

Abstract
Protein phosphorylation is a central mechanism in vertebrates for the regulation of signaling. With regard to the cardiovascular system, phosphorylation of myocyte targets is critical for the regulation of excitation contraction coupling, metabolism, intracellular calcium regulation, mitochondrial activity, transcriptional regulation, and cytoskeletal dynamics. In fact, pathways that tune protein kinase signaling have been a mainstay for cardiovascular therapies for the past 60 years. The calcium/calmodulin-dependent protein kinase II (CaMKII) is a multifunctional serine/threonine kinase with numerous roles in human physiology. Dysfunction in CaMKII-based signaling has been linked with a host of cardiovascular phenotypes including heart failure and arrhythmia, and CaMKII levels are elevated in human and animal disease models of heart disease. While nearly a decade has been invested in targeting CaMKII for the treatment of heart failure and arrhythmia phenotypes, to date, approaches to target the molecule for antiarrhythmic benefit have been unsuccessful for reasons that are still not entirely clear, although (1) lack of compound specificity and (2) the multitude of downstream targets are likely contributing factors. This review will provide an update on current pathways regulated by CaMKII with the goal of illustrating potential upstream regulatory mechanisms and downstream targets that may be modulated for the prevention of cardiac electrical defects. While the review will cover multiple aspects of CaMKII dysfunction in cardiovascular disease, we have given special attention to the potential of CaMKII-associated late Na+ current as a novel therapeutic target for cardiac arrhythmia.
Fundamental aspects of CaMKII structure/function
Calcium/calmodulin-dependent kinase II (CaMKII) is a multi-functional serine/threonine kinase with broad substrate specificity and tissue distribution. Every metazoan cell studied to date contains at least one of the four main CaMKII isoforms produced by four different genes: alpha, beta, delta, and gamma (α, β, δ, and γ). The predominant isoform in the heart is delta, with a secondary expression of gamma. Alternative splicing produces further diversity in CaMKII function and/or localization. For example, the CaMKIIδB splice variant contains a nuclear localization sequence that may regulate subcellular localization, although the precise mechanism is unclear [1–3]. There is high homology across CaMKII isoforms and the CaMKII monomer is comprised of an N-terminal catalytic domain, a regulatory domain, and a C-terminal association domain (Fig. 1) [4]. The catalytic domain is responsible for enzymatic activity of the kinase and in the baseline state is autoinhibited through interaction with the regulatory domain. The regulatory domain contains the Ca2+/calmodulin binding pocket, as well as numerous regulatory sites that confer unique activation states in response to autophosphorylation (Thr287—residue numbers correspond to location in CaMKIIδ), oxidation (Met281/282), and O-linked glycosylation (Ser280) [5–7] (Fig. 1). The association domain is responsible for assembly of the dodecameric holoenzyme. Binding of Ca2+/calmodulin to a specialized binding region in the kinase regulatory domain leads to displacement of the auto-inhibitory region from the catalytic domain, which confers the primary active state but also exposes the various regulatory sites, facilitating entry into alternative activation modes depending on the environment. CaMKII targets (phosphorylates) a large number of substrates in the cell, including ion channels, pumps, transporters, Ca2+ cycling proteins, and transcription factors (reviewed in Refs. [3,8]). Important and well-studied targets include L-type Ca2+ channels, sarcoplasmic reticulum (SR) Ca2+ release channels (RyR), phospholamban, voltage-gated Na+ channel (Nav1.5), and multiple voltage-gated K+ channels [3,9–12]. More recently, CaMKII has also been associated with the regulation of other channels potentially important for arrhythmias, including ATP-sensitive potassium channels [13–15] and chloride channels [16,17].
Fig. 1.
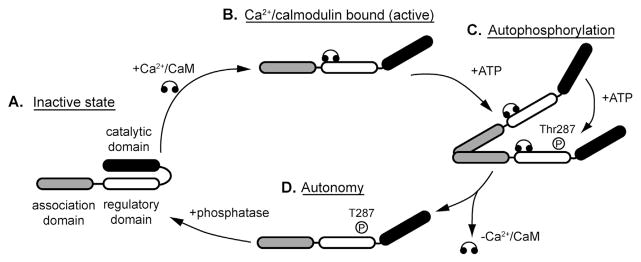
Regulation of CaMKII activity. (A) Under basal (inactive) conditions, the catalytic domain of the CaMKII subunit is autoinhibited through direct interaction with the autoregulatory domain. (B) CaMKII is activated by binding of Ca2+/calmodulin, which exposes the catalytic domain by displacing the autoregulatory domain. (C and D) Ca2+/calmodulin binding also exposes sites in the autoregulatory domain that may be subject to post-translational modification, resulting in alternative activation modes. For example, autophosphorylation of Thr287 by a neighboring active subunit (autophosphorylation) induces a high activity mode subunit that retains activity even upon dissociation of Ca2+/calmodulin (autonomy). Similar autonomy is observed with oxidation at exposed Met281 or Met282 or O-linked glycosylation at Ser280.
