but for our prevalence estimates, this was adjusted for the atherosclerosis ascertainment bias. We identified seven pLOF alleles in LDLR, but expected about one, given that the prevalence of familial hypercholesterolemia is 1/500 and about 50% of the known pathogenic alleles meet our definition of pLOF. After excluding six LDLR variants, the adjusted yield was 28/73 (38.4%, 95% CI 27.2%–49.6%).
Because our cohort was ascertained between the ages of 45 and 65, we expected to identify disorders resulting in minimal early morbidity/mortality as well as late-onset disorders of greater severity. Mild features related to identified variants included short digits with a variant in HOXD13, deafness with a variant in KCNQ4, dystonia with a variant in SGCE, and blistering of the feet with a variant in KRT16. The variants in SGCE and KRT16 segregated with the phenotype in these families even though family size was small. Although all of these individuals had clear phenotypic features of disease, they had not sought a genetic diagnosis. None of these features were reported at enrollment and were identified only during follow-up phenotyping. For less-severe phenotypic features that might be present but underdiagnosed in the general population, such as hearing loss, confirming the causation of the variant is difficult and over-interpretation is possible. Three of the identified variants resulted in biochemical phenotypes of protein S, factor XI, or TNFRSFB. Although loss-of-function variants in these genes can result in a disease phenotype, they are often non-penetrant for severe disease features. It is therefore not surprising to find these variants in a healthy population.
Some of the undiagnosed phenotypes present in our cohort presented a more significant risk of morbidity and/or mortality to our participants, but typically have later onset. Features such as lipodystrophy with a variant in PPARG, decreased lung function with a variant in SFTPC, and left ventricular non-compaction with a variant in MYH7 were identified in individuals who were unaware of their disease risk. The individual with the PPARG mutation did appreciate the presence of defined musculature in her extremities, but her clinicians had not associated this attribute with potential disease. Other relatives in the family had prominent musculature and it was assumed to be a familial trait. The mother of the proband had related health issues (including diabetes and hyperlipidemia). Nineteen of our participants had variants in genes with previously identified cancer susceptibility variants (BRCA1/2, FLCN, MSH6, PMS2, RAD51D, SDHC, XRCC2), but only six of these individuals had a clear family history of the associated cancers. Both individuals with variants in FLCN had skin findings consistent with undiagnosed fibrofolliculomas and these individuals might be at risk for associated renal cancer. For the individuals without a history of personal disease, knowing their variant status can be important in guiding appropriate screening and identifying at-risk family members. One pathogenic variant in BRCA1 (GenBank: NM_007294.3; c.68_69del [p.Glu23Valfs∗17]) was identified in three individuals, one of whom was positive and two negative for family history of associated cancers, consistent with the known reduced penetrance for this variant. Indeed, this finding raises interesting issues with respect to our determinations of a positive personal or family history of disease being subject to bias or coincidence, because breast cancer is common. In fact, our results suggest that we are underestimating the phenotypic consequences in these families. There is no debate in the literature that this BRCA1 variant is pathogenic and it has an estimated penetrance of 40%–50% per individual. By concluding that two of these three families are negative, we are being quite conservative and probably underestimating the effect of these variants. This result also has real implications for opportunistic screening—many probands with the risks of these diseases do not have a family history that allows them to be reliably identified. Genomic ascertainment might be the only way to identify such individuals.
Variants in MSH6, SDHC, and XRCC2, previously reported as pathogenic in the literature, were identified in individuals negative for personal and family histories. Of the 34 variants that were considered negative, 7 were in genes with previously identified cancer susceptibility variants where penetrance is known to be incomplete and 11 variants were identified in genes not associated with cancer but where causative variants are known to show incomplete penetrance. In contrast, nine variants considered phenotype negative were identified in genes where causative variants are thought to show high, if not complete, penetrance. For individuals without a clear family history of disease, variant identification and interpretation is more difficult because it is possible that the identified variant is not causative, and non-penetrance must always be considered. Penetrance is a major issue in predictive medicine because sufficient data are rarely available to give accurate predictions for any single individual.
Of the 82 identified variants, 15 were in genes on the ACMG list of genes to be considered for return of incidental findings (BRCA1, BRCA2, DSC2 [MIM: 125645], MSH6, MYH7, PKP2 [MIM: 602861], PMS2, SDHC, and SMAD3; Table S1). pLOF variants in these genes were identified in a total of 23 individuals, 16 of which could be evaluated for phenotypic features (Table S2). Of these 16 individuals, 7 were positive for associated phenotypic findings and/or positive family history and 9 individuals did not have associated phenotypes or a positive family history. Determining whether these individuals are non-penetrant versus the variant being non-causative is crucial in the decision to return these variants.
