

Abstract
Background
Solanum lycopersicum and Solanum habrochaites are closely related plant species; however, their cold tolerance capacities are different. The wild species S. habrochaites is more cold tolerant than the cultivated species S. lycopersicum.
Results
The transcriptomes of S. lycopersicum and S. habrochaites leaf tissues under cold stress were studied using Illumina high-throughput RNA sequencing. The results showed that more than 200 million reads could be mapped to identify genes, microRNAs (miRNAs), and alternative splicing (AS) events to confirm the transcript abundance under cold stress. The results indicated that 21 % and 23 % of genes were differentially expressed in the cultivated and wild tomato species, respectively, and a series of changes in S. lycopersicum and S. habrochaites transcriptomes occur when plants are moved from warm to cold conditions. Moreover, the gene expression patterns for S. lycopersicum and S. habrochaites were dissimilar; however, there were some overlapping genes that were regulated by low temperature in both tomato species. An AS analysis identified 75,885 novel splice junctions among 172,910 total splice junctions, which suggested that the relative abundance of alternative intron isoforms in S. lycopersicum and S. habrochaites shifted significantly under cold stress. In addition, we identified 89 miRNA sequences that may regulate relevant target genes. Our data indicated that some miRNAs (e.g., miR159, miR319, and miR6022) play roles in the response to cold stress.
Conclusions
Differences in gene expression, AS events, and miRNAs under cold stress may contribute to the observed differences in cold tolerance of these two tomato species.
Electronic supplementary material
The online version of this article (doi:10.1186/s12870-015-0521-6) contains supplementary material, which is available to authorized users.
Keywords: Cold, Transcriptome, RNA sequencing, Alternative splicing, microRNA
Background
Some plants increase their cold tolerance to deal with low temperatures; this phenomenon is termed cold acclimation. During this process, various biochemical and physiological changes occur in plants, which make plants more cold tolerant. Plants have different abilities to deal with low temperatures. Plants that have adapted to cold environments increase their cold tolerance in response to low but nonfreezing temperatures. By contrast, plants that have adapted to subtropical and tropical climates, such as maize, rice, and tomato, generally have little cold tolerance and are unable to acclimate to cold temperatures [1].
In recent years, many cold-regulated genes have been identified in plants under cold stress. The C-repeat binding factor (CBF) cold-responsive pathway is considered the best-known cold tolerance pathway in plants [2, 3]. There are three CBF/dehydration-responsive element binding factor 1 (DREB1) family members, including CBF1, CBF2, and CBF3 (DREB1b, DREB1c, and DREB1a, respectively) [4–6], encoding DNA-binding proteins of the APETALA2 (AP2)/ethylene response factor family [7]. Overexpression of CBF1, CBF2, or CBF3 of Arabidopsis thaliana caused an increase in cold tolerance in the absence of cold stress in plants, showing that the CBF genes improve cold tolerance [8–11]. Studies have shown that overexpression of CBF genes increases the cold tolerance of A. thaliana [8,9], Brassica napus [12], poplar [13], and potato [14], but not in tomato [1].
The roles of cold-regulated genes in plant cold acclimation show that differential expression of genes is related to different cold adaption abilities of plants. In A. thaliana and Chorispora bungeana, many alterations in gene expression begin within minutes of transferring plants to a low temperature [15–19]. Moreover, some studies have demonstrated that differential expression of a cold-responsive gene is caused by differences in cold tolerance in plants [20, 21]. For example, there are considerable differences in cold-responsive genes in Solanum tuberosum and Solanum commersonii, which are closely related but have different cold tolerances [22].
A large number of cold-related genes have been identified using transcript analysis techniques, and the products of cold-related genes, including regulatory proteins and functional proteins, are thought to play key roles in gene expression and signal transduction [3, 15, 17, 23–28]. The expression and alternative splicing (AS) of some serine/arginine-rich (SR) genes, which encode splicing factor proteins that are vital for splicing and constitutive expression, vary under cold stress [29–33]. Cold stress affects the expression of splicing factors; therefore, the splicing of precursor-mRNAs (pre-mRNAs) of other genes are altered under cold stress. AS of pre-mRNA is an important mechanism for increasing transcriptome and proteome variety in eukaryotes. AS has been confirmed widely at the functional level in A. thaliana, rice, and maize [34–36]. AS may be regulated spatially and developmentally under environmental stress [33, 37–39]; thus, AS could play an important role under cold stress or other abiotic stress.
MicroRNAs (miRNAs) have been discovered in plants [40–42], changing our perception of the mechanisms of gene expression, transcription, and translation. In plants, 21–24 miRNAs are negative regulators of gene expression [43]. The pool of miRNAs in plants is highly diverse [44–46]. Many studies have indicated that the key role of miRNAs is regulating organ development and biological processes [47–49]. MiRNAs are also associated with abiotic stress responses [50–52]. In accordance with their regulatory roles, many miRNAs target genes that have roles in developmental patterning and show unique development-specific, tissue-specific, and stress-induced expression patterns [47, 53, 54]. However, to date, only 44 annotated tomato miRNAs have been deposited in the miRBase database v19.0, and only a few miRNA targets have been confirmed experimentally. At present, it is unknown whether important regulators such as miRNAs play a vital role in tomato’s response to cold stress.
The cultivated tomato species (Solanum lycopersicum) suffers from cold stress at all stages of growth and development; by contrast, the wild tomato species (Solanum habrochaites) grows well at low temperatures [55–57]. To understand the molecular basis underlying why S. habrochaites can acclimate to cold and survive freezing temperatures, whereas S. lycopersicum cannot, we report the results of an RNA sequencing (RNA-seq) transcriptome and miRNA analysis of RNA populations obtained from cold-treated leaves of the two plants. The results showed that many changes in the S. lycopersicum and S. habrochaites transcriptomes occur in plants transferred from warm to cold conditions. We predicted that at least 21 % and 23 % of genes were cold responsive in S. lycopersicum and S. habrochaites, respectively. A gene ontology (GO) term enrichment analysis of the data indicated that many GO terms (“abiotic stimulus response”, “ethylene stimulus response”) were significantly enriched in the cold-responsive genes between the two species. Our data also provided an evaluation of AS between S. lycopersicum and S. habrochaites. RNA-seq identified many annotated introns, known AS, and 75,885 novel splice junctions. We identified 89 miRNA sequences and 423 targets of 83 miRNAs. Our data showed that some miRNAs (e.g., miR159, miR319, and miR6022) play important roles under cold stress in the two species. These results provided a new insight into the roles of miRNAs under cold stress in these two closely related species under cold stress. Thus, the differences in gene expression, AS events, and miRNAs under cold stress may contribute to the differences in the cold tolerance between S. lycopersicum and S. habrochaites.
Results
Phenotypic and physiological responses to cold stress
Solanum lycopersicum and S. habrochaites leaf tissue were chosen to study cold responses. The degree of cold stress was identified by malondialdehyde (MDA) content, proline content, peroxidase (POD) activity, and catalase (CAT) activity exchange in the leaves. S. habrochaites exhibited less severe wilting than S. lycopersicum after 10 days of treatment at 4 °C (Additional file 20: Figure S1A–D). Cold stress caused significant increased MDA content, proline content, POD activity, and CAT activity in these plants (Additional file 20: Figure S1E–H).
Solanum lycopersicum and S. habrochaites transcriptome analyses
The transcriptomes of S. lycopersicum (C) and S. habrochaites (Tsh) under cold stress were analyzed by RNA-seq using the Illumina Genome Analyzer II. After growth at 25 °C for 12 weeks, plants were moved to 4 °C for 0, 1, and 12 h, and the total RNA from leaves was extracted and analyzed. More than 200 million reads were produced, with approximately 33.3 million reads from each sample. We aligned the reads to the entire reference genome sequence of tomato (version SL2.40; http://solgenomics.net/) using the TopHat tool. The tolerance was set to allow two mismatches at most for reads in each alignment. Using these criteria, 96.32–97.21 % of the reads mapped uniquely to a genomic location, and 2.79–3.68 % of the reads were filtered out as multiple-mapped or low-quality reads. Alignment of the reads to tomato cDNAs demonstrated that 70 % of the tomato genome-annotated cDNAs had a sequence represented by Illumina RNA-seq reads (Table 1). Compared with the annotated tomato genome, the RNA-seq data revealed that 92.5–95 % of the reads that matched to the genome mapped to annotated genic regions, but only 5–7.5 % of the reads mapped to intergenic regions (Additional file 20: Figure S2). The depth of coverage along the length of the transcripts reduced towards the 5′ termini for RNA-seq data derived from the full-length cDNA libraries (Additional file 20: Figure S3). De novo assembly was performed using the Trinity method with default parameters [58]. Overviews of the assembly results are shown in Additional file 18 and Additional file 19. The reads were assembled into 68,051 non-redundant unigenes (>200 bp) in S. habrochaites.
Table 1.