CaMKII signaling is highly organized in the myocyte
Similar to other complex cell types such as neurons and epithelial cells, signaling pathways in the cardiomyocyte are compartmentalized to maintain both efficiency and target specificity. In fact, control of CaMKII subcellular localization is a critical task for the cardiomyocyte to maintain normal membrane excitability. Not surprisingly, CaMKII is highly localized to the transverse tubules close to L-type Ca2+ channels (Cav1.2) and SR Ca2+ release channels (RyR2), which are important targets for regulation of calcium-induced Ca2+ release.
Further, select subpopulations of CaMKII are also found at the intercalated disc, mitochondria, and nucleus [3]. An important unresolved issue for the field is the mechanism by which the cell controls temporal and spatial control of CaMKII signaling. While large families of specialized anchoring proteins have been identified for other signaling molecules (e.g., AKAPs and RACKs), no analogous group has been found to date for CaMKII. Instead, CaMKII subcellular localization appears to be determined in a heterogeneous fashion, depending on the target and membrane domain [18]. For example, CaMKII phosphorylates the L-type Ca2+ channel to increase channel open probability and mean channel open time (mode 2 gating), with identified phosphorylation sites in both alpha and beta channel subunits [3,10]. Interestingly, a phosphorylation site on the β1b and β2a subunits resides within a CaMKII-binding motif with high homology to the CaMKII auto-inhibitory region and validated binding site on the NR2B subunit of the NMDA receptor [19]. Direct binding of CaMKII to the beta subunit via this motif is required for rate-dependent facilitation of L-type Ca2+ current [19,20].
More recently, CaMKII was discovered to associate with a motif in the C-terminal region of the actin-associated protein βIV-spectrin with high homology to the binding domain found in the L-type Ca2+ channel βsubunit. Furthermore, the CaM-KII/βIV-spectrin interaction was identified as a requirement for CaMKII targeting and phosphorylation of voltage-gated Na+ channels at the cardiomyocyte intercalated disc [21,22]. An analogous macromolecular complex involving CaMKII, the MAGUK protein SAP97, and the Kv4.3 alpha subunit of transient outward K+ current (Ito) has been proposed for CaMKII-dependent regulation of early repolarization [23]. Thus, CaMKII localization appears to be at least partially dependent on sequences embedded within targets themselves (e.g., β2a subunit), as well as on cytoskeletal/adapter proteins that facilitate phosphorylation of associated targets but may also serve as targets themselves (e.g., βIV-spectrin).
Cardiovascular disease, arrhythmias, and “drugging” of CaMKII
Support for CaMKII as a critical player in the promotion of cardiovascular disease and arrhythmia phenotypes has been growing for nearly 2 decades as dysfunction in CaMKII signaling has been reported in a wide range of cardiovascular disease states. Among the most studied examples is heart failure where increased expression and activity of CaMKII has been reported in animal models and in humans, downstream of a large number of possible stimuli, including Ca2+, reactive oxygen species (ROS), β-adrenergic stimulation, angiotensin II, and aldosterone (Fig. 2) [3]. Consistent with these findings, transgenic CaMKII overexpression in the mouse leads to development of heart failure, while CaMKII deletion prevents onset of heart failure following transaortic constriction [24–26]. Beyond heart failure, dysfunction in CaMKII has now also been reported for both atrial fibrillation and sinus node disease [27]. The mechanisms and targets underlying these pathologies are likely complex [11,28,29] and will require additional investigation. Finally, CaMKII has also been linked with other cardiovascular diseases, including in the setting of diabetes, although the precise pathways are still under active investigation [30–32].
Fig. 2.