Using bioinformatic tools to predict pathogenicity is complicated and many tools show a poor correlation of predictions and actual clinical outcome. Bioinformatic tools often focus on factors known or predicted to affect protein structure and/or function including evolutionary conservation, amino acid attributes, or domain structure. Many do not assess pLOF variants. CADD scores combine many different attributes and can be calculated for pLOF variants, but these are predictions of deleteriousness, not pathogenicity.5 Correlation of CADD scores with a positive phenotype for this set of individuals was not high. For our dataset, none of the molecular attributes we tested were significantly correlated with pathogenicity. Two attributes, frameshift mutations predicting the addition of more than ten aberrant amino acids (p = 0.0234) and disruption of the major transcript (p = 0.0193), trended toward association but did not survive Bonferroni correction. These attributes should be re-evaluated in larger studies. A central position (i.e., not near the 3′ or 5′ end) of a variant in a gene has been suggested as an attribute that can be used to predict deleteriousness of a variant.1 Although variants predicted to result in premature truncation close to the ends of the gene might be less likely to disrupt protein function sufficiently to cause disease, variants in BRCA1 and SGCE identified in this study in individuals with positive phenotypes fell within the first 5% of the open reading frame (ORF) and the identified pathogenic variant in PPARG is located at the very 3′ end of the ORF.
The gold standard for predicting the pathogenicity of a variant is often thought to be variant reports in the literature, but these also need to be interpreted with care. Although the error can go in either direction, a common error is that variants are implicated in disease causation without sufficient evidence.14 It has become apparent that some reports of pathogenic variants in the literature are probably due to ascertainment bias. This is especially true for common disorders where large cohorts of individuals are screened and rare variants are identified in a subset of individuals and classified as pathogenic. When functional data exist for a variant, it can complement initial clinical reports and increase the likelihood that the effect of the variant is correctly predicted. For variants identified in our participants with a negative phenotype, 5/34 were reported in HGMD as disease-causing (DM) variants, whereas among variants found in individuals with a positive phenotype, 18/28 had this attribute (p = 0.0001, Fisher’s exact test).
From a broader perspective, these results demonstrate that abnormal phenotypes are, in aggregate, relatively common. We identified 28 out of 951 participants (2.9%, 95% CI 1.8%–4.0%, excluding 6 LDLR variants) with a rare, pLOF variant associated with an abnormal phenotype or positive family history. Because our cohort was ascertained through self-referral, it is possible that some of the individuals with more obvious phenotypes might have self-selected for this study. Although this is difficult to exclude, 18/28 positive individuals did not mention the associated condition at the time of enrollment and we believe it was therefore not likely to be related to their participation. Additionally, it is possible that some of these disorders are related either directly or indirectly to atherosclerosis (PPARG,15 BRCA1/2,16 PROS117). Employing statin use as an indicator of pre-existing disease diagnosis, 14/27 positive individuals (excluding all LDLR variants) were on statins at the time of enrollment as compared to 15/43 negative individuals. The difference between these two groups was not statistically significant, suggesting that ascertainment for cardiovascular disease does not account for the majority of our findings.
Indeed, this is likely to be an underestimate of the prevalence of these disorders. Our threshold for defining a positive phenotype might have excluded participants with subtle manifestations of disease. Because we evaluated adults, individuals with autosomal-dominant disorders mediated by haploinsufficiency that are incompatible with survival into adulthood would not be ascertained. Additionally, about 20% of rare missense variants cause haploinsufficiency18 and there are many more rare missense variants than pLOF variants in human genomes. Our 1/30 estimate is probably conservative and we conclude that the prevalence of such disorders is likely to be widely underestimated.
These results bear on Bayesian approaches to pathogenic variant identification. Making predictions of phenotypes from genomic data is difficult if the prior probability of disease is low, 1/5,000 or less. However, our data suggest that the prior probability might be better considered as the probability of the compound hypothesis of all such diseases, which is difficult to estimate by adding the prevalence estimates of individual diseases. Instead, we have utilized iterative phenotyping of a sequenced cohort to measure it directly. Our estimate of 1/30 (28/951, 95% CI 1/49 to 1/24) provides a good starting point for the aggregate probability of a phenotype attributable to a heterozygous LOF variant.
