

Abstract
Huntington’s disease (HD) is an autosomal dominant progressive neurodegenerative disorder that prominently affects the basal ganglia, leading to affective, cognitive, behavioral and motor decline. The basis of HD is a CAG repeat expansion to >35 CAG in a gene that codes for a ubiquitous protein known as huntingtin, resulting in an expanded N-terminal polyglutamine tract. The size of the expansion is correlated with disease severity, with increasing CAG accelerating the age of onset. A variety of possibilities have been proposed as to the mechanism by which the mutation causes preferential injury to the basal ganglia. The present chapter provides a basic overview of the genetics and pathology of HD.
I. Introduction
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder, characterized by affective, cognitive, behavioral, and motor dysfunctions (Albin and Tagle, 1995; Bruyn and Went, 1986; Wilson et al., 1987). HD has a prevalence of 5–10 per 100,000 in South America, North America, Australia, and most European countries and countries of European descent, but significantly lower in Africa and Asia, with an estimated prevalence of 0.5:100,000 in Japan and China, and even lower in South Africa (Walker, 2007). HD affects males and females at the same frequency, and the mean age of onset is around 40 although it can be as early as 4 and as late as 80 years of age. Epidemiologic studies show that in US, there are about 30,000 HD patients and that there are about 150,000 people at risk of developing the disease (Margolis and Ross, 2003; Walker, 2007). The primary site of neuron loss in HD is the striatal part of the basal ganglia, with striatal projection neurons being nearly completely lost in advanced HD. Early dysfunction and late loss of cortical neurons is prominent as well. Neuron loss is progressive, and the dysfunction and loss account for the cognitive and motor decline, leading to death typically about 20 years after onset in adults. The basis of HD is a CAG repeat expansion to >35 CAG in a gene that codes for a ubiquitous protein known as huntingtin, resulting in an abnormally long polyglutamine tract in the protein N-terminus (HDCRG, 1993). Many possibilities have been raised as to the means by which mutant huntingtin results in preferential destruction of the striatum and injury to cortex (Reiner et al., 2003). For example, based on the premise that mutant htt injures neurons in a cell autonomous manner, transcriptional dysregulation (Kegel et al., 2002; Luthi-Carter et al., 2002; Ross, 2002), proteosomal dysfunction (Bence et al., 2001; Chai et al., 1999), induction of autophagy (Kegel et al., 2000, Petersén et al., 2001), release of calcium from intracellular stores (Tang et al., 2009), mitochondrial failure (Bossy-Wetzel et al., 2008), induction of apoptosis (Sanchez et al., 1999; Zuccato et al., 2005), and excitotoxicity at extrasynaptic NMDA receptors (Cowan and Raymond, 2006) have been raised as possible mechanisms responsible for striatal and/or cortical neuron death. Additionally, deficient production and transport of BDNF from cortex to striatum (Cattaneo et al., 2005), excessive cortical release of glutamate, and defective glutamate uptake by glia have been invoked as possible pathogenic mechanisms involving an indirect killing action of mutant htt (Behrens et al., 2002; Cepeda et al., 2007; Joshi et al., 2009; Lievens et al., 2001; Rebec et al., 2006). The present review focuses on the genetics and pathology of HD, with comments on pathogenesis as these relate to findings on HD genetics and pathology.
II. The HD Gene
The identification of the HD gene relied strongly on the analyses of a large Venezuelan HD kindred with extremely high HD incidence, due to a high frequency of inbreeding. Using standard linkage analyses, the HD gene was mapped to the tip of the short arm of chromosome 4 in 1983 (Gusella et al., 1983, 1994), but it took scientists another 10 years to isolate it and identify the underlying mutation that causes HD (Fig. 1). In 1993, the HD gene was finally identified by The Huntington’s Disease Collaborative Research Group (HDCRG), comprising 58 researchers from six independent research groups. Using haplotype analysis of linkage disequilibrium in HD families of distinct ethnicities, they identified a small segment of 4p16.3 as the likely location of the mutation. A new gene, IT-15 (interesting transcript 15), isolated using cloned trapped exons from the target area, was shown to contain a polymorphic trinucleotide CAG repeat within the coding region of the gene that was expanded and unstable on one of the chromosomes of all 75 HD families examined (HDCRG, 1993). The HD locus was found to span 180 kb, consisting of 67 exons, and encoding a protein (huntingtin, htt) of ~350 kDa. Homologues of the human gene have been identified in several species, including but not limited to pig (Matsuyama et al., 2000), mouse (Barnes et al., 1994; Lin et al., 1995), pufferfish (Baxendale et al., 1995), zebrafish (Karlovich et al., 1998), and Drosophila (Li et al., 1999), indicating a conserved essential function of huntingtin through evolution.
Fig. 1.

Image A shows the location of the Huntington’s disease gene in band 4p16.3 of chromosome 4 (Adapted from Figure 1 of Gusella et al., 1994). Image B illustrates the huntingtin protein, showing that it contains a polyglutamine region (polyQ) and a proline-rich domain (PRD) at its N-terminus, and 10 HEAT repeats clustered in three domains in the N-terminal half of the protein (Adapted from Figs. 1 and 2 of Harjes and Wanker, 2003). Numbers indicate amino acids. Image C shows a graph depicting the relationship between CAG repeat and Huntington’s disease age of onset. Note the overall significant negative correlation between HD onset and the expanded repeat length (n = 609, r2 = 0.65, p = 0.0001). Nonetheless, the relationship is more complex than this. For example, while there is a strong correlation between CAG repeat and age of onset for adult-onset cases (>20 years) over the 35-55 repeat range, in the case of juvenile (<20 years) onset increasing CAG does not notably advance age of onset highlighted (by dark and pale shading). Moreover, this is also true for the few HD cases found with repeats >200 CAG (not shown in graph). The textured box highlights anomalous adult onset cases with expansion beyond the 60 CAG typically associated with juvenile onset (Adapted from Fig. 4 of Squitieri et al., 2006). (For color version of this figure, the reader is referred to the web version of this book.)
The promoter region of the HD gene has features in common with housekeeping genes that are expressed ubiquitously (multiple G/C rich promoter elements and no TATA box sequence (Coles et al., 1998). The CAG repeat (which encodes polyglutamine) is found within exon 1 of all vertebrate HD homologues. Downstream of the CAG repeat is a stretch of polymorphic CCG (polyproline encoding) repeats, also located within exon 1 (HDCRG, 1993). Although highly conserved across different species, with the exception of HEAT motifs, huntingtin has no homology with other proteins (Andrade and Bork, 1995). The function of huntingtin is currently unknown.
The fact that HD shows autosomal dominant inheritance had long been taken to indicate that the HD mutation acts in a “gain-of-function” manner. Discovery of the HD gene allowed further investigation of this notion, leading to several lines of evidence taken to affirm this view (Sharp and Ross, 1996; Ross, 2002). For example, hemizygous inactivation of the HD gene was found to not cause HD symptoms in humans or mice, despite a reduction in HD gene expression to half of normal (Ambrose et al., 1994; Duyao et al., 1995; Nasir et al., 1995; Zeitlin et al., 1995). Moreover, nullizygous mutant mice were found to die in utero (Duyao et al., 1995; Nasir et al., 1995; Zeitlin et al., 1995), whereas humans that are homozygous for the HD mutation are born and do not show profound differences from HD heterozygotes in disease onset or progression (Myers et al., 1989; Wexler et al., 1987).
III. Normal CAG Repeat Length
Early studies by different research groups, involving the analyses of the number of CAG repeats in ~1200 HD individuals and 2000 non-HD individuals, established that the CAG tract in the IT15 gene is polymorphic in the general population, with the normal range of repeat numbers varying from 9 to 11 at the low end and 34–37 at the high end (with an average of 17–20), and that repeat lengths longer than 37 are associated with HD (Read, 1993). Subsequent studies involving large cohorts of individuals who carried between 30 and 40 CAG repeats in the IT15 gene further refined this concept and indicated that repeats up to 35 in length do not cause HD, and that repeat lengths between 36 and 39 are associated with reduced penetrance, meaning that, within this range, some individuals develop HD within their lifetime, while others do not (McNeil et al., 1997; Rubinsztein et al., 1996). Late onset HD with as low as 29 or 34 repeats has, however, been reported (Andrich et al., 2008; Kenney et al., 2006).
IV. CAG Repeat Length and Disease Onset and Progression
The picture that eventually emerged from numerous studies is that the number of CAG repeats is inversely correlated with age of onset of the disease (Andrew et èal., 1993; Duyao et al., 1993; Snell et al., 1993; Brinkman et al., 1997). Whereas expansions of 40–50 CAG repeats in the mutant HD allele are usually associated with adult onset, juvenile-onset HD, defined as onset before 20 years of age, is usually associated with expansions above 60 CAG repeats (Fig. 1). Clinical manifestations of the disease also differ depending on the length of the CAG tract. The classical HD presentation—adult-onset with predominant chorea—has an onset of around 40 years of age, and the average repeat length is about 44 (Martin and Gusella, 1986; Kremer et al., 1994; Ross et al., 1997; Margolis and Ross, 2003). In patients displaying the reduced-penetrance repeat lengths (36–38 repeats), HD onset not only occurs late in life (60 years of age and older), but patients may present only mild chorea, and without the cognitive, psychiatric and behavioral abnormalities usually associated with longer repeat tracts (McNeil et al., 1997; Rubinsztein et al., 1996). In contrast, chorea is not a major manifestation of juvenile-onset HD, but rigidity and seizures appear to be the predominant characteristics and are often preceded by abnormal behavior (Nance and Myers, 2001; Ribaï et al., 2007). Note, however, that the rare cases with CAG repeats ranging from 60 to >200 indicate that severity does not increase as prominently with repeat expansion beyond 60 (Fig. 1) (Andresen et al., 2006; Squitieri et al., 2006).
V. CAG Repeat Instability
In the vast majority (>80%) of the hereditary transmissions from HD parents, the expanded repeat is only mildly altered by one or a few CAG repeats, usually decreasing if transmitted maternally, and increasing if transmitted paternally (Bates et al., 1997; Duyao et al., 1993). However, on occasion, paternal transmissions lead to large intergenerational expansions, causing the phenomenon of anticipation, where the age of onset tends to decrease in successive generations (Vonsattel and DiFiglia, 1998). Hence, juvenile-onset HD is associated with paternal transmission in 80–90% of the cases. So far, the longest CAG expansion reported consists of 250 repeats (Nance et al., 1999).
Due to the high rate of meiotic CAG instability during spermatogenesis, normal fathers can also have affected children. Several studies indicate that CAG repeats between 27 and 35 can also be meiotically unstable during paternal transmission, leading to descendents with HD and carrying CAG expansions of 40 or more repeats (Myers, 2004). About 10–15% of all HD cases, in fact, arise from non-affected parents whose repeat lengths fall within the high end of the normal range (Chong et al., 1997; Maat-Kievit et al., 2001; Semaka et al., 2010). Of particular interest, the highest incidence of HD among populations of European descent correlates with the higher frequency of HD alleles bearing 28–35 repeats in these populations compared to populations in either Asia or Africa (Walker, 2007).
The HD CAG repeat is also somatically unstable and undergoes progressive length increases over time. Analyses of tissues from affected individuals showed that repeat mosaicism is present in all tissues, with the greatest levels detected in sperm and in the brain, and in particular in the areas with more pronounced neuropathology (De Rooij et al., 1995; Telenius et al., 1994). Whether this plays a role in pathogenesis is yet uncertain.
VI. Genetic Modifiers of CAG Repeat Instability
Although paternal transmission has been clearly shown to increase CAG instability, other genetic factors are believed to contribute to CAG instability in HD, including cis-acting factors, such as the size of the CAG tract and HD haplotypes, and trans-acting factors.
Several studies have shown that trinucleotide repeats larger than 28 show instability during replication, and that there is a positive correlation between the instability and the size of the repeat, in particular, in the male germline. Hence, the size of CAG tract is itself a determinant of instability (Leeflang et al., 1999; MacDonald et al., 1993; Wheeler et al., 2007). A very interesting finding is that postzygotic mechanisms also may play a role in triplet repeat instability in HD (this was first observed in mouse models for HD, Kovtun et al., 2000, 2004). In any event, in maternal transmissions, the daughters will more often carry contractions of the CAG repeat, while the sons will more often carry expansions. While with paternal transmissions expansions are equally frequent in male and female offspring, the CAG repeat increases in length significantly more in sons than in daughters (Wheeler et al., 2007).
In addition, HD haplotypes also appear to influence CAG instability. In a recent study, Warby and collaborators (Warby et al., 2009) found that, in spite of the large number of single nucleotide polymorphisms (SNPs) in the HD gene, disease-associated SNPs form a cluster of similar haplotypes (termed haplogroup A) found in 95% of disease chromosomes. In addition, they found that the same haplogroup is significantly enriched (>80%) in HD genes with intermediate CAG repeats (27–35 CAGs). This finding supports the hypothesis that some variants may have a predisposition for expansion, and that would explain the origin of disease-associated haplotypes.
The availability of mouse models for HD made it possible also to analyze other potential genetic modifiers of CAG repeat instability by assessing the rate of instability in specific gene knockout backgrounds. For instance, somatic CAG instability of transgenic HD mouse models is drastically reduced in mice lacking either the mismatch repair enzyme MSH2 or the base excision repair enzyme OGG1 (Manley et al., 1999; Kovtun et al., 2007). Although the role of Msh2 in CAG repeat expansion is currently not clear, analyses of mice and cell lines lacking OGG1 provided evidence that OGG1 is responsible for initiating an escalating oxidation-excision cycle that leads to progressive age-dependent expansion of the CAG repeats in post-mitotic neurons in HD, and possibly in other trinucleotide disorders as well (Kovtun et al., 2007). Thus, at least one mechanism of CAG expansion appears to involve oxidative DNA damage and single-strand break repair.
VII. Genetic Modifiers of HD Age-of-Onset
Although the primary factor that determines whether and when a person will develop HD is the length of the expanded CAG tract, the precise manifestations of the disease and their onset are clearly affected by modifiers that include environmental and other genetic factors. While it is commonly recognized that the correlation of repeat size accounts for about 70% of the variation in age of onset (Gusella and MacDonald, 2009), there is high variation in age of onset among patients with repeat lengths <55 (Myers, 2004). Strong substantiation that heritable components account for the remaining variation in age of onset was first provided by the HD-MAPS (Modifiers of Age at onset in Pairs of Sibs) study involving >600 sibling pairs of multiple ethnicities (Djoussé et al., 2003; Li et al., 2003). These studies and their follow-ups provided strong evidence of linkage between chromosome 6q and 4q to age of onset of neurological symptoms (Li et al., 2006). Analyses of HD Venezuelan kindreds, encompassing >15,000 individuals and comprising 4500 sibships, also confirmed the association of several loci with age of onset and identified significant linkage to chromosomes 2p and 6q, among others (Gayán et al., 2008; Wexler et al., 2004). However, in both cases, the genomic regions are large and so far the specific modifier genes have not been identified. Genome-wide studies using densely spaced single-nucleotide-polymorphisms (SNPS) are currently been applied in an expanded version of the HD-MAPS collaboration to identify the modifier genes in these regions (Gusella and MacDonald, 2009).
The search for genetic modifiers among genes that are connected to pathways and processes thought to be involved in HD also led to the identification of additional candidates. GRIK2 (glutamate receptor ionotropic kainate2, also known as GLUR6) was the earliest reported genetic modifier, and multiple studies have shown that a polymorphic TAA trinucleotide repeat in its 3′ untranslated region (3′UTR) is associated with earlier HD onset (Gusella and MacDonald, 2009; MacDonald et al., 1999; Rubinsztein et al., 1997). The mechanism by which different GRIK2 alleles affect onset is still unknown.
Polymorphisms in huntingtin-associated protein 1 (HAP1) and Atg7 (autophagy-related 7 homolog) genes have also been shown to play a role in onset age in HD. By sequencing the HAP1 gene in unaffected populations, six polymorphisms have been identified, including one that substitutes methionine (M441) for threonine (T441) at amino acid 441. Analyses of 980 European HD patients revealed that patients homozygous for the HAP1 M441 genotype (that substitutes threonine by methionine) showed an 8-year delay in the onset. Functional assays demonstrated that human M441-HAP1 interacts with mutant htt more tightly than does human T441-HAP1 and protects against mutant htt-induced toxicity (Metzger et al., 2008). Using the same approach, the same group reported one polymorphism in the Atg7 gene that substitutes alanine for valine (V471A). This polymorphism showed a significant effect and was associated with an earlier disease onset of 4 years. Although the mechanism by which this polymorphism affects age of onset is unknown, it has been hypothesized that the V471A Atg7 has reduced autophagic function (Metzger et al., 2010).
The hypothesis that somatic instability of the HD CAG repeat is itself a modifier of disease age of onset gained support by the finding that somatic instability is a significant predictor of onset age, with larger repeat length gains associated with earlier disease onset (Swami et al., 2009; Veitch et al., 2007). Hence, factors that are involved in the control of repeat instability may also represent potential genetic modifiers for age of onset.
Analyses of animal models for HD also implicate several other genes as potential genetic modifiers of age of onset. For instance, age of onset is significantly earlier and pathology is exacerbated in mouse models of HD lacking either the heat shock protein Hsp70 (Wacker et al., 2009) or the neurotrophin BDNF (Canals et al., 2004), while inhibition of caspase-1 delays both the age of onset of motor symptoms and the occurrence of other behavioral and neuropathological changes (Ona et al., 1999). The role of any of these genetic factors in HD in humans, however, remains to be verified.
VIII. HD: A True Dominant Gain-of-Function Disorder?
HD is one of a group of inherited neurodegenerative disorders, commonly referred to as “trinucleotide repeat disorders,” caused by expansions of trinucleotide repeats in distinct genes. In at least nine of these diseases, including HD, these expansions involve CAG repeats that are present in the coding region of the gene and are translated into polyglutamine stretches. Although the mutant protein of the distinct disorders do not share any homology or sequence similarity, except for the presence of the polyglutamine tract, all of them have similar features (for example, repeat length—onset age correlation, and dominant inheritance) and are likely to possess some similarities in their pathogenic mechanisms. Since neuronal degeneration occurs in different areas of the brain in these different CAG repeat diseases, there clearly are also disease mechanisms specific to each disorder that impart the differential regional vulnerability. The dominant pattern of inheritance of HD strongly indicates that HD, like all other polyglutamine disorders, is caused by a gain-of-function mechanism and that the expanded polyglutamine stretch is responsible for the pathogenesis.
Homozygous HD patients are rare, and there is still controversy over whether homozygosity for the mutation in HD is associated with a more severe phenotype. Most information on homozygosity in HD has come from analyses of probable homozygous offspring within the Venezuelan kindreds (Wexler et al., 1987) or from other sporadic cases in which both parents are affected (Alonso et al., 2002; Dürr et al., 1999; Myers et al., 1989). In all these reports, the age-at-onset appeared similar in homozygotes and heterozygotes, and both progression and severity of the disease were in some cases actually worse in the heterozygotes. Together, these reports led to the conclusion that HD displays complete dominance. However, this conclusion was based on clinical evaluation of eight potential homozygous and only two confirmed cases, and did not take into account differences in CAG tract sizes between siblings, or other possible genetic modifiers. In contrast, a more detailed comparison between a large homozygous patients’ series and their heterozygous counterparts in a multicenter study revealed significant clinical and neuropathological differences between the two groups (Squitieri et al., 2003). In this study, not only the disease progression was more rapid in homozygous patients, but also homozygous patients appeared to have a wider spectrum of neurological symptoms. More recent work involving cell lines derived from heterozygous and homozygous HD patients (Mormone et al., 2006; Squitieri et al., 2010; Varani et al., 2003) and analyses of mouse models for HD (Fossale et al., 2002; Graham et al., 2006; Lin et al., 2001) also support the notion that HD is more severe in homozygosity. Thus, more recent work is consistent with the notion that, like other triplet repeat disorders, HD is not a true dominant disorder, and that gain of function is only one of the facets of this devastating disease.
