

Abstract
Background:
We previously reported increased current density through L-type voltage-gated Ca2+ (CaV1) channels in inferior colliculus (IC) neurons during alcohol withdrawal. However, the molecular correlate of this increased CaV1 current is currently unknown.
Methods:
Rats received three daily doses of ethanol every 8 hours for 4 consecutive days; control rats received vehicle. The IC was dissected at various time intervals following alcohol withdrawal, and the mRNA and protein levels of the CaV1.3 and CaV1.2 α1 subunits were measured. In separate experiments, rats were tested for their susceptibility to alcohol withdrawal–induced seizures (AWS) 3, 24, and 48 hours after alcohol withdrawal.
Results:
In the alcohol-treated group, AWS were observed 24 hours after withdrawal; no seizures were observed at 3 or 48 hours. No seizures were observed at any time in the control-treated rats. Compared to control-treated rats, the mRNA level of the CaV1.3 α1 subunit was increased 1.4-fold, 1.9-fold, and 1.3-fold at 3, 24, and 48 hours, respectively. In contrast, the mRNA level of the CaV1.2 α1 subunit increased 1.5-fold and 1.4-fold at 24 and 48 hours, respectively. At 24 hours, Western blot analyses revealed that the levels of the CaV1.3 and CaV1.2 α1 subunits increased by 52% and 32%, respectively, 24 hours after alcohol withdrawal. In contrast, the CaV1.2 and CaV1.3 α1 subunits were not altered at either 3 or 48 hours during alcohol withdrawal.
Conclusions:
Expression of the CaV1.3 α1 subunit increased in parallel with AWS development, suggesting that altered L-type CaV1.3 channel expression is an important feature of AWS pathogenesis.
Keywords: alcohol withdrawal seizures, Cacna1d mRNA, Cacna1c mRNA, Cav1.2 α1 subunit, Cav1.3 α1 subunit
Introduction
Neuronal hyperexcitability following the cessation of chronic alcohol intake (i.e. alcohol withdrawal) often increases susceptibility to generalized tonic-clonic seizures. The inferior colliculus (IC) plays a central role in auditory information processing. In rodents, the IC is critically involved in the initiation of acoustically-evoked reflex seizures (i.e. audiogenic seizures, or AGSs) during alcohol withdrawal (Frye et al., 1983; Eckardt et al., 1986; McCown and Breese, 1990, 1993; Faingold and Riaz, 1995; Chakravarty and Faingold, 1998). Electrophysiology studies revealed increased firing of IC neurons both prior to and during AGSs in rats subjected to alcohol withdrawal (Faingold and Riaz, 1995; Chakravarty and Faingold, 1998). Similarly, in vitro studies revealed elevated excitability of IC neurons following alcohol withdrawal (N’Gouemo et al., 1996; Evans et al., 2000; Faingold et al., 2000). These functional findings support the importance of IC neurons in the networks that underlie alcohol withdrawal–induced seizures (AWSs; Faingold et al., 1998).
Voltage-gated Ca2+ (CaV) channels play an important role in the mechanisms that underlie the neuronal hyperexcitability that leads to AWSs (Whittington et al., 1993). For example, blocking L-type CaV channels (i.e. the CaV1 family of Ca2+ channels) suppressed AGSs in rats subjected to alcohol withdrawal (Little et al., 1986). These findings suggest that remodeling of CaV1 channels—at least in the IC—is a key factor associated with AGS initiation during alcohol withdrawal. Consistent with this hypothesis, we found that the current density of CaV1 channels was elevated in IC neurons of rats subjected to alcohol withdrawal, and this increased current was associated with enhanced seizure susceptibility (N’Gouemo and Morad, 2003).
In the mammalian brain, CaV1 channels are comprised of two main isoforms, CaV1.3 and CaV1.2 channels; however, which isoform accounts for the increased current density in IC neurons during alcohol withdrawal is currently unknown. We previously reported that the increase in current density through CaV1 channels observed 24 hours after alcohol withdrawal (i.e. when AWS susceptibility peaks) follows a parallel time course with the occurrence of seizure susceptibility; moreover, by 48 hours after withdrawal (when the animal is no longer susceptible to AWS), CaV1 currents return to control levels (N’Gouemo and Morad, 2003). A temporal study of the onset of AGS found that seizure susceptibility is absent prior to the 7th hour of alcohol withdrawal, but progressively increases thereafter; thus, susceptibility peaks at 24 hours and is absent again at 48 hours (Faingold, 2008). Because alcohol withdrawal preferentially affects CaV1 channels, we examined whether the expression of CaV1.3 and/or CaV1.2 L-type channels is altered in IC neurons following alcohol withdrawal and whether these increases are correlated with the onset of AWS susceptibility.
Methods
Animals
Male Sprague-Dawley rats (250–300g; Taconic) were used for these experiments. The animals were maintained in a temperature- and humidity-controlled room on a 12hr light/dark cycle with food and water available ad libitum. All efforts were made to minimize the number of animals used in these experiments. All experimental procedures were approved by the Georgetown University Animal Care and Use Committee and were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Ethanol Administration
Ethanol (from a 95% stock solution) was administered by intragastric intubation in a 30% (v/v) solution in ISOMIL Infant Formula Concentrate (the ISOMIL formula was diluted 1:1 with water). Ethanol was administered as three daily doses delivered every 8 hours for 4 days, and the level of intoxication was evaluated using a standard behavior rating scale (N’Gouemo et al., 2006; Faingold, 2008). The first dose of ethanol was 5g/kg body weight, and subsequent doses were adjusted for each animal in order to achieve a moderate (i.e. accentuated staggering gait and considerable elevation of the pelvis) but not severe (i.e. lethargy without pelvic or abdominal elevation) degree of ataxia (Faingold, 2008); the amount of ethanol administered daily ranged from 9 to 15g/kg. On the fourth day, ethanol treatment was terminated after the second dose. The behavioral signs of ethanol withdrawal include hyperactivity, tremors, tail spasticity, spontaneous seizures (myoclonus and forelimb clonic seizures), and acoustically-evoked startle response and seizures (N’Gouemo et al., 2006; Faingold, 2008). The control-treated animals were maintained under similar conditions but were given the ISOMIL formula without ethanol. Weight loss and mortality were negligible in both the ethanol-treated and control-treated groups.