Number of reads sequenced and mapped with TopHat
| Sample | Raw Data | Clean Reads | Unique Mapped | Multiple Mapped | Total Mapped | Expressed Genes (Mapped Reads Number > 0) | Expressed Genes (Mapped Reads Number > 10) |
|---|---|---|---|---|---|---|---|
| C0 | 35,588,324 | 30,268,069(85.05 %) | 25,796,713(97.15 %) | 756,667(2.85 %) | 26,553,380(87.75 %) | 24,776(71.35 %) | 20,244(81.71 %) |
| C1 | 47,221,174 | 42,718,256(90.46 %) | 32,505,159(96.96 %) | 1,018,137(3.04 %) | 33,523,296(78.49 %) | 25,242(72.69 %) | 20,515(81.27 %) |
| C12 | 34,010,526 | 29,504,403(86.75 %) | 24,478,334(97.21 %) | 702,267(2.79 %) | 25,180,601(85.37 %) | 24,438(70.37 %) | 19,843(81.20 %) |
| Tsh0 | 36,781,738 | 32,613,936(88.67 %) | 23,386,815(96.45 %) | 860,942(3.55 %) | 24,247,757(74.37 %) | 25,231(72.66 %) | 20,155(79.88 %) |
| Tsh1 | 35,295,488 | 31,895,199(90.37 %) | 21,154,377(96.32 %) | 807,256(3.68 %) | 21,961,633(68.87 %) | 25,074(72.20 %) | 19,775(78.87 %) |
| Tsh12 | 37,022,418 | 33,210,743(89.70 %) | 23,125,182(96.68 %) | 793,404(3.32 %) | 23,918,586(72.04 %) | 24,845(71.54 %) | 19,698(79.28 %) |
| C0 (Small RNA) | 5,146,411 | 4,329,523(84.13 %) | 1,591,965(38.20 %) | 2,575,134(61.80 %) | 4,167,099(96.25 %) | ||
| C1 (Small RNA) | 6,430,561 | 5,210,088(81.02 %) | 2,287,038(45.28 %) | 2,764,182(54.72 %) | 5,051,220(96.95 %) | ||
| C12 (Small RNA) | 6,841,937 | 4,584,516(67.01 %) | 1,438,066(32.84 %) | 2,941,450(67.16 %) | 4,379,516(95.53 %) | ||
| Tsh0 (Small RNA) | 4,375,096 | 3,355,947(76.71 %) | 757,007 (26.23 %) | 2,128,856(73.77 %) | 2,885,863(85.99 %) | ||
| Tsh1 (Small RNA) | 4,459,693 | 3,635,142(81.51 %) | 935,282(29.92 %) | 2,190,299(70.08 %) | 3,125,581(85.98 %) | ||
| Tsh12 (Small RNA) | 5,089,855 | 4,202,130(82.56 %) | 1,011,253(27.90 %) | 2,613,036(72.10 %) | 3,624,289(86.25 %) |
The number of unique mapping reads plus multiple mapping reads equals the total number of alignments. C0, C1 and C12 represent S. lycopersicum cold treatment for 0 h, 1 h and 12 h, respectively; Tsh0, Tsh1 and Tsh12 indicate S. habrochaites cold treatment for 0 h, 1 h and 12 h, respectively
To verify the RNA-seq data, some genes whose expressions increased, some that decreased, and some that showed no change in abundance were chosen for real-time PCR (qPCR) under cold stress. The results of RNA-seq and qPCR were similar (Additional file 20: Figure S8), showing the same general expression trends by qPCR as were revealed by RNA-seq.
To identify S. lycopersicum and S. habrochaites miRNAs that affect gene regulation under cold stress, six miRNA libraries were constructed from the leaves of S. lycopersicum and S. habrochaites that were or were not treated with cold. The six miRNA libraries were sequenced using high-throughput RNA-seq and yielded approximately 5.4 million raw reads in each sample. We excluded the poor-quality reads and those whose length was smaller than 14 nucleotides from further analysis. Finally, we obtained approximate 4.2 million non-redundant reads (14–24 nucleotides) in each sample (Table 1).
Differential expression and GO enrichment
To study the impact of cold stress on gene expression in S. lycopersicum (C) and S. habrochaites (Tsh), the transcript abundance of each gene was identified by the reads per kilobase of transcript per million reads mapped method (Additional file 1, Additional file 20: Figure S4). To compare the transcriptomes in S. lycopersicum and S. habrochaites under cold stress, a heat map was generated to present the transcript abundance for all differentially expressed genes (DEGs) under cold stress at 0, 1, and 12 h (Fig. 1, Additional file 20: Figure S5–7). The results showed that a series of changes in gene expression in S. lycopersicum and S. habrochaites occur when plants are moved from warm to cold conditions. Moreover, the gene expression patterns for S. lycopersicum and S. habrochaites were dissimilar. For example, cluster A genes were little affected at 1 h in S. lycopersicum and returned to low transcript abundance levels at 12 h of cold stress; cluster B genes were unaffected in S. lycopersicum at 1 h of cold stress, but were highly increased at 12 h; cluster C or D genes were little affected after cold stress in S. lycopersicum, but were affected in S. habrochaites (Fig. 1).
Fig. 1.
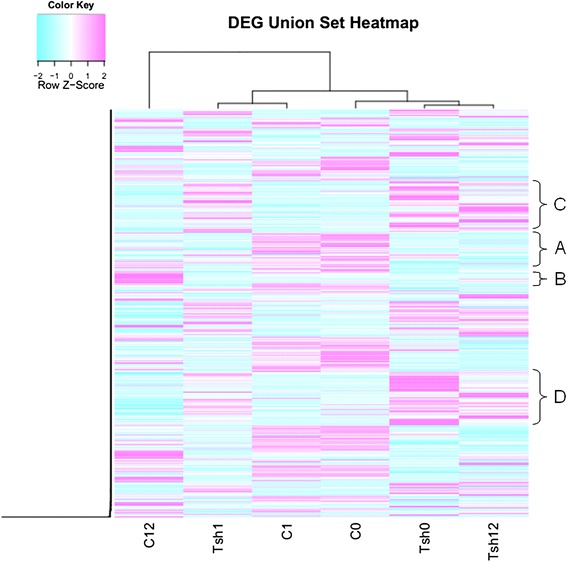
Hierarchical clustering of S. lycopersicum (C) and S. habrochaites (Tsh) transcripts at 0, 1, and 12 h of cold treatment at 4 °C
We used a threshold of a minimum 2-fold change in abundance between any two time points to define DEGs in the following analysis (Fig. 2, Table 2, Additional file 3). The results showed that ~4 % (sample C1 vs. C0), ~10 % (sample C12 vs. C0), ~5 % (sample Tsh1 vs. Tsh0), and ~8 % (sample Tsh12 vs. Tsh0) of the unigenes were cold induced; and ~2 % (sample C1 vs. C0), ~9 % (sample C12 vs. C0), ~6 % (sample Tsh1 vs. Tsh0), and ~9 % (sample Tsh12 vs. Tsh0) were cold repressed. In S. lycopersicum, transcripts for 1,256 and 3,350 unigenes increased at 1 and 12 h, respectively, and 804 unigenes increased at both time points tested; transcripts for 856 and 3,022 unigenes decreased at 1 and 12 h, respectively, and 339 unigenes decreased at both time points tested (Fig. 2, Table 2, Additional file 3, Additional file 4). In S. habrochaites, transcripts for 1,725 and 2,940 unigenes increased at 1 and 12 h, respectively, and 722 unigenes increased at both time points tested; transcripts for 1,967 and 3,126 unigenes decreased at 1 and 12 h, respectively, and 1,000 unigenes decreased at both time points tested. Moreover, in S. habrochaites, transcripts for 3,608, 2,813, and 3,549 unigenes increased at 0, 1, and 12 h, respectively, compared with S. lycopersicum at same time points; and transcripts for 3,897, 3,592, and 3,815 unigenes decreased at 0, 1, and 12 h, respectively. In sum, the gene expression profiles in S. lycopersicum and S. habrochaites changed under cold stress to different degrees; however, there were some overlapping genes that were regulated by low temperature in both tomato species.
Fig. 2.
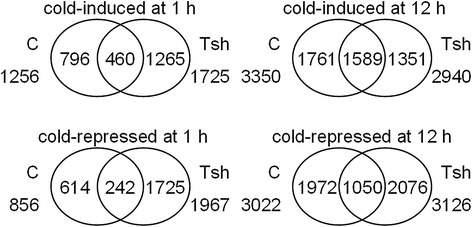
The number of total ESTs that were either cold-induced or cold-repressed by 2-fold change in S. lycopersicum (C) and S. habrochaites (Tsh). The results from 0, 1, and 12 h of cold treatment at 4 °C
Table 2.
Total number of differentially expressed genes (DEG)
| Group | Up Regulated Gene Numbe | Down Regulated Gene Number | Total |
|---|---|---|---|
| Tsh0 vs C0 | 3608 | 3897 | 7505 |
| Tsh1 vs C1 | 2813 | 3592 | 6405 |
| Tsh12 vs C12 | 3549 | 3815 | 7364 |
| C1 vs C0 | 1256 | 856 | 2112 |
| C12 vs C0 | 3350 | 3022 | 6372 |
| C12 vs C1 | 3047 | 2748 | 5795 |
| Tsh1 vs Tsh0 | 1725 | 1967 | 3692 |
| Tsh12 vs Tsh0 | 2940 | 3126 | 6066 |
| Tsh12 vs Tsh1 | 2962 | 2321 | 5283 |
C0, C1 and C12 represent S. lycopersicum cold treatment for 0 h, 1 h and 12 h, respectively; Tsh0, Tsh1 and Tsh12 indicate S. habrochaites cold treatment for 0 h, 1 h and 12 h, respectively
We analyzed the genes that we determined to be responsive to cold at 1 h. The GO terms enriched in each species were comparable (Additional file 3) and were generally related to “response to abiotic stimulus”. From the heat map, it was also obvious that S. lycopersicum was less affected by cold than S. habrochaites at 1 h. The expressions of genes that were enriched in GO categories corresponding to “cell wall metabolism” were increased under cold stress in S. lycopersicum, but the opposite result was observed in S. habrochaites. We observed a similar contrast in the GO category “response to organic substance”. In the GO categories “response to chitin”, “response to carbohydrate stimulus”, and “DNA-binding WRKY”, there was a significant enrichment in S. lycopersicum, but S. habrochaites showed no enrichment. For the categories “chloroplast”, “transit peptide”, “pentatricopeptide repeat”, “phenylpropanoid metabolic process”, “flavonoid metabolic process”, and “amino acid derivative biosynthetic process”, no significant enrichment was observed in S. lycopersicum, but enrichment was observed in S. habrochaites (Additional file 3).
We then compared responses to cold at 12 h. The analysis of GO terms for cold-regulated genes suggested that the categories “response to organic substance”, “response to endogenous stimulus”, “response to hormone stimulus”, “response to abscisic acid stimulus”, “pentatricopeptide repeat”, “response to abiotic stimulus”, “response to ethylene stimulus”, “serine/threonine-protein kinase”, “phenylpropanoid metabolic process”, “amino acid derivative biosynthetic process”, “lignin biosynthetic process”, and “flavonoid metabolic process” were enriched in both S. lycopersicum and S. habrochaites (Additional file 3). In the case of the GO category “UDP-glucuronosyl/UDP-glucosyltransferase”, there was significant enrichment for S. lycopersicum, but not for S. habrochaites.