IX. Expression of Huntingtin in Normal and HD Human brain
Huntingtin mRNA and protein are widely distributed in mammalian brain, and almost no brain region is devoid of huntingtin-containing perikarya—although glial cells typically show only low levels (Bhide et al., 1996; Fusco et al., 1999; Gutekunst et al., 1995; Landwehrmeyer et al., 1995; Li et al., 1993; Sapp et al., 1997; Sharp and Ross, 1996; Strong et al., 1993; Vonsattel and DiFiglia, 1998). Large neuronal perikarya tend to be richer in huntingtin than medium-sized or small neuronal perikarya, and huntingtin-positive neurons are especially abundant in the telencephalon and thalamus, but seemingly sparse in the hypothalamus. Within telencephalon, the highest density of huntingtin-rich neurons is in cerebral cortex, in which pyramidal neurons of layers 3 and 5 are especially rich (Fig. 2), and in hippocampus, in which the pyramidal neurons of CA2–CA3 are labeled intensely for huntingtin. The vast majority of striatal projection neurons are, however, only moderate in huntingtin, but scattered large neurons in striatum and the large neurons of globus pallidus externus, the ventral pallidum, basal nucleus of Meynert, and the globus pallidus internus are rich (Fig. 2) (Bhide et al., 1996; Fusco et al., 1999; Gutekunst et al., 1995; Landwehrmeyer et al., 1995). The disease-producing mutation in the HD gene does not appear to affect its regional expression in brain (Bhide et al., 1996; Gourfinkel-An et al., 1997; Landwehrmeyer et al., 1995; Sapp et al., 1997; Schilling et al., 1995; Vonsattel and DiFiglia, 1998). Thus, while the widespread distribution of huntingtin in brain indicates that it possesses a role in the functioning of many brain neurons, this function is not limited to the brain regions and neurons that are the major target of HD, and huntingtin expression is not obviously selectively impaired in the regions or neuron types most affected by the HD mutation. At the cellular level, huntingtin is found in the cytoplasm of neuronal perikarya, in dendrites, and to seemingly a lesser extent in axons and terminals (Vonsattel and DiFiglia, 1998). Ko et al. (2001) recently suggested, based on studies using antibodies directed against different epitopes of wild-type Htt, that Htt may play diverse roles in cellular function. Presumably as a reflection of this diversity, they found that different epitopes of huntingtin are immunohistochemically detectible in different subcellular compartments, implying differential processing or folding of Htt for its role in the different compartments. Among its functions, huntingtin appears to be a cell membrane-associated scaffolding protein involved in vesicular trafficking (DiFiglia et al., 1995; Qin et al., 2004; Sharp et al., 1995; Velier et al., 1998; Wood et al., 1996). Immunolabeling and immunoprecipitation studies indicate that huntingtin may also be involved in the endosomal-lysosomal protein degradation pathway (DiFiglia et al., 1995; Gutekunst et al., 1995; Sapp et al., 1997; Sharp et al., 1995; Velier et al., 1998; Vonsattel and DiFiglia, 1998; Wood et al., 1996). Nuclear localization of full-length wild-type Htt has also been reported (Atwal et al., 2007; Dorsman et al., 1999; Wilkinson et al., 1999).
Fig. 2.

Immunofluorescence labeling for huntingtin (Ht) in the rat striatum viewed with CLSM. Two low-magnification fields (A, B) and two high-magnification fields (C, D) show that scattered large neurons intensely labeled for huntingtin and numerous medium-sized neurons moderately labeled for huntingtin are present in striatum. Magnification in A is as in B; magnification in C is as in D. Images E and F show immunofluorescence labeling for huntingtin in the lower layers of rat cerebral cortex, at increasingly higher magnification. Both fields show intense labeling of pyramidal neurons in Layer 5 of cortex. All images are from Fusco et al. (1999).
Neuropathological studies suggest that the pathogenic HD gain of function could be the formation of ubiquitinated aggregates of the N-terminal fragment of mutated huntingtin, which is thought to occur due to enhanced cleavage and aggregation of the polyglutamine rich part of the mutant huntingtin N-terminus (DiFiglia et al., 1997; Gutekunst et al., 1999; Li and Li, 1998; Maat-Schieman et al., 1999; Martindale et al., 1998; Sieradzan et al., 1999; Vonsattel, 2008). Aggregates of mutant protein are observed in neocortex, entorhinal cortex, subiculum, hippocampal pydamidal neurons, and striatum, more so in advanced and/or juvenile onset HD (Fig. 3). Aggregates are, however, rare in globus pallidus, substantia nigra, and cerebellum. Some aggregates in HD brain possess an amyloid-like structure, suggesting parallels in aggregate formation with other amyloid-associated diseases such as Alzheimer’s and prion diseases (McGowan et al., 2000). Both cytoplasmic and intranuclear aggregation have been observed in HD brain, the latter termed neuronal intranuclear inclusions, or NIIs (Kuemmerle et al., 1999). While considerable attention has been given to the possibility that these aggregates are themselves pathogenic (Davies et al., 1997; DiFiglia et al., 1997; Kim and Tanzi, 1998; Saudou et al., 1998; Sisodia, 1998), the means by which they might lead to neuronal death remains uncertain (Cha et al., 1998; Hackham et al., 1998a,b; Sisodia, 1998). Mutant huntingtin aggregates may, in part, be pathogenic by their capacity to incorporate and thus sequester vital proteins such as the transcription factor TATA-binding protein (van Roon-Mom et al., 2002). The possibility that the aggregates may, at least in part, act by inactivating both mutant and normal huntingtin has been raised by recent evidence showing that the aggregates which form in HD can sequester normal-length polyglutamine-containing proteins, including Htt and CREB-binding protein, both of which promote BDNF production (Cattaneo et al., 2001; Narain et al., 1999; Nucifora et al., 2001; Ona et al., 1999; Preisinger et al., 1999; Shieh et al., 1998; Tao et al., 1998; Wheeler et al., 2000). Neuropathological studies, however, show that formation of NIIs in HD victims is not prominent in cerebral cortex until advanced stages of HD and is never prominent in striatum (1–4% of neurons) at any stage (DiFiglia et al., 1997; Gutekunst et al., 1999; Kuemmerle et al., 1999; Sapp et al., 1999). In fact, the striatal neurons that do possess NIIs tend to be interneurons, which survive well in HD, rather than projection neurons (Kuemmerle et al., 1999). This brings into question if NIIs are pathogenic. Neuropil aggregates (found in spines, dendrites, and axons) are far more common in HD cortex and striatum than NIIs, and thus may be pathogenic by interfering with neuronal function, particularly corticostriatal communication (DiFiglia et al., 1997; Gutekunst et al., 1999; Kuemmerle et al., 1999; Sapp et al., 1999). Regardless of the motor versus mood symptoms, there is a consistently higher number of aggregates in the superior frontal gyrus than in the motor cortex, suggesting a consistent regional difference in aggregate density that thus does not account for differing symptomatology between cases (van Roon-Mom et al., 2006).
Fig. 3.

Images showing immunolabeling for huntingtin in HD brain, revealing aggregates of mutant huntingtin in neuronal nuclei, termed intranuclear inclusions (NIIs). Image A shows the presence of numerous NIIs in cerebral cortex of juvenile HD victim at low magnification. Images B and C show immunolabeled NIIs in individual cortical pyramidal neurons in the same juvenile HD victim, using Nomarski optics to highlight the NIIs. The nucleolus in each cell is unlabeled. These images are adapted from A-C of Fig. 1 from DiFiglia et al. (1997).
X. HD Brain Pathology and the Vonsattel Grading System
Neuropathological and imaging studies reinforce the view that brain abnormalities in HD develop well before evident symptoms, are progressive, and eventually involve the entire brain to a greater or lesser extent, resulting in about 25% brain weight loss in advanced HD (Halliday et al., 1998; Sharp and Ross, 1996). Nonetheless, the most prominent neuropathology in HD occurs within the striatal part of the basal ganglia, in which gross atrophy is accompanied by extensive neuronal loss and astrogliosis, both of which become more severe as the disease progresses, with the atrophy leading to great enlargement of the lateral ventricles (Fig. 4). At least some of these dying neurons show nuclear fragmentation and marker expression characteristic of apoptotic cell death (Thomas et al., 1995; Vis et al., 2005). Reactive astrocytes are increased in HD striatum and show increased coupling by gap junctions, which may provide increased spatial buffering in an attempt to maintain a beneficial environment for neurons (Vis et al., 1998). Striatal pathology in both caudate and putamen is more prominent caudally than rostrally in early disease, and striatal degeneration proceeds, for unknown reasons, in a dorsomedial to ventrolateral direction (Roos et al., 1985; Vonsattel, 2008). Caudate atrophy as detected by MRI or CT has been shown to be correlated with CAG repeats and with a worsening of the UHDRS motor score (Culjkovic et al., 1999; Jech et al., 2007). Marked neuronal loss and shrinkage is also seen in deep layers of the cerebral cortex. Other regions, including globus pallidus, hippocampus, amygdala, thalamus, subthalamic nucleus, substantia nigra, and cerebellum, show varying degrees of atrophy and/or neuronal loss, depending on disease stage (Rosas et al., 2003). The neuron loss is reflected in regional brain atrophy. For example, late in disease, volumetric losses of the following magnitudes are observed: 20% in cortex, 30% in cerebral white matter, 60% in striatum, 55% in globus pallidus, and 30% in thalamus (de la Monte et al., 1988; Lange et al., 1976; Heinsen et al., 1994). Caudate shrinkage is significant already 10 years from estimated disease onset, while putamen and globus pallidus shrinkage is not significant until 3 years before estimated disease onset (Aylward et al., 1996). Gene expression analysis of caudate, cerebellum, prefrontal association cortex, and primary motor cortex shows the greatest number and magnitude of differentially expressed mRNAs in caudate, followed by motor cortex, then cerebellum, with no detected changes in prefrontal cortex (Hodges et al., 2006). Thus, caudate is most affected in HD, and cerebral cortex is not uniform in its response in HD. Note that caudate volume loss, overall brain volume loss, and white matter disorganization are manifest early in HD, and these HD brain abnormalities precede overt signs of disease (Aylward et al., 1994; Kassubek et al., 2004c; Paulsen et al., 2006; Squitieri et al., 2009; Reading et al., 2005; Rosas et al., 2005).
Fig. 4.
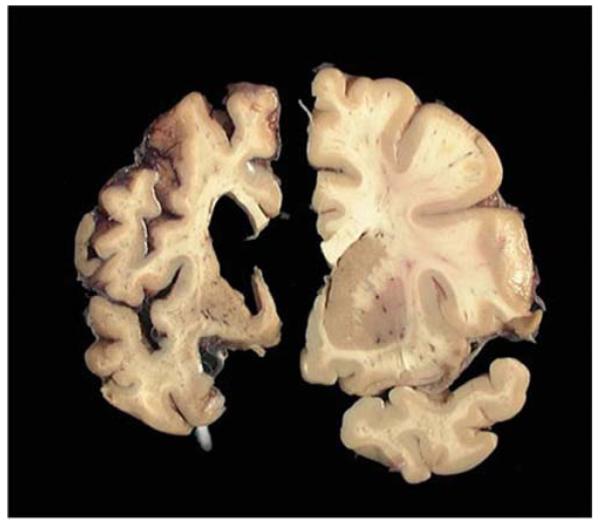
Coronal slices though human telencephalon, showing a normal brain on the right and an advanced HD brain (Grade 4) on the left. Note the profound shrinkage of cortex and caudate and the resulting ventricular expansion in the HD brain. Image courtesy of the Harvard Brain Tissue Resource Center. (For color version of this figure, the reader is referred to the web version of this book.)
A system for grading HD neuropathological severity has been developed based on macroscopic and microscopic criteria related to striatal morphology (Fig. 5) (Vonsattel et al., 1985). This system recognizes five Grades (0–4) designated in the ascending order of severity, with the grades correlating closely with the degree of clinical disability. There are no evident gross, and few microscopic abnormalities in premanifest HD striatum (Grade 0, also termed presymptomatic). The microscopic abnormalities that can be present involve increased abundance of oligodendrocytes and neurons with nuclear aggregates in the tail of caudate, and some neuron loss in head of caudate (Gómez-Tortosa et al., 2001; Vonsattel, 2008). Grade 1 cases have abnormalities that can be detected microscopically in striatum (50% neuron loss in head of caudate) but gross atrophy is not evident, as the ventricular profile of the caudate maintains its normal convex appearance. The Grade 1 changes involve neuron loss and gliosis in the medial paraventricular portions of the caudate, in the tail of the caudate, and in the dorsal part of the putamen. In Grade 2, striatal atrophy is present, but the ventricular profile of the caudate remains convex, but less so than in normal brain. The lateral half of the striatum shows relative preservation in Grades 1–2. In Grade 3, striatal atrophy is more severe, and the ventricular profile of the caudate is flat. In Grade 4, 95% of caudate neurons are lost, striatal atrophy is severe, and the ventricular surface of the caudate is concave. Astrocytes are greatly increased above normal in HD Grades 2–4. This grading system has come to be widely used in neuropathological studies of HD that seek to describe changes as disease progresses.
Fig. 5.

Schematic illustrations of caudate at HD Grades 0 through 4 according to the Vonsattel et al grading scale. Note that the ventricular profile of the caudate is diagnostic for classification, and the extent of caudate neuron loss distinguishes normal from HD, and Grade 0 versus Grade 1 HD. This illustration is adapted from Fig. 2 of Vonsattel et al. (1985).
XI. Basal Ganglia Pathology in HD
The major site of pathology in HD is the basal ganglia, which consists of striatal and pallidal subdivisions. The striatum consists of two major neuron types, projection neurons and interneurons, while globus pallidus consist mainly of projection neurons. We will detail how HD affects these various neuronal populations below. Of note, striatal neuron loss in HD largely involves projection neurons, with most striatal interneuron types highly resistant to HD.
A. Striatum—Projection Neurons
Striatal projection neurons are all GABAergic and can be subdivided into four major types based on their primary projection target: (1) those projecting only or mainly to the external segment of globus pallidus (GPe), which are typically rich in enkephalin (ENK) and poor in or devoid of substance P (SP), and located in the striatal matrix compartment; (2) those projecting mainly to the internal segment of globus pallidus (GPi), which are rich in SP and dynorphin (DYN) but poor in ENK, and located in the striatal matrix compartment; (3) those projecting mainly to the substantia nigra pars reticulata (SNr), which are also rich in SP and DYN, and typically poor in ENK, and located in the striatal matrix compartment; and (4) those projecting to the substantia nigra pars compacta (SNc), which also are rich in SP and DYN, and typically poor in ENK, and largely localized to the striatal patch compartment (Beckstead and Cruz, 1986; Feger and Crossman, 1984; Kawaguchi et al., 1990; Parent et al., 1989, 1995; Reiner and Anderson, 1990; Reiner et al., 1999; Wu et al., 2000). Because these striatal neurons are GABAergic, they all express the enzymes that convert glutamate to GABA, namely the 65kD and 67kD forms of glutamic acid decarboxylase (GAD). The perikarya of striato-GPe neurons and their terminals in GPe are also enriched in D2 dopamine and A2a adenosine receptors (Fink et al., 1992; Le Moine and Bloch, 1995; Schiffmann et al., 1991). In turn, the perikarya of striato-GPi neurons and their terminals in GPi are enriched in D1 dopamine receptors, as are the perikarya of striatonigral neurons and their terminals in substantia nigra (Fink et al., 1992; Le Moine and Bloch, 1995; Schiffmann et al., 1991). All striatal projection neuron perikarya and terminals also possess cannabinoid receptors (Glass et al., 1997; Herkenham et al., 1991; Mailleux and Vanderhaeghen, 1992). These various neurochemical traits provide markers by which the progressive effect of HD on these projection neuron populations can be characterized, either by studying the loss of terminals in the target areas or by studying loss of the perikarya. These four neuronal types play different roles in movement control, and it is thus valuable to characterize how HD affects them to better understand HD pathophysiology. As summarized below, the overall data indicate that while striatal projection neurons as a class are highly vulnerable in HD, and as a result projection neurons markers are lost from striatum as disease progresses (Goto et al., 1989; Seto-Ohshima et al., 1988), projection neuron types do exhibit differences in susceptibility. Notably, striato-GPe and striato-nigral neurons are lost more rapidly in HD than are striato-GPi neurons.
Immunohistochemical studies have indicated that ENK/GAD+ terminals in GPe and SP/GAD+ terminals in the substantia nigra are lost sooner in HD progression than are SP/GAD+ terminals in GPi (Figs. 6–8). For example, depletion of ENK+ immunostaining from GPe has been noted in premanifest HD (Albin et al., 1990b, 1992; Hedreen and Folstein, 1995), and striatal PPE expression appears reduced in premanifest HD (Albin et al., 1991; Augood et al., 1996, 1997). By Grade 1, ENK/GAD+ fibers in GPe are reduced to about 35% of control abundance and SP/GAD+ fibers in SNc and SNr are reduced to about 30% and 50%, respectively, of control abundance (Allen et al., 2009; Deng et al., 2004; Sapp et al., 1995). By contrast, the loss of striatal terminals in GPi is much less in Grade 1, with SP/GAD+ fibers being 70–80% of control abundance (Deng et al., 2004; Sapp et al., 1995). The loss of striato-GPe and striato-nigral projections remains greater than the loss of striato-GPi projections through Grades 2 and 3 (Albin et al., 1990a; Allen et al., 2009; Deng et al., 2004; Reiner et al., 1988; Sapp et al., 1995). For example, in Grade 2, striatal terminals in GPe are at 25% of normal abundance (Deng et al., 2004; Sapp et al., 1995), and in SNc and SNr are at about 35% of normal abundance (Deng et al., 2004). By contrast immunolabeled striatal terminals in GPi are at 60% of their normal abundance (Deng et al., 2004; Sapp et al., 1995). In Grade 3 HD, immunolabeled striatal fibers in GPe, SNc, and SNr are at 20% of normal abundance, but in GPi are at 50% of normal abundance (Deng et al., 2004). By Grade 4 of HD, however, profound loss in all projection systems is apparent (Albin et al., 1990a; Reiner et al., 1988), with striato-GPe and striato-GPi projections at about 5% of normal, and striato-SNc and SNr projections at 10% of normal (Deng et al., 2004). Thus, striato-GPe and striatonigral neurons appear to be lost more rapidly than striato-GPi neurons during HD progression. DTI confirms massive loss of striatal projections in HD, indicating the immunolabeling changes reflect real fiber loss and not just staining loss (Douaud et al., 2009). Direct support for this premise at the perikaryal level has come from in situ hybridization histochemistry for SP and ENK mRNA in HD striatum (Albin et al., 1991; Richfield et al., 1995a, 1995b), and from binding of D1 and D2 dopamine (Glass et al., 2000) and A2a adenosine receptors (Glass et al., 2000) in HD striatum. For example, the loss of the SP+ projection to nigra and the loss of the ENK+ projection to GPe, with the relative preservation of the SP+ projection to GPi, predict that SP+ neuron survival should be better than ENK neuron survival in HD striatum. In fact, neurons expressing mRNA for the SP precursor (i.e., preprotachykinin or PPT) are more abundant in striatum during Grades 1–3 HD than are neurons expressing mRNA for the ENK precursor PPE (Richfield et al., 1995a, 1995b).
Fig. 6.

Images of immunohistochemically labeled sections showing GPi, GPe, and substantia nigra in control, Grade 1 HD, and Grade 3 HD cases, immunostained for SP in the case of GPi and the nigra and for ENK in the case of GPe. In the control, SP+ fibers abound in GPi, ENK+ fibers abound in GPe, and SP+ fibers abound in the nigra. In Grade 1 HD, ENK+ fibers in GPe and SP+ fibers in the nigra are depleted, while SP+ fibers in GPi remain abundant. The contrast is even more evident in the Grade 3 specimen, where ENK+ fibers are markedly depleted in the atrophied GPe and SP+ fibers in the nigra are sparse and patchy, but SP+ fibers in GPi are still quite prominent. This illustration is Fig. 5 from Deng et al. (2004).
Fig. 8.

Low-power images showing GAD+ staining in both GPi and GPe of control, Grade 1 HD, and Grade 3 HD cases. Note the greater loss of GAD+ woolly fibers from GPe than from GPi. This illustration is Fig. 7 from Deng et al. (2004).
These findings for striato-GPe and striato-GPi projections in HD are also compatible with the radioimmunoassay (RIA) study of Seizinger et al. (1986), who reported that dynorphin (DYN), which is co-localized with SP in striatal terminals in GPi (Reiner et al., 1999), was undiminished in GPi in HD victims. By contrast, the PPE-derived neuropeptide MERGL was only half its normal abundance in GPe in the HD brains they studied. Biochemical studies have also shown that GABA and GAD are more greatly decreased in GPe than in GPi in symptomatic HD (Ellison et al., 1987; Spokes, 1980; Storey and Beal, 1993). Since striato-GPi and striato-GPe projection neurons are both GABAergic (Reiner and Anderson, 1990), these results too indicate a preferential loss among striatopallidal neurons of those projecting to GPe. One prior biochemical study has suggested that GABA is diminished in GPe in premanifest HD while GABA in GPi remains normal (Reynolds and Pearson, 1990).