Seizure Testing
To determine susceptibility to develop AGS, two animals from each group were randomly selected and tested for seizures 2–4 hours (the 3-hour group), 22–24 hours (the 24-hour group), and 46–48 hours (the 48-hour group) after ethanol withdrawal. The acoustic stimulus consisted of 100–110 decibel sound pressure level pure tones (Med Associates) or mixed sounds (delivered via an electrical bell) that were presented until seizure was elicited (or for 60sec if no seizure activity was observed), as described previously (Riaz and Faingold, 1994). Animals that did not exhibit a seizure during the first trial were tested again two hours later. The animals used in the molecular studies were not subjected to acoustically-evoked seizure testing because evoked seizures can induce a long-lasting increase in extracellular GABA levels (Ueda and Tsuru, 1995), which in turn can alter the expression of CaV1 channels. In addition, using animals that were not exposed to stimulus-induced seizures prevented the effects of seizure severity and/or duration on the expression of CaV1 channels.
Blood Ethanol Levels
In a separate set of experiments, blood ethanol concentration (BEC) was measured in the control group and in the ethanol-treated group 3, 24, and 48 hours during ethanol withdrawal. The rats were anesthetized (50mg/kg Nembutal; i.p.), and blood was extracted by intracardiac sampling using a 21-gauge needle. Serum ethanol concentration was measured using an Analox model GM7 analyzer (Analox Instruments).
Western Blot Analysis
At 3, 24, and 48 hours during ethanol withdrawal, the animals were deeply anesthetized with pentobarbital (100mg/kg; i.p.), and the colliculi were immediately dissected and stored at –80°C until use. Tissue homogenates from each animal were lysed in 50mM Tris-HCl (pH 7.4), 300mM NaCl, 1% octylphenoxypolyethoxyethanol (Sigma-Aldrich), 10% glycerol, 1mM ethylenediaminetetracetic acid, and 1mM Na3VO4. The homogenates were cleared by centrifugation (13 800 x g, 4°C, 30min). The supernatants were transferred to sterile microtubes and stored at -80°C until use. Protein concentration in the supernatants was measured using the bicinchoninic acid assay and a Bio-Rad Model 680 spectrophotometer. For each sample, 60 μg of total protein was separated by electrophoresis in a 7.5% sodium dodecyl sulfate-polyacrylamide gel and electro-transferred to a nitrocellulose membrane (Bio-Rad). The membranes were blocked in Odyssey blocking buffer (LI-COR Biosciences) for one hour and then probed overnight at 4°C with primary rabbit antibodies against either the α1D subunit (CaV1.3-α1; 1:200; Alomone Labs) or the α1C subunit (CaV1.2-α1; 1:200; Alomone Labs). The membranes were also incubated with anti-glyceraldehyde 3-phosphate dehydrogenase antibody (1:10 000; Thermo Fisher Scientific) overnight at 4°C as an internal control. The membranes were probed with goat anti-mouse IRDye800 (1:10 000; LI-COR Biosciences) and goat anti-rabbit IR-Dye680 (1:10 000; LI-COR Biosciences) for 1 hour at room temperature, then scanned using an Odyssey Fc imager (LI-COR Biosciences).
RNA Isolation and Quantitative Real-Time PCR
To quantify the mRNA levels of the CaV1.3 and CaV1.2 α1 subunits in the IC, rats were subjected to the 4-day ethanol intoxication paradigm (or control treatment), then anesthetized 3, 24, and 48 hours after ethanol withdrawal. The brains were removed, and the IC was immediately dissected and stored at -80°C until use. Total RNA was extracted from the IC using the RNeasy lipid tissue mini kit in accordance with the manufacturer’s instructions (Qiagen). RNA concentration was measured using an Eppendorf Biophotometer, and the samples were stored at -20°C. Complementary DNA was synthesized from 1 μg total RNA by reverse transcription using the Quantitect reverse transcription kit (Qiagen) and stored at -20°C. For each gene of interest, real-time polymerase chain reactions (PCRs) were performed in duplicate using the TaqMan fast universal PCR master kit (Applied Biosystems) in a StepOnePlus real-time PCR machine system (Applied Biosystems). The specific primers were as follows: CaV1.3 α1 subunit (rat Cacna1d); Rn.PT.49a.11686797.g, GenBank accession no. NM_017298 and CaV1.2 α1 subunit (rat Cacna1c); and Rn.PT.49a.9591539.g, GenBank accession no. NM_012517 (Integrated DNA Technologies). The amplification efficiencies of Cacna1d and Cacna1c were determined relative to a β-actin internal control using cycle conditions of denaturation at 95°C for 12sec followed by 40 cycles of 95°C for 1sec and 60°C for 12sec. After amplification, a crossing threshold (CT) value (representing the number of cycles required to reach a specific level of fluorescence) was determined for each sample.
