

Abstract
Background
Cachexia and muscle atrophy are common consequences of cancer and chemotherapy administration. The novel hormone ghrelin has been proposed as a treatment for this condition. Increases in food intake and direct effects on muscle proteolysis and protein synthesis are likely to mediate these effects, but the pathways leading to these events are not well understood.
Methods
We characterized molecular pathways involved in muscle atrophy induced by Lewis lung carcinoma (LLC) tumour implantation in c57/bl6 adult male mice and by administration of the chemotherapeutic agent cisplatin in mice and in C2C12 myotubes. The effects of exogenous ghrelin administration and its mechanisms of action were examined in these settings.
Results
Tumour implantation and cisplatin induced muscle atrophy by activating pro-inflammatory cytokines, p38-C/EBP-β, and myostatin, and by down-regulating Akt, myoD, and myogenin, leading to activation of ubiquitin-proteasome-mediated proteolysis and muscle weakness. Tumour implantation also increased mortality. In vitro, cisplatin up-regulated myostatin and atrogin-1 by activating C/EBP-β and FoxO1/3. Ghrelin prevented these changes in vivo and in vitro, significantly increasing muscle mass (P < 0.05 for LLC and P < 0.01 for cisplatin models) and grip strength (P = 0.038 for LLC and P = 0.001 for cisplatin models) and improving survival (P = 0.021 for LLC model).
Conclusion
Ghrelin prevents muscle atrophy by down-regulating inflammation, p38/C/EBP-β/myostatin, and activating Akt, myogenin, and myoD. These changes appear, at least in part, to target muscle cells directly. Ghrelin administration in this setting is associated with improved muscle strength and survival.
Keywords: Cachexia, Cancer, Muscle, Ghrelin, Growth hormone
Introduction
Cachexia (involuntary loss of muscle and fat mass) is a common complication of many chronic conditions including cancer, chronic heart failure, lung disease, and renal disease.1 In the setting of cancer, it is associated with poor tolerance to therapy, decreased quality of life, and increased mortality.2 In spite of its significance, there are currently no treatments for this condition. Nutritional supplements alone do not reverse cachexia,1 resistance training may improve muscle strength and lean body mass, but it is not commonly prescribed to patients in this setting,3 and medications such as progestational agents are often used but have been shown to increase only fat mass and can cause deep vein thrombosis, adrenal insufficiency, and hypogonadism.4 An increase in muscle proteolysis and a decrease in protein synthesis driven by activation of the ubiquitin-proteasome,5 mitogen-activated protein (MAP) kinases,6 and myostatin pathways3 among others have been identified as central processes in tumour-induced cachexia, but the pathways leading to these have not been fully characterized.7
Cisplatin is a chemotherapeutic agent that is part of the standard of care for the treatment of several cancers including lung, head and neck, ovary, testicular, and bladder cancer, and paradoxically, it is known to induce weight and muscle mass losses.8 We have recently reported the mechanisms by which cisplatin alters lipid metabolism thereby causing fat atrophy, including an increase in lipolysis and lipid oxidation and a decrease in lipogenesis.8 However, the extent to which muscle protein synthesis and degradation are affected in this setting and the mechanisms mediating these effects have not been characterized. Given that cachexia is often a dose-limiting side effect of cisplatin and other chemotherapeutic agents, treating or preventing cachexia would allow the use of higher, more effective doses of cisplatin.
Ghrelin, the endogenous ligand for the growth hormone secretagogue receptor (GHSR)-1a, has been proposed as a potential therapeutic target for cachexia, having the potential to increase appetite, fat, and muscle mass.9,10 Although its mechanisms of action have not been fully elucidated, an increase in food intake and decrease in energy expenditure via hypothalamic effects,11,12 a decrease in inflammation,13 an increase in GH,14 and direct effects in adipose tissue15 and skeletal muscle16 are thought to potentially mediate these effects.
In this study, we established the effects of ghrelin in the setting of chemotherapy- and tumour-induced muscle wasting and characterized the pathways involved. We studied these two models to determine the extent to which these mechanisms were differentially modulated, looking at gene and protein expression as well as their functional impact and their effect on survival. Because multiple pathways are likely to play a role in mediating ghrelin's effects, we postulated that several targets would be modulated by ghrelin in these settings.
Materials and methods
Animals
Adult (age 90 ± 10 days) c57bl/6 J male mice were used for all experiments. Animals were individually housed, acclimated to their cages and human handling for 1 week before the experiments, and maintained on a 12/12 light/dark cycle (lights on at 6 a.m.). All experiments were conducted with the approval of the Institutional Animal Care and Use Committee and were in compliance with the National Institutes of Health Guidelines for Use and Care of Laboratory Animals. Sample sizes of 8–10 animals/group were used for all experiments.
Acylated ghrelin was used for all in vivo and all in vitro studies. For the cisplatin-induced cachexia model, animals received vehicle (saline), cisplatin, acylated ghrelin + cisplatin, or acylated ghrelin. Cisplatin was purchased from APP Pharmaceuticals (Schaumburg, IL), and rodent acylated ghrelin was synthesized by Anaspec (Fremont, CA). The dose of cisplatin was 2.5 mg/kg given daily for 10 days at 8:30 a.m. intraperitoneally (IP), and the dose for ghrelin was 0.8 mg/kg twice daily IP at 8 a.m. and 5 p.m. for 21 days. The morning dose of acylated ghrelin was given 30 min before cisplatin. This particular dose of cisplatin and acylated ghrelin was selected based on our previous work that showed cisplatin-induced weight loss and anorexia without over toxicity, and prevention of anorexia, fat atrophy, and weight loss by ghrelin.8
For the Lewis lung carcinoma (LLC)-induced cancer cachexia model, 100 mcl (1 × 106 cells) of LLC cells (American Type Culture Collection, Manassas, VA) or an equal volume and number of heat-killed LLC cells (control) was injected subcutaneously into the right flank. Ghrelin (tumour + ghrelin, T-G group) or vehicle (saline, tumour + vehicle, T-V group) was injected subcutaneously into the left flank of mice from Day 7 after LLC implantation, when the tumour became palpable (approximately 1 cm in diameter). The dose for ghrelin was 0.8 mg/kg twice daily at 8 a.m. and 5 p.m. Mice were sacrificed on Day 28 of LLC implantation, approximately 21 days after tumour had become palpable.
Body weight and food intake were assessed daily by weighing the food and the animals before the a.m. injection. Body weight changes were expressed as change from baseline, and food intake was expressed in grammes/day. Lean body mass and fat mass were measured by nuclear magnetic resonance (NMR) with a Minispec mq NMR spectrometer (Bruker optics, The Woodlands, TX). Grip strength was measured with a grip strength metre with digital force gauge (Columbus Instruments, Columbus, OH).
Hind-leg muscles [quadriceps, gastrocnemius, soleus, tibialis anterioris (TA), and extensor digitalis longus] were collected for analyses. Comparisons between groups (i.e. muscle cross-sectional area and gene or protein expression) were always done from the same muscles. Muscle weights were normalized by lean body mass at baseline. For the LLC-induced cachexia model, body weight and lean body mass were expressed as weight after tumour weights were subtracted and expressed as change from baseline.