Biochemical studies of SP, DYN, GABA, or GAD also indicate that striatal input to nigra is severely depleted in HD (Buck et al., 1981; Beal et al., 1988; Ellison et al., 1987; Emson et al., 1980; Gale et al., 1977; Kanazawa et al., 1977, 1979; Seizinger et al., 1986; Spokes, 1980; Spokes et al., 1980; Storey and Beal, 1993). Of note, Seizinger et al. (1986) found that DYN in nigra and MERGL in GPe were halved in HD victims, but DYN in GPi was undiminished. The possibility that the striatal projection to SNc is differently affected in HD than that to SNr has been of interest because they arise from different striatal neuron types, and because Hedreen and Folstein (1995) reported that striosomal neurons, whose principal projection target is pars compacta (Gerfen, 1992), are already affected at Grade 0. Judging whether the SP+ fiber loss is greater for SNc than for SNr is difficult, however, because many dopaminergic neurons of SNc in primates are dispersed within the SNr territory, making it ambiguous to precisely define the boundaries of SNc (Arsenault et al., 1988; Hökfelt et al., 1984). Not surprisingly, the available immunolabeling data do not unambiguously support the notion that presymptomatic HD is characterized by loss in the striato-SNc projection but not in the striato-SNr projection (Deng et al., 2004). Similarly, by RIA Beal et al. (1988) observed extensive loss of SP from both SNr and SNc by Grade 1, followed by further loss in subsequent grades, with no clear differences between them at any grade. Other biochemical studies have reported varied results, however, with some observing greater loss of SP or GABA from SNr than SNc (Buck et al., 1981; Ellison et al., 1987; Emson et al., 1980; Kanazawa et al., 1977), and others the opposite (Gale et al., 1977). One study that distinguished HD cases as choreic (early to mid-HD) versus rigid (late HD) reported greater loss of GAD from SNr than SNc in both (Spokes, 1980). Tippett et al. (2007) have reported that preferential striosomal loss (i.e., striato-SNc neuron loss) is not invariably a trait of early HD but does appear associated with mood abnormality when it does occur.
The major findings in HD obtained using neuropeptides or GAD as markers have been confirmed by studies using additional markers of striatal neurons and their terminals. For example, Grade 0 HD has been found to be characterized by loss of cannabinoid, D2 and A2a receptor binding from striatum and by a large increase in GABAA binding in GPe (Glass et al., 2000). These findings are consistent with a preferential loss of ENK+ input to GPe at Grade 0. The absence of reductions in D1 receptor binding in striatum or in GPi at Grade 0 (Glass et al., 2000) suggests that striatal SP+ neurons in general and those projecting to GPi, in particular, are largely unaffected in premanifest HD. The occurrence of reduced D1 receptor binding in SNr at Grade 0 (Glass et al., 2000) and reduced striatal message for D1 receptors and PPT at Grade 0, however, suggest that defects not yet evident at the peptide level or the level of GABA/GAD production are present in presymptomatic HD in striato-SNr projection neurons.
Grade 1 HD is characterized by about 90% loss of striatal D2 dopamine and A2a adenosine receptors (localized to ENK+ neurons), 75% loss of striatal cannabinoid receptors, 50% loss of striatal D1 receptors, near complete depletion of D2 and A2a adenosine receptors from GPe, continued upregulation of GABAA receptor binding in GPe, complete preservation of D1 receptors in GPi, greater preservation of cannabinoid receptors in GPi than GPe or SNr, and 20% loss of D1 receptors from SNr (Allen et al., 2009; Glass et al., 2000; Richfield and Herkenham, 1994; Walker et al., 1984). These findings are consistent with relative preservation of the striato-GPi projection at Grade 1 concomitant with considerable loss in the striato-GPe and striatonigral projections. Grade 2 is characterized by 80–95% loss of striatal cannabinoid, D2 dopamine and A2a adenosine receptors, 50% loss of striatal D1 receptors, near complete depletion of D2 and A2a adenosine receptors from GPe, 66% loss of D1 receptors from GPi, 69% loss of D1 receptors from SNr, and greater preservation of cannabinoid receptors in GPi than GPe (Allen et al., 2009; Glass et al., 2000; Richfield and Herkenham, 1994). These findings are consistent with greater preservation of the striato-GPi than the striato-GPe projection at Grade 2, although the finding by Glass et al. (2000) of comparable preservation of D1 receptors in GPi and SNr at Grade 2 is inconsistent with greater vulnerability of the latter. At Grade 3, striatum and GPe are nearly devoid of cannabinoid, D2 and A2a receptors, but about 30% of striatal D1 receptors remain, and GPi cannabinoid receptor levels still exceed those in GPe (Allen et al., 2009; Glass et al., 2000; Richfield and Herkenham, 1994). Both GPi and SNr are, however, greatly depleted of D1 receptors by Grade 3, and substantial upregulation of GABAA receptors is evident in GPi (Allen et al., 2009; Glass et al., 2000). These findings too are consistent with greater preservation of the striato-GPi projection than the striato-GPe at Grade 3, but with significant loss of input to GPi. The data of Waeber and Palacios (1989) on 5HT-1 receptors in Grade 3 HD pallidum are also consistent with this conclusion. By Grade 4, these various receptor markers, as well as such intracellular signaling markers as calcineurin, are all greatly reduced in striatum and its targets (Goto et al., 1989b; Glass et al., 2000; Richfield and Herkenham, 1994). This is consistent with near total loss in all striatal projection systems by Grade 4, as well as the neuropathological evidence of severe striatal neuron loss by this grade (Vonsattel et al., 1985).
The attributes that make striato-GPi neurons more resistant than striato-GPe and striatonigral neurons is not known, although considerable attention has focused on the role of glutamate receptor subunit configuration, free radical defenses, calcium sequestering, and anti-apoptotic mechanisms (Beal et al., 1991; Calabresi et al., 1998; Chen et al., 1996, 1998; DiFiglia, 1990; Figueredo-Cardenas et al., 1998; Gervais et al., 2002; Hackham et al., 2000; Hedreen and Folstein, 1995; Huang et al., 1995; Medina et al., 1996; Zeron et al., 2002). Regardless, of their basis, the differential loss explains the progression of HD symptoms (Fig. 9). The early loss of striato-GPe and perhaps striato-SNc neurons accounts for the chorea seen commonly in early HD, according to the now standard direct-indirect pathway model of basal ganglia function (Albin et al., 1989; Crossman, 1987; Deng et al., 2004; Hedreen and Folstein, 1995). Given that each type of striatal projection neuron is organized into microzones that interweave with other types within striatum (Flaherty and Graybiel, 1993; Gimenez-Amaya and Graybiel, 1991), the preferential loss of some types of striatal projection neurons in early HD may be why diverse striatal projection neuron markers show patchy loss from striatum (Augood et al., 1996, 1997; Glass et al., 2000; Goto et al., 1989a; Richfield et al., 1991, 1995; Richfield and Herkenham, 1994). Loss of striato-SNr neurons by Grade 1 may cause the saccade abnormalities in early HD since SNr plays a role in saccadic eye movements (Hikosaka, 1989). By Grade 3, considerable loss of striato-GPi neurons appears to occur, and this loss may contribute to the bradykinesia that develops late in HD, while the near complete loss of this projection system by Grade 4 is likely to explain the akinesia in terminal Grade 4 HD (Albin et al., 1989). The functional implications of striato-SNc neuron loss are uncertain, but Tippett et al. (2007) indicate that loss of these neurons is associated with mood abnormalities in HD patients. Although differential loss is evident for the four main striatal projection systems, imaging studies assessing brain volume, glucose metabolism, or receptors on striatal projection neurons or their terminals emphasize that neither the striatum itself nor any striatal projection neuron type is completely normal even in premanifest HD (Antonini et al., 1996; Augood et al., 1996, 1997; Aylward et al., 1994,1996; Glass et al., 2000; Grafton et al., 1992; Kuwert et al., 1993; Weeks et al., 1996).
Fig. 9.

Schematic illustration of the preferential loss of ENK+ striato-GPe neurons compared to SP+ striato-GPi neurons during the progression of HD, and the relation of this differential loss to HD symptoms. In brief, the early loss of striato-GPe neurons, which suppress unwanted movements, explain the early appearance of chorea in HD, while the later loss of the striato-GPi neurons, which promote desired movement, explain the appearance of akinesia as a later symptom. (For color version of this figure, the reader is referred to the web version of this book.)
B. Striatum – Interneurons
Striatal interneurons include (1) very large aspiny cholinergic neurons (Bennett et al., 2000; Kawaguchi et al., 1995); (2) large aspiny neurons that contain GABA and parvalbumin (PARV) (Kawaguchi et al., 1995; Kita et al., 1990); (3) medium-sized aspiny neurons that contain GABA, somatostatin (SS), neuropeptide Y (NPY), and nitric oxide synthase (NOS) (Figueredo-Cardenas et al., 1996a; Kawaguchi et al., 1995); (4) medium-sized aspiny neurons that contain GABA and calretinin (CALR) (Bennett and Bolam, 1993; Cicchetti et al., 2000; Figueredo-Cardenas et al., 1996b; Kawaguchi et al., 1995; Kubota et al., 1993). While the roles of the SS+ and CALR+ interneurons are uncertain, cholinergic and PARV+ interneurons are known to modulate striatal projection neurons (Kawaguchi, 1993; Kawaguchi et al., 1995; Kita et al., 1990; Koos and Tepper, 1999). Cholinergic neurons mediate reward-related (i.e., dopamine-release related) alterations in projection neuron firing, while PARV+ interneurons inhibit striatal projection neurons in a feed-forward manner as part of the process of switching from one movement to the next in a sequence (Berke, 2008; Gage et al., 2010). Cholinergic, SS + , and medium-sized calretinergic striatal interneurons are resistant in HD and survive even late into the disease (Fig. 10) (Albin et al., 1990a; Beal et al., 1986, 1991; Cicchetti and Parent, 1996; Cicchetti et al., 2000; Dawbarn et al., 1985; Ferrante et al., 1985, 1986, 1987a, Ferrante et al., 1987b; Hawker and Lang, 1990; Kowall et al., 1987; Massouh et al., 2008; Norris et al., 1996; Richfield et al., 1995; Sapp et al., 1995). Existing published data, although limited, suggest that PARV+ interneurons may be lost from the striatum as HD progresses (Ferrer et al., 1994; Harrington and Kowall, 1991). This loss may contribute to the worsening motor dysfunction evident as HD progresses. Although SS+ neuron abundance does not decline in HD striatum, expression of NOS and SS in these neurons is progressively diminished (Norris et al., 1996). Similarly, the preservation of cholinergic interneurons in HD striatum is nonetheless accompanied by diminished expression of such cholinergic neuron markers as choline acetyltransferase (Aquilonius et al., 1975; Massouh et al., 2008) and the vesicular acetylcholine transporter (Smith et al., 2006).
Fig. 10.

Camera lucida reconstructions of the distributions of neuropeptide Y-immunoreactive (NPY +) neurons at comparable levels of the basal ganglia, of a normal individual (A), a choreic Grade 3HD case (B), and a rigid Grade 4 HD case (C). Although the number of NPY+ perikarya in putamen is similar, shrinkage of the putamen greatly elevates the packing density of these neurons in Grades 3 and 4 HD. Note also the progressive shrinkage of GPe and GPi in the HD cases. GPe = external globus pallidus; GPi = internal globus pallidus. This illustration is Fig. 2 from Albin et al. (1990a).
C. Globus Pallidus
In HD, significant progressive atrophy occurs in GPe and GPi, with greater atrophy and gliosis in GPe than GPi (Halliday et al., 1998; Douaud et al., 2006; Roos, 1986; Vonsattel, 2008; Vonsattel and DiFiglia, 1998). The atrophy and gliosis are evident by Grade 3, and prominent by Grade 4 (50%). The shrinkage appears to be due to both neuron loss and loss of striatal input (Lange et al., 1976). Of interest, pallidal shrinkage seems more diagnostic of symptom onset than does striatal shrinkage since imaging studies show that striatal shrinkage occurs well before symptoms are manifest, but pallidal shrinkage more immediately precedes symptom appearance (Aylward et al., 1996). The pathophysiological contribution of these pallidal changes is uncertain. In principle, preferential loss of GPe neurons would disinhibit the subthalamic nucleus and contribute to akinesia and possibly rigidity.
XII. Other Telencephalic Areas in HD
A. Cerebral Cortex
Cerebral cortex undergoes cell loss, gliosis, and shrinkage in HD, but less so and more slowly than does striatum (Byers et al., 1983; Cudkowicz and Kowall, 1990; De La Monte et al., 1988; Passani et al., 1997; Selemon et al., 2004; Vonsattel et al., 1985). The loss occurs mainly in Layers 3, 5, and 6, is evident over Grades 2–4, and is prominent in Grade 4 (Sotrel et al., 1991). For example, Hedreen et al. (1991) noted 57% loss in Layer 6 and 71% loss in Layer 5 in Grade 4 HD. The cortical neuron loss appears to involve the pyramidal projection neurons of cerebral cortex, but not interneurons (MacDonald and Halliday, 2002). For example, neither nNOS nor somatostatin mRNA are significantly decreased in the sensorimotor cortex in HD (Norris et al., 1996), indicating survival of this interneuron class in HD cortex. MRI and fMRI studies show that the cortical thinning is related to disease progress and to CAG repeat length (Kassubek et al., 2004b; Jech et al., 2007), and seems to yield loss of input to striatum (Klöppel et al., 2008; Wolf et al., 2008).
Differences in regional neuron loss and thinning in cerebral cortex occur in HD, and have been described by various authors. Primary motor and premotor cortices both consistently show 40–50% pyramidal neuron loss in late HD (MacDonald and Halliday, 2002). On the other hand, Selemon et al. (2004) reported that prefrontal cortex area 9 showed neuron loss but not prefrontal cortex area 46, but both showed shrinkage. Sotrel et al. (1993) reported that surviving pyramidal neurons of Layers 3 and 5 in prefrontal cortex showed dendritic augmentation, reflecting perhaps compensation for the loss of other neurons from those layers. MRI and CT imaging studies show results similar to these, revealing that the sensorimotor, insular, and opercular cortices show the most thinning, while frontal and temporal cortices show relatively less (Douaud et al., 2006; Kassubek et al., 2004b; Mühlau et al., 2007; Rosas et al., 2003), with thinning in these areas manifest even before overt HD motor symptoms and associated with decline in cognitive function as measured by the UHDRS (Rosas et al., 2005). Heterogeneity in HD in motor versus mood symptoms appears in part attributable to regional variation in cortical neuron loss since a significant association between motor dysfunction and neuronal loss in primary motor cortex is seen in HD, as well as between mood disturbance and neuronal loss in anterior cingulate cortex (Thu et al., 2010). Braak and Braak (1992) reported loss of entorhinal cortex neurons in advanced HD, suggesting a basis for memory deficits in late HD.
Functional alterations in neurotransmitter release also occur for neurons of cerebral cortex, and may underlie HD symptoms. For example, a loss of various presynaptic proteins, such as the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) protein, synaptosome-associated protein 25 (SNAP 25), and the vesicle docking and recycling protein rabphilin 3a, occurs in frontal cortex in HD Grades 1–4 (Morton et al., 2001; Smith et al., 2007). These losses are not due to a general loss of synapses in HD cortex (Smith et al., 2007). Similarly, Zucker et al. (2010) showed that Layer 5 motor cortex neurons in HD make less Lin7 homolog b (Lin7b, also known as veli-2 and mals2), which is a scaffold protein implicated in synaptic plasticity and neurite outgrowth. These types of changes could impair synaptic function within cortex and between cortex and striatum. In HD, uptake of glutamate was found to be reduced by 43% in prefrontal cortex, with the defect increasing in severity with CAG repeat expansion; impairment of glutamate uptake may contribute to neuronal dysfunction and pathogenesis in HD (Hassel et al., 2008).
B. Amygdala
The amygdala comprises pallial and subpallial subdivisions. Significant amygdala shrinkage has been reported in HD, based on MRI and CT (Douaud et al., 2006; Rosas et al., 2003), and Kipps et al. (2007) reported declining emotion recognition in others with amygdala volume loss in HD, possibly contributing to HD affective symptoms. Zech et al. (1986) reported that the central nucleus of the subpallial amygdala in one choreiform HD case was markedly shrunken, with considerable attenuation of immunolabeling for VIP, ENK, neurotensin, and NPY.
XIII. Brainstem Areas in HD
A. Thalamus
Thalamus and the subthalamic nucleus undergo shrinkage and cell loss in HD (Byers et al., 1973; Douaud et al., 2006; Mann et al., 1993; Vonsattel et al., 1985). The centre median, for example, shows evident neuronal loss and astrogliosis by Grade 3 (Vonsattel, 2008), and together the centromedian/parafascicular nucleus complex shows about a 25% volume loss and 50% neuron loss in advanced HD (Heinsen et al., 1996), while only 15% neuron and volume loss is seen in mediodorsal nucleus (Heinsen et al., 1999). Up to 25% volume loss is observed in subthalamic nucleus by Grade 4 (Lange et al., 1976). It is uncertain if this reflects neuron loss or loss of GPe input. Ventrobasal thalamus also shows atrophy (Dom et al., 1976). Imaging studies indicate that thalamic nuclei projecting to frontal cortex and/or striatum (dorsomedial, centre median, parafascicular, and ventrobasal) undergo considerable atrophy in HD, and their atrophy is associated with affective symptoms (Kassubek et al., 2004a).
B. Hypothalamus
Many of the nonmotor symptoms of HD, such as weight loss, sleep abnormalities, hypometabolism, and muscle atrophy are unexplained. Given the central role of hypothalamus in these functions, attention has recently focused on the impact of HD on hypothalamus. Studies using voxel-based morphometry of MR images or CT have shown hypothalamic atrophy in early HD patients (Douaud et al., 2006; Kassubek et al., 2004a,b), and significant hypothalamic atrophy (as reflected in ventricular expansion) is evident even 10 years before estimated symptom onset (Soneson et al., 2010). Among specific nuclei, atrophy of the lateral tuberal nucleus (Kremer et al., 1990), reflecting loss of somatostatinergic neurons (Timmers et al., 1996), has been seen in HD. Notably with regard to sleep disorders in HD, a 28% loss and a 27% atrophy of neurons expressing the neuropeptide orexin has been noted in the lateral hypothalamic area of HD patients (Petersén et al., 2005). The lateral hypothalamus contains neuronal populations important for regulation of sleep and wakefulness, and feeding (DiLeone et al., 2003). As loss of orexinergic neurons is associated with narcolepsy and obesity (Kok et al., 2003), their loss in HD is unlikely to be involved in HD-related weight loss but may be contributory to the sleep defects. Given that the somatostatin and orexin cell populations are small, and atrophy of the hypothalamus is prominent in HD, diverse hypothalamic populations are likely to be affected in HD. Since oxytocin and vasopressin neurons were decreased by 45% and 24%, respectively, in advanced HD, it seems likely that paraventricular and supraoptic nucleus loss contributes to HD hypothalamic shrinkage (Gabery et al., 2010). The numbers of NPY neurons (many of which are involved in feeding suppression) is, however, unchanged (Gabery et al., 2010). Detailed characterization of hypothalamic neuropathology in HD, its progression, and its relation to the nonmotor symptoms of HD is still, however, limited.
C. Substantia Nigra
Substantia nigra undergoes cell loss and shrinkage in HD, but less so than does striatum (Byers et al., 1983; Cudkowicz and Kowall, 1990; De La Monte et al., 1988; Sharp and Ross, 1996; Vonsattel et al., 1985). Shrinkage of substantia nigra as detected by CT has also been reported, which could stem from both striatal input loss and nigral neuron death (Douaud et al., 2006). Loss of both SNr GABAergic neurons, and SNc dopaminergic neurons is evident in HD (Vonsattel, 2008), and Oyanagi et al. (1989) have reported 40% loss in both populations. Yohrling et al. (2003) reported that in Grade 4 HD tyrosine hydroxylase expression by dopaminergic neurons, as detected by in situ hybridization, was decreased by 46% per surviving dopaminergic neuron. Moreover, the dopaminergic neurons were 33% smaller than normal. Neuron counts were not, however, performed to assess dopaminergic neuron loss in that study. Consistent with the reduced tyrosine hydroxylase expression, Yohrling et al. (2003) additionally found that tyrosine hydroxylase protein in the nigra was reduced by 32%. They attributed the reduced tyrosine hydroxylase expression to an effect of mutant huntingtin on the tyrosine hydroxylase promoter. As would be predicted from loss of nigral dopaminergic neurons and reduced tyrosine hydroxylase expression, terminals containing tyrosine hydroxylase appear to be reduced in abundance in advanced HD striatum (Ferrante and Kowall, 1987). Dopamine and its metabolite HVA, and VMAT2 have also been reported to be reduced in HD striatum by some authors (Bohnen et al., 1986; Bohnen et al., 2000; Kish et al., 1987; Reynolds and Garrett, 1986). Loss of dopamine input could contribute to akinesia in HD, as it does in Parkinson’s disease.