This study also pilots an approach that we believe will become a more common approach to clinical research and clinical practice in the future: iterative phenotyping or hypothesis-generating clinical research.19 Although our yield of positive phenotypes due to pLOF variants was high (43%), as expected it was not 100%. Additionally, no currently available metric was highly predictive of the presence of a phenotype for pLOF variants. For that reason, clinicians and clinical researchers will need to adopt this approach to the evaluation of individuals with novel or unexpected pLOF variants. This approach reduces a key source of ascertainment bias, which leads to inflated estimates of penetrance and skewing of phenotype spectrum assessments toward severe manifestations. These data are critical for improving our understanding of the full spectrum of genotype-phenotype correlation and gene function. Moreover, these data point to how the human genome sequence could be used in predictive medicine. Although our positive predictive rate was 43%, it should be emphasized that these data altered the risk of a rare, autosomal-dominant disorder in these 79 participants from baseline (1/500–1/500,000) to approximately half. This change in risk is enormous and can be used by a thoughtful clinician to prompt further evaluations of the proband and their family members for evidence of a discernable disorder. Although it is true that not all of the phenotypes detected here are medically actionable, this study serves as a proof of principle that there might indeed be predictive value in healthy genomes and exomes, once our mutation prediction algorithms improve and broaden to encompass all genes and many mutations.
Acknowledgments
The study described here was supported by the Intramural Research Programs of the National Human Genome Research Institute, the National Cancer Institute, the National Eye Institute, the National Institute of Deafness and Other Communication Disorders, and the National Institute of Diabetes and Digestive and Kidney Diseases of the NIH. L.G.B. is an uncompensated advisor to the Illumina Corp. and receives royalties from Genentech Corp. D.N.C. and P.D.S. acknowledge receipt of funding from BIOBASE/Qiagen through a License Agreement with Cardiff University. The authors thank Michael Collins, Julia Fekecs, Travis Hyams, Irini Manoli, and Frances Wright for support and advice.
Supplemental Data
For the purpose of this table, recessive inheritance is defined as either recessive inheritance for the disorder caused by the gene or known recessive inheritance for pLOF variants in the gene. MAF values are from ClinSeq or dbSNP. Eighty-two variants included in this study are listed as candidates in the category column. The number of individuals in ClinSeq that are heterozygous or homozygous for the variant is listed.
Web Resources
The URLs for data presented herein are as follows:
Clinical Genomic Database, http://research.nhgri.nih.gov/CGD/
Combined Annotation-Dependent Depletion (v.1.0), http://cadd.gs.washington.edu/home
Genetics Home Reference, http://ghr.nlm.nih.gov/
GraphPad, http://graphpad.com/
HGMD Professional, http://www.biobase-international.com/product/hgmd
Mann-Whitney U Test Calculator, http://elegans.som.vcu.edu/edu/∼leon/stats/utest.html
MutationTaster, http://www.mutationtaster.org/
My Family Health Portrait, https://familyhistory.hhs.gov
OMIM, http://www.omim.org/
UCSC Genome Browser, http://genome.ucsc.edu
References
- 1.MacArthur D.G., Balasubramanian S., Frankish A., Huang N., Morris J., Walter K., Jostins L., Habegger L., Pickrell J.K., Montgomery S.B., 1000 Genomes Project Consortium A systematic survey of loss-of-function variants in human protein-coding genes. Science. 2012;335:823–828. doi: 10.1126/science.1215040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xue Y., Chen Y., Ayub Q., Huang N., Ball E.V., Mort M., Phillips A.D., Shaw K., Stenson P.D., Cooper D.N., Tyler-Smith C., 1000 Genomes Project Consortium Deleterious- and disease-allele prevalence in healthy individuals: insights from current predictions, mutation databases, and population-scale resequencing. Am. J. Hum. Genet. 2012;91:1022–1032. doi: 10.1016/j.ajhg.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Biesecker L.G., Mullikin J.C., Facio F.M., Turner C., Cherukuri P.F., Blakesley R.W., Bouffard G.G., Chines P.S., Cruz P., Hansen N.F., NISC Comparative Sequencing Program The ClinSeq Project: piloting large-scale genome sequencing for research in genomic medicine. Genome Res. 2009;19:1665–1674. doi: 10.1101/gr.092841.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang N., Lee I., Marcotte E.M., Hurles M.E. Characterising and predicting haploinsufficiency in the human genome. PLoS Genet. 