

Abstract
TNFα-induced protein 8 (TNFAIP8 or TIPE) is a newly described regulator of cancer and infection. However, its precise roles and mechanisms of actions are not well understood. We report here that TNFAIP8 regulates Listeria monocytogenes infection by controlling pathogen invasion and host cell apoptosis in a RAC1 GTPase-dependent manner. TNFAIP8 knockout mice were found to be resistant to lethal L. monocytogenes infection and had reduced bacterial load in the liver and spleen. TNFAIP8-knockdown in murine liver HEPA1-6 cells increased apoptosis, reduced bacterial invasion into cells, and resulted in dysregulated RAC1 activation. TNFAIP8 could translocate to plasma membrane and preferentially associate with activated GTP-RAC1. The combined effect of reduced bacterial invasion and increased sensitivity to TNFα-induced clearance likely protected the TNFAIP8 knockout mice from lethal listeriosis. Thus, by controlling bacterial invasion and the death of infected cells through RAC1, TNFAIP8 regulates the pathogenesis of L. monocytogenes infection.
Keywords: Cell death, infection, TNF, TNFAIP8, Listeria
Introduction
TNFα-induced protein 8 (TNFAIP8 or TIPE), also known as SCC-S2, OXI-α, GG2-1, NDED, and MDC-3.13, is the first described member of the TNFAIP8 family. It is an NF-κB inducible protein upregulated in metastatic head and neck squamous cell carcinoma cell lines, and to protect cancer cells from TNFα-induced apoptosis (1, 2). Overexpression in tumor cell lines also enhanced proliferation and migration (3). TNFAIP8 has been found to be a risk factor for non-Hodgkin’s lymphoma in humans and Staphylococcus aureus infection in mice (4). A few TNFAIP8 interacting partners have been identified, including activated Gαi3 and Karyopherin α2 (5). TNFAIP8L2 (TNFAIP8 – Like 2), a closely related TNFAIP8 family protein, has been reported to interact with RAC1 and to protect against Listeria monocytogenes infection (6).
Listeria monocytogenes is a predominantly food-borne pathogen with ~10 cases per million inhabitants in most industrialized countries. Despite having a lower incidence of infection than most other food-borne pathogens, listeriosis carries a high risk of mortality ranging between 20-30%, and accounts for 19% of food-borne disease-related deaths in the USA (7, 8). L. monocytogenes is an intracellular gram-positive bacterium that infects a number of cell types including hepatocytes, neurons, and immune cells. Immune cell-mediated apoptosis of L. monocytogenes-infected cells such as hepatocytes is important for resolving infection (9, 10). One of the major mechanisms of immune defense against infectious intracellular microbes is to eliminate the infected cells by programmed cell death or apoptosis. We sought to determine the molecular pathways regulated by TNFAIP8 and their relationship with L. monocytogenes infection and clearance. In this study, we show that TNFAIP8 sensitizes mice to lethal L. monocytogenes infection, by potentially blocking apoptosis of infected cells and promoting the invasion by L. monocytogenes through RAC1. These results may provide new insights into TNFAIP8’s regulation of cell death in listeriosis and carcinogenesis.
Materials and Methods
Animals
Wild type C57BL/6 (B6) mice were purchased from The Jackson Laboratory. The Tnfaip8−/− B6 mice were generated by germline gene targeting (our unpublished data). All mice used in this study were housed under pathogen-free conditions in the University of Pennsylvania Animal Care Facilities. All animal protocols used were pre-approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
Macrophage and Neutrophil Preparations
To generate bone marrow-derived macrophages (BMDMs), bone marrow cells were flushed from the femurs and tibias of donor mice. The red blood cells were lysed with ACK solution (8.29g NH4Cl, 1g KHCO3, 37.2mg Na2EDTA in 1L of water). Cells were washed twice in ice-cold 1xDPBS and cultured for 7 days in 30% L-929 cell culture supernatant and 70% DMEM containing 10% (vol/vol) heat-inactivated FBS, 2 mM L-glutamine, and 100 units/mL penicillin/streptomycin (D10). Cells were washed twice with cold DPBS and collected with 5 mM EDTA in DPBS. After centrifugation, they were resuspended in D10 and rested for 24 h before experimentation. BMDMs were >95% CD11b+ and F4/80+ as determined by flow cytometry. Morphologically mature neutrophils were purified from murine bone marrow by Percoll gradient centrifugation. Briefly, bone marrow cells were harvested from mice using neutrophil isolation buffer (1× HBSS without Ca2+ and Mg2+ containing 0.25% BSA). After RBC lysis, cells were layered on a 62% Percoll gradient. Following centrifugation at 1,200 × g for 30 min at room temperature, pelleted cells were removed and washed once with isolation buffer before being used in the experiment. Neutrophil viability was >95% according to results from trypan blue staining. Purity was typically 75–85% as assessed by flow cytometry based on forward and side scatter and Gr1 staining.