Data Analysis
The protein levels of the CaV1.3 α1 and CaV1.2 subunits were measured using densitometry of the corresponding bands (Odyssey Image Studio v3.2 software, LI-COR Biosciences). The integrated intensity of each band was normalized to the intensity of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The mean of the control values was calculated, and the normalized intensity value of each control and alcohol-treated sample was expressed as a ratio relative to the mean control value. Fold change in mRNA expression was calculated using StepOne software with the equation 2-∆∆C T, where ∆∆CT is the relative change of a given mRNA (Livak and Schnittegen, 2001). To obtain the 2-∆∆C T value for each sample, the CT value for Cacna1c and Cacna1d mRNA was first normalized to β-actin, and the resulting ∆CT was calculated as follows: [∆CT Cacna1d = CT Cacna1d - CT β-actin] for Cacna1d (CaV1.3) and [∆CT Cacna1c = CT Cacna1c - CT β-actin] for Cacna1c (CaV1.2). Changes in Cacna1d and Cacna1c and mRNA during alcohol withdrawal were calculated relative to the control group using the following equation: [∆∆CT Cacna1d = (CT Cacna1d - CT β-actin)ethanol withdrawal – (CT Cacna1d - CT β-actin)control group] or [∆∆CT Cacna1c = (CT Cacna1c - CT β-actin)ethanol withdrawal – (CT Cacna1c - CT β-actin)control group]. The final mRNA levels were determined by calculating 2-∆∆CT. The prevalence of each type of seizure was analyzed using the chi-square (χ2) test. To evaluate differences in the protein and mRNA levels, one-way analyses of variance (ANOVA; F) followed by a Bonferroni or Student-Newman-keuls post hoc test were performed. Differences were considered significant at p < 0.05, and the summary data are presented as the mean ± standard error of the mean or as a percentage (for the prevalence of wild running seizures [WRSs] and tonic-clonic seizures [clonus]).
Results
Ethanol Withdrawal Enhances the Susceptibility to Seizures
Figure 1 summarizes the susceptibility of each group to develop AGSs following control treatment or at 3, 24, and 48 hours during ethanol withdrawal. AGSs consisted of jumping and/or WRSs, which occasionally evolved into bouncing generalized clonus; 24 hours after ethanol withdrawal, these seizure types were observed in 100% (χ2 = 196, p < 0.001 vs. control) and 60% (χ2 = 83, p < 0.001 vs. control) of ethanol-treated rats, respectively. This susceptibility to AGS was transient, as these seizures were observed only in the 24-hour group, and not in the 3-hour or 48-hour group (Figure 1). No seizures were observed in the control group. ANOVA revealed that the BEC group means were significantly different (F = 40, p < 0.001). The BECs were significantly (t = 9, p < 0.05) elevated in the 3-hour group (0.26±0.04g/dL, n = 4) compared with those of the 24 (0.01±0.001g/dL, n = 4) and 48 (0.01±0.001g/dL, n = 4) hour ethanol groups and the control group (0.005±0.001g/dL, n = 4).
Figure 1.

Prevalence of acoustically-evoked generalized seizures following ethanol withdrawal. These seizures consisted of wild running seizures (WRSs) or bouncing generalized tonic-clonic seizures (clonus [C]). At 24 hours after ethanol withdrawal, 100% and 60% of animals (n = 10) exhibited WRSs and clonus, respectively. No seizures were observed at 3 (n = 6) or 48 hours (n = 6) following ethanol withdrawal, and no seizures also were observed in the control group (n = 6, *p < 0.001 versus control; chi-square test).
Ethanol Withdrawal Upregulates CaV1 Channels at the mRNA Level
Figure 2 summarizes the mRNA levels of the CaV1.3 (Figure 2A) and CaV1.2 (Figure 2B) α1 subunits (Cacna1c and Cacna1d mRNA, respectively) in IC tissues obtained from the control and ethanol-treated groups. The expression of both Cacna1d (F = 8, p < 0.001) and Cacna1c (F = 6, p < 0.01) mRNA were increased significantly in the IC following ethanol withdrawal. Interestingly, the expression of Cacna1d mRNA was significantly increased at the 24-hour time point (2-∆∆CT = 1.9±0.2, n = 6, t = 4, P < 0.05), but not at the 3-hour time points (2-∆∆CT = 1.4±0.1, n = 6)- or 48 (2-∆∆CT = 1.3±0.2, n = 6) during ethanol withdrawal, in comparison to the the control group (2-∆∆CT = 1.0±0.1, n = 6; Figure 2A). The level of Cacna1d mRNA was also significantly increased 24 hours during ethanol withdrawal compared to the 3-hour (t = 3, p < 0.05) and 48-hour (t = 4, p < 0.05) groups.
Figure 2.