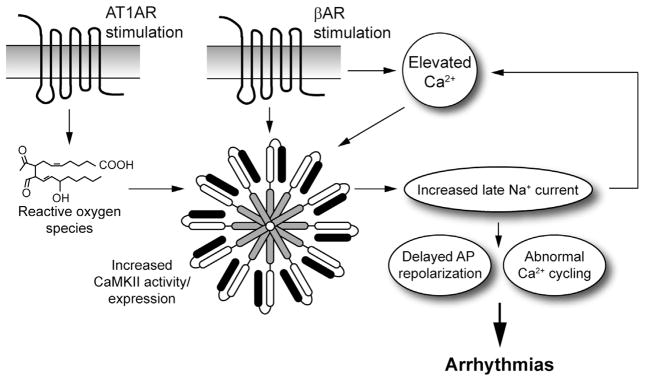
Role for CaMKII and late Na+ current in arrhythmia. CaMKII resides downstream of several second messengers and/or neurohumoral factors relevant for cardiovascular disease, including Ca2+, oxidative stress, beta-adrenergic receptor (βAR), and angiotensin receptor (AT1AR) stimulation (result in defects in Ca2+ and/or reactive oxygen species). Hyperactive CaMKII in turn produces defects in activity of multiple ion channels, pumps, and transporters, including the voltage-gated Na+ channel. Specifically, increased CaMKII activity in disease has been linked to increased inappropriate persistent (“late”) Na+ current that not only promotes arrhythmias by altering cell excitability and Ca2+ handling, but also “feed backs” on CaMKII to exacerbate the signaling defect. Thus, the late Na+ current may serve as a viable alternative therapeutic target to reduce arrhythmia burden in cardiovascular disease patients.
Aside from acquired disease, CaMKII dysregulation contributes to pathology in a number of inherited arrhythmia syndromes, including catecholaminergic polymorphic ventricular tachycardia, long QT type 3, ankyrin-B syndrome (long QT type 4), and Timothy syndrome (long QT type 8) [33–36]. These findings raise the question of when/if will a therapeutic agent be available that specifically targets CaM-KII? Of course, a large and often insurmountable chasm resides between identifying a potential target and introduction of an approved therapeutic agent to the market [37]. So, what is the state of the field with regards to development of a CaMKII drug and what have been the challenges? There are currently a variety of CaMKII inhibitors available for research purposes, including the commonly used KN-93, which lack potency and/or specificity required for a viable therapeutic agent [37]. For example, KN-93 not only blocks CaMKII activation but also has direct effects on several ion channels, including multiple voltage-gated K+ channel family members and the L-type Ca2+ channel. Other peptide inhibitors have been developed that mimic the CaMKII auto-inhibitory region without the Ca2+/calmodulin binding motif (AIP and AC3-I); however, these agents also have important limitations, including specificity and off-target effects related to delivery. Perhaps the most promising of the tool inhibitors is the endogenous inhibitor CaMKIIN and its derivatives (CaMKII-Ntides). CaMKIIN binds to the active kinase in a region (B/C sites) that also may prevent protein–protein interactions involving CaMKII and cytoskeletal/adapter proteins important for targeting [37]. In light of the difficulties and uncertainties associated with “drugging” CaMKII, it may be logical at this juncture to consider additional downstream elements in the CaMKII signaling pathway that may serve as effective therapeutic targets.
Late Na+ current as a novel therapeutic target for cardiac arrhythmia
Precise regulation of voltage-gated Na+ channel (Nav) activity is essential for normal cell membrane excitability. During a normal cardiac action potential, Na+ channels open rapidly to generate the phase 0 AP upstroke. This opening is followed by almost instantaneous inactivation of INa, allowing for a delicate balance of voltage-gated Ca2+ current and delayed rectifier K+ currents to define the plateau and repolarization phases. While voltage-dependent inactivation rapidly turns off the Na+ current, a small persistent (late) component is apparent even under normal conditions. Increased late current is characteristic of cardiomyocytes from failing hearts, where elevated CaMKII activity is also a common finding (Fig. 2) [38–40]. CaMKII phosphorylates voltage-gated Na+ channels to regulate INa gating, with reported effects on steady-state inactivation, recovery from inactivation, and magnitude of this late component [12,21,41]. Studies in heterologous cells and primary myocytes have shown an increase in inappropriate late Na+ current with CaMKII activation. Mechanistically, several potential sites for CaMKII phosphorylation have been identified in the DI–DII linker of Nav1.5 [21,34,42,43]. Nav1.5 Ser571 was first identified as a potential phosphorylation site for CaMKII through functional screening in heterologous cells of a library of mutants created by ablating putative CaMKII sites in the intracellular regions of Nav1.5 [21]. Studies using a Nav1.5 pS571-specific antibody showed increased CaMKII-dependent phosphorylation of this site in disease [34]. A subsequent study using a phosphorylation assay followed by mass spectrometry identified additional sites, including Ser516 and Thr594, which may also be important for CaMKII in the myocyte [42]. It will be important in the future to evaluate these mutants in parallel using in vivo models to define their relative and potentially integrative roles. Finally, an unbiased mass spectrometry approach identified 11 potential sites, including Ser571, as targets for CaMKII phosphorylation [43]. Additional studies will be required to sort out the specific roles of these sites in vivo. Regardless, agents that selectively block late Na+ current (e.g., ranolazine approved as an antianginal medication) have demonstrated antiarrhythmic potential across species and preparations [44].