Bone Marrow Chimeras
Bone marrow cells were flushed from the femurs and tibias of donor mice. The red blood cells were lysed with ACK solution (8.29g NH4Cl, 1g KHCO3, 37.2mg Na2EDTA in 1L of water). Cells were washed twice and re-suspended in cold PBS. Recipient mice were sub-lethally irradiated with 500 rads twice separated by 4 hours. The irradiated mice received a total of 10x106 donor bone marrow cells by tail vein injection one or two hours after irradiation. Mice were used seven to eight weeks later for experiments.
Cell lines and plasmids
The HEK293T and Hepa1-6 cells were cultured in D10. Full-length TNFAIP8 cDNA was generated by PCR and cloned in frame with an N-terminal Flag into vector pRK5. Human wild-type RAC1, RAC1 T17N, RAC1 Q61L cDNAs were obtained from Addgene and subcloned into pRK5 with Myc or HA tag at the N terminus. Truncated forms of Rac1 lacking the N-terminal amino acids 1–47 and C-terminal amino acids 162–192 or 189–192 were generated by PCR and cloned in-frame with an N-terminal HA tag into vector pRK5. cDNAs encoding TNFAIP8, wild-type RAC1, RAC1 T17N and RAC1 Q61L were subcloned into the pEGFP-N3 plasmid.
Plasmid DNA transfection and viral infection
293T cells were transfected with plasmid DNA using Fugene 6 (Promega) reagent according to the manufacturer’s instructions. For virus production, pLKO.1 (with puromycin resistance) with shRNA-Tnfaip8 (purchased from Open Biosystems) or shScr (a non-specific scramble shRNA purchased from Addgene) fragments and packaging constructs were co-transfected into 293T cells. After 24 and 48 hrs, virus-containing medium was filtered and used to infect Hepa1-6 cell lines in the presence of 6.5 mg/ml of polybrene (Millipore). Infected cells were selected using puromycin (Sigma) to establish shRNA-Scr, shRNA-Tnfaip8-1 (SH1), and shRNA-Tnfaip8-2 (SH2) cell lines. Viral vectors were produced from the pLKO.1 plasmid containing SH1 (AAAGGGATTGTACAAGGCAGC), SH2 (TTGAACTGATTGTTCCTGTGG), or shRNA-scr (CGAGGGCGACTTAACCTTAGG).
TNFα stimulation and death assays
Hepa1-6 cell lines were serum starved for 16 hours in DMEM containing 2mM L-glutamine, and 100 units/mL penicillin/streptomycin. Cells were treated with 10 ng/ml of TNFα for 0, 1, 5, 15, 30, 60 minutes for protein lysates, or with 5 ng/ml TNFα plus 20 ng/ml cyclohexamide (CHX) for 7 or 16 hours. Death was measured by flow cytometry using Annexin V PE and 7-AAD (BD Pharmingen) staining. For measuring death following RAC1 mutant transfections, EGFP positive cells were gated on for comparison. To inhibit RAC1, cells were incubated with inhibitor Z62954982 (Millipore) at 100μM for 30 min prior to experimental treatment.
Western blot
For Western blot analysis, cells were lysed for 20 min at 4°C in 1% Triton X-100 lysis buffer containing protease and phosphatase inhibitors. Cell debris was removed by centrifugation at 14,000 × g for 20 min at 4°C. For subcellular fractionation, membrane and cytoplasmic proteins were separated using Qproteome Cell Compartment Kit (QIAGEN) according to the manufacturer’s instructions. The protein concentration of the lysates was determined by Bradford assay. Equal amounts of total protein were resolved by SDS-PAGE, transferred to membranes, immunoblotted with specific primary and secondary antibodies, and the signals were detected by chemiluminescence (Pierce). Primary antibodies against p-AKT(S473), AKT, Myc, as well as anti-Myc conjugated to HRP, were purchased from Cell Signaling Technology and used according to the manufacturer’s instructions. Anti-TNFAIP8 (1:500) was purchased from ProteinTech Group and Anti-RAC1 (1:500) was purchased from Millipore. Anti-Flag (1:2000), anti-Flag-M2-HRP, anti-β-actin (1:3000) and anti-GAPDH antibody (1:3000) were purchased from Sigma. HRP-conjugated secondary anti-mouse and anti-rabbit IgG (1:1500) were purchased from GE Healthcare. The densitometric quantification of Western blot signals was performed using ImageJ software. Paired t-test was used to evaluate the statistical significance of the results.