Cell culture
C2C12 (ATCC, Manassas, VA) myoblasts were maintained in Dulbecco's modified Eagle medium (DMEM) (Life Technologies, Grand Island, NY) with 10% foetal bovine serum (HyClone, Logan, UT), penicillin (200 units/mL), and streptomycin (50 µg/mL) (Invitrogen, Carlsbad, CA). At 90% confluence, the media was changed to DMEM plus 2% horse serum (Life Technologies, Grand Island, NY) to induce myotube formation. Cells were treated with cisplatin (50 μM), acylated ghrelin (1 μM) or both on Day 3 when myotube formed and were harvested 24 h after.
Real-time quantitative polymerase chain reaction
RNA from gastrocnemii or cultured C2C12 myotubes was isolated (Trizol reagent, Life Technologies, Grand Island, NY). Transcript levels were measured by real-time PCR (7000 Sequence Detection System; Applied Biosystems). Total RNA was reverse transcribed (QuantiTect Reverse Transcription Kit, Qiagen, Valencia, CA) to cDNA. Primers for real-time PCR amplification used are from Sigma-Aldrich (St. Louis, MO; see Supporting Information, Table S1). Signals are expressed relative to glyceraldehyde-3-phosphate dehydrogenase using the standard 2-ΔCT method.
Western blot
Protein from gastrocnemii or myotubes was isolated using tissue lysis/extraction reagent or cell lysis reagent (Sigma-Aldrich, St. Louis, MO) with protease inhibitor and phosphorylation protease inhibitor cocktails (Roche, Nutley, NJ). Protein concentration was measured using Pierce BCA Protein Assay (Thermo Scientific, Logan, UT). Nuclear protein was isolated using EpiQuik Nuclear Extraction Kit (Epigentek Farmingdale, NY). After Phosphate Buffered Saline (PBS) washing, cells were scraped and centrifuged. Cell pellets were re-suspended in 100 μL of diluted NE1 [1X, 1:1000 ratio dithiothreitol (DTT) solution and Protease Inhibitor Cocktail (PIC)] and incubated on ice for 10 min, then vortexed and centrifuged for 1 min at 24000 g. The cytoplasmic extract was removed from the nuclear pellet, and two volumes of NE2 containing DTT and PIC (1:1000 ratio) were added to the nuclear pellet. The extract was incubated on ice for 15 min, vortexing samples for 5 s every 3 min. The transcription factors C/EBP-β and FOXO1 were assessed in nuclear extracts given that they bind to specific DNA sequences controlling transcription to mRNA in the nuclei. We separated protein extracts on 4–12% NuPAGE gels (Invitrogen, Carlsbad, CA) and blotted them onto Immobilon Polyvinylidene Fluoride (PVDF) (Millipore, Billerica, MA). Membranes were blocked at room temperature (RT) for 1 h in 5% Bovine serumalbumin (BSA) and incubated in 1ry antibodies overnight at 4°C. DyLight 680/800 anti-rabbit/mouse/goat IgG 2ry antibodies were used (Thermo Scientific, Waltham, MA) for 1 h at RT. After three washes in TBST, blots were scanned with LI-COR Odyssey (LI-COR, Lincoln, NE) and quantified with Image-Pro plus.
The following antibodies were used to assess by western blots protein levels representing key steps in the pathways of interest: p-Akt (Ser 473, cat#4060), p-p38 (T180, cat#9212), p38 (cat#4511), FOXO3a (cat#9464), C/EBP-β (cat#3084), and glyceraldehyde-3-phosphate dehydrogenase (cat#2118) from Cell Signaling (Beverly MA); myoD (cat#12344), myosin (cat#M4276), myostatin (GDF8, cat#1403860), and A/C lamin (cat#L9393) from Sigma-Aldrich; and Akt (cat#8312) and atrogin-1/MAFbx (cat#33782) from Santa Cruz Biotechnology (Santa Cruz, CA).
Protein synthesis and degradation
Protein synthesis was measured from the incorporation of l-[3,5-3H]tyrosine (5 μCi/mL; PerkinElmer, Waltham, MA) into cellular proteins during a 24 h incubation as described elsewhere.7,8 Myotubes were then washed three times with ice-cold PBS before adding 10% trichloroacetic acid (TCA) to precipitate proteins. After three additional PBS washings, the precipitate was rinsed twice with TCA and solubilized by sonication in lysis solution (1% Triton X-100 and 1 N NaOH). The incorporation of radiolabeled tyrosine radioactivity was measured using liquid scintillation (Beckman Coulter, Brea, CA), and protein concentration was measured using Pierce BCA Protein Assay.
l-[3,5-3H]tyrosine (5 μCi/mL; PerkinElmer, USA) was added to C2C12 myotubes and incubated 24 h. After rinsing by fresh 2% horse serum DMEM, the myotubes were treated with vehicle (PBS), cisplatin (50 μM), and cisplatin (50 μM) + ghrelin (1 μM). Aliquots (200 μL) of culture media were taken at specified times for quantization of l-[3,5-3H]tyrosine release. Proteins were precipitated at 4°C with 10% TCA and centrifuged at 37000 g for 5 min. The precipitate was rinsed twice with TCA and solubilized by sonication in lysis solution (1% Triton X-100 and 1 N NaOH). Radioactivity in the TCA-soluble supernatant and the proteins (TCA-insoluble fraction) were measured using liquid scintillation counting. At the end of the chase period, cells were rinsed twice in PBS and precipitated at 4°C in 10% TCA, and the radioactivity in cell protein was measured as described above. Total radioactivity is the sum of the residual radioactivity in cell proteins and the TCA-soluble radioactivity at different time points. Protein degradation was expressed as l-[3,5-3H]tyrosine released as a percentage of total l-[3,5-3H]tyrosine incorporated.
Immunofluorescence staining
Frozen TA muscles sections or C2C12 myotubes were fixed with 4% paraformaldehyde. After three Tris Buffered Saline Tween-20 (TBST) washings, slides were blocked for 1 h (Dako North America, Inc., Carpentaria, CA). The primary antibody (1:100) dilution was incubated overnight at 4°C. Alexa Fluor 488 or 546 secondary antibodies (Life technologies, Grand Island, NY) were used with 1:1000 dilutions and incubated 1 h at RT. 4'6-Diamidino-2-phenylindole (Cell Signaling) was used to stain nuclei.
To assess differences in cross-sectional areas of myofibers, 8 µm sections of TA muscles were stained with an anti-laminin antibody (Sigma-Aldrich). The areas of at least 500 myofibers (x100 magnification) per TA muscle were measured. In studies of C2C12 myotubes, after being stained with anti-myosin/myosin heavy chain (Skeletal, Fast, Sigma-Aldrich), the areas of 200 myotubes were measured in at least 10 fields (×100 magnification).