Alternative splicing in S. lycopersicum and S. habrochaites
To study the effect of cold stress on AS in S. lycopersicum and S. habrochaites, we compared splicing events between the two tomato genotypes. We identified splice junctions using the TopHat software [59]. Collectively, using RNA-seq data, we identified 105,663, 109,251, 102,316, 106,690, 104,440, and 105,323 splice junctions in samples C0, C1, C12, Tsh0, Tsh1, and Tsh12 with 21,548, 25,492, 22,870, 20,909, 19,957, and 23,179 novel junctions, respectively (Additional file 5). We categorized each AS event using the primary known types of AS and the sequencing data (Table 3, Additional file 6). As previously reported [60–62], we found that intron retention was the primary type of AS.
Table 3.
Classification of all detected alternative splicing events in tomato
| Samples | 3pMXE | 5pMXE | A3SS | A3SS and ES | A5SS | A5SS and ES | Cassette Exon | ESx | IntronR | MXE | Total | Detected novel junctionx | AS/100 novel SJ |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C0 | 187 | 206 | 1220 | 35 | 941 | 35 | 136 | 328 | 3079 | 24 | 6191 | 21548 | 28.73 |
| C1 | 190 | 204 | 1116 | 33 | 919 | 30 | 104 | 376 | 3361 | 18 | 6351 | 25492 | 24.91 |
| C12 | 187 | 208 | 1138 | 43 | 933 | 38 | 86 | 1071 | 3983 | 17 | 7704 | 22870 | 33.69 |
| Tsh0 | 201 | 249 | 1055 | 24 | 846 | 46 | 99 | 317 | 1931 | 16 | 4784 | 20909 | 22.88 |
| Tsh1 | 183 | 213 | 1007 | 28 | 798 | 35 | 60 | 326 | 1970 | 5 | 4625 | 19957 | 23.17 |
| Tsh12 | 188 | 233 | 1096 | 50 | 858 | 47 | 57 | 1143 | 2951 | 10 | 6633 | 23179 | 28.62 |
| Total | 396 | 506 | 2953 | 102 | 2230 | 94 | 317 | 2091 | 8211 | 57 | 16957 | 75885 | 22.35 |
C0, C1 and C12 represent S. lycopersicum cold treatment for 0 h, 1 h and 12 h, respectively; Tsh0, Tsh1 and Tsh12 indicate S. habrochaites cold treatment for 0 h, 1 h and 12 h, respectively
Illuminative examples, including intron retention in the LOB domain protein 38 (Solyc01g107190.2) (Fig. 3a) and receptor-like protein (RLK) (Solyc01g007980.2) (Fig. 3b), are shown in Fig. 3. The TopHat-generated S. lycopersicum LOB domain protein 38 mRNA model predicted a distinct AS event yielding a splice isoform that retains full-length intron 1 (Fig. 3a, Additional file 7). An analysis of RLK, a putative resistance protein with an antifungal domain, provides another example of an IntronR event in plants. The depth of coverage of the intron 3 splice junction was confirmed by RNA-seq (Fig. 3b, Additional file 7). Dense micro-read coverage of intron 3 in the RLK transcript contrasted with the low coverage of other introns, indicating that intron 3 may be retained in some mature RLK transcripts (Fig. 3b).
Fig. 3.
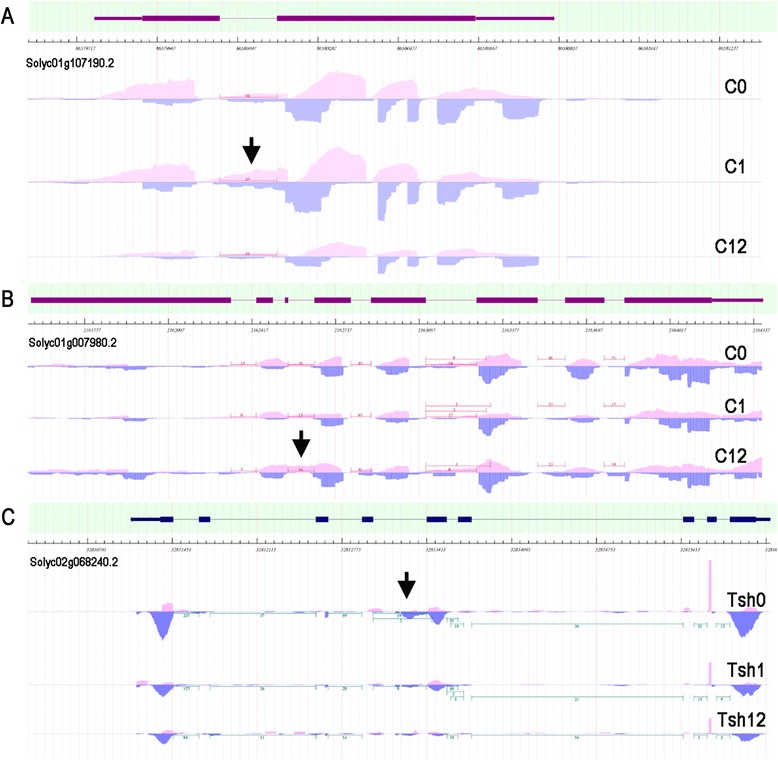
Identification of alternative splicing in the LOB domain-containing protein 37 (Solyc01g107190.2) (a), cysteine-rich RLK 2 (Solyc01g007980.2) (b), and diacylglycerol acyltransferase 2 (Solyc02g068240.2) (c) transcripts. Changes in read density coverage are indicated by pink (forward reads) and blue (reverse reads). The intron retention events are indicated by an arrow
We tried to identify differences in altered AS events between the two species at 0, 1, and 12 h of cold treatment at 4 °C. Then, 270 (sample Tsh0 vs. C0), 241 (sample Tsh1 vs. C1), 474 (sample Tsh12 vs. C12), 131 (sample C1 vs. C0), 575 (sample C12 vs. C0), 114 (sample Tsh1 vs. Tsh0), and 606 (sample Tsh12 vs. Tsh0) AS events were increased under cold stress; 204 (sample Tsh0 vs. C0), 237 (sample Tsh1 vs. C1), 412 (sample Tsh12 vs. C12), 119 (sample C1 vs. C0), 152 (sample C12 vs. C0), 122 (sample Tsh1 vs. Tsh0), and 130 (sample Tsh12 vs. Tsh0) events were decreased (Table 4). Table 4 shows that AS occurred more frequently in genes regulated in response to cold at 12 h than in genes at 1 h.
Table 4.
Altered alternative splicing events between S. lycopersicum and S. habrochaites
| Type | Total AS Number | Tsh0 | Tsh1 | Tsh12 | C1 | C12 | C12 | Tsh1 | Tsh12 | Tsh12 |
|---|---|---|---|---|---|---|---|---|---|---|
| Vs | Vs | Vs | Vs | Vs | Vs | Vs | Vs | Vs | ||
| C0 | C1 | C12 | C0 | C0 | C1 | Tsh0 | Tsh0 | Tsh1 | ||
| Increased under cold stress | ||||||||||
| 3pMXE | 396 | 18 | 22 | 20 | 2 | 12 | 13 | 9 | 15 | 12 |
| 5pMXE | 506 | 33 | 33 | 38 | 3 | 15 | 11 | 13 | 23 | 16 |
| A3SS | 2953 | 88 | 75 | 95 | 35 | 88 | 80 | 33 | 91 | 77 |
| A3SSandES | 102 | 2 | 1 | 3 | 0 | 2 | 1 | 1 | 6 | 7 |
| A5SS | 2230 | 53 | 35 | 64 | 18 | 66 | 59 | 23 | 70 | 54 |
| A5SSandES | 94 | 1 | 2 | 5 | 2 | 4 | 5 | 1 | 4 | 5 |
| CassetteExon | 317 | 16 | 11 | 8 | 12 | 14 | 11 | 4 | 12 | 13 |
| ES | 2091 | 27 | 35 | 206 | 34 | 300 | 291 | 19 | 358 | 324 |
| MXE | 57 | 5 | 2 | 3 | 5 | 5 | 4 | 1 | 3 | 3 |
| IntronR | 8211 | 27 | 25 | 32 | 20 | 69 | 86 | 10 | 24 | 32 |
| Total | 16957 | 270 | 241 | 474 | 131 | 575 | 561 | 114 | 606 | 543 |
| Decreased under cold stress | ||||||||||
| 3pMXE | 396 | 7 | 10 | 13 | 7 | 2 | 4 | 6 | 6 | 8 |
| 5pMXE | 506 | 6 | 11 | 18 | 3 | 3 | 6 | 13 | 17 | 12 |
| A3SS | 2953 | 68 | 60 | 72 | 32 | 49 | 35 | 41 | 41 | 34 |
| A3SSandES | 102 | 2 | 0 | 1 | 0 | 1 | 0 | 0 | 1 | 1 |
| A5SS | 2230 | 39 | 54 | 54 | 15 | 42 | 27 | 14 | 22 | 24 |
| A5SSandES | 94 | 2 | 3 | 5 | 1 | 1 | 1 | 2 | 0 | 1 |
| CassetteExon | 317 | 19 | 21 | 15 | 18 | 18 | 19 | 18 | 20 | 11 |
| ES | 2091 | 31 | 48 | 182 | 19 | 17 | 17 | 17 | 14 | 13 |
| MXE | 57 | 10 | 5 | 5 | 5 | 8 | 6 | 2 | 1 | 1 |
| IntronR | 8211 | 20 | 25 | 47 | 19 | 11 | 22 | 9 | 8 | 6 |
| Total | 16957 | 204 | 237 | 412 | 119 | 152 | 137 | 122 | 130 | 111 |
C0, C1 and C12 represent S. lycopersicum cold treatment for 0 h, 1 h and 12 h, respectively; Tsh0, Tsh1 and Tsh12 indicate S. habrochaites cold treatment for 0 h, 1 h and 12 h, respectively
Next, 121 (sample C1 vs. C0), 522 (sample C12 vs. C0), 112 (sample Tsh1 vs. Tsh0), and 553 (sample Tsh12 vs. Tsh0) of the AS genes were increased under cold stress; 110 (sample C1 vs. C0), 140 (sample C12 vs. C0), 111 (sample Tsh1 vs. Tsh0), and 122 (sample Tsh12 vs. Tsh0) of the genes were decreased (Fig. 4, Additional file 7). Certain AS events are associated with specific abiotic stress conditions [34]. An observation of individual events under cold stress showed that certain AS genes are cold associated (Additional file 8).