While CaMKII regulation is now widely considered to be central for the modulation of Nav1.5 function, recent data support that all cardiac Nav1.5 channels may not be identically targeted by the kinase. Work over the past 5 years demonstrates not only that there are multiple membrane populations of Nav1.5 in the cardiac myocyte, but also that these populations are differentially regulated and have unique biophysical properties for myocyte function [45]. To date, three defined populations of Nav1.5 channels have been identified, each with their own select group of targe ting, scaffolding, and regulatory proteins. For example, the intercalated disc is the primary site of myocyte Nav1.5 populations, where it is targeted, retained, and regulated by ankyrin-G, βIV spectrin, and CaMKII, as described above. Human SCN5A variants that block ankyrin-G/Nav1.5 targeting alter Nav1.5 trafficking, resulting in reduced INa and Brugada syndrome arrhythmia phenotypes [46]. More recently, work by Makara et al. [22] showed that mice selectively lacking ankyrin-G expression in the heart display defects in Nav1.5, βIV spectrin, and CaMKII intercalated disc expression as well as defects in CaMKII regulation of Nav1.5-dependent late current. Notably, loss of ankyrin-G did not alter sarcolemmal membrane Nav1.5 channels [22].
Interestingly, the second population of Nav1.5 at the peripheral sarcolemma is targeted and retained by a unique cellular pathway dependent on alpha1-syntrophin [47,48], a gene product previously linked with congenital long QT syndrome [49]. More specifically, Nav1.5 associates via its C-terminal (S-I-V motif) with the PDZ domain of alpha1-syntrophin [48]. Recent work by Hughes et al. showed that mice harboring mutant alpha1-syntrophin lacking the C-terminal motif (ΔSIV) showed altered lateral membrane targeting and reduced INa [48]. Notably, in line with the above findings from ankyrin-G knockout mice, intercalated disc Nav1.5 targeting is retained in the alpha1-syntrophin ΔSIV mouse line [48]. Thus, two unique pathways are utilized for Nav1.5 targeting and regulation in the same cell. Based on the role of CaMKII in the regulation of Nav1.5-dependent late current, it will be critical in the future to define if the ankyrin-G-based pathway may be tuned to modulate late Na+ current, while protecting critical upstroke and repolarization. Finally, it remains to be determined whether precise molecular information about how CaMKII regulates Nav1.5 will be useful in designing new therapeutic strategies for preventing arrhythmia and/or maladaptive remodeling in cardiovascular disease patients.
Mathematical modeling as a tool to define CaMKII roles in cardiac excitability
Mathematical modeling has been very useful in trying to understand a number of issues related to CaMKII signaling [50,51]. Early models demonstrated a potential role for CaMKII in rate-dependent regulation of cell membrane excitability and calcium handling [52,53]. Subsequent theoretical studies have been critical in shaping our understanding of how CaMKII hyperactivity promotes dysfunction in disease [20,34,54–57] and the complex cross talk with other signaling pathways important for disease (protein kinase A) [58]. More recently, elegant modeling work has demonstrated the positive feedback loop between CaMKII and the often pro-arrhythmogenic late Na+ current, with CaMKII causing an increase in late current, which in turn further activates CaMKII through elevations in Na+ and Ca2+ [59]. In the future, a major challenge for modeling relates to the deleterious effects of chronic CaMKII activation involving changes in gene transcription, apoptosis, and/or metabolic remodeling. For example, recent combined experimental and modeling work has demonstrated the importance of CaMKII-mediated cell loss in sinus node dysfunction in the setting of heart failure and diabetes [60,61]. It will be important for future efforts to account for both acute and chronic CaMKII effects in disease. Furthermore, modeling will be instrumental in our efforts to understand local control of CaMKII signaling and implications in disease [62].