Immunoprecipitation
Immunoprecipitation was performed using Dynabeads protein G (Invitrogen). In brief, 1.5 mg protein-G Dynabeads was coated with 5 μg specific antibodies or Ig control for 1 h at room temperature with rotation. After removing unbound antibody, the bead–antibody complex was incubated with 500 μL cell lysate for 4 h at 4 °C with rotation. The captured Dynabead-Ab-Ag complex was washed four times with PBS and boiled in 2× Laemmli buffer. The eluted proteins were fractionated by SDS-PAGE and detected by Western blot.
Loading of RAC1 with GDP and GTP
The 293T cells were transiently transfected with Myc-tagged RAC1 and Flag-tagged TNFAIP8 for 18 h. Cells were lysed in cell lysis buffer (50 mM Tris, pH 7.5, 10 mM EDTA, 0.2 M NaCl, 0.5% Nonidet P-40, and 1× protease inhibitors mixture) (Roche). A total of 1 mM GDPβS or 0.2 mM GTPγS (Enzo Life Sciences) was loaded to transfected cell extracts. After 20 min incubation at 30 °C, samples were placed on ice immediately and MgCl2 was added to a final concentration of 10 mM to stop nucleotide exchange.
PAK Pull-Down Assay
Hepa1-6 cells were serum starved for 16 hours, and then treated with 10 ng/ml TNFα for 5 minutes. The cells were washed in PBS and lysed in PBD lysis buffer (50mMTris, pH 7.5, 10 mM MgCl2, 0.2 M NaCl, 2% Nonidet P-40, and 1× protease inhibitors mixture) (Roche). The lysate was incubated with 20 μg of p21-activated kinase (PAK)-GST protein beads (Cytoskeleton) for 30 min at 4 °C. After washing, protein on beads and in total cell lysates was subjected to Western blot to determine the level of active Rac1.
F-actin Assay
Hepa1-6 cells were plated on 12-well Nunc Multidishes with UpCell Surface (Thermo Scientific) and incubated overnight at 37 °C. Cells were then serum-starved overnight and washed with PBS before resuspending in ice cold PBS according to the manufacturer’s protocol. Cells were fixed with 4% paraformaldehyde for 10 minutes and permeabilized for 5 minutes in 0.1% TRITON-X 100. The cells were subsequently stained with phalloidin-FITC (SIGMA) for 30 minutes. Samples were analyzed by flow cytometry and mean fluorescence intensity was calculated using FlowJo software.
Microbial Strains and Infection
Wild-type L. monocytogenes (10403s) were provided by H. Shen and Y. Paterson (University of Pennsylvania). L. monocytogenes and Staphylococcus aureus (ATCC 29213) were grown at 37 °C in brain-heart-infusion medium (Becton Dickinson), and Columbia medium with 2% NaCl, respectively. For all assays, mid-log-phase bacteria were used. Bacterial in vitro infection was assayed in 6-well plates. A total of 7.5 × 105 Hepa1-6 cells were seeded in each well, followed by culturing overnight in DMEM with 10% FBS. Cells were the serum starved for 18-24 hours. Cells were infected with L. monocytogenes at multiplicity of infection (MOI) of 50 in antibiotic-free media. Sixty minutes after the inoculation, cells were washed three times with PBS, and fresh medium containing gentamycin (150 μg/mL) was added. At 1.5 hours after infection, cells were lysed with 0.1% Triton in PBS and serial dilutions of the homogenate were plated on brain-heart infusion agar plates (BHI) (Becton Dickinson). The colonies were counted 24 hours later. For in vivo bacterial infection, L. monocytogenes was grown in brain-heart-infusion medium until the absorbance at 600 nm reached 0.1 of optical density. S. aureus was grown in Columbia media with 2% NaCl. Six- to 7-wk old WT and TNFAIP8 KO mice were infected intravenously with 2 × 105 L. monocytogenes in 200 μL PBS or 2 × 107 S. aureus in 200 μL saline. For measurement of the bacterial burden in liver and spleen, mice were sacrificed 72 h after inoculation, organs were homogenized in 0.1%Triton in PBS, and serial dilutions of the homogenate were plated on BHI agar plates. The colonies were counted 24 h later. Serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were determined using the Infinity ALT or AST liquid stable reagent (Thermo).