D. Cerebellum
Volumetric loss and sporadic Purkinje cell loss is evident in HD Grades 3 and 4, notably in juvenile onset victims (Castaigne et al., 1976; Hattori et al., 1984; Jeste et al., 1984; Rodda, 1981; Vonsattel, 2008). Amino acid and neuropeptide neurotransmitter levels, and GABA receptor levels appear to be largely normal in HD cerebellum (Beal et al., 1988; Kish et al., 1983).
E. Brainstem
Significant loss of neurons from diverse brainstem regions and overall brainstem shrinkage has also been reported in HD (Hattori et al., 1984). For example, Koeppen (1989) reported about 30% loss from the midline pontine region controlling saccades, and linked this loss to saccadic defects in HD.
XIV. HD and Neurogenesis
In response to striatal injury, the subventricular zone (SVZ) of the caudate increases the production of progenitor cells that migrate toward the site of the injury where they can differentiate into mature neurons and glia as part of a restorative process (Curtis et al., 2007). Curtis et al. (2003) showed an increase in cell proliferation in the SVZ in HD caudate, progressive with HD grade and CAG repeat, using the cell cycle marker proliferating cell nuclear antigen (PCNA). Proliferating cells were shown to express the neuronal marker beta III-tubulin or the glial cell marker GFAP, demonstrating generation of neurons and glial cells in the SVZ of HD caudate. The SVZ of HD caudate is 2.8-fold thicker than normal at Grade 2/3, with thickness increasing with grade. An increase in glial cells is mainly responsible for the large increase, but neuroblasts and progenitor cells are also increased in abundance (Curtis et al., 2005a,b).
XV. Neuroinflammatory Neuropathology in HD
Microglial activation and the associated neuroinflammation appear to be a prominent pathological feature of HD, evident from early in the disease process (Tai et al., 2007). For example, activated microglia are greatly increased in abundance in HD cortex, striatum, and globus pallidus, and their abundance increases with grade and neuron loss (Pavese et al., 2006; Sapp et al., 2001a). Similarly, the expression of the neuroinflammation mediators, CCL2 and IL-10, is increased specifically in the striatum in HD, presumably in activated microglia, but not in cortex or cerebellum (Silvestroni et al., 2009). By contrast, an upregulation of the neuroinflammation mediators IL-6, IL-8, and MMP9 is seen in cortex and the cerebellum. The activated microglia may be neurotrophic and act to combat the HD pathogenic process, or their sustained activation may exacerbate the HD injury process (Möller, 2010).
Fig. 7.

High-power images showing SP+ fibers in GPi (B, D, F) and ENK+ fibers (A, C, D) in GPe. In the control case, abundant woolly fibers can be seen in both GPi and GPe. In Grade 1, loss of ENK+ fibers in GPe is apparent, while the SP+ fibers in GPi are indistinguishable from that in control. In Grade 3, ENK+ woolly fibers are completely absent, while the SP+ fibers in GPi are relatively preserved, although a decrease in terminal density is apparent. This illustration is Fig. 6 from Deng et al. (2004).
Acknowledgments
Our research on Huntington’s disease has been supported by Cure HD Contracts from the Hereditary Disease and High Q Foundations (AR/ID), the Hereditary Disease Foundation (AR), and NIH grants NS19620 (AR), NS28721 (AR).
References
- Albin RL, Qin Y, Young AB, Penney JB, Chesselet MF. Preproenkephalin messenger RNA-containing neurons in striatum of patients with symptomatic and presymptomatic Huntington’s disease: an in situ hybridization study. Ann. Neurol. 1991;30:542–549. doi: 10.1002/ana.410300406. [DOI] [PubMed] [Google Scholar]
- Albin RL, Reiner A, Anderson KD, Dure LSI, Handelin B, Balfour R, Whetsell WO, Jr., Penney JB, Young AB. Preferential loss of striato-external pallidal projection neurons in presymptomatic Huntington’s disease. Ann. Neurol. 1992;31:425–430. doi: 10.1002/ana.410310412. [DOI] [PubMed] [Google Scholar]
- Albin RL, Reiner A, Anderson KD, Penney JB, Young AB. Striatal and nigral neuron subpopulations in rigid Huntington’s disease: implications for the functional anatomy of chorea and rigidity-akinesia. Ann. Neurol. 1990a;27:357–365. doi: 10.1002/ana.410270403. [DOI] [PubMed] [Google Scholar]
- Albin RL, Tagle DA. Genetics and molecular biology of Huntington’s disease. Trends Neurosci. 1995;18:11–14. doi: 10.1016/0166-2236(95)93943-r. [DOI] [PubMed] [Google Scholar]
- Albin RL, Young AB, Penney JB, Handelin B, Balfour R, Anderson KD, Markel DS, Tourtellotte WW, Reiner A. Abnormalities of striatal projection neurons and N-methyl-D-aspartate receptors in presymptomatic Huntington’s disease. N. Engl. J. Med. 1990b;332:1923–1298. doi: 10.1056/NEJM199005033221807. [DOI] [PubMed] [Google Scholar]
- Albin RL, Young AB, Penney JB. The functional anatomy of basal ganglia disorders. Trends Neurosci. 1989;12:366–375. doi: 10.1016/0166-2236(89)90074-x. [DOI] [PubMed] [Google Scholar]
- Allen KL, Waldvogel HJ, Glass M, Faull RL. Cannabinoid (CB1), GABA(A) and GABA(B) receptor subunit changes in the globus pallidus in Huntington’s disease. J Chem Neuroanat. 2009;37:266–281. doi: 10.1016/j.jchemneu.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Alonso ME, Yescas P, Rasmussen A, Ochoa A, Macías R, Ruiz I, Suástegui R. Homozygosity in Huntington’s disease: new ethical dilemma caused by molecular diagnosis. Clin Gen. 2002;61:437–442. doi: 10.1034/j.1399-0004.2002.610607.x. [DOI] [PubMed] [Google Scholar]
- Andresen JM, Javiar G, Djousse L, Roberts S, Brocklebank D, Cherny SS, The US-Venezuela-Collaborative Research Group. The HD-MAPS Collaborative Research Group. The HD-MAPS Collaborative Research Group. Cardon LR, Gusella JF, MacDonald MF, Myers RH, Houseman DE, Wexler NS. The relationship between CAG repeat length and age of onset differs for Huntington’s disease patients with juvenile onset or adult onset. Ann. Hum. Gen. 2006;71:295–301. doi: 10.1111/j.1469-1809.2006.00335.x. [DOI] [PubMed] [Google Scholar]
- Andrew SE, Goldberg YP, Kremer B, Telenius H, Theilmann J, Adam S, Starr E, Squitieri F, Lin B, Kalchman MA, Graham RK, Hayden MR. The relationship between trinucleotide (CAG) repeat length and the clinical features of Huntington’s disease. Nat. Gen. 1993;4:398–403. doi: 10.1038/ng0893-398. [DOI] [PubMed] [Google Scholar]
- Andrich J, Arning L, Wieczorek S, Kraus PH, Gold R, Saft C. Huntington’s disease as caused by 34 repeats. Movement Dis. 2008;23:879–881. doi: 10.1002/mds.21958. [DOI] [PubMed] [Google Scholar]
- Antonini A, Leenders KL, Spiegel R, Meier D, Vontobel P, Weigell-Weber M, Sanchez-Pernaute R, de Yebenez JG, Boesinger P, Weindl A, Maguire RP. Striatal glucose metabolism and dopamine D2 receptor binding in asymptomatic gene carriers and patients with Huntington’s disease. Brain. 1996;11:2085–2095. doi: 10.1093/brain/119.6.2085. [DOI] [PubMed] [Google Scholar]
- Aquilonius AM, Eckernas SA, Sundwall A. Regional distribution of choline acetyltransferase in the human brain: changes in Huntington’s chorea. J. Neurol. Neurosurg. Psychiatry. 1975;38:669–677. doi: 10.1136/jnnp.38.7.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arsenault MY, Parent A, Seguela P, Descarries L. Distribution and morphological characteristics of dopamine-immunoreactive neurons in the midbrain of the Squirrel monkey (Saimiri sciureus) J. Comp. Neurol. 1988;267:489–506. doi: 10.1002/cne.902670404. [DOI] [PubMed] [Google Scholar]
- Atwal RS, Xia J, Pinchev D, Taylor J, Epand RM, Truant R. Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Hum. Mol. Evol. 2007;16:2600–2615. doi: 10.1093/hmg/ddm217. [DOI] [PubMed] [Google Scholar]
- Augood SJ, Faull RLM, Emson PC. Dopamine D1 and D2 receptor gene expression in the striatum in Huntington’s disease. Ann. Neurol. 1997;42:215–221. doi: 10.1002/ana.410420213. [DOI] [PubMed] [Google Scholar]
- Augood SJ, Faull RLM, Love DR, Emson PC. Reduction in enkephalin and substance P mRNA in the striatum of early grade Huntington’s disease:a detailed cellular in situ hybridization study. Neuroscience. 1996;72:1023–1036. doi: 10.1016/0306-4522(95)00595-1. [DOI] [PubMed] [Google Scholar]
- Aylward EH, Brandt J, Codori AM, Mangus RS, Barta PE, Harris GJ. Reduced basal ganglia volume associated with the gene for Huntington’s disease in asymptomatic at-risk persons. Neurology. 1994;44:823–828. doi: 10.1212/wnl.44.5.823. [DOI] [PubMed] [Google Scholar]
- Aylward EH, Codori AM, Barta PE, Pearlson GD, Harris GJ, Brandt J. Basal ganglia volume and proximity to onset in presymptomatic Huntington disease. Arch. Neurol. 1996;53:1293–1296. doi: 10.1001/archneur.1996.00550120105023. [DOI] [PubMed] [Google Scholar]
- Barnes GT, Duyao MP, Ambrose CM, McNeil S, Persichetti F, Srinidhi J, Gusella JF, MacDonald ME. Mouse Huntington’s disease gene homolog (Hdh) Somat. Cell Mol. Gen. 1994;20:87–97. doi: 10.1007/BF02290678. [DOI] [PubMed] [Google Scholar]
- Bates GP, Mangiarini L, Mahal A, Davies SW. Transgenic models of Huntington’s disease. Hum. Mol. Gen. 1997;6:1633–1637. doi: 10.1093/hmg/6.10.1633. [DOI] [PubMed] [Google Scholar]
- Baxendale S, Abdulla S, Elgar G, Buck D, Berks M, Micklem G, Durbin R, Bates G, Brenner S, Beck S. Comparative sequence analysis of the human and pufferfish Huntington’s disease genes. Nature Gen. 1995;10:67–76. doi: 10.1038/ng0595-67. [DOI] [PubMed] [Google Scholar]
- Beal MF, Ferrante RJ, Swartz KJ, Kowall NW. Chronic quinolinic acid lesions in rats closely resemble Huntington’s disease. J. Neurosci. 1991;11:1649–1659. doi: 10.1523/JNEUROSCI.11-06-01649.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF, Kowall NW, Ellison DW, Mazurek MF, Swartz KJ, Martin JB. Replication of the neurochemical characteristics of Huntington’s disease by quinolinic acid. Nature. 1986;321:168–171. doi: 10.1038/321168a0. [DOI] [PubMed] [Google Scholar]
- Beal MF, Ellison DW, Mazurek MF, Swartz KJ, Malloy JR, Bird ED, Martin JB. A detailed examination of substance P in pathologically graded cases of Huntington’s disease. J. Neurol. Sci. 1988;84:51–61. doi: 10.1016/0022-510x(88)90173-6. [DOI] [PubMed] [Google Scholar]
- Beckstead RM, Cruz CJ. Striatal axons to the globus pallidus, entopeduncular nucleus and substantia nigra come mainly from separate cell populations in cat. Neuroscience. 1986;19:147–158. doi: 10.1016/0306-4522(86)90012-6. [DOI] [PubMed] [Google Scholar]
- Behrens PF, Franz P, Woodman B, Lindenberg KS, Landwehrmeyer GB. Impaired glutamate transport and glutamate-glutamine cycling:downstream effects of the Huntington mutation. Brain. 2002;125:1908–1922. doi: 10.1093/brain/awf180. [DOI] [PubMed] [Google Scholar]
- Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteosome system by protein aggregation. Science. 2001;292:1552–1555. doi: 10.1126/science.292.5521.1552. [DOI] [PubMed] [Google Scholar]
- Bennett BD, Bolam JP. Characterization of calretinin-immunoreactive structures in the striatum of the rat. Brain Res. 1993;609:137–148. doi: 10.1016/0006-8993(93)90866-l. [DOI] [PubMed] [Google Scholar]
- Bennett BD, Callaway JC, Wilson CJ. Intrinsic membrane properties underlying spontaneous tonic firing in neostriatal cholinergic interneurons. J. Neurosci. 2000;20:8493–8503. doi: 10.1523/JNEUROSCI.20-22-08493.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berke JD. Uncoordinated firing rate changes of striatal fast-spiking interneurons during behavioral task performance. J. Neurosci. 2008;40:10075–10080. doi: 10.1523/JNEUROSCI.2192-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhide PG, Day M, Sapp E, Schwarz C, Sheth A, Kim J, Young AB, Penney J, Golden J, Aronin N, DiFiglia M. Expression of normal and mutant huntingtin in the developing brain. J. Neurosci. 1996;17:5523–5535. doi: 10.1523/JNEUROSCI.16-17-05523.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohnen NI, Koeppe RA, Meyer P, Ficaro E, Wernette K, Kilbourn MR, Kuhl DE, Frey KA, Albin RL. Decreased striatal monoaminergic terminals in Huntington disease. Neurology. 2000;54:1753–1759. doi: 10.1212/wnl.54.9.1753. [DOI] [PubMed] [Google Scholar]
- Bossy-Wetzel E, Petrilli A, Knott AB. Mutant huntingtin and mitochondrial dysfunction. Trends Neurosci. 2008;31:609–616. doi: 10.1016/j.tins.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Allocortical involvement in Huntington’s disease. Neuropathol Appl Neurobiol. 1992;18:539–547. doi: 10.1111/j.1365-2990.1992.tb00824.x. [DOI] [PubMed] [Google Scholar]
- Brinkman RR, Mezei MM, Theilmann J, Almqvist E, Hayden MR. The likelihood of being affected with Huntington disease by a particular age, for a specific CAG size. Am J Hum. Gen. 1997;60:1202–1210. [PMC free article] [PubMed] [Google Scholar]
- Bruyn GW, Went LN. Huntington’s chorea. In: Vinken PJ, Bruyn LW, Klawans HL, editors. Handbook of clinical neurology. Vol. 5. Elsevier; Amsterdam: 1986. pp. 267–313. [Google Scholar]
- Buck SH, Burks TF, Brown MR, Yamamura HI. Reduction in basal ganglia and substantia nigra substance P level in Huntington’s disease. Brain Res. 1981;209:464–469. doi: 10.1016/0006-8993(81)90171-2. [DOI] [PubMed] [Google Scholar]
- Byers RK, Gilles FH, Fung C. Huntington’s disease in children. Neuropathological study of four cases. Neurology. 1983;23:561–569. doi: 10.1212/wnl.23.6.561. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Pisani A, Sancesario G, Gubellini P, Marfia GA, Bernardi G. Striatal spiny neurons and cholinergic interneurons express differential ionotropic glutamatergic responses and vulnerability:implications for ischemia and Huntington’s disease. Ann. Neurol. 1998;43:586–597. doi: 10.1002/ana.410430506. [DOI] [PubMed] [Google Scholar]
- Canals JM, Pineda JR, Torres-Peraza JF, Bosch M, Martín-Ibanez R, Munoz MT, Mengod G, Ernfors P, Alberch J. Brain-derived neurotrophic factor regulates the onset and severity of motor dysfunction associated with enkephalinergic neuronal degeneration in Huntington’s disease. J. Neurosci. 2004;24:7727–7739. doi: 10.1523/JNEUROSCI.1197-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaneo E, Zuccato C, Tartari M. Normal Huntingtin function: An alternative approach to Huntington’s disease. Nat Rev Neurosci. 2005;6:919–930. doi: 10.1038/nrn1806. [DOI] [PubMed] [Google Scholar]
- Cattaneo E, Rigamonti D, Goffredo D, Zuccato C, Squitieri F, Sipione S. Loss of normal huntingtin function: new developments in Huntington’s disease research. Trends Neurosci. 2001;24:182–188. doi: 10.1016/s0166-2236(00)01721-5. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Wu N, Andre VM, Cummings DM, Levine MS. The corticostriatal pathway in Huntington’s disease. Prog Neurobiol. 2007;81:253–271. doi: 10.1016/j.pneurobio.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha JHJ, Kosinski CM, Kerner JA, Alsdorf SA, Mangiarini L, Davies SW, Penney JB, Bates GP, Young AB. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human huntington disease gene. J. Neurosci. 1998;19:1189–1202. doi: 10.1073/pnas.95.11.6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai Y, Koppenhafer SL, Shoesmith SJ, Perez MK, Paulson HL. Evidence for proteosome involvement in polyglutamine disease: localization to nuclear inclusions in SCA3/MJD and suppression of polyglutamine aggregation in vitro. Hum. Mol. Gen. 1999;8:673–682. doi: 10.1093/hmg/8.4.673. [DOI] [PubMed] [Google Scholar]
- Chen Q, Veenman CL, Reiner A. Cellular expression of ionotropic glutamate receptor subunits on specific striatal neuron types and its implication for striatal vulnerability in glutamate receptor-mediated excitotoxicity. Neuroscience. 1996;73:715–731. doi: 10.1016/0306-4522(96)00011-5. [DOI] [PubMed] [Google Scholar]
- Chen Q, Veenman L, Knopp K, Yan Z, Medina L, Song WJ, Surmeier DJ, Reiner A. Evidence for the preferential localization of glutamate receptor-1 subunits of AMPA receptors to the dendritic spines of medium spiny neurons in rat striatum. Neuroscience. 1998;83:749–761. doi: 10.1016/s0306-4522(97)00452-1. [DOI] [PubMed] [Google Scholar]
- Chong SS, Almqvist E, Telenius H, LaTray L, Nichol K, Bourdelat-Parks B, Goldberg YP, Haddad BR, Richards F, Sillence D, Greenberg CR, Ives E, Van den Engh G, Hughes MR, Hayden MR. Contribution of DNA sequence and CAG size to mutation frequencies of intermediate alleles for Huntington disease: evidence from single sperm analyses. Hum. Mol. Gen. 1997;6:301–309. doi: 10.1093/hmg/6.2.301. [DOI] [PubMed] [Google Scholar]
- Cicchetti F, Parent A. Striatal interneurons in Huntington’s disease: selective increase in the density of calretinin-immunoreactive medium-sized neurons. Mov Disord. 1996;11:619–626. doi: 10.1002/mds.870110605. [DOI] [PubMed] [Google Scholar]
- Cicchetti F, Prensa L, Wu Y, Parent A. Chemical anatomy of striatal interneurons in normal individuals and in patients with Huntington’s disease. Brain Res. Rev. 2000;34:80–101. doi: 10.1016/s0165-0173(00)00039-4. [DOI] [PubMed] [Google Scholar]
- Coles R, Caswell R, Rubinsztein DC. Functional analysis of the Huntington’s disease (HD) gene promoter. Hum. Mol. Gen. 1998;7:791–800. doi: 10.1093/hmg/7.5.791. [DOI] [PubMed] [Google Scholar]