Conclusion
CaMKII resides at the center of a vast signaling network with major implications for human health and disease. The kinase targets a large number of substrates important for Ca2+ cycling, cell excitability, and cell function and is responsive to multiple cues relevant for disease, including Ca2+, reactive oxygen species, and neurohumoral factors. While an impressive collection of experimental inhibitor tools have been developed to study CaMKII function, targeting CaMKII for therapeutic benefit has not yet proved successful. As noted above, CaMKII is ubiquitously expressed in humans. Therefore, CaMKII-based therapies to treat cardiac arrhythmia must balance therapeutic benefit versus potential off-target effects on key noncardiac pathways (e.g., neuronal or metabolic). Thus, in parallel with efforts to discover new compounds and optimize existing ones, it is important to consider downstream/upstream nodes in the CaMKII pathway that may serve as alternative targets, particularly pathways that are specific to cardiac myocytes. In fact, it may be beneficial to design therapies against select CaMKII targets that are expressed in specific cellular subpopulations (e.g., mitochondria and intercalated disc). Among the most promising of these candidates is the late Na+ current that is upregulated by CaMKII commonly in disease. It is anticipated that a greater understanding of this pathway may yield important advances in the overall effort to develop new and improved therapies for arrhythmia patients.
Acknowledgments
This work was funded by NIH, USA Grants HL114893 (T.J.H.), HL084583, HL083422, and HL114383 (P.J.M.), the James S. McDonell Foundation (T.J.H.), and the American Heart Association, USA (P.J.M.).
Footnotes
The authors have indicated there are no conflicts of interest.
References
- 1.Srinivasan M, Edman CF, Schulman H. Alternative splicing introduces a nuclear localization signal that targets multi-functional CaM kinase to the nucleus. J Cell Biol. 1994;126:839–52. doi: 10.1083/jcb.126.4.839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mishra S, Gray CB, Miyamoto S, Bers DM, Brown JH. Location matters: clarifying the concept of nuclear and cytosolic CaMKII subtypes. Circ Res. 2011;109:1354–62. doi: 10.1161/CIRCRESAHA.111.248401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swaminathan PD, Purohit A, Hund TJ, Anderson ME. Calmodulin-dependent protein kinase II: linking heart failure and arrhythmias. Circ Res. 2012;110:1661–77. doi: 10.1161/CIRCRESAHA.111.243956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hoelz A, Nairn AC, Kuriyan J. Crystal structure of a tetradecameric assembly of the association domain of Ca2+/calmodulin-dependent kinase II. Mol Cell. 2003;11:1241–51. doi: 10.1016/s1097-2765(03)00171-0. [DOI] [PubMed] [Google Scholar]
- 5.Hanson PI, Meyer T, Stryer L, Schulman H. Dual role of calmodulin in autophosphorylation of multifunctional CaM kinase may underlie decoding of calcium signals. Neuron. 1994;12:943–56. doi: 10.1016/0896-6273(94)90306-9. [DOI] [PubMed] [Google Scholar]
- 6.Erickson JR, Joiner ML, Guan X, Kutschke W, Yang J, Oddis CV, et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. 2008;133:462–74. doi: 10.1016/j.cell.2008.02.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, et al. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature. 2013;502:372–6. doi: 10.1038/nature12537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bers DM, Grandi E. Calcium/calmodulin-dependent kinase II regulation of cardiac ion channels. J Cardiovasc Pharmacol. 2009;54:180–7. doi: 10.1097/FJC.0b013e3181a25078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mustroph J, Maier LS, Wagner S. CaMKII regulation of cardiac K channels. Front Pharmacol. 2014;5:20. doi: 10.3389/fphar.2014.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bers DM, Morotti S. Ca2+ current facilitation is CaMKII-dependent and has arrhythmogenic consequences. Front Pharmacol. 2014;5:144. doi: 10.3389/fphar.2014.00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li N, Chiang DY, Wang S, Wang Q, Sun L, Voigt N, et al. Ryanodine receptor- mediated calcium leak drives progressive development of an atrial fibrillation substrate in a transgenic mouse model. Circulation. 2014;129:1276–85. doi: 10.1161/CIRCULATIONAHA.113.006611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, et al. Ca/calmodulin-dependent protein kinase II regulates cardiac Na channels. J Clin Invest. 