Fig. 4.
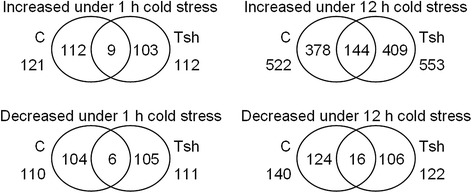
The total number of differentially alternative splicing genes (DASG) in S. lycopersicum (C) and S. habrochaites (Tsh) at 0, 1, and 12 h of cold treatment
The AS event of diacylglycerol acyltransferase (Solyc02g068240.2) under cold stress is shown in Fig. 3c. The TopHat-generated S. habrochaites diacylglycerol acyltransferase mRNA model predicted a distinct AS event that yielded a splice isoform that retains intron 4 (Fig. 3c). Accumulation of the no IntronR 4-containing transcripts decreased approximately three-fold under cold treatment. Other examples of cold stress-associated AS genes (SR45a, SR30) are provided in Additional file 20: Figure S9.
We compared the functions of the AS genes that were regulated in response to cold at 1 h and 12 h with the DEGs. Cold-regulated differentially expressed AS genes overlapped with DEGs in S. lycopersicum (C) and S. habrochaites (Tsh), and these genes were in the GO categories “dephosphorylation” and “phosphoprotein phosphatase activity” (Additional file 8), suggesting these activities were present in both plants. In the case of the GO categories “detection of light stimulus”, “phenylpropanoid metabolic process”, “response to cadmium ion”, “phosphoinositide binding”, and “heat shock protein binding”, there was significant enrichment for S. lycopersicum, but S. habrochaites showed no enrichment. For the categories “carboxylic acid catabolic process”, “proteolysis”, “cell death”, “reproductive developmental process”, and “ethylene mediated signaling pathway”, no enrichment was observed in S. lycopersicum, but significant enrichment was observed in S. habrochaites (Additional file 8).
Identification of single nucleotide polymorphisms (SNPs)
In comparison to the tomato reference genome, we identified 5,344 SNPs in S. lycopersicum ‘glamor’, and 3,625 of these SNPs were specific SNPs; and 154,870 SNPs were identified in S. habrochaites ‘LA1777’, and 153,157 of these SNPs were specific SNPs (Table 5, Additional file 9). We identified 696 (sample C1 vs. C0), 2,330 (sample C12 vs. C0), 1,157 (sample Tsh1 vs. Tsh0), and 2,183 (sample Tsh12 vs. Tsh0) genes that contained SNPs and were also cold induced; 463 (sample C1 vs. C0), 2,060 (sample C12 vs. C0), 1,452 (sample Tsh1 vs. Tsh0), and 2,311 (sample Tsh12 vs. Tsh0) genes that contained SNPs and were cold repressed (Additional file 10, Additional file 11). Genes that contained SNPs that were enriched in GO categories corresponding to “response to chitin” were increasingly expressed under cold stress at 1 h in S. lycopersicum, but not in S. habrochaites. Other examples of a similar contrast in GO categories are provided in Additional file 10.
Table 5.
Statistical analysis of SNPs
| Sample | SNP NO. | gene NO. | Specific SNP NO. | Specific gene NO. | Total SNP NO. | Total gene NO. |
|---|---|---|---|---|---|---|
| C | 5344 | 2509 | 3625 | 1857 | 160214 | 19273 |
| Tsh | 154870 | 19090 | 153157 | 18905 |
C stand for S. lycopersicum; Tsh stand for S. habrochaites
Impact of cold stress on miRNAs in tomato
To identify miRNAs in tomato, we analyzed miRNAs by BLAST searches against the tomato genome sequence by BOWTIE (Additional file 20: Figure. S10). The novel miRNAs were then identified by the miRDeep2 tool. The sequences corresponding to the known non-coding RNAs (tRNAs, rRNAs, small nucleolar and small nuclear RNAs) were filtered out using BLASTn to search the Rfam database (http://rfam.xfam.org/) (Additional file 20: Figure S11). The remaining sequences were assigned as either other endogenous small RNAs or miRNA candidates and used for a fold-back structure prediction. We compared the unique miRNAs with the miRBase database (version 19.0). In this analysis, the miRNAs were required to show a perfect or nearly perfect match (mismatch ≤ 1) to known miRNAs. After these analyses, 112 unique miRNA were obtained as novel miRNA candidates (Additional file 12).
A large number of miRNA sequences were produced by Illumina sequencing, permitting us to confirm the relative abundance of miRNAs in tomato. To study the expression dynamics of miRNAs and their potential roles in gene expression regulation in S. lycopersicum and S. habrochaites, the transcript abundance of each miRNA was evaluated by transcripts per million (TPM). The TPM of the miRNAs varied from 0 to 27,670 (miR396a, sample C1), suggesting that the expression of miRNAs varied greatly in tomato (Additional file 13). MiR159 and miR396a were the most abundant miRNAs in the six sequencing datasets. According to the TPM, some miRNAs (miR159, miR396a, miR396b, miR482b, and miR6022) were highly expressed in tomato, with a TPM of greater than 100 each. MiR6027, miR171a, miR482, miR319, and miR1919a were moderately expressed and had a TPM between 10 and 100. MiR5303, miR169b, miR1916, miR171c, and miR395a represent miRNAs with low expression and a TPM of less than 10 (Additional file 13). Sequence analysis showed that the relative abundance of some members in one miRNA family changed considerably in tomato. For example, the TPM for miR396a was 9,433, whereas the TPM for miR396b was only 4,347 (Additional file 13).
To detect the effect of cold stress on miRNAs, the expression of miRNAs in S. lycopersicum and S. habrochaites seedlings with and without cold treatment was examined. Fourteen (sample C1 vs. C0), eight (sample C12 vs. C0), two (sample Tsh1 vs. Tsh0), and four (sample Tsh12 vs. Tsh0) of the miRNAs were cold induced; seven (sample C1 vs. C0), six (sample C12 vs. C0), five (sample Tsh1 vs. Tsh0), and eight (sample Tsh12 vs. Tsh0) of the miRNAs were cold repressed (Fig. 5, Additional file 14). In response to cold treatment, the most significant change was observed for miR169c, whose expression level increased approximately 35-fold in sample C1 compared with C0. The expressions of some miRNAs in S. habrochaites were opposite to those in S. lycopersicum under cold stress. For example, miR1919a–miR1919c, and miR396b were upregulated under cold stress for 1 h in S. lycopersicum, whereas they were downregulated in S. habrochaites (Additional file 14). In contrast, miR172a and miR172b were downregulated by cold stress for 1 h in S. lycopersicum, while they upregulated in S. habrochaites by cold stress for 12 h.
Fig. 5.
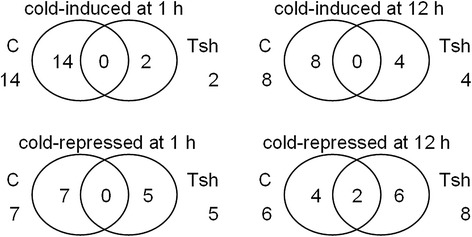
The total number of microRNAs (miRNAs) that were either cold-induced or cold-repressed in S. lycopersicum (C) and S. habrochaites (Tsh)
We used psRNATarget (http://plantgrn.noble.org/psRNATarget/) to predict targets for the miRNAs. For the miRNAs that were annotated as described above, we identified 423 mRNA targets (Fig. 6, Additional file 15). From Fig. 6 it was also evident that S. lycopersicum was more affected by 1 h of cold than S. habrochaites. To further characterize the role of the miRNAs in response to cold, we examined the target list for genes that could be related to the cold response and that were either induced or repressed by cold, based on our Illumina results (Additional file 17). For example, one of the predicted targets was the transcript of the homeodomain leucine zipper class I (HD-Zip I) protein (ATHB13, AT1G69780, Solyc02g087840.2) (Additional file 17). ATHB13 is induced in S. lycopersicum after cold treatment for 12 h based on our sequencing data (Additional file 3). The miRNA predicted to target ATHB13 is miR6022. Our sequencing data showed that miR6022 was downregulated in S. lycopersicum after cold stress for 12 h (Additional file 14). Based on our sequencing data, we did not find differential expression of miR6022 after cold treatment for 1 h in S. lycopersicum. The induction of ATHB13 under cold stress for 12 h correlates with miR6022 repression by cold, suggesting that ATHB13 levels are post-transcriptionally regulated by this miRNA in response to cold. Thus, miR6022/ATHB13 represents an abiotic stimulus module that could be important for the cold response in S. lycopersicum leaves. Other examples of cold stress-associated miRNAs (miR159, miR319) are provided in Additional file 17.
Fig. 6.
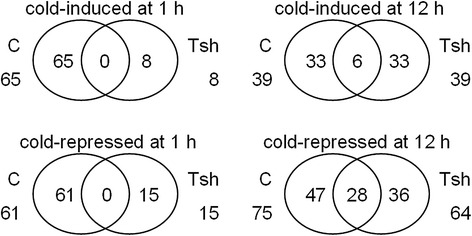
The total number of targets of differentially expressed microRNAs (DEmiRNAs) that were either cold-induced or cold-repressed in S. lycopersicum (C) and S. habrochaites (Tsh)
For comprehensive annotation, all putative target genes in each sample were analyzed by GO terms using the DAVID program. An analysis of GO enrichment for the targets revealed that target functions were enriched in many different biological processes (Additional file 16). Among the mRNA targets that were upregulated in response to cold at 1 h and 12 h, comparable cold-regulated mRNA target expression was observed between S. lycopersicum (C) and S. habrochaites (Tsh) in relation to the GO terms (Additional file 16), which included “ATP binding” and “nucleotide binding” in both species. In the case of the GO categories “leaf development”, “shoot development”, “CCAAT-binding factor”, “CBF”, “regulation of RNA metabolic process”, “cell death”, “gene silencing”, “immune response”, “flower development”, “ATPase activity”, and “leucine-rich repeat”, there was significant enrichment in S. lycopersicum, but not in S. habrochaites. For the categories “response to cold” and “hormone stimulus”, no enrichment was observed in S. lycopersicum, but significant enrichment was observed in S. habrochaites (Additional file 16).