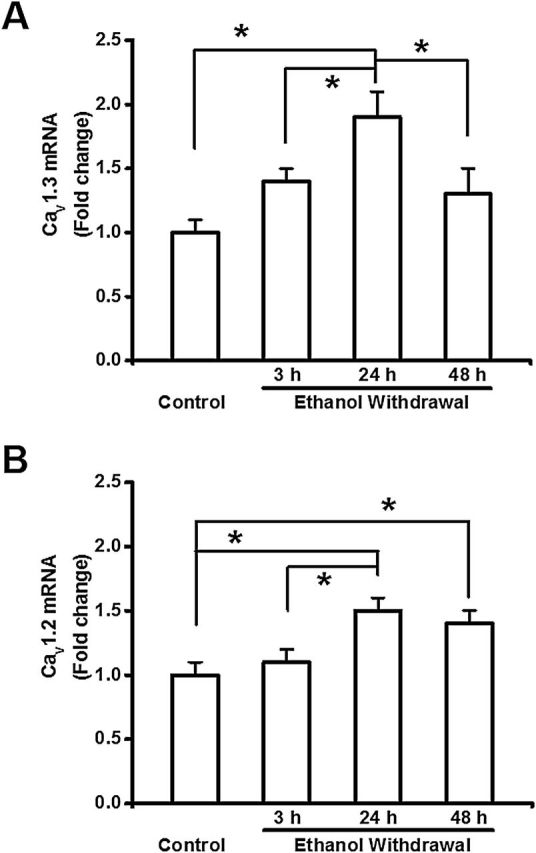
Ethanol withdrawal upregulates Cacna1d and Cacna1c mRNA expression in the inferior colliculus (IC). The levels of Cacna1d (CaV1.3 α1 subunit; panel A) and Cacna1c (CaV1.2 α1 subunit; panel B) mRNA were measured in the IC; the data are expressed as the fold change relative to the control group. (A) The level of Cacna1d mRNA was significantly higher in the IC 24 hours after alcohol withdrawal compared to the control group and the other alcohol-treated groups. (B) The level of Cacna1c CaV1.2 mRNA was significantly higher in the IC 24 and 48 hours after alcohol withdrawal compared to the control group. The level of Cacna1c CaV1.2 mRNA also was significantly elevated 24 hours after alcohol withdrawal compared to the 3-hour group. The data are shown as mean ± standard error of the mean. n = 6 rats per group. *p < 0.05 versus control (ANOVA followed by a Bonferroni test or Student-Newman-Keuls).
The expression of Cacna1c mRNA was significantly (t = 4, p < 0.05) increased 24 hours (2-∆∆CT = 1.5±0.1, n = 6) and 48 (2-∆∆CT = 1.4±0.1, n = 6) during ethanol withdrawal compared to the control group (2-∆∆CT = 1.0±0.1, n = 6, Figure 2B). The expression of Cacna1c mRNA also was significantly increased in the 24-hour group compared to the 3-hour group during ethanol withdrawal (2-∆∆CT = 1.1±0.1, n = 6, t = 3, p < 0.05). No change in the Cacna1c mRNA expression was observed 3 hours during ethanol withdrawal compared with the control group (Figure 2B). During ethanol withdrawal, the ratios of the mRNA expression levels of the Cacna1d to those of Cacna1c were 1.4:1.1, 1.9:1.5, and 1.3:1.4 at 3, 24, and 48 hours, respectively. Thus, a dominant expression of Cacna1d mRNA occurred at 3 and 24 hours following ethanol withdrawal.
Ethanol Withdrawal Upregulates CaV1 Channels at the Protein Level
We next examined whether the increased expression of CaV1 channels was reflected in a change in protein levels. Figure 3 summarizes the protein levels of CaV1.3 (Figure 3A) and CaV1.2 (Figure 3B) α1 subunits. Ethanol withdrawal significantly increased the protein levels of the CaV1.3 (F = 7, p < 0.001) but not of CaV1.2 α1 subunit in the IC. Consistent with the Cacna1d mRNA levels, the protein level of CaV1.3 α1 subunits was increased significantly 24 hours (152±8%, n = 8, t = 4, p < 0.05) after ethanol withdrawal, but not at the earlier (3-hour group: 102±10%, n = 8) or later (48-hour group: 116±12%, n = 8) time points compared to the control group (100±8%, n = 8, Figure 3A). Comparison between ethanol-treated groups reaveled that the protein expression level for the CaV1.3-α1 subunit was significantly elevated in the 24-hour group compared to the 3-hour (t = 4, p < 0.05) and 48-hour (t = 3, p < 0.05) groups during the withdrawal.
Figure 3.