- Cowan CM, Raymond LA. Selective neuronal degeneration in Huntington’s disease. Curr Top Dev Biol. 2006;75:25–71. doi: 10.1016/S0070-2153(06)75002-5. [DOI] [PubMed] [Google Scholar]
- Crossman AR. Primate models of dyskinesia: the experimental approach to the study of basal ganglia-related involuntary movement disorders. Neuroscience. 1987;21:1–40. doi: 10.1016/0306-4522(87)90322-8. [DOI] [PubMed] [Google Scholar]
- Cudkowicz M, Kowall NS. Degeneration of pyramidal projection neurons in Huntington’s disease cortex. Ann. Neurol. 1990;27:200–204. doi: 10.1002/ana.410270217. [DOI] [PubMed] [Google Scholar]
- Culjkovic B, Stojkovic O, Vojvodic N, Svetel M, Rakic L, Romac S, Kostic V. Correlation between triplet repeat expansion and computed tomography measures of caudate nuclei atrophy in Huntington’s disease. J Neurol. 1999;246:1090–1093. doi: 10.1007/s004150050518. [DOI] [PubMed] [Google Scholar]
- Curtis MA, Penney EB, Pearson AG, van Roon-Mom WM, Butterworth NJ, Dragunow M, Connor B, Faull RL. Increased cell proliferation and neurogenesis in the adult human Huntington’s disease brain. Proc. Natl. Acad. Sci. USA. 2003;100:9023–9027. doi: 10.1073/pnas.1532244100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MA, Waldvogel HJ, Synek B, Faull RL. A histochemical and immunohistochemical analysis of the subependymal layer in the normal and Huntington’s disease brain. J Chem Neuroanat. 2005a;30:55–66. doi: 10.1016/j.jchemneu.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Curtis MA, Penney EB, Pearson J, Dragunow M, Connor B, Faull RL. The distribution of progenitor cells in the subependymal layer of the lateral ventricle in the normal and Huntington’s disease human brain. Neuroscience. 2005b;132:777–788. doi: 10.1016/j.neuroscience.2004.12.051. [DOI] [PubMed] [Google Scholar]
- Curtis MA, Eriksson PS, Faull RL. Progenitor cells and adult neurogenesis in neurodegenerative diseases and injuries of the basal ganglia. Clin Exp Pharmacol Physiol. 2007;34:528–532. doi: 10.1111/j.1440-1681.2007.04609.x. [DOI] [PubMed] [Google Scholar]
- Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- Dawbarn D, DeQuidt ME, Emson PC. Survival of basal ganglia neuropeptide Y-somatostatin neurons in Huntington’s disease. Brain Res. 1985;340:251–260. doi: 10.1016/0006-8993(85)90921-7. [DOI] [PubMed] [Google Scholar]
- De La Monte SM, Vonsattel JP, Richardson EP., Jr. Morphometric demonstrations of atrophic changes in the cerebral cortex, white matter, and neostriatum in Huntington’s disease. J. Neuropathol. Exp. Neurol. 1988;44:516–525. doi: 10.1097/00005072-198809000-00003. [DOI] [PubMed] [Google Scholar]
- Deng YP, Penney JB, Young AB, Albin RL, Anderson KD, Reiner A. Differential loss of striatal projection neurons in Huntington’s disease: A quantitative immunohistochemical study. J Chem Neur. 2004;27:143–164. doi: 10.1016/j.jchemneu.2004.02.005. [DOI] [PubMed] [Google Scholar]
- De Rooij KE, De Koning Gans PA, Roos RA, Van Ommen GJ, Den Dunnen JT. Somatic expansion of the (CAG)n repeat in Huntington disease brains. Hum. Gen. 1995;95:270–274. doi: 10.1007/BF00225192. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase K, Schwarts C, Meloni A, Young C, Martin E, Vonsattel JP, Carraway R, Reeves SA, Boyce FM, Aronin N. Huntingtin is a cytoplasmatic protein associated with vesicles in human and rat brain neurons. Neuron. 1995;14:1075–1081. doi: 10.1016/0896-6273(95)90346-1. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin JP. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- DiFiglia M. Excitotoxic injury of the neostriatum: a model for Huntington’s disease. Trends Neurosci. 1990;13:286–289. doi: 10.1016/0166-2236(90)90111-m. [DOI] [PubMed] [Google Scholar]
- DiLeone RJ, Georgescu D, Nestler EJ. Lateral hypothalamic neuropeptides in reward and drug addiction. Life Sci. 2003;73:759–768. doi: 10.1016/s0024-3205(03)00408-9. [DOI] [PubMed] [Google Scholar]
- Djoussé L, Knowlton B, Hayden M, Almqvist EW, Brinkman R, Ross C, Margolis R, Rosenblatt A, Dürr A, Dode C, Morrison PJ, Novelletto A, Frontali M, Trent RJ, McCusker E, Gómez-Tortosa E, Mayo D, Jones R, Zanko A, Nance M, Abramson R, Suchowersky O, Paulsen J, Harrison M, Yang Q, Cupples LA, Gusella JF, MacDonald ME, Myers RH. Interaction of normal and expanded CAG repeat sizes influences age at onset of Huntington disease. Am J Med Gen A. 2003;119A:279–282. doi: 10.1002/ajmg.a.20190. [DOI] [PubMed] [Google Scholar]
- Dom R, Malfroid M, Baro F. Neuropathology of Huntington’s chorea. Studies of the ventrobasal complex of the thalamus. Neurology. 1976;26:64–68. doi: 10.1212/wnl.26.1.64. [DOI] [PubMed] [Google Scholar]
- Dorsman JC, Smoor MA, Maat-Schieman MLC, Bout M, Siesling S, van Duinen SG, Verschuuren JJ, den Dunnen JT, Roos RA, van Ommen GJ. Analysis of the subcellular localization of huntingtin with a set of rabbit polyclonal antibodies in cultured mammalian cells of neuronal origin: comparison with distribution of huntingtin in Huntington’s disease autopsy brain. Phil Trans R Soc Lond. 1999;354:1061–1067. doi: 10.1098/rstb.1999.0459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douaud G, Behrens TE, Poupon C, Cointepas Y, Jbabdi S, Gaura V, Golestani N, Krystkowiak P, Verny C, Damier P, Bachoud-Lévi AC, Hantraye P, Remy P. In vivo evidence for the selective subcortical degeneration in Huntington’s disease. NeuroImage. 2009;46:958–966. doi: 10.1016/j.neuroimage.2009.03.044. [DOI] [PubMed] [Google Scholar]
- Douaud G, Gaura V, Riveiro MJ, Lethimonnier F, Maroy R, Vemy C, Krystkowiak P, Damier P, Bachoud-Levi AC, Hantraye P, Remy P. Distribution of grey matter atrophy in Huntington’s disease patients: a combined ROI-based and voxel-based morphometric study. Neuroimage. 2006;32:1562–1575. doi: 10.1016/j.neuroimage.2006.05.057. [DOI] [PubMed] [Google Scholar]
- Dürr A, Hahn-Barma V, Brice A, Pêcheux C, Dodé C, Feingold J. Homozygosity in Huntington’s disease. J Med Gen. 1999;36:172–173. [PMC free article] [PubMed] [Google Scholar]
- Duyao M, Ambrose C, Myers R, Novelletto A, Persichetti F, Frontali M, Folstein S, Ross C, Franz M, Abbott M, Gray J, Conneally P, Young A, Penney J, Hollingsworth Z, Shoulson I, Lazzarini A, Falek A, Koroshetz W, Sax D, Bird E, Vonsattel J, Bonilla E, Alvir J, Bickham Conde J, Cha JH, Dure L, Gomez F, Ramos M, Sanchez-Ramos J, Snodgrass S, de Young M, Wexler N, Moscowitz C, Penchaszadeh G, MacFarlane H, Anderson M, Jenkins B, Srinidhi J, Barnes G, Gusella J, MacDonald M. Trinucleotide repeat length instability and age of onset in Huntington’s disease. Nat. Gen. 1993;4:387–392. doi: 10.1038/ng0893-387. [DOI] [PubMed] [Google Scholar]
- Ellison DW, Beal MF, Mazurek MF, Malloy JR, Bird ED, Martin JB. Amino acid neurotransmitter abnormalities in Huntington’s disease and the quinolinic acid animal model of Huntington’s disease. Brain. 1987;110:1657–1673. doi: 10.1093/brain/110.6.1657. [DOI] [PubMed] [Google Scholar]
- Emson PC, Arregui A, Clement-Jones V, Sandberg BEB, Rossor M. Regional distribution of methionine-enkephalin and substance P-like immunoreactivity in normal human brain and in Huntington’s disease. Brain Res. 1980;199:147–160. doi: 10.1016/0006-8993(80)90237-1. [DOI] [PubMed] [Google Scholar]
- Feger J, Crossman AR. Identification of different subpopulations of neostriatal neurons projecting to globus pallidus or substantia nigra in the monkey: a retrograde fluorescence double-labeling study. Neurosci. Lett. 1984;49:7–12. doi: 10.1016/0304-3940(84)90127-7. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Beal MF, Kowall NW, Richardson EP, Martin JB., Jr. Sparing of acetylcholinesterase-containing striatal neurons in Huntington’s disease. Brain Res. 1987b;411:162–166. doi: 10.1016/0006-8993(87)90694-9. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Kowall NW, Beal MF, Martin JB, Bird ED, Richardson EP., Jr. Morphological and histochemical characteristics of a spared subset of striatal neurons in Huntington’s disease. J. Neuropathol. Exp. Neurol. 1987 a;46:12–27. doi: 10.1097/00005072-198701000-00002. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Kowall NW, Beal MF, Richardson EP, Jr., Bird ED, Martin JB. Selective sparing of a class of striatal neurons in Huntington’s disease. Science. 1985;230:561–563. doi: 10.1126/science.2931802. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Kowall NW, Richardson EP, Jr., Bird ED, Martin JB. Topography of enkephalin, substance P, and acetylcholinesterase staining in Huntington’s disease striatum. Neurosci. Lett. 1986;71:283–288. doi: 10.1016/0304-3940(86)90634-8. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Kulisevsky J, Gonzalez G, Escartin A, Chivite A, Casas R. Parvalbumin-immunoreactive neurons in the cerebral cortex and striatum in Huntington’s disease. Neurodegeneration. 1994;3:169–173. [Google Scholar]
- Figueredo-Cardenas G, Morello M, Sancesario G, Bernardi G, Reiner A. Colocalization of somatostatin, neuropeptide Y, neuronal nitric oxide synthase and NADPH-diaphorase in striatal interneurons in rats. Brain Res. 1996a;735:317–324. doi: 10.1016/0006-8993(96)00801-3. [DOI] [PubMed] [Google Scholar]
- Figueredo-Cardenas G, Medina L, Reiner A. Calretinin is localized to a unique population of striatal interneurons in rats. Brain Res. 1996 b;709:145–150. doi: 10.1016/0006-8993(95)01392-x. [DOI] [PubMed] [Google Scholar]
- Figueredo-Cardenas G, Harris C, Anderson KD, Reiner A. Relative resistance of striatal neurons containing calbindin or parvalbumin to quinolinic acid-mediated excitotoxicity compared to other striatal neuron types. Exp. Neurol. 1998;149:356–372. doi: 10.1006/exnr.1997.6724. [DOI] [PubMed] [Google Scholar]
- Fink JS, Weaver DR, Rivkees SA, Peterfreund RA, Pollack AE, Adler EM, Reppert SM. Molecular cloning of the rat A2 adenosine receptor: selective co-expression with D2 dopamine receptors in rat striatum. Mol. Brain Res. 1992;14:186–195. doi: 10.1016/0169-328x(92)90173-9. [DOI] [PubMed] [Google Scholar]
- Flaherty AW, Graybiel AM. Output architecture of the primate putamen. J. Neurosci. 1993;13:3222–3237. doi: 10.1523/JNEUROSCI.13-08-03222.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fossale E, Wheeler VC, Vrbanac V, Lebel LA, Teed A, Mysore JS, Gusella JF, MacDonald ME, Persichetti F. Identification of a presymptomatic molecular phenotype in Hdh CAG knock-in mice. Hum. Mol. Gen. 2002;11:2233–2241. doi: 10.1093/hmg/11.19.2233. [DOI] [PubMed] [Google Scholar]
- Fusco FR, Chen Q, Lamoreaux WJ, Figueredo-Cardenas G, Jiao Y, Coffman J, Surmeier DJ, Honig MG, Carlock LR, Reiner A. Cellular localization of huntingtin in striatal and cortical neurons in rats: Lack of correlation with neuronal vulnerability in Huntington’s disease. J. Neurosci. 1999;19:1189–1202. doi: 10.1523/JNEUROSCI.19-04-01189.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabery S, Murphy K, Schultz K, Loy CT, McCusker E, Kirik D, Halliday G, Petersén A. Changes in key hypothalamic neuropeptide populations in Huntington disease revealed by neuropathological analyses. Acta Neuropathol. 2010;120:777–788. doi: 10.1007/s00401-010-0742-6. [DOI] [PubMed] [Google Scholar]
- Gage GJ, Stoetzner CR, Wiltschko AB, Berke JD. Selective activation of striatal fast-spiking interneurons during choice execution. Neuron. 2010;67:466–479. doi: 10.1016/j.neuron.2010.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale GS, Bird ED, Spokes EG, Iversen LL, Jessell T. Human brain substance P: distribution in controls and Huntington’s chorea. J Neurochem. 1977;30:633–634. doi: 10.1111/j.1471-4159.1978.tb07818.x. [DOI] [PubMed] [Google Scholar]
- Gayán J, Brocklebank D, Andresen JM, Alkorta-Aranburu G, US-Venezuela Collaborative Research Group. Zameel Cader M, Roberts SA, Cherny SS, Wexler NS, Cardon LR, Housman DE. Genomewide linkage scan reveals novel loci modifying age of onset of Huntington’s disease in the Venezuelan HD kindreds. Gen Epidemiol. 2008;32:445–453. doi: 10.1002/gepi.20317. [DOI] [PubMed] [Google Scholar]
- Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization. Trends Neurosci. 1992;15:133–139. doi: 10.1016/0166-2236(92)90355-c. [DOI] [PubMed] [Google Scholar]
- Gervais FG, Singaraja R, Xanthoudakis S, Gutekunst CA, Leavitt BR, Metzler M, Hackam AS, Tam J, Vaillancourt JP, Houtzager V, Rasper DM, Roy S, Hayden MR, Nicholson DW. Recruitment and activation of caspase-8 by the Huntingtin-interacting protein Hip-1 and a novel partner Hippi. Nat Cell Biol. 2002;4:95–105. doi: 10.1038/ncb735. [DOI] [PubMed] [Google Scholar]
- Gimenez-Amaya JM, Graybiel AM. Modular organization of projection neurons in the matrix compartment of the primate striatum. J. Neurosci. 1991;11:779–791. doi: 10.1523/JNEUROSCI.11-03-00779.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass M, Dragunow M, Faull RLM. Cannabinoid receptors in the human brain: a detailed anatomical and quantitative autoradiographic study in the fetal, neonatal and adult human brain. Neuroscience. 1997;77:299–318. doi: 10.1016/s0306-4522(96)00428-9. [DOI] [PubMed] [Google Scholar]
- Glass M, Dragunow M, Faull RLM. The pattern of neurodegeneration in Huntington’s disease: a comparative study of cannabinoid, dopamine, adenosine and GABAA receptor alterations in the human basal ganglia in Huntington’s disease. Neuroscience. 2000;97:505–519. doi: 10.1016/s0306-4522(00)00008-7. [DOI] [PubMed] [Google Scholar]
- Gómez-Tortosa E, MacDonald ME, Friend JC, Taylor SA, Weiler LJ, Cupples LA, Srinidhi J, Gusella JF, Bird ED, Vonsattel JP, Myers RH. Quantitative neuropathological changes in presymptomatic Huntington’s disease. Ann. Neurol. 2001;49:29–34. [PubMed] [Google Scholar]
- Goto S, Hirano A, Rojas-Corona RR. Immunohistochemical visualization of afferent nerve terminals in human globus pallidus and its alteration in neostriatal neurodegenerative disorders. Acta Neuropathol. 1989 a;78:543–550. doi: 10.1007/BF00687717. [DOI] [PubMed] [Google Scholar]
- Goto S, Hirano A, Rojas-Corona RR. An immunohistochemical investigation of the human neostriatum in Huntington’s disease. Annals Neurol. 1989 b;25:298–304. doi: 10.1002/ana.410250315. [DOI] [PubMed] [Google Scholar]
- Grafton ST, Mazziotta JC, Pahl JJ, St George-Hyslop P, Haines JL, Gusella J, Hoffman JM, Baxter LR, Phelps ME. Serial changes of cerebral glucose metabolism and caudate size in persons at risk for Huntington’s disease. Arch Neurol. 1992;49:1161–1167. doi: 10.1001/archneur.1992.00530350075022. [DOI] [PubMed] [Google Scholar]
- Graham RK, Slow EJ, Deng Y, Bissada N, Lu G, Pearson J, Shehadeh J, Leavitt BR, Raymond LA, Hayden MR. Levels of mutant huntingtin influence the phenotypic severity of Huntington disease in YAC128 mouse models. Neurobiol Dis. 2006;21:444–455. doi: 10.1016/j.nbd.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Gusella JF, MacDonald ME. Huntington’s disease: the case for genetic modifiers. Genome Med. 2009;1:80.1–80.6. doi: 10.1186/gm80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusella JF, Wexler NS, Conneally PM, Naylor SL, Anderson MA, Tanzi RE, Watkins PC, Ottina K, Wallace MR, Sakaguchi AY, Young AB, Shoulson I, Bonilla E, Martin JB. A polymorphic DNA marker genetically linked to Huntington’s disease. Nature. 1983;306:234–238. doi: 10.1038/306234a0. [DOI] [PubMed] [Google Scholar]
- Gusella JF, MacDonald ME, Ambrose CM, Duyao MP. Molecular Genetics of Huntington’s Disease. Arch Neurol. 1994;50:1157–1163. doi: 10.1001/archneur.1993.00540110037003. [DOI] [PubMed] [Google Scholar]
- Gutekunst CA, Levey AI, Heilman CJ, Whaley WL, Yi H, Nash NR, Rees HD, Madden JJ, Hersh SM. Identification and localization on huntingtin in brain and human lymphoblastoid cell lines with anti-fusion protein antibodies. Proc. Natl. Acad. Sci. USA. 1995;92:8710–8714. doi: 10.1073/pnas.92.19.8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ. Nuclear and neuropil aggregates in Huntington’s disease: relationship to neuropathology. J. Neurosci. 1999;19:2522–2534. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackham AS, Yassa AS, Singaraja R, Metzler M, Gutekunst CA, Gan L, Warby S, Wellington CL, Vaillancourt J, Chen N, Gervais FG, Raymond L, Nicholson DW, Hayden MR. Huntingtin interacting protein 1 induces apoptosis via novel caspase-dependent death effector domain. J Biol Chem. 2000;275:41299–41308. doi: 10.1074/jbc.M008408200. [DOI] [PubMed] [Google Scholar]
- Hackham AS, Singaraja R, Wellington CL, Metzler M, Zhang Z, Kalchman M, Hayden MR. The influence of huntingtin protein size on nuclear localization and cellular toxicity. J Cell Biol. 1998 b;141:1097–1105. doi: 10.1083/jcb.141.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackham AS, Wellington CL, Hayden MR. The fatal attraction of polyglutamine-containing proteins. Clin Gen. 1998a;53:233–242. doi: 10.1111/j.1399-0004.1998.tb02687.x. [DOI] [PubMed] [Google Scholar]
- Halliday GM, McRitchie DA, Macdonald V, Double KL, Trent RJ, McCusker E. Regional specificity of brain atrophy in Huntington’s disease. Exp. Neurol. 1998;154:663–672. doi: 10.1006/exnr.1998.6919. [DOI] [PubMed] [Google Scholar]