Among the targets that were determined as downregulated in response to cold at 1 h and 12 h, an analysis of GO enrichment showed “defense response”, “ATP binding”, and “nucleotide binding” in both S. lycopersicum and S. habrochaites. In the case of the GO categories “leucine-rich repeat”, “reproductive structure development”, “meristem development”, “intracellular signaling cascade”, and “response to hormone stimulus”, there was no significant enrichment in S. habrochaites, but S. lycopersicum showed enrichment.
Discussion
Plants have different abilities to deal with low temperatures. The cultivated tomato (S. lycopersicum) suffers from cold stress, but the wild species (S. habrochaites) does not [55–57]. RNA-seq of cold-stressed S. lycopersicum leaves and a comparison with the transcriptome of S. habrochaites are presented here. The results revealed the effects of cold stress on transcript abundance in S. lycopersicum and S. habrochaites; 21 % and 23 % of transcripts in S. lycopersicum and S. habrochaites, respectively, are cold regulated. There is a large overlap in the genes that were cold responsive in both plant species, but the results indicated many differences in the cold-responsive genes of the two species (Figs. 1, 2). The diversity of GO categories that were enriched in cold-stressed S. lycopersicum and S. habrochaites (Additional file 3) indicated the complexity of the response.
For cold-regulated DEGs of S. lycopersicum and S. habrochaites, some similar clusters of GO categories “response to abiotic stimulus” was found in both plants (Additional file 3), confirming earlier observations [20]. However, in response to cold stress in S. lycopersicum at 1 h, many genes encoding proteins associated with the abiotic stimulus response showed increased transcript abundance, and a few genes showed decreased transcript abundance (Additional file 3).
The data also suggested that some GO terms overlap in cold-treated S. habrochaites, but not in S. lycopersicum (Additional file 3). Some photosynthesis-related GO terms were significantly enriched among the upregulated genes in S. habrochaites under cold stress at 1 h. The results indicated that photosynthesis-related genes in cold regulatory programs might contribute to transient cold tolerance of S. habrochaites. In 2012, Liu et al. studied the transcriptomes of S. lycopersicum and S. habrochaites under cold stress at 3 days using GeneChip [20]. Their experiments indicated that S. lycopersicum showed more severe inhibition of photosynthesis than S. habrochaites during chilling stress.
The data showed that the GO category “cell wall metabolism” was enriched with genes with increased expression in cold-treated S. lycopersicum at 1 h, but the opposite result was observed for cold-treated S. habrochaites at both 1 and 12 h (Additional file 3). A GO term enrichment analysis indicated that “cell wall metabolism” was significantly depressed in the long term by cold stress in S. habrochaites, but was transiently induced by cold stress in S. lycopersicum. The present findings in S. habrochaites are similar to those of Fowler and Thomashow [15], who used a microarray chip to analyze the transcriptome of Arabidopsis under cold stress.
Hormones are signaling molecules that play key roles in regulating gene expression under cold stress [15, 20]. RNA-seq analysis showed that many genes related to abscisic acid, ethylene, auxin, jasmonic acid, and gibberellin were regulated by cold stress in S. lycopersicum and S. habrochaites (Additional file 3). Two hormone-related GO terms, “response to abscisic acid stimulus” and “response to ethylene stimulus”, were significantly enriched among the differentially expressed genes in S. lycopersicum and S. habrochaites under cold stress (Additional file 3). In S. lycopersicum, our data show that ABA-related GO terms were significantly enriched among the upregulated genes under cold stress at 12 h and were not enriched at 1 h. In contrast, ABA-related GO terms were significantly enriched among the upregulated and downregulated genes under cold stress at multiple time points in S. habrochaites. The production of ethylene also has been associated with cold stress [63–65]. In S. lycopersicum, our data show that ethylene-related GO terms were significantly enriched among the upregulated genes under cold stress at multiple time points. In contrast, ethylene-related GO terms were only significantly enriched among the upregulated genes under cold stress at 12 h, but not at 1 h, in S. habrochaites.
Transcription factors (TFs) play a key role in the regulation of gene expression under abiotic and biotic stresses in plants. The RNA-seq results showed that many TFs were regulated under cold stress in S. lycopersicum and S. habrochaites, and some members of the CBF were upregulated under cold stress at 1 h or 12 h (Fig. 7; Additional file 3). Two additional cold-regulated genes were found that encode transcription factors: a homolog of Cys2/His2-type zinc-finger protein (ZAT10) and a zinc finger protein involved in high light and cold stress (ZAT12). Indeed, the RNA-seq results showed that the transcript levels of a MYC-type bHLH transcription factor (ICE1) increased under cold stress at 1 h, and decrease after 12 h.
Fig. 7.
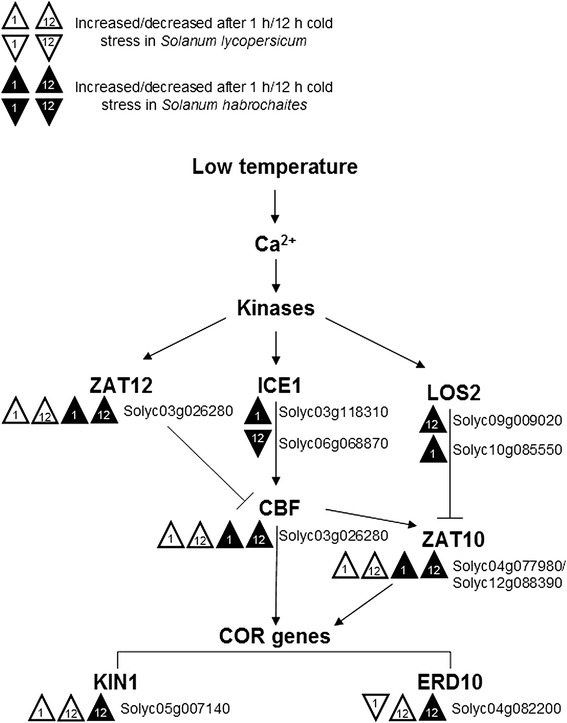
Diagram of cold-responsive transcriptional network in plant. Solid arrows indicate activation, whereas lines ending with a bar show negative regulation. Abbreviations: CBF, C-repeat binding factor (an AP2-type transcription factor); ICE1, inducer of CBF expression 1 (a MYC-type bHLH transcription factor); LOS2, low expression of osmotically responsive genes 2 (a bifunctional enolase with transcriptional repression activity); ZAT12, a zinc finger protein involved in high light and cold acclimation; ZAT10, related to Cys2/His2-type zinc-finger proteins found in higher plants; KIN1, encodes protein kinase APK2a; ERD10, encodes a gene induced by low temperature and dehydration
AS of pre-mRNA is an important mechanism to increase transcriptome and proteome variation in eukaryotes. Previous examinations of AS events under abiotic stress showed that some AS events are stress related [34, 66, 67]. Different coverage of mRNA isoforms in RNA-seq was observed under abiotic stress, likely reflecting the regulation of AS events. Here, we used RNA-seq to identify the AS events in S. lycopersicum and S. habrochaites that differed under normal conditions and cold stress treatment. Compared with other methods, RNA-seq supplies a wide and deep sequencing of the transcriptome, providing experimental confirmation of splice junctions and AS events with low false-positive rates. Our data provide an exceptional and impartial evaluation of AS in S. lycopersicum and S. habrochaites. The results were similar to those of Filichkin et al. [34], who used A. thaliana RNA-seq data to compare the specific abiotic stress transcriptomes of A. thaliana. The authors identified 6,000 novel AS events within the introns of 3,120 genes.
Our RNA-seq analysis of the S. lycopersicum and S. habrochaites transcriptomes suggests numerous genes with AS may be associated with cold stress (Additional file 7). The expressions of 121 (sample C1 vs. C0), 522 (sample C12 vs. C0), 112 (sample Tsh1 vs. Tsh0), and 553 (sample Tsh12 vs. Tsh0) AS genes were increased under cold stress; and the expressions of 110 (sample C1 vs. C0), 140 (sample C12 vs. C0), 111 (sample Tsh1 vs. Tsh0), and 122 (sample Tsh12 vs. Tsh0) genes decreased (Fig. 4, Additional file 7). Most of the genes identified as undergoing cold-induced AS were transcripts whose levels remained constant under cold stress. Thus, despite the lack of change in transcript expression level, their coding ability could be very different.
Recently, miRNAs have been identified as new players in plant tolerance to abiotic stress, such as cold, heat, high salinity, drought, oxidative, hypoxia, and UV B [68]. Many studies have attempted to understand the roles of miRNAs in the response to cold in several plants, including A. thaliana [69], Brachypodium distachyon [70], Oryza sativa [71], and Populus trichocarpa [72]. In this study, sequencing was used to confirm the genome-wide miRNA expression patterns of S. lycopersicum and S. habrochaites under cold stress.
Some miRNAs in different plant species present different expression patterns under cold stress. For example, the expression of miR172 was inhibited after cold stress at 1 h in S. lycopersicum, but was induced in S. habrochaites after cold stress at 12 h (Additional file 14). Additionally, miR172 was upregulated in A. thaliana [69] and B. distachyon [70]. Similarly, miR170/171 expression was upregulated under cold stress at 1 h in S. lycopersicum, while the transcript level of miR170/171 was decreased under cold stress at 12 h in S. habrochaites. The transcript level of miR170/171 was upregulated in A. thaliana [69] and downregulated in Oryza sativa [71] and Populus trichocarpa [72] in response to cold.