Proteasome assay
As previously described,17 gastrocnemius was homogenized in lysis buffer (20 mM Tris–HCl [pH7.2], 0.1 mM EDTA, 1 mM 2-mercaptoethanol, 1 mM DTT, 5 mM ATP, 20% glycerol, 0.04% (v/v) Triton X-100). The lysate was centrifuged, and the supernatant was collected. Protein concentration was determined using the Pierce BCA Protein Assay Kit. The chymotrypsin-like proteasome activity was determined fluorometrically by incubating 40 µg protein with 0.167 µg/μL N-succinyl-Leu-Leu-Val-Try-7-amido-4-methylcoumarin (N-Suc-LLVY-AMC, Sigma-Aldrich) in incubation buffer (100 mM Tris–HCl [pH7.4], 50 mM HEPES [pH8.0], and 5 mM ethleneglycoltetraacetic-acid) for 1 h at 37°C.
Luciferase assay
The myostatin luciferase reporter gene recombinant plasmid was provided by Dr Allen.18 The Tranfectin Lipid Reagent (Bio-Rad, Hercules, CA) was used for the recombinant plasmid transfection to C2C12 myoblasts. When the cells are 80% confluent, the complexes of DNA (recombinant plasmid and Renilla internal reference, phRLTK- luc, Promega, Madison, WI) and transfection solutions were added to cells. After a 6 h incubation, myoblasts were treated with vehicle (PBS), ghrelin (1 μM), cisplatin (50 μM), and cisplatin (50 μM) + ghrelin (1 μM). For transient expression, assay for reporter gene activity was measured 24 h after transfection. The dual-luciferase reporter assay system (Promega) was used for detecting luciferase activity.
Statistical analysis
SPSS 18.00 software for Windows (SPSS Inc., Chicago, IL) was used for all statistical analysis. Parameters are expressed as mean ± standard error of the mean. Statistical comparisons were performed using one-way ANOVA followed by Tukey test adjustments for multiple comparisons. P values of 0.05 or smaller were considered statistically significant. The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle.19
Results
Effect of ghrelin on body weight, muscle mass, and myofiber size changes caused by LLC tumour or cisplatin
Both LLC and cisplatin induced a significant decrease in body weight compared with control animals, whereas ghrelin administration prevented these changes (Figure A and B). Lean body mass was also decreased by LLC, and cisplatin and ghrelin also prevented these changes (for the LLC model, change from baseline for heat-killed + vehicle [HK + V] 95.70 ± 4.30%, tumour + vehicle [T + V] 83.80 ± 1.52%, and tumour + ghrelin [T + G] 96.47 ± 2.04%, P < 0.05; for the cisplatin model, change from baseline for vehicle [V] 100.39 ± 0.41%, cisplatin [C] 83.41 ± 0.78%, cisplatin + ghrelin [C + G] 93.39 ± 0.61%, and ghrelin [G] 110.20 ± 0.92%, P < 0.01) All muscles in the hind leg showed significant atrophy, and this was also prevented by ghrelin (Figure 1C and D). Paralleling the changes in muscle mass, grip strength was significantly decreased by tumour implantation and cisplatin, and these changes were also prevented by ghrelin, highlighting the functional impact of these interventions (Figure 1E and F). Cachexia in these two models was associated with a decrease in daily food intake that was also prevented by ghrelin (for the LLC model, HK + V 3.55 ± 0.13 g/day, T + V 2.74 ± 0.16 g/day, and T + G 3.23 ± 0.08 g/day, P < 0.05; for the cisplatin model, V 3.67 ± 0.05 g/day, C 2.55 ± 0.09 g/day, C + G 3.12 ± 0.05 g/day, and G 4.05 ± 0.06 g/day, P < 0.01), suggesting that one of the mechanisms mediating ghrelin's effects is by decreasing anorexia. LLC implantation and cisplatin administration induced a significant decrease in myocyte cross-sectional area in TA muscles, and this was prevented by ghrelin (see Supporting Information, Figure S1A–D, for LLC model, P < 0.05; for the cisplatin model, P < 0.05). LLC implantation and cisplatin also decreased fat mass significantly as measured by NMR. These changes were prevented by ghrelin (for the LLC model change from baseline for HK + V 85.25 ± 0.75%, T + V 32.80 ± 5.13%, and T + G 57.53 ± 5.01%, P < 0.01; for the cisplatin model change from baseline for V 101.70 ± 1.34%, C 60.92 ± 2.51%, C + G 85.75 ± 0.93%, and G 116.66 ± 1.45%, P < 0.01).
Figure 1.

Ghrelin improves tumour and cisplatin-induced cachexia. (A) Changes in body weight in the Lewis lung carcinoma (LLC)-induced cachexia model. HK-V represents the group injected with heat-killed (HK) LLC and vehicle. T-V (tumour + vehicle) and T-G (tumour + ghrelin) groups represent animals inoculated with LLC (106 cells) receiving vehicle (saline) or ghrelin (0.8 mg/kg twice daily), respectively. The change is expressed as % from baseline. The carcass weight represents the weight after subtracting the tumour mass. (B) Changes in body weight in cisplatin-induced cachexia. V, vehicle-treated group; C, cisplatin-treated group; CG, cisplatin + ghrelin-treated group; G, ghrelin-treated group. The change expressed as % from baseline. (C) Quadriceps, gastrocnemius, soleus, tibialis anterioris (TA), and extensor digitalis longus (EDL) muscle mass normalized to baseline lean body mass (LBM) weight by nuclear magnetic resonance in the LLC-induced cachexia model. (D) Muscle mass in cisplatin-induced cachexia. (E) Grip strength % change from baseline compared to HK-V group in LLC-induced cachexia. (F) Grip strength change in cisplatin-induced cachexia. Unadjusted muscle weight changes in both models (not shown) follow the same pattern as adjusted muscle weight changes shown in Figure 1C and D.
Proteolysis and proteasome activity
Tumour implantation and cisplatin administration were associated with an increase in the expression of the ubiquitin ligases MAFbx/Atrogin-1 and muscle ring finger-1 (MuRF-1) (Figure 2A–D); whereas, the markers of muscle differentiation MyoD and myogenin were decreased by LLC and cisplatin. These changes were prevented by ghrelin suggesting that ghrelin-decreased proteolysis and increased muscle mass are mediated at least in part through these pathways. Also, to confirm the relevance of the changes in atrogin-1/MuRF-1 seen in these models, we measured the proteasome activity directly. As shown in Figure 2E and F, cisplatin and LLC tumour implantation increased proteasome activation, and this was also prevented by ghrelin. The transcription of Atrogin-1 and MuRF-1 is increased in part by dephosphorylation of the transcriptional factor Fox-O1-3, which is in turn regulated by the central mediator Akt. LLC inoculation and cisplatin decreased the phosphorylation of Akt (Figure 2C and D), suggesting that this pathway is involved in LLC and cisplatin-induced muscle atrophy.
Figure 2.