- Harjes P, Wanker EE. The hunt for huntingtin function: interaction partners tell many different stories. Trends Biochem Sci. 2003;28:225–233. doi: 10.1016/S0968-0004(03)00168-3. [DOI] [PubMed] [Google Scholar]
- Harrington KM, Kowall NW. Parvalbumin immunoreactive neurons resist degeneration in Huntington’s disease striatum. J. Neuropathol. Exp. Neurol. 1991;50:309. doi: 10.1097/00005072-199107000-00006. [DOI] [PubMed] [Google Scholar]
- Hassel B, Tessler S, Faull RL, Emson PC. Glutamate uptake is reduced in prefrontal cortex in Huntington’s disease. Neurochem Res. 2008;33:232–237. doi: 10.1007/s11064-007-9463-1. [DOI] [PubMed] [Google Scholar]
- Hawker K, Lang AE. Hypoxic-ischemic damage of the basal ganglia: case reports and a review of the literature. Mov Disord. 1990;5:219–224. doi: 10.1002/mds.870050306. [DOI] [PubMed] [Google Scholar]
- Hedreen JC, Folstein SE. Early loss of neostriatal striosome neurons in Huntington’s disease. J. Neuropathol. Exp. Neurol. 1995;54:105–120. doi: 10.1097/00005072-199501000-00013. [DOI] [PubMed] [Google Scholar]
- Hedreen JC, Peyser CE, Folstein SE, Ross CA. Neuronal loss in layers V and VI of cerebral cortex in Huntingon’s disease. Neurosci. Lett. 1991;133:257–261. doi: 10.1016/0304-3940(91)90583-f. [DOI] [PubMed] [Google Scholar]
- Heinsen H, Strik M, Bauer M, Luther K, Ulmar G, Gangnus D, Jungkunz G, Eisenmenger W, Götz M. Cortical and striatal neurone number in Huntington’s disease. Acta Neuropatho. 1994;88:320–333. doi: 10.1007/BF00310376. [DOI] [PubMed] [Google Scholar]
- Heinsen H, Rüb U, Gangnus D, Jungkunz G, Bauer M, Ulmar G, Bethke B, Schüler M, Böcker F, Eisenmenger W, Götz M, Strik M. Nerve cell loss in the thalamic centromedian-parafascicular complex in patients with Huntington’s disease. Acta Neuropathol. 1996;91:161–168. doi: 10.1007/s004010050408. [DOI] [PubMed] [Google Scholar]
- Heinsen H, Rüb U, Bauer M, Ulmar G, Bethke B, Schüler M, Böcker F, Eisenmenger W, Götz M, Korr H, Schmitz C. Nerve cell loss in the thalamic mediodorsal nucleus in Huntington’s disease. Acta Neuropathol. 1999;97:613–622. doi: 10.1007/s004010051037. [DOI] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, de Costa BR, Richfield EK. Neuronal localization of cannabinoid receptors in the basal ganglia of the rat. Brain Res. 1991;547:267–274. doi: 10.1016/0006-8993(91)90970-7. [DOI] [PubMed] [Google Scholar]
- Hikosaka O. Role of basal ganglia in saccades. Rev Neurol (Paris) 1989;145:580–586. [PubMed] [Google Scholar]
- Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G, Elliston LA, Hartog C, Goldstein DR, Thu D, Hollingsworth ZR, Collin F, Synek B, Holmans PA, Young AB, Wexler NS, Delorenzi M, Kooperberg C, Augood SJ, Faull RL, Olson JM, Jones L, Luthi-Carter R. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum. Mol. Gen. 2006;15:965–977. doi: 10.1093/hmg/ddl013. [DOI] [PubMed] [Google Scholar]
- Hökfelt T, Martensson R, Björklund A, Kleinau S, Goldstein M. Distributional maps of tyrosine hydroxylase-immunoreactive neurons in the rat brain. In: Björklund A, Hökfelt T, editors. Handbook of Chemical Neuroanatomy, vol. 2. Classical Transmitter in the CNS. Part I. Elsevier; Amsterdam, The Netherlands: 1984. pp. 277–379. [Google Scholar]
- Huang Q, Zhou D, Sapp E, Aizawa H, Ge P, Bird ED, Vonsattel JP, DiFiglia M. Quinolinic acid induced increases in calbindin D28k immunoreactivity in rat striatal neurons in vivo and in vitro mimic the pattern seen in Huntington’s disease. Neuroscience. 1995;65:397–407. doi: 10.1016/0306-4522(94)00494-p. [DOI] [PubMed] [Google Scholar]
- Huntington’s Disease Collaborative Research Group A novel gene containing a trinucleotide repeat that is expanded and unstable on the HD chromosome. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Jech R, Klempír J, Vymazal J, Zidovská J, Klempírová O, Ruzicka E, Roth J. Variation of selective gray and white matter atrophy in Huntington’s disease. Movement Dis. 2007;22:1783–1789. doi: 10.1002/mds.21620. [DOI] [PubMed] [Google Scholar]
- Joshi PR, Wu NP, Andre VM, Cummings DM, Cepeda C, Joyce JA, Carroll JB, Leavitt BR, Hayden MR, Levine MS, Bamford NS. Age-dependent alterations of corticostriatal activity in the YAC128 mouse model of Huntington disease. J. Neurosci. 2009;29:2414–2427. doi: 10.1523/JNEUROSCI.5687-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanazawa I, Bird E, O’Connell R, Powell D. Evidence for a decrease in substance P content of substantia nigra in Huntington’s chorea. Brain Res. 1977;120:387–392. doi: 10.1016/0006-8993(77)90919-2. [DOI] [PubMed] [Google Scholar]
- Kanazawa I, Bird ED, Gale JS, Iversen LL, Jessell TM, Muramoto O, Spokes EG, Sutoo D. Substance P: decrease in substantia nigra and globus pallidus in Huntington’s disease. Advances in Neurology. 1979;23:495–504. [Google Scholar]
- Karlovich CA, John RM, Ramirez L, Stainier DY, Myers RM. Characterization of the Huntington’s disease (HD) gene homologue in the zebrafish Danio rerio. Gene. 1998;217:117–125. doi: 10.1016/s0378-1119(98)00342-4. [DOI] [PubMed] [Google Scholar]
- Kassubek J, Juengling FD, Ecker D, Landwehrmeyer GB. Thalamic atrophy in Huntington’s disease co-varies with cognitive performance: a morphometric MRI analysis. Cerebral Cortex. 2004a;15:846–853. doi: 10.1093/cercor/bhh185. [DOI] [PubMed] [Google Scholar]
- Kassubek J, Juengling FD, Kioschies T, Henkel K, Karitzky J, Kramer B, Ecker D, Andrich J, Saft C, Kraus P, Aschoff AJ, Ludolph AC, Landwehrmeyer GB. Topography of cerebral atrophy in early Huntington’s disease: a voxel based morphometric MRI study. J Neurol Neurosurg Psychiatry. 2004b;75:213–220. [PMC free article] [PubMed] [Google Scholar]
- Kassubek J, Bernhard Landwehrmeyer G, Ecker D, Juengling FD, Muche R, Schuller S, Weindl A, Peinemann A. Global cerebral atrophy in early stages of Huntington’s disease: quantitative MRI study. Neuroreport. 2004c;15:363–365. doi: 10.1097/00001756-200402090-00030. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Wilson CJ, Augood SJ, Emson PC. Striatal interneurons: chemical, physiological and morphological characterization. Trends Neurosci. 1995;18:527–535. doi: 10.1016/0166-2236(95)98374-8. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y. Physiological, morphological, and histochemical characterization of three classes of interneurons in rat neostriatum. J. Neurosci. 1993;13:4908–4923. doi: 10.1523/JNEUROSCI.13-11-04908.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Wilson CJ, Emson PC. Projection subtypes of rat neostriatal matrix cells revealed by intracellular injection of biocytin. J. Neurosci. 1990;10:3421–3438. doi: 10.1523/JNEUROSCI.10-10-03421.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kegel KB, Kim M, Sapp E, Mclntyre C, Castano JG, Aronin N, DiFiglia M. Huntingtin expression stimulates endosomal-lysosomal activity, endosome tubulation, and autophagy. J. Neurosci. 2000;20:7268–7278. doi: 10.1523/JNEUROSCI.20-19-07268.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kegel KB, Meloni AR, Yi Y, Kim YJ, Doyle E, Cuiffo BG, Sapp E, Wang Y, Qin ZH, Chen JD, Nevins JB, Aronin N, DiFiglia M. Huntingtin is present in the nucleus,interacts with the transcriptional co-repressor C-terminal binding protein, and represses transcription. J Biol Chem. 2002;277:7466–7476. doi: 10.1074/jbc.M103946200. [DOI] [PubMed] [Google Scholar]
- Kenney C, Powell S, Jankovic J. Autopsy proven Huntington’s disease with 29 trinucleotide repeats. Movement Dis. 2006;22:127–130. doi: 10.1002/mds.21195. [DOI] [PubMed] [Google Scholar]
- Kim TW, Tanzi RE. Neuronal intranuclear inclusions in polyglutamine diseases: nuclear weapons or nuclear fallout. Neuron. 1998;21:657–659. doi: 10.1016/s0896-6273(00)80581-4. [DOI] [PubMed] [Google Scholar]
- Kipps CM, Duggins AJ, McCusker EA, Calder AJ. Disgust and happiness recognition correlate with anteroventral insula and amygdala volume respectively in preclinical Huntington’s disease. J Cog Neurosci. 2007;19:1206–1217. doi: 10.1162/jocn.2007.19.7.1206. [DOI] [PubMed] [Google Scholar]
- Kish SJ, Shannak K, Hornykiewicz O. Elevated serotonin and reduced dopamine in subregionally divided Huntington’s disease striatum. Ann. Neurol. 1987;22:386–389. doi: 10.1002/ana.410220318. [DOI] [PubMed] [Google Scholar]
- Kita H, Kosaka T, Heizmann CW. Parvalbumin-immunoreactive neurons in the rat neostriatum: a light and electron microscopic study. Brain Res. 1990;536:1–15. doi: 10.1016/0006-8993(90)90002-s. [DOI] [PubMed] [Google Scholar]
- Klöppel S, Stonnington CM, Barnes J, Chen F, Chu C, Good CD, Mader I, Mitchell LA, Patel AC, Roberts CC, Fox NC, Jack CR, Jr., Ashburner J, Frackowiak RS. Accuracy of dementia diagnosis: a direct comparison between radiologists and a computerized method. Brain. 2008;131:2969–2974. doi: 10.1093/brain/awn239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J, Ou S, Patterson PH. New antihuntingtin monoclonal antibodies: Implications for huntingtin conformation and its binding proteins. Brain Res. Bull. 2001;56:319–329. doi: 10.1016/s0361-9230(01)00599-8. [DOI] [PubMed] [Google Scholar]
- Koeppen AH. The nucleus pontis centralis caudalis in Huntington’s disease. J Neuro Sci. 1989;91:126–141. doi: 10.1016/0022-510x(89)90082-8. [DOI] [PubMed] [Google Scholar]
- Kok SW, Overeem S, Visscher TL, Lammers GJ, Seidell JC, Pijl H, Meinders AE. Hypocretin deficiency in narcoleptic humans is associated with abdominal obesity. Obes Res. 2003;11:1147–1154. doi: 10.1038/oby.2003.156. [DOI] [PubMed] [Google Scholar]
- Koos T, Tepper JM. Inhibitory control of neostriatal projection neurons by GABAergic interneurons. Nat. Neurosci. 1999;2:467–472. doi: 10.1038/8138. [DOI] [PubMed] [Google Scholar]
- Kovtun IV, Therneau TM, McMurray CT. Gender of the embryo contributes to CAG instability in transgenic mice containing a Huntington’s disease gene. Hum. Mol. Gen. 2000;9:2767–2775. doi: 10.1093/hmg/9.18.2767. [DOI] [PubMed] [Google Scholar]
- Kovtun IV, Welch G, Guthrie HD, Hafner KL, McMurray CT. CAG repeat lengths in X-and Y-bearing sperm indicate that gender bias during transmission of Huntington’s disease gene is determined by the embryo. J Biol Chem. 2004;279:9389–9391. doi: 10.1074/jbc.M313080200. [DOI] [PubMed] [Google Scholar]
- Kovtun IV, Liu Y, Bjoras M, Klungland A, Wilson SH, McMurray CT. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature. 2007;447:447–452. doi: 10.1038/nature05778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowall NW, Ferrante RJ, Martin JB. Pattern of cell loss in Huntington’s disease. Trends Neurosci. 1987;10:24–29. [Google Scholar]
- Kremer HP, Roos RA, Dingjan G, Marani E, Bots GT. Atrophy of the hypothalamic lateral tuberal nucleus in Huntington’s disease. J. Neuropathol. Exp. Neurol. 1990;49:371–382. doi: 10.1097/00005072-199007000-00002. [DOI] [PubMed] [Google Scholar]
- Kremer B, Goldberg P, Andrew SE, Theilmann J, Telenius H, Zeisler J, Squitieri F, Lin B, Bassett A, Almqvist E, Bird TD, Hayden MR. A worldwide study of the Huntington’s disease mutation. The sensitivity and specificity of measuring CAG repeats. N. Engl. J. Med. 1994;330:1401–1406. doi: 10.1056/NEJM199405193302001. [DOI] [PubMed] [Google Scholar]
- Kubota Y, Mikawa S, Kawaguchi Y. Neostriatal GABAergic interneurons contain NOS, calretinin or parvalbumin. NeuroReport. 1993;5:205–208. doi: 10.1097/00001756-199312000-00004. [DOI] [PubMed] [Google Scholar]
- Kuemmerle S, Gutekunst CA, Klein AM, Li XJ, Li SH, Beal MF, Hersch SM, Ferrante RJ. Huntingtin aggregates may not predict neuronal death in Huntington’s disease. Ann. Neurol. 1999;46:842–849. [PubMed] [Google Scholar]
- Kuwert T, Lange HW, Boecker H, Titz H, Herzog H, Aulich A, Wang BC, Nayak U, Feinendegen LE. Striatal glucose consumption in chorea-free subjects at risk of Huntington’s disease. J Neurol. 1993;241:31–36. doi: 10.1007/BF00870669. [DOI] [PubMed] [Google Scholar]
- Landwehrmeyer GB, McNeil SM, Dure IVLS, Ge P, Aizawa H, Huang Q, Ambrose CM, Duyao MP, Bird ED, Bonilla E, deYoung M, Avila-Gonzales AJ, Wexler NS, DiFiglia M, Gusella JF, MacDonald ME, Penney JB, Young AB, Vonsattel JP. Huntington’s disease gene: regional and cellular expression in brain of normal and affected individuals. Ann. Neurol. 1995;37:218–230. doi: 10.1002/ana.410370213. [DOI] [PubMed] [Google Scholar]
- Lange H, Thörner G, Hopf A, Schröder KF. Morphometric studies of the neuropathological changes in choreatic diseases. J Neurol Sci. 1976;28:401–425. doi: 10.1016/0022-510x(76)90114-3. [DOI] [PubMed] [Google Scholar]
- Leeflang EP, Tavaré S, Marjoram P, Neal CO, Srinidhi J, MacFarlane H, MacDonald ME, Gusella JF, de Young M, Wexler NS, Arnheim N. Analysis of germline mutation spectra at the Huntingtons disease locus supports a mitotic mutation mechanism. Hum. Mol. Gen. 1999;8:173–183. doi: 10.1093/hmg/8.2.173. [DOI] [PubMed] [Google Scholar]
- Le Moine C, Bloch B. D1 and D2 dopamine receptor gene expression in the rat striatum: sensitive cRNA probes demonstrate prominent segregation of D1 and D2 mRNAs in distinct neuronal populations of the dorsal and ventral striatum. J Comp Neurol. 1995;355:418–426. doi: 10.1002/cne.903550308. [DOI] [PubMed] [Google Scholar]
- Li JL, Hayden MR, Almqvist EW, Brinkman RR, Dürr A, Dodé C, Morrison PJ, Suchowersky O, Ross CA, Margolis RL, Rosenblatt A, Gómez-Tortosa E, Cabrero DM, Novelletto A, Frontali M, Nance M, Trent RJ, McCusker E, Jones R, Paulsen JS, Harrison M, Zanko A, Abramson RK, Russ AL, Knowlton B, Djoussé L, Mysore JS, Tariot S, Gusella MF, Wheeler VC, Atwood LD, Cupples LA, Saint-Hilaire M, Cha JH, Hersch SM, Koroshetz WJ, Gusella JF, MacDonald ME, Myers RH. A genome scan for modifiers of age at onset in Huntington disease: The HD MAPS study. Am J Hum. Gen. 2003;73:682–687. doi: 10.1086/378133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JL, Hayden MR, Warby SC, Dürr A, Morrison PJ, Nance M, Ross CA, Margolis RL, Rosenblatt A, Squitieri F, Frati L, Gómez-Tortosa E, García CA, Suchowersky O, Klimek ML, Trent RJ, McCusker E, Novelletto A, Frontali M, Paulsen JS, Jones R, Ashizawa T, Lazzarini A, Wheeler VC, Prakash R, Xu G, Djoussé L, Mysore JS, Gillis T, Hakky M, Cupples LA, Saint-Hilaire MH, Cha JH, Hersch SM, Penney JB, Harrison MB, Perlman SL, Zanko A, Abramson RK, Lechich AJ, Duckett A, Marder K, Conneally PM, Gusella JF, MacDonald ME, Myers RH. Genome-wide significance for a modifier of age at neurological onset in Huntington’s disease at 6q23-24: the HD-MAPS study. BMC Med Gen. 2006;7:71. doi: 10.1186/1471-2350-7-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SH, Schilling G, Young WS, 3rd, Li XJ, 3rd, Margolis RL, Stine OC, Wagster MV, Abbott MH, Franz ML, Ranen NG, Folstein SE, Hedreen JC, Ross CA. Huntington’s disease gene (IT15) is widely expressed in human and rat tissues. Neuron. 1993;11:985–993. doi: 10.1016/0896-6273(93)90127-d. [DOI] [PubMed] [Google Scholar]
- Li SH, Li XJ. Aggregation of N-terminal huntingtin is dependent on the length of its glutamine repeats. Hum. Mol. Gen. 1998;7:777–782. doi: 10.1093/hmg/7.5.777. [DOI] [PubMed] [Google Scholar]
- Li Z, Karlovich CA, Fish MP, Scott MP, Myers RM. A putative Drosophila homolog of the Huntington’s disease gene. Hum. Mol. Gen. 1999;8:1807–1815. doi: 10.1093/hmg/8.9.1807. [DOI] [PubMed] [Google Scholar]
- Lievens JC, Woodman B, Mahal A, Spasic-Boscovic O, Samuel D, Kerkerian-Le GL, Bates GP. Impaired glutamate uptake in the R6 Huntington’s disease transgenic mice. Neurobiol. Dis. 2001;8:807–821. doi: 10.1006/nbdi.2001.0430. [DOI] [PubMed] [Google Scholar]
- Lin B, Nasir J, Kalchman MA, McDonald H, Zeisler J, Goldberg YP, Hayden MR. Structural analysis of the 5′ region of the mouse and human Huntington’s disease genes reveals conservation of putative promoter region and di-and trinucleotide polymorphisms. Genomics. 1995;25:707–715. doi: 10.1016/0888-7543(95)80014-d. [DOI] [PubMed] [Google Scholar]
- Lin CH, Tallaksen-Greene S, Chien WM, Cearley JA, Jackson WS, Crouse AB, Ren S, Li XJ, Albin RL, Detloff PJ. Neurological abnormalities in a knock-in mouse model of Huntington’s disease. Hum. Mol. Gen. 2001;10:137–144. doi: 10.1093/hmg/10.2.137. [DOI] [PubMed] [Google Scholar]
- Luthi-Carter R, Hanson SA, Strand AD, Bergstrom DA, Chun W, Peters NL, Woods AM, Chan Kooperberg EY, Krainc CD, Young AB, Tapscott SJ, Olson JM. Dysregulation of gene expression in the R6/2 model of polyglutamine disease: parallel changes in muscle and brain. Hum. Mol. Gen. 2002;11:1911–1926. doi: 10.1093/hmg/11.17.1911. [DOI] [PubMed] [Google Scholar]
- Maat-Kievit A, Helderman-van den Enden P, Losekoot M, de Knijff P, Belfroid R, Vegter-van der Vlis M, Roos R, Breuning M. Using a roster and haplotyping is useful in risk assessment for persons with intermediate and reduced peetrance alleles in Huntington disease. Am. J. Med. Gen. 2001;105:737–744. doi: 10.1002/ajmg.1610. [DOI] [PubMed] [Google Scholar]
- Maat-Schieman ML, Dorsman JC, Smoor MA, Siesling S, Van Duinen SG, Verschuuren JJ, den Dunnen JT, Van Ommen GJ, Roos RA. Distribution of inclusions in neuronal nuclei and dystrophic neurites in Huntington disease brain. J. Neuropathol. Exp. Neurol. 1999;58:129–137. doi: 10.1097/00005072-199902000-00003. [DOI] [PubMed] [Google Scholar]