2006;116:3127–38. doi: 10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kline CF, Wright PJ, Koval OM, Zmuda EJ, Johnson BL, Anderson ME, et al. betaIV-Spectrin and CaMKII facilitate Kir6. 2 regulation in pancreatic beta cells. Proc Natl Acad Sci U S A. 2013;110:17576–81. doi: 10.1073/pnas.1314195110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sierra A, Zhu Z, Sapay N, Sharotri V, Kline CF, Luczak ED, et al. Regulation of cardiac ATP-sensitive potassium channel surface expression by calcium/calmodulin-dependent protein kinase II. J Biol Chem. 2013;288:1568–81. doi: 10.1074/jbc.M112.429548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J, Kline CF, Hund TJ, Anderson ME, Mohler PJ. Ankyrin-B regulates Kir6. 2 membrane expression and function in heart. J Biol Chem. 2010;285:28723–30. doi: 10.1074/jbc.M110.147868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sellers ZM, De Arcangelis V, Xiang Y, Best PM. Cardiomyocytes with disrupted CFTR function require CaMKII and Ca (2+)-activated Cl(−) channel activity to maintain contraction rate. J Physiol. 2010;588:2417–29. doi: 10.1113/jphysiol.2010.188334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duan DY, Liu LL, Bozeat N, Huang ZM, Xiang SY, Wang GL, et al. Functional role of anion channels in cardiac diseases. Acta Pharmacol Sin. 2005;26:265–78. doi: 10.1111/j.1745-7254.2005.00061.x. [DOI] [PubMed] [Google Scholar]
- 18.Anderson ME. Sticky fingers: CaMKII finds a home on another ion channel. Circ Res. 2009;104:712–4. doi: 10.1161/CIRCRESAHA.109.195503. [DOI] [PubMed] [Google Scholar]
- 19.Grueter CE, Abiria SA, Dzhura I, Wu Y, Ham AJ, Mohler PJ, et al. L-type Ca2+ channel facilitation mediated by phosphorylation of the beta subunit by CaMKII. Mol Cell. 2006;23:641–50. doi: 10.1016/j.molcel.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 20.Koval OM, Guan X, Wu Y, Joiner ML, Gao Z, Chen B, et al. CaV1. 2 beta-subunit coordinates CaMKII-triggered cardiomyocyte death and afterdepolarizations. Proc Natl Acad Sci U S A. 2010;107:4996–5000. doi: 10.1073/pnas.0913760107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hund TJ, Koval OM, Li J, Wright PJ, Qian L, Snyder JS, et al. A betaIV spectrin/CaMKII signaling complex is essential for membrane excitability in mice. J Clin Invest. 2010;120:3508–19. doi: 10.1172/JCI43621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Makara MA, Curran J, Little S, Musa H, Polina I, Smith SA, et al. Ankyrin-G coordinates intercalated disc signaling platform to regulate cardiac excitability in vivo. Circ Res. 2014 doi: 10.1161/CIRCRESAHA.115.305154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.El-Haou S, Balse E, Neyroud N, Dilanian G, Gavillet B, Abriel H, et al. Kv4 potassium channels form a tripartite complex with the anchoring protein SAP97 and CaMKII in cardiac myocytes. Circ Res. 2009;104:758–69. doi: 10.1161/CIRCRESAHA.108.191007. [DOI] [PubMed] [Google Scholar]
- 24.Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J, Jr, Bers DM, et al. The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res. 2003;92:912–9. doi: 10.1161/01.RES.0000069686.31472.C5. [DOI] [PubMed] [Google Scholar]
- 25.Backs J, Backs T, Neef S, Kreusser MM, Lehmann LH, Patrick DM, et al. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc Natl Acad Sci U S A. 2009;106:2342–7. doi: 10.1073/pnas.0813013106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ling H, Zhang T, Pereira L, Means CK, Cheng H, Gu Y, et al. Requirement for Ca2+/calmodulin-dependent kinase II in the transition from pressure overload- induced cardiac hypertrophy to heart failure in mice. J Clin Invest. 2009;119:1230–40. doi: 10.1172/JCI38022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu Y, Anderson ME. CaMKII in sinoatrial node physiology and dysfunction. Front Pharmacol. 2014;5:48. doi: 10.3389/fphar.2014.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Christ T, Rozmaritsa N, Engel A, Berk E, Knaut M, Metzner K, et al. Arrhythmias, elicited by catecholamines and serotonin, vanish in human chronic atrial fibrillation. Proc Natl Acad Sci U S A. 2014;111:11193–8. doi: 10.1073/pnas.1324132111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yaniv Y, Maltsev VA. Numerical Modeling Calcium and CaMKII Effects in the SA Node. Front Pharmacol. 2014;5:58. doi: 10.3389/fphar.2014.00058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Santos GJ, Ferreira SM, Ortis F, Rezende LF, Li C, Naji A, et al. Metabolic memory of ss-cells controls insulin secretion and is mediated by CaMKII. Mol Metab. 