Conclusions
S. lycopersicum and S. habrochaites are closely related plant species, but their cold tolerances are different. In recent years, our research group has investigated the cold-tolerance mechanisms of these plants at the physiological and molecular levels. Here, we studied the transcriptomes of cold stressed leaves of S. lycopersicum and S. habrochaites. We obtained 68,051 assembled unigenes, and many cold-regulated genes were detected, representing useful resources for gene cloning to improve cold tolerance of crops. Furthermore, the comparison of the functional networks of cold-regulated genes in S. lycopersicum and S. habrochaites provided information that could help us to identify the differences in cold-tolerance mechanisms between S. lycopersicum and S. habrochaites. We found that 21 % and 23 % of genes were differentially expressed between the cultivated and wild tomato species, respectively, when plants were transferred from warm to cold temperatures. An AS analysis suggested that the relative abundance of isoforms of S. lycopersicum and S. habrochaites significantly shifted under cold stress. In addition, certain miRNAs (e.g., miR159, miR319, and miR6022) play roles in the response to cold stress. Thus, differences in cold regulatory mechanisms may contribute to the differences in cold tolerance of these two tomato species.
Methods
Plant material and cold stress conditions
S. habrochaites LA1777 was supplied by Tomato Genetics Research Center (University of California, Davis, USA). S. lycopersicum ‘glamor’ and S. habrochaites were grow at 25 °C with 16-h light and 8-h dark cycles for 8 weeks before harvesting. To avoid changes caused by the circadian rhythm, all cold stress treatments were started at 4 °C at 12 PM under light and continued for 0 (untreated control), 1, and 12 h.
Physiological responses to cold stress
The MDA content was assayed as described by Campos [73]. The free proline content was determined according to the method described by Zhang et al., [70]. POD activities were determined as described by Quiroga [74]. CAT activity was assayed as described by Yao [75].
Total RNA extraction and library preparation
The total RNA from leaves was extracted using the TRIzol reagent (Invitrogen) and digested with RQ1 DNase (Promega) to remove genomic DNA. The quality and integrity of the total RNA were detected using a SmartSpec plus Spectrophotometer (Bio-Rad) and 1.5 % agarose gel electrophoresis. Polyadenylated mRNAs were purified and concentrated using oligo(dT)-conjugated magnetic beads before being used for directional RNA-seq library preparation. Purified mRNAs were fragmented at 95 °C and subjected to end repair and 5′ adaptor ligation, followed by reverse transcription using randomized hexamers and an RT primer with a 3′ adaptor. Purified cDNAs were amplified, and 200–500 bp PCR products were quantified and purified. RNA-seq libraries were prepared and applied to an Illumina Genome Analyzer IIx system for 80 nt single-end sequencing by ABlife Inc. (Wuhan, China).
For small RNAs, 3 μg of total RNA was used for small RNA library preparation using the Balancer NGS Library Preparation Kit for small/microRNA (GnomeGen), following the manufacturer’s instructions. The purified small RNA libraries were quantified using a Qubit Fluorometer (Invitrogen) and used for cluster generation and 36 nt single-end sequencing analysis using the Illumina Genome Analyzer IIx system.
Processing, mapping of Illumina reads, detection of alternative splicing, and de novo assembly
The adapter sequences and low-quality bases at the 3′ ends were removed from the RNA-seq reads generated by the Illumina Genome Analyzer. Reads whose lengths were more than 20 bp were used for further analysis. The reads were mapped to the tomato genome using TopHat [59], which allows the confirmation of AS events. In total, 172,910 junction sites were identified. We categorized the AS events into different types according to the exon structures, using the ABLas-1 package (ABlife Inc., Wuhan, China). These categories included exon skipping, intron retention, alternative 5′ splice site, alternative 3′ splice site, and mutually exclusive exons, as described by Wang et al. [76]. Reads were assembled separately from each S. lycopersicoides library using the Trinity method [58]. There are three software modules in Trinity: Inchworm, Chrysalis, and Butterfly, which were used to process the RNA-seq reads sequentially. First, the Inchworm program assembled the reads to contigs. Second, the Chrysalis program clustered the minimal overlapping contigs. Third, the Butterfly program constructed the transcripts. Finally, the multiple sequence alignment tool BLAST was used to cluster the transcripts by similarity of the right match length [49]. The coding sequences (CDS) of the unigenes were predicted using EMBOSS (http://emboss.sourceforge.net/) and the longest CDS was considered as the complete CDS of a unigene.
Differential expression and GO enrichment
The expression level of genes were evaluated and normalized using the reads per kilobase of transcript per million reads mapped method [77]. Unigene expression was analyzed using the Bioconductor package with the edgeR and Bayseq methods. A very stringent cutoff, normalized fold change ≥2 or ≤0.5 and a P-value (P ≤0.01), was used to identify cold-regulated DEGs. A GO analysis was employed to predict gene function and calculate the functional distribution frequency, using the DAVID package (ABlife Inc., Wuhan, China).
qPCR analysis
To validate the transcript abundance of genes measured by RNA-seq, we performed qPCR using Power SYBR Green Mastermix in an Applied Biosystems 7500 Real-Time PCR System. The RNAs from S. habrochaites and S. lycopersicum used in RNA-seq were reverse transcribed into cDNAs. The 14 primer pairs used are listed in Additional file 2. The ACTIN gene was used as a reference in these experiments. Three technical replicates were used for qPCR. The single amplicons were confirmed by melting curve analysis and gel electrophoresis of the final product. The cycle threshold (CT) value of each gene was normalized to the reference gene to detect the relative fold changes in each sample, which was calculated using the ΔΔCT method, as described previously [78].
Identification of miRNAs in tomato
High-quality small RNA reads ranging from 14 to 24 nucleotides were acquired from the raw data. Adaptor sequences and low-quality tags were removed to detect known and novel miRNAs in tomato. Small RNA reads were used to search the Rfam database and NCBI database to remove non-coding RNAs, such as rRNA, tRNA, snRNA, and snoRNA. The remaining sequences were searched in the miRBase database v19.0, with no mismatch permitted, to identify conserved mature miRNA orthologs. Small RNAs that did not map to any miRNAs in miRBase database were analyzed as novel miRNAs using miRDeep2 (developed by ABlife Inc.).
Availability of supporting data
The raw RNA-seq data supporting the result of this article is available in the Sequence Read Archive (SRA), with accession numbers SRX1013429 and SRX1014317.
Acknowledgements
This work was supported by the grants from the the National Science Foundation of China (J1210069) and the Innovation Team Project of Heilongjiang Provincial Education Department to A.X. Wang, the Returned Oversea Scholar Foundation by Heilongjiang Provincial Education Department (1252HQ011), the National Science Foundation of China (31301780), and the Heilongjiang Postdoctoral Fund (LBH-Z12044) to X.L. Chen. The authors appreciated professor Michael Deyholos of British Columbia University for critically reading and reviewing the manuscripts.
Abbreviations
- A3SS
Alternative 3’ splice site
- A5SS
Alternative 5’ splice site
- ABA
Abscisic acid
- AS
Alternative splicing events
- CAT
Catalase
- DEG
Differentially expressed gene
- ES
Exon skipping
- GO
Gene ontology
- IR
Intron retention
- JA
Jasmonic acid
- MDA
Malondialdehyde
- miRNA
microRNA
- MXE
Mutually exclusive exons
- PCR
Polymerase chain reaction
- POD
Peroxidase
- RPKM
Reads per kilobase of a gene per million reads
- RLK
Receptor-like protein
- SNP
Single nucleotide polymorphism
- SR
Serine/arginine-rich gene
- TPM
Transcripts per million
- UV
Ultraviolet
Additional files
List of genes used to identify differentially expressed genes (DEG).
Specify primers used in expression analysis of selected genes.
GO term analysis of differentially expressed genes (DEG).
GO term analysis of co-differentially expressed genes (co-DEG).
All alternative splicing events in tomato.
Distribution of each class of alternative splicing events.
GO term analysis of differentially expressed alternative splicing genes (DASG).
Differentially expressed alternative splicing genes (DASG) overlap with differentially expressed genes (DEG).
GO term analysis of genes with SNPs.
SNP overlap with differentially expressed genes (DEG).
SNP overlap with co-differentially expressed genes (co-DEG).
List of novel miRNAs.
List of miRNAs to identify differentially expressed miRNAs.
Differentially expressed miRNAs.
List of differentially expressed miRNA targets.
GO term analysis of differentially expressed miRNA targets.
Differentially expressed miRNA targets overlap with differentially expressed genes (DEG).
Complete list of unigenes in S. habrochaites with BLASTX hits.
Unigene sequences were assembled from S. habrochaites reads.
Figure S1-11. Figure S1: Phenotypic and physiological responses of the two tomato genotypes under cold stress. Figure S2: Reads distribution across genomic regions. Figure S3: The mRNA coverage analysis. Figure S4: mRNA expression profile. Figure S5: Analysis of scatterplots comparing the gene expression correlation. Figure S6: Analysis of the sample correlation according to gene expression. Figure S7: Identification of the differentially expressed genes. Figure S8: qRT-PCR validation of differentially expressed genes. Figure S9: Identification of alternative splicing. Figure S10: Small RNA reads distribution across the genomic regions. Figure S11: Clean reads mapping classification results.
Footnotes
Hongyu Chen and Xiuling Chen contributed equally to this work.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AW, YZ, HC, XC and JL designed the experiments and drafted the manuscript. HC and DC performed the RNA sequencing and bioinformatics data processing. HC and XC prepared plant materials and carried out qPCR analysis. All authors read and approved the final manuscript.
Contributor Information
Hongyu Chen, Email: chy810501@163.com.
Xiuling Chen, Email: xiuling.chen@ymail.com.
Dong Chen, Email: dongchen@ablife.cc.
Jingfu Li, Email: lijf_2005@126.com.
Yi Zhang, Email: yizhang@ablife.cc.
Aoxue Wang, Email: axwang@neau.edu.cn.