- MacDonald ME, Barnes G, Srinidhi J, Duyao MP, Ambrose CM, Myers RH, Gray J, Conneally PM, Young A, Penney J. Gametic but not somatic instability of CAG repeat length in Huntington’s disease. J. Med. Gen. 1993;30:982–986. doi: 10.1136/jmg.30.12.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald ME, Vonsattel JP, Shrinidhi J, Couropmitree NN, Cupples LA, Bird ED, Gusella JF, Myers RH. Evidence for the GluR6 gene associated with younger onset age of Huntington’s disease. Neurology. 1999;53:1330–1332. doi: 10.1212/wnl.53.6.1330. [DOI] [PubMed] [Google Scholar]
- MacDonald V, Halliday G. Pyramidal cell loss in motor cortices in Huntington’s disease. Neurobiol. Dis. 2002;10:378–386. doi: 10.1006/nbdi.2002.0528. [DOI] [PubMed] [Google Scholar]
- Mailleux P, Vanderhaeghen JJ. Localization of cannabinoid receptor in the human developing and adult basal ganglia. Higher levels in the striatonigral neurons. Neurosci. Lett. 1992;148:173–176. doi: 10.1016/0304-3940(92)90832-r. [DOI] [PubMed] [Google Scholar]
- Manley K, Shirley TL, Flaherty L, Messer A. Msh2 deficiency prevents in vivo somatic instability of the CAG repeat in Huntington disease transgenic mice. Nat. Gen. 1999;23:471–473. doi: 10.1038/70598. [DOI] [PubMed] [Google Scholar]
- Mann DM, Oliver R, Snowden JS. The topographic distribution of brain atrophy in Huntington’s disease and progressive supranuclear palsy. Acta Neuropathol. 1993;85:553–559. doi: 10.1007/BF00230496. [DOI] [PubMed] [Google Scholar]
- Margolis RL, Ross CA. Diagnosis of Huntington disease. Clin. Chem. 2003;49:1726–1732. doi: 10.1373/49.10.1726. [DOI] [PubMed] [Google Scholar]
- Martindale D, Hackam A, Wieczorek A, Ellerby L, Wellington C, McCutcheon K, Singaraja R, Kazemi-Esfarjani P, Devon R, Kim SU, Bredesen DE, Tufaro F, Hayden MR. Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nature Gen. 1998;18:150–154. doi: 10.1038/ng0298-150. [DOI] [PubMed] [Google Scholar]
- Massouh M, Wallman MJ, Pourcher E, Parent A. The fate of the large striatal interneurons expressing calretinin in Huntington’s disease. Neurosci. Res. 2008;62:216–224. doi: 10.1016/j.neures.2008.08.007. [DOI] [PubMed] [Google Scholar]
- Matsuyama N, Hadano S, Onoe K, Osuga H, Showguchi-Miyata J, Gondo Y, Ikeda JE. Identification and characterization of the miniature pig Huntington’s disease gene homolog: evidence for conservation and polymorphism in the CAG triplet repeat. Genomics. 2000;69:72–85. doi: 10.1006/geno.2000.6317. [DOI] [PubMed] [Google Scholar]
- McGowan DP, van Roon-Mom W, Holloway H, Bates GP, Mangiarini L, Cooper GJ, Faull RL, Snell RG. Amyloid-like inclusions in Huntington’s disease. Neuroscience. 2000;100:677–680. doi: 10.1016/s0306-4522(00)00391-2. [DOI] [PubMed] [Google Scholar]
- McNeil SM, Novelletto A, Srinidhi J, Barnes G, Kornbluth I, Altherr MR, Wasmuth JJ, Gusella JF, MacDonald ME, Myers RH. Reduced penetrance of the Huntington’s disease mutation. Hum. Mol. Gen. 1997;6:775–779. doi: 10.1093/hmg/6.5.775. [DOI] [PubMed] [Google Scholar]
- Medina L, Figueredo-Cardenas G, Reiner A. Differential abundance of superoxide dismutase in interneurons versus projection neurons in patch versus matrix neurons in monkey striatum. Brain Res. 1996;708:59–70. doi: 10.1016/0006-8993(95)01320-2. [DOI] [PubMed] [Google Scholar]
- Metzger S, Rong J, Nguyen HP, Cape A, Tomiuk J, Soehn AS, Propping P, Freudenberg-Hua Y, Freudenberg J, Tong L, Li SH, Li XJ, Riess O. Huntingtin-associated protein-1 is a modifier of the age-at-onset of Huntington’s disease. Hum. Mol. Gen. 2008;17:1137–1146. doi: 10.1093/hmg/ddn003. [DOI] [PubMed] [Google Scholar]
- Metzger S, Saukko M, Van Che H, Tong L, Puder Y, Riess O, Nguyen HP. Age at onset in Huntington’s disease is modified by the autophagy pathway: implication of the V471A polymophism in Atg7. Hum. Gen. 2010;128:453–459. doi: 10.1007/s00439-010-0873-9. [DOI] [PubMed] [Google Scholar]
- Möller T. Neuroinflammation in Huntington’s disease. J. Neural Transm. 2010;117:1001–1008. doi: 10.1007/s00702-010-0430-7. [DOI] [PubMed] [Google Scholar]
- Mormone E, Matarrese P, Tinari A, Cannella M, Maglione V, Farrace MG, Piacentini M, Frati L, Malorni W, Squitieri F. Genotype-dependent priming of self-and xenocanibalism in heterozygous lymphoblasts from patients with Huntington’s disease. J. Neurochem. 2006;98:1090–1099. doi: 10.1111/j.1471-4159.2006.03998.x. [DOI] [PubMed] [Google Scholar]
- Morton AJ, Faull RL, Edwardson JM. Abnormalities in the synaptic vesicle fusion machinery in Huntington’s disease. Brain Res. Bull. 2001;56:111–117. doi: 10.1016/s0361-9230(01)00611-6. [DOI] [PubMed] [Google Scholar]
- Mühlau M, Weindl A, Wohlschläger AM, Gaser C, Städtler M, Valet M, Zimmer C, Kassubek J, Peinemann A. Voxel-based morphometry indicates relative preservation of the limbic prefrontal cortex in early Huntington disease. J. Neural Transm. 2007;114:367–372. doi: 10.1007/s00702-006-0571-x. [DOI] [PubMed] [Google Scholar]
- Myers RH. Huntington’s disease genetics. NeuroRx. 2004;1:255–262. doi: 10.1602/neurorx.1.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers RH, Leavitt J, Farrer LA, Jagadeesh J, McFarlane H, Mastromauro CA, Mark RJ, Gusella JF. Homozygote for Huntington disease. Am. J. Hum. Gen. 1989;45:615–618. [PMC free article] [PubMed] [Google Scholar]
- Nance MA, Mathias-Hagen V, Breningstall G, Wick MJ, McGlennen RC. Analysis of a very large trinucleotide repeat in a patient with juvenile Huntington’s disease. Neurology. 1999;52:392–394. doi: 10.1212/wnl.52.2.392. [DOI] [PubMed] [Google Scholar]
- Nance MA, Myers RH. Juvenile onset Huntington’s disease - clinical and research perspectives. Ment. Retard. Dev. Disabil. Res. Rev. 2001;7:153–157. doi: 10.1002/mrdd.1022. [DOI] [PubMed] [Google Scholar]
- Narain Y, Wyttenbach A, Rankin J, Furlong RA, Rubinsztein DC. A molecular investigation of true dominance in Huntington’s disease. J. Med. Gen. 1999;36:739–746. doi: 10.1136/jmg.36.10.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris PJ, Waldvogel HJ, Faull RL, Love DR, Emson PC. Decreased neuronal nitric oxide synthase messenger RNA and somatostatin messenger RNA in the striatum of Huntington’s disease. Neuroscience. 1996;72:1037–1047. doi: 10.1016/0306-4522(95)00596-x. [DOI] [PubMed] [Google Scholar]
- Nucifora FC, Jr., Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA. Interference by huntingtin and atrophin-1 with CBP-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- Ona VO, Li M, Vonsattel JP, Andrews LJ, Khan SQ, Chung WM, Frey AS, Menon AS, Li XJ, Stieg PE, Yuan J, Penney JB, Young AB, Cha JH, Friedlander RM. Inhibition of caspase-1 slows disease progression in a mouse model of Huntington’s disease. Nature. 1999;399:263–267. doi: 10.1038/20446. [DOI] [PubMed] [Google Scholar]
- Oyanagi K, Takeda S, Takahashi H, Ohama E, Ikuta F. A quantitative investigation of the substantia nigra in Huntington’s disease. Ann. Neurol. 1989;26:13–19. doi: 10.1002/ana.410260103. [DOI] [PubMed] [Google Scholar]
- Parent A, Charara A, Pinault D. Single striatofugal axons arborizing in both pallidal segments and in the substantia nigra in primates. Brain Res. 1995;698:280–284. doi: 10.1016/0006-8993(95)01017-p. [DOI] [PubMed] [Google Scholar]
- Parent A, Smith Y, Filion M, Dumas J. Distinct afferents to the internal and external pallidal segments in the squirrel monkey. Neurosci. Lett. 1989;96:140–144. doi: 10.1016/0304-3940(89)90047-5. [DOI] [PubMed] [Google Scholar]
- Passani LA, Vonsattel JP, Coyle JT. Distribution of N-acetylaspartylglutamate immunoreactivity in human brain and its alteration in neurodegenerative disease. Brain Res. 1997;772:9–22. doi: 10.1016/s0006-8993(97)00784-1. [DOI] [PubMed] [Google Scholar]
- Paulsen JS, Magnotta VA, Mikos AE, Paulson HL, Penziner E, Andreasen NC, Nopoulos PC. Brain structure in preclinical Huntington’s disease. Biol. Psychiatry. 2006;59:57–63. doi: 10.1016/j.biopsych.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Pavese N, Gerhard A, Tai YF, Ho AK, Turkheimer F, Barker RA, Brooks DJ, Piccini P. Microglial activation correlates with severity in Huntington disease: a clinical and PET study. Neurology. 2006;66:1638–1643. doi: 10.1212/01.wnl.0000222734.56412.17. [DOI] [PubMed] [Google Scholar]
- Petersén Å, Larsen KE, Behr GG, Romero N, Przedborski S, Brundin P, Sulzer D. Expanded CAG repeats in exon 1 of the Huntington’s disease gene stimulate dopamine-mediated striatal neuron autophagy and degeneration. Hum. Mol. Gen. 2001;10:1243–1254. doi: 10.1093/hmg/10.12.1243. [DOI] [PubMed] [Google Scholar]
- Petersén Å, Gil J, Maat-Schieman ML, Bjorkqvist M, Tanila H, Araujo IM, Smith R, Popovic N, Wierup N, Norlen P, Li JY, Roos RA, Sundler F, Mulder H, Brundin P. Orexin loss in Huntington’s disease. Hum. Mol. Gen. 2005;14:39–47. doi: 10.1093/hmg/ddi004. [DOI] [PubMed] [Google Scholar]
- Preisinger E, Jordan BM, Kazantsev A, Housman D. Evidence for a recruitment and sequestration mechanism in Huntinton’s disease. Phil. Tran. Royal Soc. Lond. 1999;354:1029–1034. doi: 10.1098/rstb.1999.0455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin ZH, Wang Y, Sapp E, Cuiffo B, Wanker E, Hayden MR, Kegel KB, Aronin N, DiFiglia M. Huntingtin bodies sequester vesicle-associated proteins by a polyproline-dependent interaction. J. Neurosci. 2004;24:269–281. doi: 10.1523/JNEUROSCI.1409-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read AP. Huntington’s disease. Nat. Gen. 1993;4:329–330. doi: 10.1038/ng0893-329. [DOI] [PubMed] [Google Scholar]
- Reading SA, Yassa MA, Bakker A, Dziorny AC, Gourley LM, Yallapragada V, Rosenblatt A, Margolis RL, Aylward EH, Brandt J, Mori S, van Zijl P, Bassett SS, Ross CA. Regional white matter change in pre-symptomatic Huntington’s disease: a diffusion tensor imaging study. Psychiat. Res: Neuroimaging. 2005;140:55–62. doi: 10.1016/j.pscychresns.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Rebec GV, Conroy SK, Barton SJ. Hyperactive striatal neurons in symptomatic Huntington R6/2 mice: variations with behavioral state and repeated ascorbate treatment. Neuroscience. 2006;137:327–336. doi: 10.1016/j.neuroscience.2005.08.062. [DOI] [PubMed] [Google Scholar]
- Reiner A, Albin RL, Anderson KD, D’Amato CJ, Penney JB, Young AB. Differential loss of striatal projection neurons in Huntington’s disease. Proc. Natl. Acad. Sci. USA. 1988;85:5733–5737. doi: 10.1073/pnas.85.15.5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiner A, Anderson KD. The patterns of neurotransmitter and neuropeptide co-occur-rence among striatal projection neurons: conclusions based on recent findings. Brain Res. Rev. 1990;15:251–265. doi: 10.1016/0165-0173(90)90003-7. [DOI] [PubMed] [Google Scholar]
- Reiner A, Medina L, Haber SN. The distribution of dynorphinergic terminals in striatal target regions in comparison to the distribution of substance P-containing and enkephalinergic terminals in monkeys and humans. Neuroscience. 1999;88:775–793. doi: 10.1016/s0306-4522(98)00254-1. [DOI] [PubMed] [Google Scholar]
- Reiner A, Dragatsis I, Zeitlin SO, Goldowitz D. Wild-type huntingtin plays a role in brain development and neuronal survival. Mol. Neurobiol. 2003;28:259–275. doi: 10.1385/MN:28:3:259. [DOI] [PubMed] [Google Scholar]
- Reynolds GP, Garrett NJ. Striatal dopamine and homovanillic acid in Huntington’s Disease. J. Neural Transmission. 1986;65:151–155. doi: 10.1007/BF01256491. [DOI] [PubMed] [Google Scholar]
- Reynolds GP, Pearson SJ. Brain GABA levels in asymptomatic Huntington’s disease. N. Engl. J. Med. 1990;323:682. doi: 10.1056/NEJM199009063231014. [DOI] [PubMed] [Google Scholar]
- Ribaï P, Nguyen K, Hahn-Barma V, Gourfinkel-An I, Vidailhet M, Legout A, Dodé C, Brice A, Dürr A. Psychiatric and cognitive difficulties as indicators of juvenile Huntington disease onset in 29 patients. Arch. Neurol. 2007;64:813–819. doi: 10.1001/archneur.64.6.813. [DOI] [PubMed] [Google Scholar]
- Richfield EK, Herkenham M. Selective vulnerability in Huntington’s disease: preferential loss of cannabinoid receptors in lateral globus pallidus. Ann. Neurol. 1994;36:577–584. doi: 10.1002/ana.410360406. [DOI] [PubMed] [Google Scholar]
- Richfield EK, Maguire-Zeiss KA, Cox C, Gilmore J, Voorn P. Reduced expression of preproenkephalin in striatal neurons from Huntington’s disease patients. Ann. Neurol. 1995 a;37:335–343. doi: 10.1002/ana.410370309. [DOI] [PubMed] [Google Scholar]
- Richfield EK, Maguire-Zeiss KA, Vonkeman HE, Voorn P. Preferential loss of preproenkephalin versus preprotachykinin neurons from the striatum of Huntington’s disease patients. Ann. Neurol. 1995 b;38:852–861. doi: 10.1002/ana.410380605. [DOI] [PubMed] [Google Scholar]
- Richfield EK, O’Brien CF, Eskin T, Shoulson I. Heterogeneous dopamine receptor changes in early and late Huntington’s disease. Neurosci. Lett. 1991;132:121–126. doi: 10.1016/0304-3940(91)90448-3. [DOI] [PubMed] [Google Scholar]
- Roos RA, Pruyt JF, de Vries J, Bots GT. Neuronal distribution in the putamen in Huntington’s disease. J. Neurol. Neurosurg. Psychiat. 1985;48:422–425. doi: 10.1136/jnnp.48.5.422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos RAC. Neuropathology of Huntington’s chorea. In: Vinken PI, Bruyn GW, Klawans HL, editors. Hand Book of Clinical Neurology. Vol. 49. Elsevier; New York: 1986. pp. 315–326. [Google Scholar]
- Rosas HD, Hevelone ND, Zaleta AK, Greve DN, Salat DH, Fischl B. Regional cortical thinning in preclinical Huntington disease and its relationship to cognition. Neurology. 2005;65:745–747. doi: 10.1212/01.wnl.0000174432.87383.87. [DOI] [PubMed] [Google Scholar]
- Rosas HD, Koroshetz WJ, Chen YI, Skeuse C, Vangel M, Cudkowicz ME, Caplan K, Marek K, Seidman LJ, Makris N, Jenkins BG, Goldstein JM. Evidence for more widespread cerebral pathology in early HD: an MRI-based morphometric analysis. Neurology. 2003;60:1615–1620. doi: 10.1212/01.wnl.0000065888.88988.6e. [DOI] [PubMed] [Google Scholar]
- Ross CA, Margolis RL, Rosenblatt A, Ranen NG, Becher MW, Aylward E. Huntington disease and the related disorder, dentatorubral-pallidoluysian atrophy (DRPLA) Medicine (Baltimore) 1997;76:305–338. doi: 10.1097/00005792-199709000-00001. [DOI] [PubMed] [Google Scholar]
- Ross CA. Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington’s disease and related disorders. Neuron. 2002;35:819–822. doi: 10.1016/s0896-6273(02)00872-3. [DOI] [PubMed] [Google Scholar]
- Rubinsztein DC, Leggo J, Coles R, Almqvist E, Biancalana V, Cassiman JJ, Chotai K, Connarty M, Crauford D, Curtis A, Curtis D, Davidson MJ, Differ AM, Dode C, Dodge A, Frontali M, Ranen NG, Stine OC, Sherr M, Abbott MH, Franz ML, Graham CA, Harper PS, Hedreen JC, Hayden MR, et al. Phenotypic characterization of individuals with 30-40 CAG repeats in the Huntington disease (HD) gene reveals HD cases with 36 repeats and apparently normal elderly individuals with 36–39 repeats. Am. J. Hum. Gen. 1996;59:16–22. [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC, Leggo J, Chiano M, Dodge A, Norbury G, Rosser E, Craufurd D. Genotypes at the GluR6 kainate receptor locus are associated with variation in the age of onset in Huntington disease. Proc. Natl. Acad. Sci. USA. 1997;94:3872–3876. doi: 10.1073/pnas.94.8.3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez I, Xu CJ, Juo P, Kakizaka A, Bienis J, Yuan J. Caspase-8 is required for cell death induced by expanded polyglutamine repeats. Neuron. 1999;22:623–633. doi: 10.1016/s0896-6273(00)80716-3. [DOI] [PubMed] [Google Scholar]
- Sapp E, Kegel KB, Aronin N, Hashikawa T, Uchiyama Y, Tohyama K, Bhide PG, Vonsattel JP, DiFiglia M. Early and progressive accumulation of reactive microglia in the Huntington disease brain. J. Neuropath. Exp. Neurol. 2001a;60:161–172. doi: 10.1093/jnen/60.2.161. [DOI] [PubMed] [Google Scholar]
- Sapp E, Penney J, Young A, Aronin N, Vonsattel JP, DiFiglia M. Axonal transport of N-terminal huntingtin suggests early pathology of corticostriatal projections in Huntington’s disease. J. Neuropathol. Exp. Neurol. 1999;58:165–173. doi: 10.1097/00005072-199902000-00006. [DOI] [PubMed] [Google Scholar]
- Sapp E, Ge P, Aizawa H, Bird E, Penny J, Young AB, Vonsattel JP, DiFiglia M. Evidence for a preferential loss of enkephalin immunoreactivity in the external globus pallidus in low grad Huntington’s disease using high resolution image analysis. Neuroscience. 1995;64:397–404. doi: 10.1016/0306-4522(94)00427-7. [DOI] [PubMed] [Google Scholar]
- Sapp E, Schwarz C, Chase K, Bhide PG, Young AB, Penney J, Vonsattel JP, Aronin N, DiFiglia M. Huntingtin localization in brains of normal and Huntington’s disease patients. Ann. Neurol. 1997;42:604–612. doi: 10.1002/ana.410420411. [DOI] [PubMed] [Google Scholar]
- Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- Schiffmann SN, Jacobs O, Vanderhaeghen JJ. Striatal restricted adenosine A2 receptor (RDC8) is expressed by enkephalin but not by substance P neurons: an in situ hybridization histochemistry study. J. Neurochem. 1991;57:1062–1067. doi: 10.1111/j.1471-4159.1991.tb08257.x. [DOI] [PubMed] [Google Scholar]