2010;6:e1001154. doi: 10.1371/journal.pgen.1001154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kircher M., Witten D.M., Jain P., O’Roak B.J., Cooper G.M., Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014;46:310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Facio F.M., Feero W.G., Linn A., Oden N., Manickam K., Biesecker L.G. Validation of My Family Health Portrait for six common heritable conditions. Genet. Med. 2010;12:370–375. doi: 10.1097/GIM.0b013e3181e15bd5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Teer J.K., Bonnycastle L.L., Chines P.S., Hansen N.F., Aoyama N., Swift A.J., Abaan H.O., Albert T.J., Margulies E.H., Green E.D., NISC Comparative Sequencing Program Systematic comparison of three genomic enrichment methods for massively parallel DNA sequencing. Genome Res. 2010;20:1420–1431. doi: 10.1101/gr.106716.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teer J.K., Green E.D., Mullikin J.C., Biesecker L.G. VarSifter: visualizing and analyzing exome-scale sequence variation data on a desktop computer. Bioinformatics. 2012;28:599–600. doi: 10.1093/bioinformatics/btr711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schwarz J.M., Rödelsperger C., Schuelke M., Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods. 2010;7:575–576. doi: 10.1038/nmeth0810-575. [DOI] [PubMed] [Google Scholar]
- 10.Thomas A.Q., Lane K., Phillips J., 3rd, Prince M., Markin C., Speer M., Schwartz D.A., Gaddipati R., Marney A., Johnson J. Heterozygosity for a surfactant protein C gene mutation associated with usual interstitial pneumonitis and cellular nonspecific interstitial pneumonitis in one kindred. Am. J. Respir. Crit. Care Med. 2002;165:1322–1328. doi: 10.1164/rccm.200112-123OC. [DOI] [PubMed] [Google Scholar]
- 11.Gerrits M.C., Foncke E.M., Koelman J.H., Tijssen M.A. Pediatric writer’s cramp in myoclonus-dystonia: maternal imprinting hides positive family history. Eur. J. Paediatr. Neurol. 2009;13:178–180. doi: 10.1016/j.ejpn.2008.03.007. [DOI] [PubMed] [Google Scholar]
- 12.Jamsheer A., Sowińska A., Kaczmarek L., Latos-Bieleńska A. Isolated brachydactyly type E caused by a HOXD13 nonsense mutation: a case report. BMC Med. Genet. 2012;13:4. doi: 10.1186/1471-2350-13-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Johnston J.J., Rubinstein W.S., Facio F.M., Ng D., Singh L.N., Teer J.K., Mullikin J.C., Biesecker L.G. Secondary variants in individuals undergoing exome sequencing: screening of 572 individuals identifies high-penetrance mutations in cancer-susceptibility genes. Am. J. Hum. Genet. 2012;91:97–108. doi: 10.1016/j.ajhg.2012.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bell C.J., Dinwiddie D.L., Miller N.A., Hateley S.L., Ganusova E.E., Mudge J., Langley R.J., Zhang L., Lee C.C., Schilkey F.D. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci. Transl. Med. 2011;3:ra4. doi: 10.1126/scitranslmed.3001756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahituv N., Kavaslar N., Schackwitz W., Ustaszewska A., Martin J., Hebert S., Doelle H., Ersoy B., Kryukov G., Schmidt S. Medical sequencing at the extremes of human body mass. Am. J. Hum. Genet. 2007;80:779–791. doi: 10.1086/513471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh K.K., Shukla P.C., Quan A., Al-Omran M., Lovren F., Pan Y., Brezden-Masley C., Ingram A.J., Stanford W.L., Teoh H., Verma S. BRCA1 is a novel target to improve endothelial dysfunction and retard atherosclerosis. J. Thorac. Cardiovasc. Surg. 2013;146:949–960.e4. doi: 10.1016/j.jtcvs.2012.12.064. [DOI] [PubMed] [Google Scholar]
- 17.Suleiman L., Négrier C., Boukerche H. Protein S: A multifunctional anticoagulant vitamin K-dependent protein at the crossroads of coagulation, inflammation, angiogenesis, and cancer. Crit. Rev. Oncol. Hematol. 2013;88:637–654. doi: 10.1016/j.critrevonc.2013.07.004. [DOI] [PubMed] [Google Scholar]
- 18.Kryukov G.V., Pennacchio L.A., Sunyaev S.R. Most rare missense alleles are deleterious in humans: implications for complex disease and association studies. Am. J. Hum. Genet. 2007;80:727–739. doi: 10.1086/513473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Biesecker L.G. Hypothesis-generating research and predictive medicine. Genome Res. 2013;23:1051–1053. doi: 10.1101/gr.157826.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
For the purpose of this table, recessive inheritance is defined as either recessive inheritance for the disorder caused by the gene or known recessive inheritance for pLOF variants in the gene. MAF values are from ClinSeq or dbSNP. Eighty-two variants included in this study are listed as candidates in the category column. The number of individuals in ClinSeq that are heterozygous or homozygous for the variant is listed.