References
- 1.Zhang X, Fowler SG, Cheng H, Lou Y, Rhee SY, Stockinger EJ. Freezing-sensitive tomato has a functional CBF cold response pathway, but a CBF regulon that differs from that of freezing-tolerant Arabidopsis. Plant J. 2004;39:905–19. doi: 10.1111/j.1365-313X.2004.02176.x. [DOI] [PubMed] [Google Scholar]
- 2.Van-Buskirk HA, Thomashow MF. Arabidopsis transcription factors regulating cold acclimation. Physiol Plant. 2006;126:72–80. doi: 10.1111/j.1399-3054.2006.00625.x. [DOI] [Google Scholar]
- 3.Chinnusamy V, Zhu J, Zhu JK. Cold stress regulation of gene expression in plants. Trends Plant Sci. 2007;12:444–51. doi: 10.1016/j.tplants.2007.07.002. [DOI] [PubMed] [Google Scholar]
- 4.Stockinger EJ, Gilmour SJ, Thomashow MF. Arabidopsis thaliana CBF1 encodes an AP2 domain-containing transcriptional activator that binds to the C-repeat/DRE, a cis-acting DNA regulatory element that stimulates transcription in response to low temperature and water deficit. Proc Natl Acad Sci U S A. 1997;94:1035–40. doi: 10.1073/pnas.94.3.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gilmour SJ, Zarka DG, Stockinger EJ, Salazar MP, Houghton JM, Thomashow MF. Low temperature regulation of the Arabidopsis CBF family of AP2 transcriptional activators as an early step in cold-induced COR gene expression. Plant J. 1998;16:433–42. doi: 10.1046/j.1365-313x.1998.00310.x. [DOI] [PubMed] [Google Scholar]
- 6.Medina J, Bargues M, Terol J, Pérez-Alonso M, Salinas J. The Arabidopsis CBF gene family is composed of three genes encoding AP2 domain-containing proteins whose expression Is regulated by low temperature but not by abscisic acid or dehydration. Plant Physiol. 1999;119:463–70. doi: 10.1104/pp.119.2.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Riechmann JL, Meyerowitz EM. The AP2/EREBP family of plant transcription factors. Biol Chem. 1998;379:633–46. doi: 10.1515/bchm.1998.379.6.633. [DOI] [PubMed] [Google Scholar]
- 8.Jaglo-Ottosen KR, Gilmour SJ, Zarka DG, Schabenberger O, Thomashow MF. Arabidopsis CBF1 overexpression induces COR genes and enhances freezing tolerance. Science. 1998;280:104–6. doi: 10.1126/science.280.5360.104. [DOI] [PubMed] [Google Scholar]
- 9.Liu Q, Kasuga M, Sakuma Y, Abe H, Miura S, Yamaguchi-Shinozaki K, et al. Two transcription factors, DREB1 and DREB2, with an EREBP/AP2 DNA binding domain separate two cellular signal transduction pathways in drought- and low-temperature-responsive gene expression, respectively, in Arabidopsis. Plant Cell. 1998;10:1391–406. doi: 10.1105/tpc.10.8.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gilmour SJ, Fowler SG, Thomashow MF. Arabidopsis transcriptional activators CBF1, CBF2, and CBF3 have matching functional activities. Plant Mol Biol. 2004;54:767–81. doi: 10.1023/B:PLAN.0000040902.06881.d4. [DOI] [PubMed] [Google Scholar]
- 11.Kasuga M, Miura S, Shinozaki K, Yamaguchi-Shinozaki K. A combination of the Arabidopsis DREB1A gene and stress-inducible rd29A promoter improved drought- and low-temperature stress tolerance in tobacco by gene transfer. Plant Cell Physiol. 2004;45:346–50. doi: 10.1093/pcp/pch037. [DOI] [PubMed] [Google Scholar]
- 12.Jaglo KR, Kleff S, Amundsen KL, Zhang X, Haake V, Zhang JZ, et al. Components of the Arabidopsis C-repeat/dehydration-responsive element binding factor cold-response pathway are conserved in Brassica napus and other plant species. Plant Physiol. 2001;127:910–7. doi: 10.1104/pp.010548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benedict C, Skinner JS, Meng R, Chang Y, Bhalerao R, Huner NP, et al. The CBF1-dependent low temperature signalling pathway, regulon and increase in freeze tolerance are conserved in Populus spp. Plant Cell Environ. 2006;29:1259–72. doi: 10.1111/j.1365-3040.2006.01505.x. [DOI] [PubMed] [Google Scholar]
- 14.Pino MT, Skinner JS, Park EJ, Jeknić Z, Hayes PM, Thomashow MF, et al. Use of a stress inducible promoter to drive ectopic AtCBF expression improves potato freezing tolerance while minimizing negative effects on tuber yield. Plant Biotechnol J. 2007;5:591–604. doi: 10.1111/j.1467-7652.2007.00269.x. [DOI] [PubMed] [Google Scholar]
- 15.Fowler S, Thomashow MF. Arabidopsis transcriptome profiling indicates that multiple regulatory pathways are activated during cold acclimation in addition to the CBF cold response pathway. Plant Cell. 2002;14:1675–90. doi: 10.1105/tpc.003483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maruyama K, Sakuma Y, Kasuga M, Ito Y, Seki M, Goda H, et al. Identification of cold-inducible downstream genes of the Arabidopsis DREB1A/CBF3 transcriptional factor using two microarray systems. Plant J. 2004;38:982–93. doi: 10.1111/j.1365-313X.2004.02100.x. [DOI] [PubMed] [Google Scholar]
- 17.Lee BH, Henderson DA, Zhu JK. The Arabidopsis cold-responsive transcriptome and its regulation by ICE1. Plant Cell. 2005;17:3155–75. doi: 10.1105/tpc.105.035568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vogel JT, Zarka DG, Van-Buskirk HA, Fowler SG, Thomashow MF. Roles of the CBF2 and ZAT12 transcription factors in configuring the low temperature transcriptome of Arabidopsis. Plant J. 2005;41:195–211. doi: 10.1111/j.1365-313X.2004.02288.x. [DOI] [PubMed] [Google Scholar]
- 19.Zhao Z, Tan L, Dang C, Zhang H, Wu Q, An L. Deep-sequencing transcriptome analysis of chilling tolerance mechanisms of a subnival alpine plant, Chorispora bungeana. BMC Plant Biol. 2012;12:222. doi: 10.1186/1471-2229-12-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu H, Ouyang B, Zhang J, Wang T, Li H, Zhang Y, et al. Differential modulation of photosynthesis, signaling, and transcriptional regulation between tolerant and sensitive tomato genotypes under cold stress. PLoS One. 2012;7 doi: 10.1371/journal.pone.0050785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dong XS, Im SB, Lim YP, Nou IS, Hur Y. Comparative transcriptome profiling of freezing stress responsiveness in two contrasting Chinese cabbage genotypes, Chiifu and Kenshin. Genes Genom. 2014;36:215–27. doi: 10.1007/s13258-013-0160-y. [DOI] [Google Scholar]
- 22.Carvallo MA, Pino MT, Jeknic Z, Zou C, Doherty CJ, Shiu SH, et al. A comparison of the low temperature transcriptomes and CBF regulons of three plant species that differ in freezing tolerance: Solanum commersonii, Solanum tuberosum, and Arabidopsis thaliana. J Exp Bot. 2011;62:3807–19. doi: 10.1093/jxb/err066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kreps JA, Wu Y, Chang HS, Zhu T, Wang X, Harper JF. Transcriptome changes for Arabidopsis in response to salt, osmotic, and cold stress. Plant Physiol. 2002;130:2129–41. doi: 10.1104/pp.008532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chinnusamy V, Ohta M, Kanrar S, Lee BH, Hong X, Agarwal M, et al. ICE1: a regulator of cold-induced transcriptome and freezing tolerance in Arabidopsis. Genes Dev. 2003;17:1043–54. doi: 10.1101/gad.1077503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee BH, Kapoor A, Zhu J, Zhu JK. STABILIZED1, a stress-upregulated nuclear protein, is required for pre-mRNA splicing, mRNA turnover, and stress tolerance in Arabidopsis. Plant Cell. 2006;18:1736–49. doi: 10.1105/tpc.106.042184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xin Z, Mandaokar A, Chen J, Last RL, Browse J. Arabidopsis ESK1 encodes a novel regulator of freezing tolerance. Plant J. 2007;49:786–99. doi: 10.1111/j.1365-313X.2006.02994.x. [DOI] [PubMed] [Google Scholar]
- 27.Zhu J, Dong CH, Zhu JK. Interplay between cold-responsive gene regulation, metabolism and RNA processing during plant cold acclimation. Curr Opin Plant Biol. 2007;10:290–5. doi: 10.1016/j.pbi.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 28.Matsui A, Ishida J, Morosawa T, Mochizuki Y, Kaminuma E, Endo TA, et al. Arabidopsis transcriptome analysis under drought, cold, high-salinity and ABA treatment conditions using a tiling array. Plant Cell Physiol. 2008;49:1135–49. doi: 10.1093/pcp/pcn101. [DOI] [PubMed] [Google Scholar]
- 29.Zahler AM, Lane WS, Stolk JA, Roth MB. SR proteins: a conserved family of pre-mRNA splicing factors. Genes Dev. 1992;6:837–47. doi: 10.1101/gad.6.5.837. [DOI] [PubMed] [Google Scholar]
- 30.Seki M, Narusaka M, Ishida J, Nanjo T, Fujita M, Oono Y, et al. Monitoring the expression profiles of 7000 Arabidopsis genes under drought, cold and high-salinity stresses using a full-length cDNA microarray. Plant J. 2002;31:279–92. doi: 10.1046/j.1365-313X.2002.01359.x. [DOI] [PubMed] [Google Scholar]
- 31.Iida K, Seki M, Sakurai T, Satou M, Akiyama K, Toyoda T, et al. Genome-wide analysis of alternative pre-mRNA splicing in Arabidopsis thaliana based on full-length cDNA sequences. Nucleic Acids Res. 2004;32:5096–103. doi: 10.1093/nar/gkh845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kilian J, Whitehead D, Horak J, Wanke D, Weinl S, Batistic O, et al. The AtGenExpress global stress expression data set: protocols, evaluation and model data analysis of UV-B light, drought and cold stress responses. Plant J. 2007;50:347–63. doi: 10.1111/j.1365-313X.2007.03052.x. [DOI] [PubMed] [Google Scholar]
- 33.Palusa SG, Ali GS, Reddy AS. Alternative splicing of pre-mRNAs of Arabidopsis serine/arginine-rich proteins: regulation by hormones and stresses. Plant J. 2007;49:1091–107. doi: 10.1111/j.1365-313X.2006.03020.x. [DOI] [PubMed] [Google Scholar]