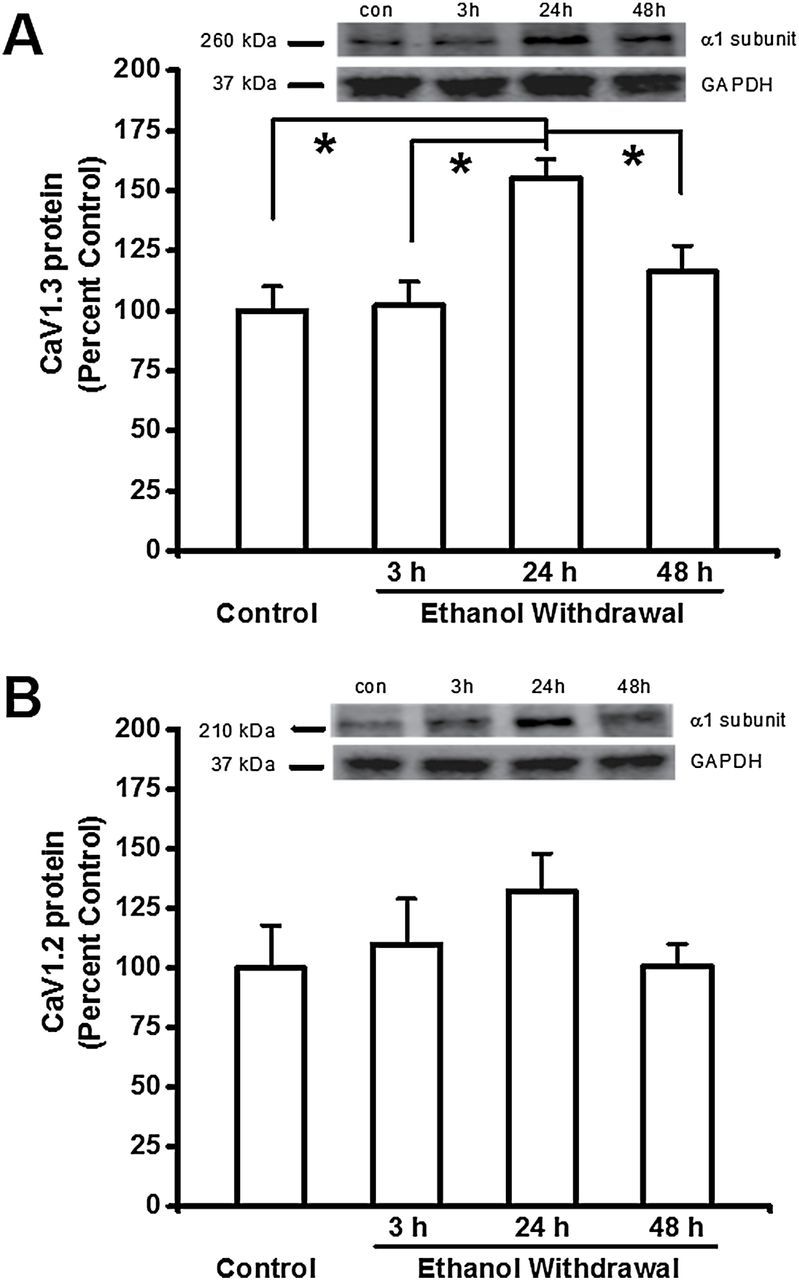
Ethanol withdrawal increases the protein levels of CaV1.3 α1 subunits in the inferior colliculus (IC). Shown in insets are representative immunoblots of the (A<B) GAPDH, (A) CaV1.3 and (B) CaV1.2 α1 subunits measured from control IC samples and IC samples obtained at the indicated times after ethanol withdrawal. The bar graphs summarize the relative protein levels of CaV1.3 α1 and CaV1.2 subunits in the IC, expressed as a percentage of the control group. The density of the 260kDa immunoreactive band (i.e. the CaV1.3 α1 subunit; panel A) increased significantly in the IC 24 hours after ethanol withdrawal. The density of the 210kDa immunoreactive bands (i.e. the CaV1.2 α1 subunit; panel B) did not change significantly following ethanol withdrawal. The summary data are shown as the mean ± standard error of the mean. n = 8 rats per group. *p < 0.05 versus control (ANOVA followed by a Bonferroni test or Student-Newman-Keuls).
In contrast, the protein expression levels of the CaV1.2-α1 subunit were increased by 6% at 3 hours (106±19%, n = 8) and 32% at 24 hours (132±16%, n = 8) during ethanol withdrawal, and then returned to control levels (100±19%, n = 8) at 48 hours (100±10, n = 8, Figure 3B) later. The protein expression ratios of the CaV1.3-α1 and CaV1.2-α1 subunits were 100:106, 152:132, and 116:100 at 3, 24, and 48h during ethanol withdrawal, respectively. Thus, ethanol withdrawal favored the protein expression of CaV1.3-α1 subunit at 24 hours during ethanol withdrawal when the susceptibility to seizures peaked.
Discussion
Here, we report that the mRNA and protein levels of the CaV1.3 and CaV1.2 α1 subunits increase in the IC during alcohol withdrawal, a condition that is associated with increased seizure susceptibility. In particular, expression of the CaV1.3 α1 subunit was significantly increased 24 hours after alcohol withdrawal, when seizure susceptibility peaked. In contrast, expression of the CaV1.2 α1 subunit increased 24 and 48 hours after alcohol withdrawal, when seizure susceptibility peaked and then resolved, respectively. Thus, expression of the CaV1.3 α1 subunit changed in parallel with the occurrence of AWS, whereas upregulation of the CaV1.2 1 subunit outlasted the period of AWS susceptibility. At the protein level, Western blot analyses revealed that the CaV1.3 α1 subunit—but not the CaV1.2 α1 subunit—was significantly increased in the IC 24 hours after alcohol withdrawal. A significant increase in CaV1.3 α1 subunit proteins was also observed 24 hours following alcohol withdrawal, as compared to the 3-hour group. These findings suggest that upregulation of CaV1.3 L-type Ca2+ channels might play an important role in the pathogenesis of AWS.
In the brain, CaV1 channels are located primarily in the dendrites and cell bodies; 80% and 20% of these channels are CaV1.2 and CaV1.3 channels, respectively (Hell et al., 1993). This predominance of CaV1.2 channels may also occur in the IC. Evidence suggests that CaV1.3 channels activate at more negative voltages and inactivate more slowly than CaV1.2 channels (Koschak et al., 2001; Xu and Lipscombe, 2001). Given their biophysical properties, CaV1.3 channels may play an important role in the control of Ca2+-dependent firing, and these channels can help maintain the Ca2+ influx at membrane potentials at which CaV1.2 channels would remain closed. Here, we found that increased susceptibility to AWS was accompanied by an increase in CaV1.3 channels in the IC, consistent with a role for CaV1.3 channels in mediating neuronal hyperexcitability, leading to seizures. Analysis of the ratio of CaV1.3 to CaV1.2 channels reveals that alcohol withdrawal preferentially upregulates CaV1.3 channels (at 24 but not 3 hours during alcohol withdrawal when the susceptibility to seizures peaked) in IC neurons, consistent with the role of these channels in the pathogenesis of AWSs.