Phagocytosis Assay
To prepare apoptotic cells for phagocytosis assay, thymocytes were harvested from 3- to 4-wk-old C57BL/6 mice, loaded with 10 uM carboxyfluorescein succinimidyl ester (CFSE) (Molecular Probes) for 10 minutes at 37°C and washed 2X in PBS. Apoptosis was induced by incubation at 37°C in 5% CO2 for 5 h in the presence of 5 μM dexamethasone (Sigma-Aldrich). After dexamethasone treatment, cells were washed three times with PBS and resuspended in DMEM with 2% FBS. This treatment routinely yielded over 70% apoptotic thymocytes as measured by annexin V staining. Phagocytosis assay was performed in a 12-well nontissue culture-treated plate (BD Falcon). A total of 1 × 106 BMDMs were seeded in each well, followed by culturing overnight in DMEM with 2% FBS. CFSE+ apoptotic thymocytes were then added at a ratio of 5:1, and centrifugation was performed at 500 × g for 2 min to synchronize binding and internalization. After 30 min of incubation at 37 °C, plates were rapidly washed two times with ice-cold PBS, and cells collected with 5 mM EDTA-PBS. Cells were then fixed with 2% paraformalhyde in PBS, stained with APC-conjugated anti-F4/80, and analyzed by flow cytometry. Gates were set for macrophages in FSC/SSC dot blots. Experiments using fluorescent latex beads (2 μm; Sigma-Aldrich) were performed in a similar fashion. For bacterial phagocytosis assay, L. monocytogenes were washed twice with sterile PBS and incubated (at 1 × 109/mL bacteria) in 2 μM CFSE for 20–30 min under constant shaking at 37 °C. CFSE-labeled bacteria were washed three times with PBS before being used. Live bacteria were fed to BMDMs at a ratio of 10:1 in DMEM with 10% serum.
Chemotaxis assay
Primary neutrophils were loaded with 5 μM Calcein AM in PBS supplemented with 2% FBS for 30 minutes at 37°C. After 3 washes with PBS, neutrophils were resuspended to with 2 × 106 cells per ml of PBS. ChemoTX plates (Neuro Probe) were loaded with 50 μl of cell over each 5.7mm diameter chamber with 5 μm pores. The lower chambers were loaded with 30 μl of PBS containing the specified concentration of IL-8. The Neutrophils were incubated for one hour, and then the top chambers were washed off and the plates were read in a fluorescent plate reader. Chemotactic index was calculated as the number of neutrophils in the lower chambers of chemokine containing wells over the wells with no chemokines.
ELISA
Sera were collected from WT and TNFAIP8 KO mice 72 hours after L. monocytogenes infection and kept at −80°C. Antibodies used in ELISA were purchased from BD Pharmingen and eBioscience, including purified and biotinylated rat anti-mouse TNFα. Quantitative ELISA was performed using paired mAbs specific for corresponding cytokines according to the manufacturer’s recommendations.
Statistical analysis
Quantitative data are presented as means±SEM of two or three experiments. Survival curves were plotted via the Kaplan-Meier method and compared by the log-rank test. Two-tailed Student t test was used for all other results and p<0.05 was considered statistically significant. All statistical analysis was performed using Graphpad Prism Software.
Results
TNFAIP8-knockout mice are resistant to lethal Listeria monocytogenes infection
It has been previously reported that TIPE2-deficient mice are resistant to L. monocytogenes infection (6). We hypothesized that TNFAIP8-knockout mice might have a similar phenotype. To test this, TNFAIP8 knockout and wild type C57BL/6 mice were intravenously injected with a lethal dose of L. monocytogenes. As expected, TNFAIP8 knockout mice were found to be resistant to death (Figure 1A), and experienced reduced weight-loss during infection (Figure 1B). Next we examined the bacterial load in the liver and spleen 3 days post infection, and found reduced bacterial load in the knockout mice (Figure 1C). We assessed liver damage by measuring serum levels of ALT and AST, but we found no significant differences between wild type and knockout mice (Figure 1D). Serum TNFα and IL-6 levels were also assayed. TNFα and IL-6 are important for controlling L. monocytogenes, and mice deficient in either of them have increased bacterial burden (11). Although the knockout mice had less TNFα and more IL-6, the differences were not statistically significant (Figure 1E). Therefore TNFAIP8 KO mice are resistant to lethal L. monocytogenes infection, independent of serum cytokines and hepatocellular toxicity.