- 34.Filichkin SA, Priest HD, Givan SA, Shen R, Bryant DW, Fox SE, et al. Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res. 2010;20:45–58. doi: 10.1101/gr.093302.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li P, Ponnala L, Gandotra N, Wang L, Si Y, Tausta SL, et al. The developmental dynamics of the maize leaf transcriptome. Nat Genet. 2010;42:1060–7. doi: 10.1038/ng.703. [DOI] [PubMed] [Google Scholar]
- 36.Lu T, Lu G, Fan D, Zhu C, Li W, Zhao Q, et al. Function annotation of the rice transcriptome at single-nucleotide resolution by RNA-seq. Genome Res. 2010;20:1238–49. doi: 10.1101/gr.106120.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brett D, Pospisil H, Valcárcel J, Reich J, Bork P. Alternative splicing and genome complexity. Nat Genet. 2002;30:29–30. doi: 10.1038/ng803. [DOI] [PubMed] [Google Scholar]
- 38.Mazzucotelli E, Mastrangelo AM, Crosatti C, Guerra D, Stanca AM, Cattivelli L. Abiotic stress response in plants: When post-transcriptional and post-translational regulations control transcription. Plant Sci. 2008;174:420–31. doi: 10.1016/j.plantsci.2008.02.005. [DOI] [Google Scholar]
- 39.Reddy AS. Alternative splicing of pre-messenger RNAs in plants in the genomic era. Annu Rev Plant Biol. 2007;58:267–94. doi: 10.1146/annurev.arplant.58.032806.103754. [DOI] [PubMed] [Google Scholar]
- 40.Llave C, Kasschau KD, Rector MA, Carrington JC. Endogenous and silencing-associated small RNAs in plants. Plant Cell. 2002;14:1605–19. doi: 10.1105/tpc.003210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mette MF, Van-der-Winden J, Matzke M, Matzke AJ. Short RNAs can identify new candidate transposable element families in Arabidopsis. Plant Physiol. 2002;130:6–9. doi: 10.1104/pp.007047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park W, Li J, Song R, Messing J, Chen X. CARPEL FACTORY, a Dicer homolog, and HEN1, a novel protein, act in microRNA metabolism in Arabidopsis thaliana. Curr Biol. 2002;12:1484–95. doi: 10.1016/S0960-9822(02)01017-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Voinnet O. Origin, biogenesis, and activity of plant microRNAs. Cell. 2009;136:669–87. doi: 10.1016/j.cell.2009.01.046. [DOI] [PubMed] [Google Scholar]
- 44.Lu C, Tej SS, Luo S, Haudenschild CD, Meyers BC, Green PJ. Elucidation of the small RNA component of the transcriptome. Science. 2005;309:1567–9. doi: 10.1126/science.1114112. [DOI] [PubMed] [Google Scholar]
- 45.Kasschau KD, Fahlgren N, Chapman EJ, Sullivan CM, Cumbie JS, Givan SA, et al. Genome-wide profiling and analysis of Arabidopsis siRNAs. PLoS Biol. 2007;5 doi: 10.1371/journal.pbio.0050057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Johnson C, Kasprzewska A, Tennessen K, Fernandes J, Nan GL, Walbot V, et al. Clusters and superclusters of phased small RNAs in the developing inflorescence of rice. Genome Res. 2009;19:1429–40. doi: 10.1101/gr.089854.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jones-Rhoades MW, Bartel DP, Bartel B. MicroRNAS and their regulatory roles in plants. Annu Rev Plant Biol. 2006;57:19–53. doi: 10.1146/annurev.arplant.57.032905.105218. [DOI] [PubMed] [Google Scholar]
- 48.Mallory AC, Vaucheret H. Functions of microRNAs and related small RNAs in plants. Nat Genet. 2006;38(Suppl):S31–6. doi: 10.1038/ng1791. [DOI] [PubMed] [Google Scholar]
- 49.Chen X. Small RNAs and their roles in plant development. Annu Rev Cell Dev Biol. 2009;25:21–44. doi: 10.1146/annurev.cellbio.042308.113417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sunkar R, Chinnusamy V, Zhu J, Zhu JK. Small RNAs as big players in plant abiotic stress responses and nutrient deprivation. Trends Plant Sci. 2007;12:301–9. doi: 10.1016/j.tplants.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 51.Leung AK, Sharp PA. MicroRNA functions in stress responses. Mol Cell. 2010;40:205–15. doi: 10.1016/j.molcel.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sunkar R. MicroRNAs with macro-effects on plant stress responses. Semin Cell Dev Biol. 2010;21:805–11. doi: 10.1016/j.semcdb.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 53.Juarez MT, Kui JS, Thomas J, Heller BA, Timmermans MC. MicroRNA-mediated repression of rolled leaf1 specifies maize leaf polarity. Nature. 2004;428:84–8. doi: 10.1038/nature02363. [DOI] [PubMed] [Google Scholar]
- 54.Sunkar R, Zhu JK. Novel and stress-regulated microRNAs and other small RNAs from Arabidopsis. Plant Cell. 2004;16:2001–19. doi: 10.1105/tpc.104.022830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Venema JH, Posthumus F, De-Vries M, Van-Hasselt PR. Differential response of domestic and wild Lycopersicon species to chilling under low light: growth, carbohydrate content, photosynthesis and the xanthophyll cycle. Physiol Plant. 1999;105:81–8. doi: 10.1034/j.1399-3054.1999.105113.x. [DOI] [Google Scholar]
- 56.Foolad MR, Lin GY. Relationship between cold tolerance during seed germination and vegetative growth in tomato: germplasm evaluation. J Am Soc Horticultural Sci. 2000;125:679–83. [Google Scholar]
- 57.Venema JH, Linger P, Van-Heusden AW, Van-Hasselt PR, Bru¨ggemann W. The inheritance of chilling tolerance in tomato (Lycopersicon spp.) Plant Biology. 2005;7:118–30. doi: 10.1055/s-2005-837495. [DOI] [PubMed] [Google Scholar]
- 58.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–52. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ner-Gaon H, Halachmi R, Savaldi-Goldstein S, Rubin E, Ophir R, Fluhr R. Intron retention is a major phenomenon in alternative splicing in Arabidopsis. Plant J. 2004;39:877–85. doi: 10.1111/j.1365-313X.2004.02172.x. [DOI] [PubMed] [Google Scholar]
- 61.Wang BB, Brendel V. Genomewide comparative analysis of alternative splicing in plants. Proc Natl Acad Sci. 2006;103:7175–80. doi: 10.1073/pnas.0602039103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang G, Guo G, Hu X, Zhang Y, Li Q, Li R, et al. Deep RNA sequencing at single base-pair resolution reveals high complexity of the rice transcriptome. Genome Res. 2002;20:646–54. doi: 10.1101/gr.100677.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ciardi JA, Deikman J, Orzolek MD. Increased ethylene synthesis enhances chilling tolerance in tomato. Physiol Plant. 1997;101:333–40. doi: 10.1111/j.1399-3054.1997.tb01005.x. [DOI] [Google Scholar]
- 64.Morgan PW, Drew MC. Ethylene and plant responses to stress. Physiol Plant. 1997;100:620–30. doi: 10.1111/j.1399-3054.1997.tb03068.x. [DOI] [Google Scholar]
- 65.Yu XM, Griffith M, Wiseman SB. Ethylene induces antifreeze activity in winter rye leaves. Plant Physiol. 2001;126:1232–40. doi: 10.1104/pp.126.3.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gulledge AA, Roberts AD, Vora H, Patel K, Loraine AE. Mining Arabidopsis thaliana RNA-seq data with Integrated Genome Browser reveals stress-induced alternative splicing of the putative splicing regulator SR45a. Am J Bot. 2012;99:219–31. doi: 10.3732/ajb.1100355. [DOI] [PubMed] [Google Scholar]
- 67.Leviatan N, Alkan N, Leshkowitz D, Fluhr R. Genome-wide survey of cold stress regulated alternative splicing in Arabidopsis thaliana with tiling microarray. PLoS One. 2013;8 doi: 10.1371/journal.pone.0066511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sunkar R, Li YF, Jagadeeswaran G. Functions of microRNAs in plant stress responses. Trends Plant Sci. 2012;17:196–203. doi: 10.1016/j.tplants.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 69.Liu HH, Tian X, Li YJ, Wu CA, Zheng CC. Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA. 2008;14:836–43. doi: 10.1261/rna.895308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang J, Xu Y, Huan Q, Chong K. Deep sequencing of Brachypodium small RNAs at the global genome level identifies microRNAs involved in cold stress response. BMC Genomics. 2009;10:449. doi: 10.1186/1471-2164-10-449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lv DK, Bai X, Li Y, Ding XD, Ge Y, Cai H, et al. Profiling of cold-stress-responsive miRNAs in rice by microarrays. Gene. 2010;459:39–47. doi: 10.1016/j.gene.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 72.Lu S, Sun YH, Chiang VL. Stress-responsive microRNAs in Populus. Plant J. 2008;55:131–51. doi: 10.1111/j.1365-313X.2008.03497.x. [DOI] [PubMed] [Google Scholar]
- 73.Campos PS, Quartin V, Ramalho JC, Nunes MA. Electrolyte leakage and lipid degradation account for cold sensitivity in leaves of Coffea sp. plants. J Plant Physiol. 2003;160:283–92. doi: 10.1078/0176-1617-00833. [DOI] [PubMed] [Google Scholar]
- 74.Quiroga M, Guerrero C, Botella MA, Barceló A, Amaya I, Medina MI, et al. A tomato peroxidase involved in the synthesis of lignin and suberin. Plant Physiol. 2000;122:1119–27. doi: 10.1104/pp.122.4.1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yao YX, Dong QL, Zhai H, You CX, Hao YJ. The functions of an apple cytosolic malate dehydrogenase gene in growth and tolerance to cold and salt stresses. Plant Physiol Biochem. 2011;49:257–64. doi: 10.1016/j.plaphy.2010.12.009. [DOI] [PubMed] [Google Scholar]
- 76.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–6. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5:621–8. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 78.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]