During alcohol withdrawal, the IC plays an important role in initiating AGSs (Fyre et al., 1983; Riaz and Faingold, 1994; Faingold and Riaz, 1995; Chakravarty and Faingold, 1998). Pharmacological studies found that blocking CaV1 channels during alcohol withdrawal suppresses AGSs, leading to the hypothesis that these channels undergo remodeling, at least in IC neurons (Little et al., 1986). Consistent with this hypothesis, we previously reported increased currents through CaV1 channels in IC neurons during alcohol withdrawal at the time point at which susceptibility to seizures peaks (N’Gouemo and Morad, 2003). These findings suggest a possible causal relationship between changes in the composition of CaV1 channels in IC neurons and the occurrence of AWS. Here, we present evidence that the increase in CaV1 channels in IC neurons might not be a consequence of AWS, as CaV1.3 α1 mRNA and protein levels increased in parallel with the occurrence of AWS. Thus, altered CaV1.3 channel expression may predispose the animals to develop AWS.
Alcohol withdrawal–induced clonus (i.e. bouncing generalized clonic-tonic seizures) originate primarily in the brainstem (Faingold et al., 1998). Whether the changes in CaV1.3 α1 channels that we observed in the IC also occur in other epileptogenic sites in the forebrain (for example, in the hippocampus) is currently unknown. Increased levels of CaV1.3 α1 subunits have been found in cortical neurons during alcohol withdrawal and are associated with increased seizure susceptibility (Katsura et al., 2005). Similarly, CaV1 channels are upregulated throughout the brain following alcohol withdrawal, and this upregulation occurs in parallel with the occurrence of seizures (Watson and Little, 1999). Generalized tonic-clonic seizures during alcohol withdrawal resemble seizures observed in the genetically epilepsy-prone rat (Faingold et al., 1998; Faingold, 1999). In these rats, CaV1.3—but not CaV1.2—α1 subunits were increased in IC neurons of both seizure-naive and seizure-experienced animals (N’Gouemo et al., 2010). These findings suggest that the upregulation of CaV1.3 channels in IC neurons may be the molecular correlate of the enhanced susceptibility to generalized tonic-clonic seizures originating in the brainstem. In a Mongolian gerbil model of generalized tonic-clonic seizures, no changes in CaV1.3 or CaV1.2 α1 subunits were found in pyramidal cells in the CA1 or dentate gyrus of seizure-prone animals (Kang et al., 2004). However, in the CA2 and CA3 regions of the hippocampus, both CaV1.3 and CaV1.2 α1 subunits were significantly increased in pyramidal cells during the pre-seizure state, as well as postictally in seizure-sensitive gerbils (Kang et al., 2004). Whether these changes also occur in the brainstem is currently unknown. The increase in both CaV1.3 and CaV1.2 channels in the hippocampus may represent the molecular correlates of increased Ca2+-dependent excitability in an inherited model of generalized tonic-clonic seizures. Nevertheless, upregulation of CaV1.3 channels was reported in cerebellar Purkinje cells of seizure-sensitive gerbils (Park et al., 2003). In a kindling model of epileptogenesis, the mRNA level of CaV1.3—but not CaV1.2—channels was increased in the hippocampus during the non-convulsive limbic phase (Hendriksen et al., 1997). In contrast, no change in CaV1.3 mRNA levels was found in the hippocampus during generalized seizures (Hendriksen et al., 1997). Altered levels of CaV1 channels have also been reported in the thalamus. Notably, significant increases in the mRNA and protein levels of the CaV1.3 α1 subunit were found in the dorsal part of the lateral geniculate nucleus of epileptic Wistar Albino Glaxo from Rijswijk rats; this brain section is a thalamic nucleus that is relevant in the pathophysiology of absence epilepsy (Kanyshkova et al., 2014; Dilger et al., 2011; Lo et al., 2002). Evidence suggests that altered CaV1.2 channels in hippocampal pyramidal cells may play a role in the pathogenesis of generalized tonic-clonic seizures in a model of febrile seizures (Radzicki et al., 2013). Finally, we posit that upregulation of both CaV1.3 and CaV1.2 channels in the forebrain is associated primarily with non-convulsive seizures, whereas upregulation of CaV1.3 channels in the brainstem and cortex is associated with increased susceptibility to generalized tonic-clonic seizures.
Although we did not specifically examine the mechanism underlying the upregulation of CaV1 channels in the IC following alcohol withdrawal, the consequence of this upregulation is likely an increase in Ca2+ influx. Alcohol exposure can suppress the activity of Ca2+ channels in IC neurons (N’Gouemo, unpublished observations). Thus, an increase in the number of CaV1 channels following alcohol withdrawal might represent a compensatory mechanism for changes in the activity of IC neuronal circuits in response to decreased CaV1 channel activity during alcohol exposure. Among a myriad of other key functions, CaV1 channels control neuronal excitability, in part via their coupling with Ca2+-dependent K+ (KCa) channels (Morisset and Nagy, 1999). Notably, Ca2+ influx via CaV1.3 channels can induce a slow afterhyperpolarization by activating KCa channels, which in turn control neuronal firing (Shah and Haylett, 2000; Bowden et al., 2001; Goldberg and Wilson, 2005). Our finding of CaV1.3 channel upregulation suggests a role for currents that drive a slow afterhyperpolarization. However, both the abundance and current density of the large-conductance KCa channels that contribute to the slow afterhyperpolarization are decreased in IC neurons following alcohol withdrawal (N’Gouemo and Morad, 2014). Thus, the combination of CaV1.3 channel upregulation and large-conductance KCa channel downregulation might contribute to IC neuronal hyperexcitability and increase seizure susceptibility during alcohol withdrawal.