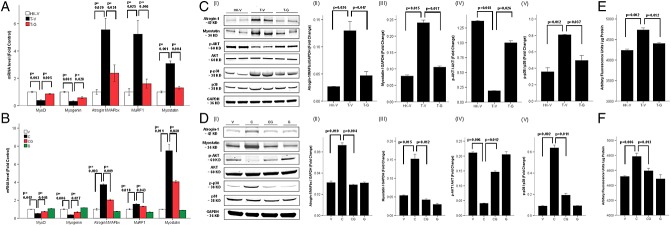
Ghrelin increases protein synthesis markers and decreases proteolytic markers and proteasome activity induced by tumour or cisplatin administration. mRNA levels of MyoD, myogenin, atrogin-1/MAFbx, MuRF-1, and myostatin in Lewis lung carcinoma (LLC)-induced cachexia (A) and in cisplatin-induced cachexia (B). mRNA levels were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and expressed as fold change from controls. Protein levels measured by western blot (I) in LLC-induced cachexia (C) and in cisplatin-induced cachexia (D). Quantification of Atrogin-1/MAFbx (II) and Myostatin (III) was normalized to GAPDH, p-Akt (IV) to total Akt, and p-p38 (V) to total p38. Proteasome activity in LLC-induced cachexia (E) and in cisplatin-induced cachexia (F) expressed as arbitrary fluorescence units/µg protein. HK-V, heat-killed (HK) LLC + vehicle. T-V (tumour + vehicle) and T-G (tumour + ghrelin) groups represent animals inoculated with LLC (106 cells) receiving vehicle (saline) or ghrelin (0.8 mg/kg twice daily), respectively. V, vehicle-treated group; C, cisplatin-treated group; CG, cisplatin + ghrelin-treated group; G, ghrelin-treated group.
Myostatin was up-regulated by LLC inoculation or cisplatin, and these changes were also abolished by ghrelin. Also, the MAP kinase p38 is believed to mediate protein degradation in cancer cachexia by activating atrogin-1 directly and through up-regulation of myostatin.7 Phosphorylated p38 levels were increased by LLC inoculation or cisplatin administration, and this was prevented by ghrelin.
Pro-inflammatory cytokines
Inflammation plays a role in muscle wasting through activation of p38 and down-regulation of Akt among other mechanisms,20,21 and ghrelin has been proposed to have anti-inflammatory effects in non-cancer settings.13,22 Serum levels of the pro-inflammatory cytokines interleukin (IL)-6, tumour necrosis factor (TNF)-α, and IL-1β were significantly increased in tumour-bearing and cisplatin-treated animals, and this was prevented by ghrelin co-administration (see Supporting Information, Figure S2).
Effect of ghrelin on myotube breakdown, protein synthesis, and proteolysis changes induced by cisplatin in vitro
To further characterize the mechanisms mediating the effects of cisplatin and ghrelin in cachexia, C2C12 myotubes were treated with vehicle, cisplatin, ghrelin, or ghrelin + cisplatin. Cisplatin induced a significant decrease in myotube size and myosin heavy chain content, and these changes were prevented by ghrelin (Figure 3A–D). Cisplatin increased the expression of atrogin-1, MuRF-1, p38, and myostatin and decreased the expression of Akt, myoD, and myogenin, and these changes were prevented by ghrelin (Figure 4). Direct measurements of protein synthesis and degradation in myotubes treated with cisplatin or cisplatin + ghrelin followed the same pattern confirming the relevance of these changes (protein synthesis after 24 h measured by l-[3,5-3H]tyrosine incorporation compared to control samples: cisplatin 53.00 ± 2.00%, C + G 75.50 ± 3.50%, ghrelin 114.00 ± 2.00%, P < 0.01; protein degradation after 24 h measured by l-[3,5-3H]tyrosine release compared to control: cisplatin 127.98 ± 2.14%, C + G 102.57 ± 2.86%, ghrelin 76.57 ± 4.52%, P < 0.01).
Figure 3.
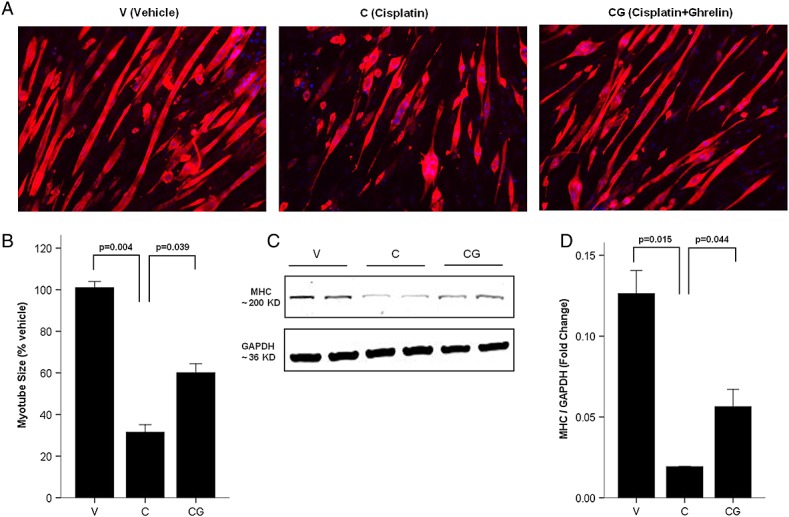
Ghrelin improves cisplatin-induced myotubes breakdown. (A) Immunofluorescence staining for anti-myosin/myosin heavy chain (MHC) antibody in C2C12 myotubes. MHC staining outlines the myotubes (red). 4'6-Diamidino-2-phenylindole was used to stain the nuclei (blue). (B) Myotubes size expressed as % from vehicle. (C–D) Western blot of MHC and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in C2C12 myotubes. V, vehicle-treated group; C, cisplatin-treated group; CG, cisplatin + ghrelin-treated group.
Figure 4.
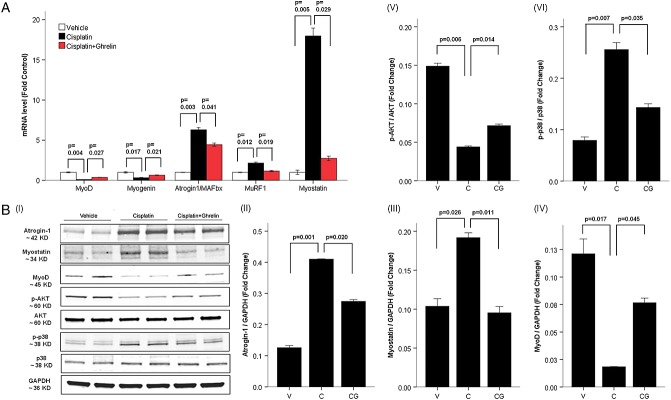
Ghrelin prevents cisplatin-induced changes in protein synthesis and degradation in vitro. (A) mRNA levels for myoD, myogenin, atrogin-1/MAFbx, MuRF-1, and myostatin normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and expressed as fold change form vehicle. (B) Western blot of cisplatin- and ghrelin-treated C2C12 myotubes (I). Atrogin-1/MAFbx (II), Myostatin (III), and MyoD (IV) are normalized to GAPDH, p-Akt (V) to total Akt and p-p38 (VI) to total p38. V, vehicle-treated group; C, cisplatin-treated group; CG, cisplatin + ghrelin-treated group.
Nuclear C/EBP-β and FoxO1/3 were significantly increased by cisplatin and prevented by ghrelin in C2C12 myotubes (Figure 5A). We then confirmed the importance of these pathways on myostatin expression using a construct expressing luciferase alongside the FoxO1/3, SMAD 2/3, and C/EBP-β binding sites in the myostatin promoter. As shown in Figure 5B, cisplatin induced activation of the myostatin promoter, and this was prevented by ghrelin. We also used a different construct with intact or mutated FoxO1/3 and C/EBP-β binding sites in the atrogin-1 promoter to test the relative contribution of these two transcriptional factors to atrogin-1 activity. As shown in Figure 5C, FoxO1/3 and C/EBP-β both significantly contribute to the activation of the atrogin-1 promoter similarly.