- Schilling G, Sharp AH, Loev SJ, Wagster MV, Li SH, Stine OC, Ross CA. Expression of the Huntington’s disease (IT15) protein product in HD patients. Hum. Mol. Gen. 1995;4:1365–1371. doi: 10.1093/hmg/4.8.1365. [DOI] [PubMed] [Google Scholar]
- Seizinger BR, Liebisch DC, Kish SJ, Arendt RM, Hornykiewicz O, Herz A. Opioid peptides in Huntington’s disease: alterations in prodynorphin and proenkephalin system. Brain Res. 1986;378:405–408. doi: 10.1016/0006-8993(86)90946-7. [DOI] [PubMed] [Google Scholar]
- Selemon LD, Rajkowska G, Goldman-Rakic PS. Evidence for progression in frontal cortical pathology in late-stage Huntington’s disease. J. Comp. Neurol. 2004;468:190–204. doi: 10.1002/cne.10938. [DOI] [PubMed] [Google Scholar]
- Semaka A, Collins JA, Hayden MR. Unstable familial transmissions of Huntington disease alleles with 27-35 CAG repeats (intermediate alleles) Am. J. Med. Gen. B Neuropsychiatr. Gen. 2010;153B:314–320. doi: 10.1002/ajmg.b.30970. [DOI] [PubMed] [Google Scholar]
- Seto-Ohshima A, Emson PC, Lawson E, Mountjoy CQ, Carrasco LH. Loss of matrix calcium binding protein containing neurons in Huntington’s disease. Lancet. 1988;1:1252–1255. doi: 10.1016/s0140-6736(88)92073-9. [DOI] [PubMed] [Google Scholar]
- Sharp AH, Loev SJ, Schiling G, Li SH, Li XJ, Bao J, Wagster MV, Kotzuk JA, Steiner JP, Lo A, Hedreen J, Sisodia S, Snyder SH, Dawson TM, Ryugo DDK, Ross CA. Widespread expression of Huntington’s disease gene (IT15) protein product. Neuron. 1995;14:1065–1074. doi: 10.1016/0896-6273(95)90345-3. [DOI] [PubMed] [Google Scholar]
- Sharp AH, Ross CA. Neurobiology of Huntington’s disease. Neurobiol. Dis. 1996;3:3–15. doi: 10.1006/nbdi.1996.0002. [DOI] [PubMed] [Google Scholar]
- Shieh PB, Hu SC, Bobb K, Timmusk T, Ghosh A. Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron. 1998;20:727–740. doi: 10.1016/s0896-6273(00)81011-9. [DOI] [PubMed] [Google Scholar]
- Sieradzan KA, Mechan AO, Jones L, Wanker EE, Nukina N, Mann DM. Huntington’s disease intranuclear inclusions contain truncated, ubiquitinated huntingtin protein. Exp. Neurol. 1999;156:92–99. doi: 10.1006/exnr.1998.7005. [DOI] [PubMed] [Google Scholar]
- Silvestroni A, Faull RL, Strand AD, Möller T. Distinct neuroinflammatory profile in post-mortem human Huntington’s disease. Neuroreport. 2009;20:1098–1103. doi: 10.1097/WNR.0b013e32832e34ee. [DOI] [PubMed] [Google Scholar]
- Sisodia SS. Nuclear inclusion in glutamine repeat disorders: are they pernicious, coincidental or beneficial? Cell. 1998;95:1–4. doi: 10.1016/s0092-8674(00)81743-2. [DOI] [PubMed] [Google Scholar]
- Smith R, Chung H, Rundquist S, Maat-Schieman ML, Colgan L, Englund E, Liu YJ, Roos RA, Faull RL, Brundin P, Li JY. Cholinergic neuronal defect without cell loss in Huntington’s disease. Hum. Mol. Gen. 2006;15:3119–3131. doi: 10.1093/hmg/ddl252. [DOI] [PubMed] [Google Scholar]
- Smith R, Klein P, Koc-Schmitz Y, Waldvogel HJ, Faull RL, Brundin P, Plomann M, Li JY. Loss of SNAP-25 and rabphilin 3a in sensory-motor cortex in Huntington’s disease. J. Neurochem. 2007;103:115–123. doi: 10.1111/j.1471-4159.2007.04703.x. [DOI] [PubMed] [Google Scholar]
- Snell RG, MacMillan JC, Cheadle JP, Fenton I, Lazarou LP, Davies P, MacDonald ME, Gusella JF, Harper PS, Shaw DJ. Relationship between trinucleotide repeat expansion and phenotypic variation in Huntington’s disease. Nat. Gen. 1993;4:393–397. doi: 10.1038/ng0893-393. [DOI] [PubMed] [Google Scholar]
- Soneson C, Fontes M, Zhou Y, Denisov V, Paulsen JS, Kirik D, Petersén, AHuntington Study Group PREDICT-HD investigators Early changes in the hypothalamic region in prodromal Huntington disease revealed by MRI analysis. Neurobiol. Dis. 2010;40:531–543. doi: 10.1016/j.nbd.2010.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotrel A, Paskevich PA, Kiely DK, Bird ED, Williams RS, Myers RH. Morphometric analysis of the prefrontal cortex in Huntington’s disease. Neurology. 1991;41:1117–1123. doi: 10.1212/wnl.41.7.1117. [DOI] [PubMed] [Google Scholar]
- Sotrel A, Williams RS, Kaufmann WE, Myers RH. Evidence for neuronal degeneration and dendritic plasticity in cortical pyramidal neurons of Huntington’s disease: a quantitative Golgi study. Neurology. 1993;43:2088–2096. doi: 10.1212/wnl.43.10.2088. [DOI] [PubMed] [Google Scholar]
- Spokes EGS. Neurochemical alterations in Huntington’s chorea: a study of post-mortem brain tissue. Brain. 1980;103:179–210. doi: 10.1093/brain/103.1.179. [DOI] [PubMed] [Google Scholar]
- Spokes EGS, Garrett NJ, Rossor MN, Iversen LL. Distribution of GABA in postmortem brain tissue from control, psychotic and Huntington’s chorea subjects. J. Neurol. Sci. 1980;48:303–313. doi: 10.1016/0022-510x(80)90103-3. [DOI] [PubMed] [Google Scholar]
- Squitieri F, Gellera C, Cannella M, Mariotti C, Ciskaghi G, Rubinsztein DC, Almqvist EW, Turner D, Bachoud-Levi AC, Simpson SA, Delatycki M, Maglione V, Hayden MR, DiDonato S. Homozygosity for CAG mutation in Huntington disease is associated with a more severe clinical course. Brain. 2003;126:946–955. doi: 10.1093/brain/awg077. [DOI] [PubMed] [Google Scholar]
- Squitieri F, Frati L, Ciarmiello A, Lastoria S, Quarrell O. Juvenile Huntington’s disease: does a dosage-effect pathogenic mechanism differ from the classical adult disease? Mech. Aging Dev. 2006;127:208–212. doi: 10.1016/j.mad.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Squitieri F, Cannella M, Simonelli M, Sassone J, Martino T, Venditti E, Ciammola A, Colonnese C, Frati L, Ciarmiello A. Distinct brain volume changes correlating with clinical stage, disease progression rate, mutation size, and age at onset prediction as early biomarkers of brain atrophy in Huntington’s disease. CNS Neurosci. Ther. 2009;15:1–11. doi: 10.1111/j.1755-5949.2008.00068.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Squitieri F, Falleni A, Cannella M, Orobello S, Fulceri F, Lenzi P, Fornai F. Abnormal morphology of peripheral cell tissues from patients with Huntington disease. J. Neural Transm. 2010;117:77–83. doi: 10.1007/s00702-009-0328-4. [DOI] [PubMed] [Google Scholar]
- Storey E, Beal MF. Neurochemical substrates of rigidity and chorea in Huntington’s disease. Brain. 1993;116:1201–1222. doi: 10.1093/brain/116.5.1201. [DOI] [PubMed] [Google Scholar]
- Strong TV, Tagle DA, Valdes JM. Widespread expression of the human and rat Huntington’s disease gene in brain and nonneural tissues. Nat. Gen. 1993;5:259–265. doi: 10.1038/ng1193-259. [DOI] [PubMed] [Google Scholar]
- Swami M, Hendricks AE, Gillis T, Massood T, Mysore J, Myers RH, Wheeler VC. Somatic expansion of the Huntington’s disease CAG repeat in the brain is associated with an earlier age of disease onset. Hum. Mol. Gen. 2009;18:3039–3047. doi: 10.1093/hmg/ddp242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai YF, Pavese N, Gerhard A, Tabrizi SJ, Barker RA, Brooks DJ, Piccini P. Imaging microglial activation in Huntington’s disease. Brain Res. Bull. 2007;72:148–151. doi: 10.1016/j.brainresbull.2006.10.029. [DOI] [PubMed] [Google Scholar]
- Tang TS, Guo C, Wang H, Chen X, Bezprozvanny I. Neuroprotective effects of inositol 1,4,5-trisphosphate receptor C-terminal fragment in a Huntington’s disease mouse mode. J. Neurosci. 2009;29:1257–1266. doi: 10.1523/JNEUROSCI.4411-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao X, Finkbeiner S, Arnold DB, Shaywitz A, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- Telenius H, Kremer B, Goldberg YP, Theilmann J, Andrew SE, Zeisler J, Adam S, Greenberg C, Ives EJ, Clarke LA, Hayden MR. Somatic and gonadal mosaicism of the Huntington disease gene CAG repeat in the brain and sperm. Nat. Gen. 1994;6:409–414. doi: 10.1038/ng0494-409. [DOI] [PubMed] [Google Scholar]
- Thomas LB, Gates DJ, Richfield EK, O’Brien TF, Schweitzer JB, Steindler DA. DNA end labeling (TUNEL) in Huntington’s disease and other neuropathological conditions. Exp. Neurol. 1995;133:265–272. doi: 10.1006/exnr.1995.1029. [DOI] [PubMed] [Google Scholar]
- Thu DC, Oorschot DE, Tippett LJ, Nana AL, Hogg VM, Synek BJ, Luthi-Carter R, Waldvogel HJ, Faull RL. Cell loss in the motor and cingulate cortex correlates with symptomatology in Huntington’s disease. Brain. 2010;133:1094–1110. doi: 10.1093/brain/awq047. [DOI] [PubMed] [Google Scholar]
- Timmers HJ, Swaab DF, van de Nes JA, Kremer HP. Somatostatin 1-12 immuno-reactivity is decreased in the hypothalamic lateral tuberal nucleus of Huntington’s disease patients. Brain Res. 1996;728:141–148. doi: 10.1016/0006-8993(96)00080-7. [DOI] [PubMed] [Google Scholar]
- Tippett LJ, Waldvogel HJ, Thomas SJ, Hogg VM, van Roon-Mom W, Synek BJ, Graybiel AM, Faull RL. Striosomes and mood dysfunction in Huntington’s disease. Brain. 2007;130:206–221. doi: 10.1093/brain/awl243. [DOI] [PubMed] [Google Scholar]
- van Roon-Mom WM, Hogg VM, Tippett LJ, Faull RL. Aggregate distribution in frontal and motor cortex in Huntington’s disease brain. Neuroreport. 2006;17:667–670. doi: 10.1097/00001756-200604240-00022. [DOI] [PubMed] [Google Scholar]
- van Roon-Mom WM, Reid SJ, Jones AL, MacDonald ME, Faull RL, Snell RG. Insoluble TATA-binding protein accumulation in Huntington’s disease cortex. Mol. Brain Res. 2002;109:1–10. doi: 10.1016/s0169-328x(02)00450-3. [DOI] [PubMed] [Google Scholar]
- Varani K, Abbracchio MP, Cannella M, Cislaghi G, Giallonardo P, Mariotti C, Cattabriga E, Cattabeni F, Borea PA, Squitieri F, Cattaneo E. Aberrant A2A receptor function in peripheral blood cells in Huntington’s disease. FASEB J. 2003;17:2148–2150. doi: 10.1096/fj.03-0079fje. [DOI] [PubMed] [Google Scholar]
- Veitch NJ, Ennis M, McAbney JP, Shelbourne PF, Monckton DG. Inherited CAG. CTG allele length is a major modifier of somatic length variability in Huntington’s disease. DNA Repair. 2007;6:789–796. doi: 10.1016/j.dnarep.2007.01.002. [DOI] [PubMed] [Google Scholar]
- Velier J, Kim M, Schwarz C, Kim TW, Sapp E, Chase K, Aronin N, DiFiglia M. Wild-type and mutant huntingtins function in vesicle trafficking in the secretory and endocytic pathways. Exp. Neurol. 1998;152:34–40. doi: 10.1006/exnr.1998.6832. [DOI] [PubMed] [Google Scholar]
- Vis JC, Nicholson LF, Faull RL, Evans WH, Severs NJ, Green CR. Connexin expression in Huntington’s diseased human brain. Cell. Biol. Int. 1998;11–12:837–847. doi: 10.1006/cbir.1998.0388. [DOI] [PubMed] [Google Scholar]
- Vis JC, Schipper E, de Boer-van Huizen RT, Verbeek MM, de Waal RM, Wesseling P, ten Donkelaar HJ, Kremer B. Expression pattern of apoptosis-related markers in Huntington’s disease. Acta Neuropath. 2005;109:321–328. doi: 10.1007/s00401-004-0957-5. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP. Neuropathological classification of Huntington’s disease. J. Neuropathol. Exp. Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP. Huntington disease models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115:55–69. doi: 10.1007/s00401-007-0306-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol. Exp. Neurol. 1998;57:369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- Wacker JL, Huang SY, Steele AD, Aron R, Lotz GP, Nguyen Q, Giorgini F, Roberson ED, Lindquist S, Masliah E, Muchowski PJ. Loss of Hsp70 exacerbates pathogenesis but not levels of fibrillar aggregates in a mouse model of Huntington’s disease. J. Neurosci. 2009;29:9104–1914. doi: 10.1523/JNEUROSCI.2250-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker FO, Young AB, Penney JB, Dovorini-Zis K, Shoulson I. Benzodiazepine and GABA receptors in early Huntington’s disease. Neurology. 1984;34:1237–1240. doi: 10.1212/wnl.34.9.1237. [DOI] [PubMed] [Google Scholar]
- Walker FO. Huntington’s disease. Lancet. 2007;369:118–218. doi: 10.1016/S0140-6736(07)60111-1. [DOI] [PubMed] [Google Scholar]
- Warby SC, Montpetit A, Hayden AR, Carroll JB, Butland SL, Visscher H, Collins JA, Semaka A, Hudson TJ, Hayden MR. CAG expansion in the Huntington disease gene is associated with a specific and targetable predisposing haplogroup. Am. J. Hum. Gen. 2009;84:351–366. doi: 10.1016/j.ajhg.2009.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks RA, Piccini P, Harding AE, Brooks DJ. Striatal D1 and D2 dopamine receptor loss in asymptomatic mutation carriers of Huntington’s disease. Ann. Neurol. 1996;40:49–54. doi: 10.1002/ana.410400110. [DOI] [PubMed] [Google Scholar]
- Wexler NS, Young AB, Tanzi RE, Travers H, Starosta-Rubinstein S, Penney JB, Snodgrass SR, Shoulson I, Gomez F, Ramos Arroyo MA, Penchaszadeh GK, Moreno H, Gibbons K, Faryniarz A, Hobbs W, Anderson MA, Bonilla E, Conneally PM, Gusella JF. Homozygotes for Huntington disease. Nature. 1987;326:194–197. doi: 10.1038/326194a0. [DOI] [PubMed] [Google Scholar]
- Wexler NS, Lorimer J, Porter J, Gomez F, Moskowitz C, Shackell E, Marder K, Penchaszadeh G, Roberts SA, Gayán J, Brocklebank D, Cherny SS, Cardon LR, Gray J, Dlouhy SR, Wiktorski S, Hodes ME, Conneally PM, Penney JB, Gusella J, Cha JH, Irizarry M, Rosas D, Hersch S, Hollingsworth Z, MacDonald M, Young AB, Andresen JM, Housman DE, De Young MM, Bonilla E, Stillings T, Negrette A, Snodgrass SR, Martinez-Jaurrieta MD, Ramos-Arroyo MA, Bickham J, Ramos JS, Marshall F, Shoulson I, Rey GJ, Feigin A, Arnheim N, Acevedo-Cruz A, Acosta L, Alvir J, Fischbeck K, Thompson LM, Young A, Dure L, O’Brien CJ, Paulsen J, Brickman A, Krch D, Peery S, Hogarth P, Higgins DS, Jr., Landwehrmeyer BUS, Venezuela Collaborative Research Project Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington’s disease age of onset. Proc. Natl. Acad. Sci. USA. 2004;101:3498–3503. doi: 10.1073/pnas.0308679101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler VC, White JK, Gutekunst CA, Vrbanac V, Weaver M, Li XJ, Li SH, Yi H, Vonsattel JP, Gusella JF, Hersch S, Auerbach W, Joyner AL, MacDonald ME. Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Hum. Mol. Gen. 2000;9:503–513. doi: 10.1093/hmg/9.4.503. [DOI] [PubMed] [Google Scholar]
- Wheeler VC, Persichetti F, McNeil SM, Mysore JS, Mysore SS, MacDonald ME, Myers RH, Gusella JF, Wexler N, SUS-Venezuela Collaborative Research Group Factors associated with HD CAG repeat instability in Huntington disease. J. Med. Gen. 2007;44:695–701. doi: 10.1136/jmg.2007.050930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson FL, Man NT, Manilal SB, Thomas P, Neal JW, Harper PS, Jones AL, Morris GE. Localization of rabbit huntingtin using a new panel of monoclonal antibodies. Mol. Brain Res. 1999;69:10–20. doi: 10.1016/s0169-328x(99)00097-2. [DOI] [PubMed] [Google Scholar]
- Wilson RS, Como PG, Garron DC, Klawans HL, Barr A, Klawans D. Memory failure in Huntington’s disease. J. Clin. Exp. Neuropsychol. 1987;9:147–154. doi: 10.1080/01688638708405354. [DOI] [PubMed] [Google Scholar]
- Wolf RC, Sambataro F, Vasic N, Schönfeldt-Lecuona C, Ecker D, Landwehrmeyer B. Aberrant connectivity of lateral prefrontal networks in presymptomatic Huntington’s disease. Exp. Neurol. 2008;213:137–144. doi: 10.1016/j.expneurol.2008.05.017. [DOI] [PubMed] [Google Scholar]
- Wood JD, McLaughlin JC, Harper PS, Lowenstein PR, Jones AL. Partial characterization of murine huntingtin and apparent variations in the subcellular localization of huntingtin in human, mouse and rat brain. Hum. Mol. Gen. 1996;5:481–487. doi: 10.1093/hmg/5.4.481. [DOI] [PubMed] [Google Scholar]
- Wu Y, Richard S, Parent A. The organization of the striatal output system: a single-cell juxtacellular labeling study in the rat. Neurosci. Res. 2000;38:49–62. doi: 10.1016/s0168-0102(00)00140-1. [DOI] [PubMed] [Google Scholar]
- Yohrling GJ, 4th, Jiang GC, DeJohn MM, Miller DW, Young AB, Vrana KE, Cha JH. Analysis of cellular, transgenic and human models of Huntington’s disease reveals tyrosine hydroxylase alterations and substantia nigra neuropathology. Brain Res. Mol. Brain Res. 2003;119:28–36. doi: 10.1016/j.molbrainres.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Zech M, Roberts GW, Bogerts B, Crow TJ, Polak JM. Neuropeptides in the amygdala of controls, schizophrenics and patients suffering from Huntington’s chorea: an immunohistochemical study. Acta Neuropathol. 1986;71:259–266. doi: 10.1007/BF00688048. [DOI] [PubMed] [Google Scholar]
- Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P, Hayden MR, Raymond LA. Increased sensitivity to N-methyl-d-aspartate receptor-mediated exciitotoxicity in a mouse model of Huntington’s disease. Neuron. 2002;33:849–860. doi: 10.1016/s0896-6273(02)00615-3. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Liber D, Ramos C, Tarditi A, Rigamonti D, Tartari M, Valenza M, Cattaneo E. Progressive loss of BDNF in a mouse model of Huntington’s disease and rescue by BDNF delivery. Pharmacol. Res. 2005;52:133–139. doi: 10.1016/j.phrs.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Zucker B, Kama JA, Kuhn A, Thu D, Orlando LR, Dunah AW, Gokce O, Taylor DM, Lambeck J, Friedrich B, Lindenberg KS, Faull RL, Weiller C, Young AB, Luthi-Carter R. Decreased Lin7b expression in layer 5 pyramidal neurons may contribute to impaired corticostriatal connectivity in huntington disease. J. Neuropathol. Exp. Neurol. 2010;69:880–895. doi: 10.1097/NEN.0b013e3181ed7a41. [DOI] [PMC free article] [PubMed] [Google Scholar]