Our study has some limitations that bear mentioning. Notably, the relatively small sample size used in this study could, in principle, favor type II errors (i.e. a false negative result) over type I errors (i.e. a false positive result). For example, our finding that CaV1.3 α1 subunits are not upregulated 3 hours after ethanol withdrawal may have been due to a type II error. We also found a lack of positive correlation between the expression of Cacna1d mRNA and the levels of CaV1.3 α1 subunit proteins, which could be due to different mRNA and protein degradation rates. In the present study, total (cell suface and intracellular) expression of CaV1 protein was measured in IC neurons, which doesn’t represent the functional (at the cell surface) CaV1 channels.
In summary, alcohol withdrawal differentially affects the expression of CaV1 channel subtypes in the rat IC neurons. The transient increase in the expression of CaV1.3 channels occurred in parallel with the occurrence of AWSs, whereas the increase in CaV1.2 channels outlasted the enhanced susceptibility to seizures. These findings suggest that CaV1.3 channels in IC neurons play an important role in the pathogenesis of AWSs.
Statement of Interest
The authors have no conflicts of interest.
Acknowledgments
This work was supported by National Institutes of Health Public Health Service Grants (AA020073 and AA020073-03S1, to Dr N’Gouemo) and the NIAAA Division of Intramural Clinical and Biological Research (to Dr Lovinger). The authors would like to thank Dr Katherine Conant and Guoxiang Luo for technical assistance in the early stages of this project and Dr George Luta for help with statistics.
References
- Bowden SE, Fletcher S, Loane DJ, Marrion NV. (2001). Somatic colocalization of rat SK1 and Dclass (Ca(V)1.3) L-type calcium channels in rat CA1 hippocampal pyramidal neurons. J Neurosci 21:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarty DN, Faingold CL. (1998). Comparison of neuronal response patterns in the external and central nuclei of inferior colliculus during ethanol administration and ethanol withdrawal. Brain Res 783:102–108. [DOI] [PubMed] [Google Scholar]
- Dilger EK, Shin HS, Guido W. (2011). Requirements for synaptically evoked plateau potentials in relay cells of the dorsal lateral geniculate nucleus of the mouse. J Physiol 589(Pt 4):919–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckardt MJ, Campbell GA, Marietta CA, Majchrowicz E, Wixon HN, Weight FF. (1986). Cerebral 2-deoxyglucose uptake in rats during ethanol withdrawal and post-withdrawal. Brain Res 366:1–9. [DOI] [PubMed] [Google Scholar]
- Evans MS, Li Y, Faingold CL. (2000). Inferior colliculus intracellular response abnormalities in vitro associated with susceptibility to ethanol withdrawal seizures. Alcohol Clin Exp Res 24:1180–1186. [PubMed] [Google Scholar]
- Faingold CL. (1999). Neuronal networks in the genetically epilepsy-prone rat. Adv Neurol 79:311–321. [PubMed] [Google Scholar]
- Faingold CL. (2008). The Majchrowicz binge alcohol protocol: an intubation technique to study alcohol dependence in rats. Curr Protoc Neurosci 44:9.28:9.28.1-9.28.12. [DOI] [PubMed] [Google Scholar]
- Faingold CL, Riaz A. (1995). Ethanol withdrawal induces increased firing in the inferior colliculus neurons associated with audiogenic seizure susceptibility. Exp Neurol 132:91–98. [DOI] [PubMed] [Google Scholar]
- Faingold CL, N’Gouemo P, Riaz A. (1998). Ethanol and neurotransmitter interaction-from molecular to integration effects. Prog Neurobiol 55:509–535. [DOI] [PubMed] [Google Scholar]
- Faingold CL, Li Y, Evans MS. (2000). Decreased GABA and increase glutamate-mediated activity on inferior colliculus neurons in vitro are associated with susceptibility to ethanol withdrawal seizures. Brain Res 868:287–295. [DOI] [PubMed] [Google Scholar]
- Frye GD, McCown TJ, Breese GR. (1983). Characterization of susceptibility to audiogenic seizures in ethanol-dependent rats after microinjection of gamma-aminobutyric acid (GABA) agonists into the inferior colliculus, substantia nigra or medial septum. J Pharm Exp Ther 227:663–670. [PMC free article] [PubMed] [Google Scholar]
- Goldberg JA, Wilson CJ. (2005). Control of spontaneous firing patterns by selectivecoupling of Ca2+ currents to Ca2+-activated potassium currents in striatal cholinergic interneurons. J Neurosci 25:10230–10238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, Snutch TP, Catterall WA. (1993). Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J Cell Biol 123:949–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendriksen H, Kamphuis W, Lopes da Silva FH. (1997). Changes in voltage-dependent calcium channel alpha1-subunit mRNA levels in the kindling model of epileptogenesis. Brain Res Mol Brain Res 50:257–266. [DOI] [PubMed] [Google Scholar]
- Kang TC, Kim DS, Yoo KY, Hwang IK, Kwak SE, Kim JE, Jung JY, Won MH, Suh JG, Oh YS. (2004). Elevated voltage-gated Ca2+ channel immunoreactivities in the hippocampus of seizure-prone gerbil. Brain Res 1029:168–178. [DOI] [PubMed] [Google Scholar]