Figure 5.

Mechanism of action of ghrelin in C2C12 myotubes. (A) Western blot of transcriptional factor in nuclear extracts. (I) Blot and quantification of C/EBP-β (II) and FoxO1 (III) in nuclear protein, Lamin A was used as the reference. (B) Luciferase assay of myostatin promoter. Map of myostatin promoter (top), which includes the FoxO, drosophila mothers against decapentaplegic protein (SMAD), and C/EBP-β DNA-binding sites and linked to a vector containing luciferase. The relative luciferase activity (bottom) is expressed as fold change from vehicle. (C) Luciferase assay of atrogin-1/MAFbx promoter. Top is the map of atrogin-1/MAFbx promoter, pA includes the C/EBP-β and FoxO DNA-binding sites. pA-C/EBP-β-M includes a mutant C/EBP-β site and a normal FoxO site. pA-FOXO-M includes a mutant FoxO site and a normal C/EBP-β site. The relative luciferase activity expressed as fold change from vehicle (bottom).V, vehicle-treated group; C, cisplatin-treated group; CG, cisplatin + ghrelin-treated group; G, ghrelin-treated group.
Survival
Tumour mass was no different between animals treated with vehicle and ghrelin (tumour mass for T + V 5.87 ± 1.08 g, and for T + G 6.54 ± 1.89 g, P =0.756). Survival was decreased in tumour-bearing animals, and this was significantly improved by ghrelin administration (Figure 6). The cisplatin regimen used was not lethal.
Figure 6.
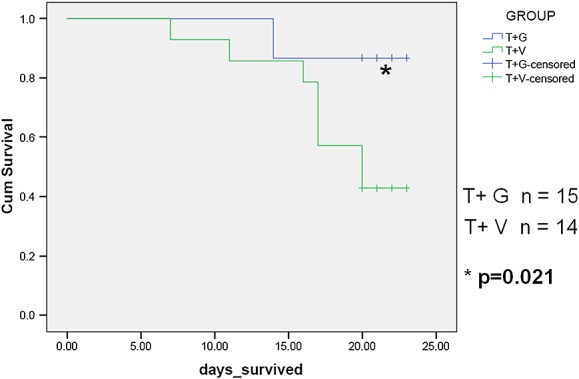
Ghrelin increases survival in Lewis lung carcinoma (LLC)-induced cachexia (Kaplan–Meyer curve). Days survived from when tumours were noted and ghrelin and vehicle injections started (approximately 7 days after LLC cell inoculation). T+V, tumour + vehicle; T+G, tumour + ghrelin.
Discussion
Cachexia is a devastating complication of cancer that contributes to a decrease in quality of life and early demise in individuals with this condition.2 Paradoxically, it is exacerbated by the administration of chemotherapeutic drugs such as cisplatin, and cachexia is often a dose-limiting side effect of these agents. Muscle atrophy contributes the most to a decrease in functionality in cancer patients, and it is associated with an increased risk of chemotherapy-induced toxicity23 and poor outcome. In spite of its significance, the mechanisms underlying cancer- or chemotherapy-induced muscle atrophy are incompletely understood.
The gastric hormone ghrelin causes weight gain by increasing food intake and by food intake-independent mechanisms8,24–26, and ghrelin or ghrelin receptor agonists have been proposed as potential therapies for cancer cachexia as they may improve anorexia, muscle mass and strength, and weight loss in patients with cancer, particularly those receiving cisplatin-based chemotherapy.27,11 However, their mechanisms of action in muscle remain to be fully elucidated.
In this study, tumour-bearing and cisplatin-treated animals developed marked muscle atrophy and weakness that were associated with activation of the ubiquitin-proteasome pathway. Phosphorylated Akt, which is known to down-regulate this pathway through phosphorylation of the transcriptional factor FoxO1-3,28 was decreased by tumour or cisplatin. These changes were prevented by ghrelin administration in both models suggesting that ghrelin's effects are mediated through the ubiquitin-proteasome pathway in these settings. It is noteworthy that LLC cells from different suppliers behave somewhat differently in terms of their capacity in inducing muscle catabolism and activating related signalling pathways. For example, in this study, we used LLC cells from ATCC, which take 28 days to fully develop cachexia and induce MuRF1 up-regulation through Akt-mediated FoxO activation. However, LLC cells from National Cancer Institute (NCI) require only half of the time period to develop cachexia, yet, without activating FoxOs and up-regulating MuRF1.29 It is also important to notice that the relative contribution of this pathway to cancer cachexia in humans is not well understood with some30 but not all studies31 showing an increase in protein degradation through this pathway.
The inflammatory cytokine IL-6 is increased in the setting of cancer, and it may decrease Akt phosphorylation through activation of the suppressor of cytokine signalling-3.21 Other inflammatory cytokines such as IL-1β and TNF-α may play an important role in inducing cachexia by activating nuclear factor kappa-B and MAP kinases including p38.32 Ghrelin has been shown to down-regulate inflammation in the setting of LPS-induced sepsis and chronic kidney disease.13 Whether this is also true in the setting of cancer is not known. Here, we show that tumour implantation and cisplatin administration increased circulating cytokine levels, and these changes were prevented by ghrelin co-administration. These results suggest that the effects of ghrelin in this setting may be mediated through its anti-inflammatory effects, at least in part.
The MAP kinase p38 has been shown to play a role in the development of cachexia and to be activated by inflammation.32–36 However, its specific role is controversial with reports of p38 increasing in the setting of cancer cachexia29 or decreasing in the setting of dexamethasone-induced muscle wasting.16 Moreover, ghrelin was shown to decrease p38 activation in C2C12 cells.37 We found the phosphorylated levels of p38 to increase with tumour or cisplatin administration and that ghrelin prevented these changes. The increase in p38 activation we describe is consistent with previous tumour models, and the down-regulation of this pathway with ghrelin suggests that its effects in the setting of muscle wasting may depend in part on the model used, down-regulating it in models where inflammation plays a role (i.e. cancer-induced cachexia) but not in other non-inflammatory models of wasting (i.e. dexamethasone-induced muscle atrophy).
Muscle wasting in cancer cachexia also has been associated recently with up-regulation of myostatin. Myostatin, a member of the Transforming growth factor beta (TGF-β) family made in muscle, is known to activate the activin receptor IIB and to reduce Akt signalling.38,39 Moreover, targeting this pathway has been shown to ameliorate cachexia.17 In our study, myostatin was significantly up-regulated by tumour implantation and by cisplatin, and this was prevented by ghrelin administration. This is in contrast to a recent report of ghrelin not preventing the increase in myostatin induced by dexamethasone.16 Given that p38 mediates its effects through C/EBP-β and that C/EBP-β activation, in turn, induces the expression of myostatin,40 we then tested the relevance of this pathway by using different constructs. Cisplatin induced an increase in nuclear C/EBP-β and myostatin in C2C12 cells confirming activation of the p38/C/EBP-β/myostatin pathway in this setting. Moreover, ghrelin prevented these changes. Taken together, these data suggest that ghrelin may prevent myostatin activation through inactivation of p38 and C/EBP-β. This could potentially explain why ghrelin previously failed to decrease the expression of myostatin induced by dexamethasone, a model where stimulation of p38 was not seen.