2014;3:484–9. doi: 10.1016/j.molmet.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ozcan L, Cristina de Souza J, Harari AA, Backs J, Olson EN, Tabas I. Activation of calcium/calmodulin-dependent protein kinase II in obesity mediates suppression of hepatic insulin signaling. Cell Metab. 2013;18:803–15. doi: 10.1016/j.cmet.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jain SS, Paglialunga S, Vigna C, Ludzki A, Herbst EA, Lally JS, et al. High-fat diet-induced mitochondrial biogenesis is regulated by mitochondrial-derived reactive oxygen species activation of CaMKII. Diabetes. 2014;63:1907–13. doi: 10.2337/db13-0816. [DOI] [PubMed] [Google Scholar]
- 33.DeGrande S, Nixon D, Koval O, Curran JW, Wright P, Wang Q, et al. CaMKII inhibition rescues pro-arrhythmic phenotypes in model of human ankyrin-B syndrome. Heart Rhythm. 2012;9:2034–41. doi: 10.1016/j.hrthm.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koval OM, Snyder JS, Wolf RM, Pavlovicz RE, Glynn P, Curran J, et al. Ca2+/calmodulin-dependent protein kinase II-based regulation of voltage-gated Na+ channel in cardiac disease. Circulation. 2012;126:2084–94. doi: 10.1161/CIRCULATIONAHA.112.105320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu N, Ruan Y, Denegri M, Bachetti T, Li Y, Colombi B, et al. Calmodulin kinase II inhibition prevents arrhythmias in RyR2(R4496C+/−) mice with catecholaminergic polymorphic ventricular tachycardia. J Mol Cell Cardiol. 2011;50:214–22. doi: 10.1016/j.yjmcc.2010.10.001. [DOI] [PubMed] [Google Scholar]
- 36.Thiel WH, Chen B, Hund TJ, Koval OM, Purohit A, Song LS, et al. Proarrhythmic defects in Timothy syndrome require calmodulin kinase II. Circulation. 2008;118:2225–34. doi: 10.1161/CIRCULATIONAHA.108.788067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pellicena P, Schulman H. CaMKII inhibitors: from research tools to therapeutic agents. Front Pharmacol. 2014;5:21. doi: 10.3389/fphar.2014.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Toischer K, Hartmann N, Wagner S, Fischer TH, Herting J, Danner BC, et al. Role of late sodium current as a potential arrhythmogenic mechanism in the progression of pressure-induced heart disease. J Mol Cell Cardiol. 2013;61:111–22. doi: 10.1016/j.yjmcc.2013.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Undrovinas AI, Maltsev VA, Sabbah HN. Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: role of sustained inward current. Cell Mol Life Sci. 1999;55:494–505. doi: 10.1007/s000180050306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maltsev VA, Sabbah HN, Higgins RS, Silverman N, Lesch M, Novel Undrovinas AI. ultraslow inactivating sodium current in human ventricular cardiomyocytes. Circulation. 1998;98:2545–52. doi: 10.1161/01.cir.98.23.2545. [DOI] [PubMed] [Google Scholar]
- 41.Aiba T, Hesketh GG, Liu T, Carlisle R, Villa-Abrille MC, O’Rourke B, et al. Na+ channel regulation by Ca2+/calmodulin and Ca2+/calmodulin-dependent protein kinase II in guinea-pig ventricular myocytes. Cardiovasc Res. 2010;85:454–63. doi: 10.1093/cvr/cvp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ashpole NM, Herren AW, Ginsburg KS, Brogan JD, Johnson DE, Cummins TR, et al. Ca2+/calmodulin-dependent protein kinase II (CaMKII) regulates cardiac sodium channel NaV1. 5 gating by multiple phosphorylation sites. J Biol Chem. 2012;287:19856–69. doi: 10.1074/jbc.M111.322537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marionneau C, Lichti CF, Lindenbaum P, Charpentier F, Nerbonne JM, Townsend RR, et al. Mass spectrometry-based identification of native cardiac Nav1. 5 channel alpha subunit phosphorylation sites. J Proteome Res. 2012;11:5994–6007. doi: 10.1021/pr300702c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Antzelevitch C, Nesterenko V, Shryock JC, Rajamani S, Song Y, Belardinelli L. The role of late INa in development of cardiac arrhythmias. Handb Exp Pharmacol. 2014;221:137–68. doi: 10.1007/978-3-642-41588-3_7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin X, Liu N, Lu J, Zhang J, Anumonwo JM, Isom LL, et al. Subcellular heterogeneity of sodium current properties in adult cardiac ventricular myocytes. Heart Rhythm. 2011;8:1923–30. doi: 10.1016/j.hrthm.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mohler PJ, Rivolta I, Napolitano C, LeMaillet G, Lambert S, Priori SG, et al. Nav1.5 E1053K mutation causing Brugada syndrome blocks binding to ankyrin-G and expression of Nav1. 5 on the surface of cardiomyocytes. Proc Natl Acad Sci U S A. 2004;101:17533–8. doi: 10.1073/pnas.0403711101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gavillet B, Rougier JS, Domenighetti AA, Behar R, Boixel C, Ruchat P, et al. Cardiac sodium channel Nav1. 5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circ Res. 2006;99:407–14. doi: 10.1161/01.RES.0000237466.13252.5e. [DOI] [PubMed] [Google Scholar]