- Kanyshkova T, Ehling P, Cerina M, Meuth P, Zobeiri M, Meuth SG, Pape HC, Budde T. (2014). Regionally specific expression of high-voltage-activated calcium channels in thalamic nuclei of epileptic and non-epileptic rats. Mol Cell Neurosci 61:110–122. [DOI] [PubMed] [Google Scholar]
- Katsura M, Torigoe F, Hayashida S, Honda T, Tsujimura A, Ohkuma S. (2005). Ethanol physical dependence is accompanied by up-regulated expression of L-type high voltage-gated calcium channel alpha1 subunits in mouse brain. Brain Res 1039:211–215. [DOI] [PubMed] [Google Scholar]
- Koschak A, Reimer D, Huber I, Grabner M, Glossmann H, Engel J, Striessnig J. alpha 1D (Cav1.3) subunits can form L-type Ca2+ channels activating at negative voltages (2001). J Biol Chem 276:22100–22106. [DOI] [PubMed] [Google Scholar]
- Little HJ, Dolin SJ, Halsey MJ. (1986). Calcium channel antagonists decrease the ethanol withdrawal syndrome. Life Sci 39:2059–2065. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25:402–408. [DOI] [PubMed] [Google Scholar]
- Lo FS, Ziburkus J, Guido W.(2002). Synaptic mechanisms regulating the activation of a Ca2+-mediated plateau potential in developing relay cells of the LGN. J Neurophysiol 87:1175–1185. [DOI] [PubMed] [Google Scholar]
- McCown TJ, Breese GR. (1990). Multiple withdrawals from chronic ethanol “kindles” inferior collicular seizure activity: evidence for kindling seizures associated with alcoholism. Alcohol Clin Exp Res 14:394–399. [DOI] [PubMed] [Google Scholar]
- McCown TJ, Breese GR. (1993). A potential contribution to ethanol withdrawal kindling: reduced GABA function in inferior collicular cortex. Alcohol Clin Exp Res 17:1290–1294. [DOI] [PubMed] [Google Scholar]
- Morisset L, Nagy F. (1999). Ionic basis for plateau potentials in deep dorsal horn neurons of the rat spinal cord. J Neurosci 19:17309–17316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- N’Gouemo P, Morad M. (2003). Ethanol withdrawal seizure susceptibility is associated with upregulation of L- and P-type Ca2+ channels currents in rat inferior colliculus neurons. Neuropharmacology 45:429–437. [DOI] [PubMed] [Google Scholar]
- N’Gouemo P, Morad M. (2014). Alcohol withdrawal is associated with a downregulation of large-conductance Ca2⁺-activated K⁺ channels in rat inferior colliculus neurons. Psychopharmacology (Berl) 231:2009–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- N’Gouemo P, Caspary DM, Faingold CL. (1996). Decreased GABA effectiveness in the inferior colliculus neurons during ethanol withdrawal in rat susceptible to audiogenic seizures. Brain Res 724:200–204. [DOI] [PubMed] [Google Scholar]
- N’Gouemo P, Yasuda RP, Morad M. (2006). Ethanol withdrawal is accompanied by downregulation of calcium channel alpha 1B subunit in rat inferior colliculus neurons. Brain Res 1108:216–220. [DOI] [PubMed] [Google Scholar]
- N’Gouemo P, Yasuda R, Faingold CL. (2010). Seizure susceptibility is associated with altered protein expression of voltage-gated calcium channel subunits in inferior colliculus neurons of the genetically epilepsy-prone rat. Brain Res 1308:153–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SK, Hwang IK, An SJ, Won MH, Kang TC. (2003). Elevated P/Q-type (a1A) and L2 type (a1D) Purkinje cell voltage-gated calcium channels in the cerebella of seizure prone gerbils. Mol Cells 16:297–301. [PubMed] [Google Scholar]
- Radzicki D, Yau HJ, Pollema-Mays SL, Mlsna L, Cho K, Koh S, Martina M. (2013). Temperature-sensitive Cav1.2 calcium channels support intrinsic firing of pyramidal neurons and provide a target for the treatment of febrile seizures. J Neurosci 33:9920–9931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riaz A, Faingold CL. (1994). Seizures during ethanol withdrawal are blocked by focal microinjection of excitant amino acid antagonists in the inferior colliculus and pontine reticular formation. Alcohol Clin Exp Res 18:1456–1462. [DOI] [PubMed] [Google Scholar]
- Shah M, Haylett DC. (2000). Ca2+ channels involved in the generation of the slow afterhyperpolarization in cultured rat hippocampal pyramidal neurons. J Neurosci 83:2554–2561. [DOI] [PubMed] [Google Scholar]
- Ueda Y, Tusru N. (1995). Simultaneous monitoring of the seizure related change in extracellular glutamate and gamma-aminobutyric acid concentration in bilateral hippocampi following development of amygdaloid kindling. Epilepsy Res 20:213–219. [DOI] [PubMed] [Google Scholar]
- Xu W, Lipscombe D. (2001). Neuronal Ca(V)1.3alpha(1) L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J Neurosci 21:5944–5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson WP, Little HJ. (1999). Correlation between increases in dihydropyridine binding in vivo and behavioural signs of ethanol withdrawal in mice. Alcohol Alcohol 34:35–42. [DOI] [PubMed] [Google Scholar]
- Whittington MA, Lambert JD, Little HJ. (1993). Increases in synaptic activation of calcium current as a mechanism for generation of alcohol withdrawal seizures. Alcohol Alcohol (Suppl) 2:391–394. [PubMed] [Google Scholar]