The myogenic regulatory factors MyoD and myogenin have been shown to play an important role in tumour-induced cachexia by regulating muscle regeneration and are regulated by Akt phosphorylation, p38, myostatin and TNF-α.41,42 Their expression levels decreased in both models suggesting that satellite cell proliferation and muscle cell differentiation and regeneration may be impaired. These changes were also prevented by ghrelin administration in vivo in both models. This points out to a potential new mechanism for ghrelin to ameliorate cachexia.43
We also developed a model of cisplatin toxicity in C2C12 cells to determine if the effects of cisplatin were due to a direct effect on muscle cells. Administration of cisplatin to C2C12 cells induced myotube atrophy, and this was associated with activation of p38, myostatin, and the ubiquitin ligases atrogin-1 and MuRF-1 along with down-regulation of Akt and the regenerative pathway (MyoD and myogenin) as we had demonstrated in vivo. This suggests that cisplatin exerts at least part of its effects directly on muscle and is in agreement with a previous report of cisplatin inducing myotube atrophy through Akt down-regulation.44 The effects of cisplatin treatment on C2C12 cells also may represent disruption of the process of differentiation, and it has been suggested before that ghrelin may restore this process.37 Ghrelin increased food intake, and this is thought to mediate some but not all of its effects in this setting as we8 and others45 have recently shown. These in vitro studies suggest that ghrelin exerts its effects at least in part directly on muscle cells, independently of its orexigenic effects as suggested by these previous studies.
The only identified receptor for ghrelin to this date is the GHSR-1a, and this receptor is not expressed in muscle tissue or C2C12 cells.46,16 Nevertheless, recent reports suggest that some of ghrelin's effects are not mediated through this receptor.47,16 Our in vitro studies show that ghrelin may prevent atrophy induced by cisplatin in the absence of GHSR-1a. Very recently, the corticotrophin-releasing factor receptor 2 has been postulated to mediate some of ghrelin's effect on glucose metabolism.48 Future studies should focus on this or other alternative pathways that could explain ghrelin's effects in the absence of GHSR-1a.
Lastly, ghrelin administration lead to increased survival in tumour-bearing animals, indicating that the benefits of improving muscle mass go beyond simply an increase in functionality. This is also relevant given the potential concerns for an anabolic agent like ghrelin in inducing tumour growth and worsening outcomes. Although in vitro studies have given conflicting results with some showing an increase and some showing a decrease in cell proliferation with ghrelin administration,49,50 all in vivo models and human studies where ghrelin or ghrelin mimetics were used have not shown an increase in tumour proliferation, although none of these studies previously reported survival.51,52
In summary, we show here how multiple pathways interact and are involved in the development of cachexia, either induced by a tumour or, paradoxically, by the chemotherapeutic agent cisplatin. Activation of p38/C/EBP-β, myostatin, and inflammatory cytokines, and a decrease in Akt and Myogenin/myoD ultimately lead to increased proteolysis, decreased muscle mass, and strength. The novel hormone ghrelin prevents muscle atrophy by decreasing inflammation, by down-regulating the p38/C/EBP-β/myostatin pathway, and by increasing Akt phosphorylation and activating myogenin and myoD. The increase in muscle strength and survival induced by ghrelin in tumour-bearing animals highlights the clinical relevance of these findings.
Acknowledgments
We thank Dr David Allen (Univ. of Colorado, Boulder) for providing us the myostatin promoter and Dr Pradip Saha for his help with the measurements of protein synthesis and degradation.
Acknowledgement
The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia and Muscle (von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. J Cachexia Sarcopenia Muscle. 2010;1:7-8).
Funding
US Department of Veterans Affairs (Merit grants I01-BX000507 and I01 CX000174, Merit Review Entry Program and a South-central healthcare network career development award); Caroline Wiess Law Fund for Molecular Medicine; National Institutes of Health (AG040583 to J.M.G. and AR063786 to Y.P.L); National Institute of aging (T32AG000183) to B.G.; National Natural Science Foundation of China (81072262 and 81372944) to J.A.C; Vanderbilt MMPC (supported in part by U24 DK59637); Vanderbilt University Medical Center Hormone Assay and Analytical Services Core (supported by National Institutes of Health grants DK059637 and DK020593); University of Virginia (DK076037); and Baylor Diabetes & Endocrinology Research Center (P30 DK079638).
Conflicts of interests
Jose M Garcia receives research support and is a consultant for Aeterna Zentaris, Inc and Helsinn Therapeutics, Inc.
Supporting Information
Supporting Information is available at Journal of Cachexia, Sarcopenia and Muscle online.
Figure S1. Ghrelin prevents the decrease in myofiber size caused by tumour or cisplatin. (A) Immunofluorescence staining for anti-laminin antibody of TA muscles in LLC-induced cachexia. Laminin staining (green) outlines myofibers. Dapi was used to stain the nuclei (blue). (B) Immunofluorescence staining for anti-laminin antibody of TA muscle in cisplatin-induced cachexia. (C) Average cross-sectional area of myofibers in LLC-induced cachexia. (D) Average cross-sectional area of myofibers in cisplatin-induced cachexia. HK-V represents the group injected with heat-killed (HK) LLC and vehicle. T-V and T-G group represent animals inoculated with LLC (106 cells) receiving vehicle (saline) or ghrelin (0.8 mg/kg twice daily), respectively. V, vehicle-treated group, C, cisplatin-treated group; CG, cisplatin + ghrelin-treated group; G, ghrelin-treated group. *P < 0.05, **P < 0.01, compared to HK-V or vehicle group; §P < 0.05, §§P < 0.01 compared to T-V or C group.
Figure S2. Serum IL-6, TNF-α, and IL-1β levels in LLC-induced cachexia (A) and cisplatin-induced cachexia (B). Ghrelin prevents the increase in these pro-inflammatory cytokines induced by tumour- and cisplatin-induced cachexia. * P < 0.05 comparing to HK-V or vehicle group; §P < 0.05 comparing to T-V or C group.