- 48.Shy D, Gillet L, Ogrodnik J, Albesa M, Verkerk AO, Wolswinkel R, et al. PDZ domain-binding motif regulates cardiomyocyte compartment-specific NaV1. 5 channel expression and function. Circulation. 2014;130:147–60. doi: 10.1161/CIRCULATIONAHA.113.007852. [DOI] [PubMed] [Google Scholar]
- 49.Ueda K, Valdivia C, Medeiros-Domingo A, Tester DJ, Vatta M, Farrugia G, et al. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc Natl Acad Sci U S A. 2008;105:9355–60. doi: 10.1073/pnas.0801294105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Greenstein JL, Foteinou PT, Hashambhoy-Ramsay YL, Winslow RL. Modeling CaMKII-mediated regulation of L-type Ca (2+) channels and ryanodine receptors in the heart. Front Pharmacol. 2014;5:60. doi: 10.3389/fphar.2014.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Onal B, Unudurthi SD, Hund TJ. Modeling CaMKII in cardiac physiology: from molecule to tissue. Front Pharmacol. 2014;5:9. doi: 10.3389/fphar.2014.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hund TJ, Rudy Y. Rate dependence and regulation of action potential and calcium transient in a canine cardiac ventricular cell model. Circulation. 2004;110:3168–74. doi: 10.1161/01.CIR.0000147231.69595.D3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Iribe G, Kohl P, Noble D. Modulatory effect of calmodulin-dependent kinase II (CaMKII) on sarcoplasmic reticulum Ca2+ handling and interval-force relations: a modelling study. Philos Transact A Math Phys Eng Sci. 2006;364:1107–33. doi: 10.1098/rsta.2006.1758. [DOI] [PubMed] [Google Scholar]
- 54.Christensen MD, Dun W, Boyden PA, Anderson ME, Mohler PJ, Hund TJ. Oxidized calmodulin kinase II regulates conduction following myocardial infarction: a computational analysis. PLoS Comput Biol. 2009;5:e1000583. doi: 10.1371/journal.pcbi.1000583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hund TJ, Decker KF, Kanter E, Mohler PJ, Boyden PA, Schuessler RB, et al. Role of activated CaMKII in abnormal calcium homeostasis and INa remodeling after myocardial infarction: insights from mathematical modeling. J Mol Cell Cardiol. 2008;45:420–8. doi: 10.1016/j.yjmcc.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hashambhoy YL, Winslow RL, Greenstein JL. CaMKII-induced shift in modal gating explains L-type Ca2+ current facilitation: a modeling study. Biophys J. 2009;96:1770–85. doi: 10.1016/j.bpj.2008.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Koivumaki JT, Korhonen T, Takalo J, Weckstrom M, Tavi P. Regulation of excitation-contraction coupling in mouse cardiac myocytes: integrative analysis with mathematical modelling. BMC Physiol. 2009;9:16. doi: 10.1186/1472-6793-9-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Soltis AR, Saucerman JJ. Synergy between CaMKII substrates and beta-adrenergic signaling in regulation of cardiac myocyte Ca2+ handling. Biophys J. 2010;99:2038–47. doi: 10.1016/j.bpj.2010.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morotti S, Edwards AG, McCulloch AD, Bers DM, Grandi E. A novel computational model of mouse myocyte electro-physiology to assess the synergy between Na+ loading and CaMKII. J Physiol. 2014;592:1181–97. doi: 10.1113/jphysiol.2013.266676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Luo M, Guan X, Di L, Kutschke W, Gao Z, Yang J, et al. Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. J Clin Invest. 2013;123:1262–74. doi: 10.1172/JCI65268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Swaminathan PD, Purohit A, Soni S, Voigt N, Singh MV, Glukhov AV, et al. Oxidized CaMKII causes sinus node dysfunction in mice. J Clin Invest. 2011;121:3277–88. doi: 10.1172/JCI57833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saucerman JJ, Bers DM. Calmodulin mediates differential sensitivity of CaMKII and calcineurin to local Ca2+ in cardiac myocytes. Biophys J. 2008;95:4597–612. doi: 10.1529/biophysj.108.128728. [DOI] [PMC free article] [PubMed] [Google Scholar]