Supporting info item
Supporting info item
Supporting info item
References
- Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol. 2011;12:489–495. doi: 10.1016/S1470-2045(10)70218-7. [DOI] [PubMed] [Google Scholar]
- Utech AE, Tadros EM, Hayes TG, Garcia JM. Predicting survival in cancer patients: the role of cachexia and hormonal, nutritional and inflammatory markers. J Cachexia Sarcopenia Muscle. 2012;3:245–251. doi: 10.1007/s13539-012-0075-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little JP, Phillips SM. Resistance exercise and nutrition to counteract muscle wasting. Appl Physiol Nutr Metab. 2009;34:817–828. doi: 10.1139/H09-093. [DOI] [PubMed] [Google Scholar]
- Loprinzi CL, Kugler JW, Sloan JA, Mailliard JA, Krook JE, Wilwerding MB, et al. Randomized comparison of megestrol acetate versus dexamethasone versus fluoxymesterone for the treatment of cancer anorexia/cachexia. J Clin Oncol. 1999;17:3299–3306. doi: 10.1200/JCO.1999.17.10.3299. [DOI] [PubMed] [Google Scholar]
- Lecker SH, Solomon V, Mitch WE, Goldberg AL. Muscle protein breakdown and the critical role of the ubiquitin-proteasome pathway in normal and disease states. J Nutr. 1999;129:227S–237S. doi: 10.1093/jn/129.1.227S. [DOI] [PubMed] [Google Scholar]
- Cai D, Frantz JD, Tawa NE, Jr, Melendez PA, Oh BC, Lidov HG, et al. IKKbeta/NF-kappaB activation causes severe muscle wasting in mice. Cell. 2004;119:285–298. doi: 10.1016/j.cell.2004.09.027. [DOI] [PubMed] [Google Scholar]
- Fearon KC, Glass DJ, Guttridge DC. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab. 2012;16:153–166. doi: 10.1016/j.cmet.2012.06.011. [DOI] [PubMed] [Google Scholar]
- Garcia JM, Scherer T, Chen JA, Guillory B, Nassif A, Papusha V, et al. Inhibition of cisplatin-induced lipid catabolism and weight loss by ghrelin in male mice. Endocrinology. 2013;154:3118–3129. doi: 10.1210/en.2013-1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisse BE, Frayo RS, Schwartz MW, Cummings DE. Reversal of cancer anorexia by blockade of central melanocortin receptors in rats. Endocrinology. 2001;142:3292–3301. doi: 10.1210/endo.142.8.8324. [DOI] [PubMed] [Google Scholar]
- Garcia JM, Garcia-Touza M, Hijazi RA, Taffet G, Epner D, Mann D, et al. Active ghrelin levels and active to total ghrelin ratio in cancer-induced cachexia. J Clin Endocrinol Metab. 2005;90:2920–2926. doi: 10.1210/jc.2004-1788. [DOI] [PubMed] [Google Scholar]
- Garcia JM, Friend J, Allen S. Therapeutic potential of anamorelin, a novel, oral ghrelin mimetic, in patients with cancer-related cachexia: a multicenter, randomized, double-blind, crossover, pilot study. Support Care Cancer. 2013;21:129–137. doi: 10.1007/s00520-012-1500-1. [DOI] [PubMed] [Google Scholar]
- Murphy MG, Plunkett LM, Gertz BJ, He W, Wittreich J, Polvino WM, et al. MK-677, an orally active growth hormone secretagogue, reverses diet-induced catabolism. J Clin Endocrinol Metab. 1998;83:320–325. doi: 10.1210/jcem.83.2.4551. [DOI] [PubMed] [Google Scholar]
- Dixit VD, Schaffer EM, Pyle RS, Collins GD, Sakthivel SK, Palaniappan R, et al. Ghrelin inhibits leptin- and activation-induced proinflammatory cytokine expression by human monocytes and T cells. J Clin Invest. 2004;114:57–66. doi: 10.1172/JCI21134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia JM, Polvino WJ. Pharmacodynamic hormonal effects of anamorelin, a novel oral ghrelin mimetic and growth hormone secretagogue in healthy volunteers. Growth Horm IGF Res. 2009;19:267–273. doi: 10.1016/j.ghir.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Kos K, Harte AL, O'Hare PJ, Kumar S, McTernan PG. Ghrelin and the differential regulation of des-acyl (DSG) and oct-anoyl ghrelin (OTG) in human adipose tissue (AT) Vol. 70. Clin Endocrinol (Oxf); 2009. pp. 383–389. [DOI] [PubMed] [Google Scholar]
- Porporato PE, Filigheddu N, Reano S, Ferrara M, Angelino E, Gnocchi VF, et al. Acylated and unacylated ghrelin impair skeletal muscle atrophy in mice. J Clin Invest. 2013;123:611–622. doi: 10.1172/JCI39920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell. 2010;142:531–543. doi: 10.1016/j.cell.2010.07.011. [DOI] [PubMed] [Google Scholar]
- Allen DL, Unterman TG. Regulation of myostatin expression and myoblast differentiation by FoxO and SMAD transcription factors. Am J Physiol Cell Physiol. 2007;292:C188–C199. doi: 10.1152/ajpcell.00542.2005. [DOI] [PubMed] [Google Scholar]
- von Haehling S, Morley JE, Coats AJ, Anker SD. Ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. J Cachexia Sarcopenia Muscle. 2010;1:7–8. doi: 10.1007/s13539-010-0003-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul PK, Gupta SK, Bhatnagar S, Panguluri SK, Darnay BG, Choi Y, et al. Targeted ablation of TRAF6 inhibits skeletal muscle wasting in mice. J Cell Biol. 2010;191:1395–1411. doi: 10.1083/jcb.201006098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Du J, Hu Z, Han G, Delafontaine P, Garcia G, et al. IL-6 and serum amyloid A synergy mediates angiotensin II-induced muscle wasting. J Am Soc Nephrol. 2009;20:604–612. doi: 10.1681/ASN.2008060628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deboer MD, Zhu X, Levasseur PR, Inui A, Hu Z, Han G, et al. Ghrelin treatment of chronic kidney disease: improvements in lean body mass and cytokine profile. Endocrinology. 2008;149:827–835. doi: 10.1210/en.2007-1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado CM, Baracos VE, McCargar LJ, Mourtzakis M, Mulder KE, Reiman T, et al. Body composition as an independent determinant of 5-fluorouracil-based chemotherapy toxicity. Clin Cancer Res. 2007;13:3264–3268. doi: 10.1158/1078-0432.CCR-06-3067. [DOI] [PubMed] [Google Scholar]
- Tschop M, Smiley DL, Heiman ML. Ghrelin induces adiposity in rodents. Nature. 2000;407:908–913. doi: 10.1038/35038090. [DOI] [PubMed] [Google Scholar]
- Granado M, Priego T, Martin AI, Villanua MA, Lopez-Calderon A. Ghrelin receptor agonist GHRP-2 prevents arthritis-induced increase in E3 ubiquitin-ligating enzymes MuRF1 and MAFbx gene expression in skeletal muscle. Am J Physiol Endocrinol Metab. 2005;289:E1007–E1014. doi: 10.1152/ajpendo.00109.2005. [DOI] [PubMed] [Google Scholar]
- Sugiyama M, Yamaki A, Furuya M, Inomata N, Minamitake Y, Ohsuye K, et al. Ghrelin improves body weight loss and skeletal muscle catabolism associated with angiotensin II-induced cachexia in mice. Regul Pept. 2012;178:21–28. doi: 10.1016/j.regpep.2012.06.003. [DOI] [PubMed] [Google Scholar]
- Hiura Y, Takiguchi S, Yamamoto K, Kurokawa Y, Yamasaki M, Nakajima K, et al. Fall in plasma ghrelin concentrations after cisplatin-based chemotherapy in esophageal cancer patients. Int J Clin Oncol. 2012;17:316–323. doi: 10.1007/s10147-011-0289-0. [DOI] [PubMed] [Google Scholar]
- Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. 2004;117:399–412. doi: 10.1016/s0092-8674(04)00400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Jin B, Li YP. C/EBPbeta mediates tumour-induced ubiquitin ligase atrogin1/MAFbx upregulation and muscle wasting. EMBO J. 2011;30:4323–4335. doi: 10.1038/emboj.2011.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YS, Ye ZY, Qian ZY, Xu XD, Hu JF. Expression of TRAF6 and ubiquitin mRNA in skeletal muscle of gastric cancer patients. J Exp Clin Cancer Res. 2012;31:81. doi: 10.1186/1756-9966-31-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens NA, Gallagher IJ, Rooyackers O, Skipworth RJ, Tan BH, Marstrand T, et al. Using transcriptomics to identify and validate novel biomarkers of human skeletal muscle cancer cachexia. Genome medicine. 2010;2:1. doi: 10.1186/gm122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YP, Chen Y, John J, Moylan J, Jin B, Mann DL, et al. TNF-alpha acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J. 2005;19:362–370. doi: 10.1096/fj.04-2364com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavel S, Siffroi-Fernandez S, Coldefy AS, Boulukos K, Pisani DF, Derijard B. Regulation of the intracellular localization of Foxo3a by stress-activated protein kinase signaling pathways in skeletal muscle cells. Mol Cell Biol. 2010;30:470–480. doi: 10.1128/MCB.00666-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadrado A, Nebreda AR. Mechanisms and functions of p38 MAPK signalling. Biochem J. 2010;429:403–417. doi: 10.1042/BJ20100323. [DOI] [PubMed] [Google Scholar]
- McClung JM, Judge AR, Powers SK, Yan Z. p38 MAPK links oxidative stress to autophagy-related gene expression in cachectic muscle wasting. Am J Physiol Cell Physiol. 2010;298:C542–C549. doi: 10.1152/ajpcell.00192.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Won KJ, Lee HM, Hwang BY, Bae YM, Choi WS, et al. p38 MAPK participates in muscle-specific ring finger 1-mediated atrophy in cast-immobilized rat gastrocnemius muscle. The Korean Journal of Physiology & Pharmacology: Official Journal of the Korean Physiological Society and the Korean Society of Pharmacology. 2009;13:491–496. doi: 10.4196/kjpp.2009.13.6.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filigheddu N, Gnocchi VF, Coscia M, Cappelli M, Porporato PE, Taulli R, et al. Ghrelin and des-acyl ghrelin promote differentiation and fusion of C2C12 skeletal muscle cells. Mol Biol Cell. 2007;18:986–994. doi: 10.1091/mbc.E06-05-0402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morissette MR, Cook SA, Buranasombati C, Rosenberg MA, Rosenzweig A. Myostatin inhibits IGF-I-induced myotube hypertrophy through Akt. Am J Physiol Cell Physiol. 2009;297:C1124–C1132. doi: 10.1152/ajpcell.00043.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trendelenburg AU, Meyer A, Rohner D, Boyle J, Hatakeyama S, Glass DJ. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am J Physiol Cell Physiol. 2009;296:C1258–C1270. doi: 10.1152/ajpcell.00105.2009. [DOI] [PubMed] [Google Scholar]
- Allen DL, Cleary AS, Hanson AM, Lindsay SF, Reed JM. CCAAT/enhancer binding protein-delta expression is increased in fast skeletal muscle by food deprivation and regulates myostatin transcription in vitro. Am J Physiol Regul Integr Comp Physiol. 2010;299:R1592–R1601. doi: 10.1152/ajpregu.00247.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layne MD, Farmer SR. Tumor necrosis factor-alpha and basic fibroblast growth factor differentially inhibit the insulin-like growth factor-I induced expression of myogenin in C2C12 myoblasts. Exp Cell Res. 1999;249:177–187. doi: 10.1006/excr.1999.4465. [DOI] [PubMed] [Google Scholar]
- Penna F, Costamagna D, Fanzani A, Bonelli G, Baccino FM, Costelli P. Muscle wasting and impaired myogenesis in tumor bearing mice are prevented by ERK inhibition. PLoS One. 2010;5:e13604. doi: 10.1371/journal.pone.0013604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Zhao L, Mulholland MW. Ghrelin stimulates myocyte development. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2007;20:659–664. doi: 10.1159/000107549. [DOI] [PubMed] [Google Scholar]
- Fanzani A, Zanola A, Rovetta F, Rossi S, Aleo MF. Cisplatin triggers atrophy of skeletal C2C12 myotubes via impairment of Akt signalling pathway and subsequent increment activity of proteasome and autophagy systems. Toxicol Appl Pharmacol. 2011;250:312–321. doi: 10.1016/j.taap.2010.11.003. [DOI] [PubMed] [Google Scholar]
- Sakai H, Sagara A, Arakawa K, Sugiyama R, Hirosaki A, Takase K, et al. Mechanisms of cisplatin-induced muscle atrophy. Toxicol Appl Pharmacol. 2014;278:190–199. doi: 10.1016/j.taap.2014.05.001. [DOI] [PubMed] [Google Scholar]
- Sun Y, Garcia JM, Smith RG. Ghrelin and growth hormone secretagogue receptor expression in mice during aging. Endocrinology. 2007;148:1323–1329. doi: 10.1210/en.2006-0782. [DOI] [PubMed] [Google Scholar]
- Sheriff S, Kadeer N, Joshi R, Friend LA, James JH, Balasubramaniam A. Des-acyl ghrelin exhibits pro-anabolic and anti-catabolic effects on C2C12 myotubes exposed to cytokines and reduces burn-induced muscle proteolysis in rats. Mol Cell Endocrinol. 2012;351:286–295. doi: 10.1016/j.mce.2011.12.021. [DOI] [PubMed] [Google Scholar]
- Gershon E, Vale WW. CRF type 2 receptors mediate the metabolic effects of ghrelin in C2C12 cells. Obesity (Silver Spring) 2014;22:380–389. doi: 10.1002/oby.20535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassoni P, Papotti M, Catapano F, Ghe C, Deghenghi R, Ghigo E, et al. Specific binding sites for synthetic growth hormone secretagogues in non-tumoral and neoplastic human thyroid tissue. J Endocrinol. 2000;165:139–146. doi: 10.1677/joe.0.1650139. [DOI] [PubMed] [Google Scholar]
- Cassoni P, Papotti M, Ghe C, Catapano F, Sapino A, Graziani A, et al. Identification, characterization, and biological activity of specific receptors for natural (ghrelin) and synthetic growth hormone secretagogues and analogs in human breast carcinomas and cell lines. J Clin Endocrinol Metab. 2001;86:1738–1745. doi: 10.1210/jcem.86.4.7402. [DOI] [PubMed] [Google Scholar]
- Hanada T, Toshinai K, Kajimura N, Nara-Ashizawa N, Tsukada T, Hayashi Y, et al. Anti-cachectic effect of ghrelin in nude mice bearing human melanoma cells. Biochem Biophys Res Commun. 2003;301:275–279. doi: 10.1016/s0006-291x(02)03028-0. [DOI] [PubMed] [Google Scholar]
- Wang W, Andersson M, Iresjo BM, Lonnroth C, Lundholm K. Effects of ghrelin on anorexia in tumor-bearing mice with eicosanoid-related cachexia. Int J Oncol. 2006;28:1393–1400. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting info item
Supporting info item
Supporting info item