Figure 1. TNFAIP8-knockout mice are resistant to lethal Listeria monocytogenes infection.

Wild type (n=5) and TNFAIP8 knockout mice (n=7) were infected with 2x105 CFUs of L. monocytogenes through the tail vein. (A) Mouse Kaplan-Meier survival curve after L. monocytogenes injection (P = 0.01). (B) Changes in body weight following infection (P = 0.04). Mice were sacrificed at day 3 post infection and their livers, spleens, and sera collected. (C) Spleen and liver bacterial load was measured by a colony-forming assay. (D) Serum levels of aspartate aminotransferase (AST) and alanine aminotransferase (ALT). (E) Serum TNFα and IL-6 levels were measured by ELISA. The results are representative of at least two independent experiments. N.S., not significant; * P < 0.05.
Immune system function does not sufficiently explain the resistance to lethal L. monocytogenes in TNFAIP8-knockout mice
To test whether TNFAIP8 knockout immune cells are responsible for the resistance to L. monocytogenes infection, we produced bone marrow chimeric mice using both wild type and knockout irradiated mice as recipients of wild type bone marrow. After their immune cells were replenished, the recipient mice were challenged with a lethal dose of L. monocytogenes. We once again found that TNFAIP8 knockout mice still exhibited significant resistance to death (Figure 2A). Next we tested whether there was a difference in mouse survival with another gram-positive pathogen. We chose Staphylococcus aureus since the Tnfaip8 gene was reported to be a risk factor for its infection, and it is an extracellular pathogen (4). TNFAIP8 knockout (KO) and wild type (WT) mice were infected intravenously with a lethal dose of S. aureus, but there was no difference in mouse survival (Figure 2B). To investigate if there were any differences in innate immune cell function, we isolated primary neutrophils from bone marrow and checked if there was a difference in neutrophil chemotaxis. We found no significant difference between WT and KO cells in chemotaxis towards IL-8 in a Boyden chamber assay (Figure 2C). Although IL-8 is not expressed in mice, murine neutrophils express the functional IL-8 receptor beta (CXCR2), and IL-8 has been previously used to induce chemotaxis in mice (12, 13). We next studied the phagocytic capacity of bone marrow-derived macrophages and found that there were no differences between WT and KO cells in the phagocytosis of killed CFSE-labeled thymocytes, fluorescently labeled beads, or L. monocytogenes (Figure 2D). To determine whether TNFAIP8 deficiency affects the production of TNFα and IL-6 in response to peptidoglycan (PGN), a major component of Gram-positive bacteria, we injected PGN into WT and TNFAIP8 KO mice, and checked the serum levels of the cytokines. Again, we did not find any significant differences between the two groups (Figure 2E). Similarly, bone marrow-derived macrophages did not show a difference in the production of TNFα and IL-6 upon stimulation with PGN in vitro (Figure 2F). Taken together, these results suggest that the immune system does not play a primary role in the resistance to lethal L. monocytogenes infection in TNFAIP8 knockout mice.
Figure 2. Immune system function does not sufficiently explain the resistance to lethal L. monocytogenes of TNFAIP8-knockout mice.

(A) Bone marrow chimeras were produced by irradiating wild type (N=6) and TNFAIP8 (N=6) knockout mice and transferring wild type bone morrow into them. Mice were given 8 weeks to reconstitute their immune system before being infected with 2x105 CFUs of L. monocytogenes. The difference in survival between the two groups is statistically significant (P = 0.02). (B) Wild type (N=5) and TNFAIP8 knockout mice (N=7) were challenged with 2x107 CFUs of Staphylococcus aureus. The difference in survival between the two groups is not statistically significant. (C) Calcein AM-labeled primary neutrophils were incubated for 1 hour in a Boyden chamber filled with or without 50 ng/ml or 500 ng/ml of IL-8. The rate of chemotaxis was determined using a fluorescent plate reader. (D) Bone marrow-derived macrophages (BMDMs) were incubated with 2-μm fluorescent beads at a ratio of 1:20, apoptotic CFSE-labeled thymocytes at a ratio of 1:5, or CFSE-labeled L. monocytogenes at a ratio of 1:10 for 30 minutes. The rate of phagocytosis was measured by flow cytometry. (E) Wild type and TNFAIP8 knockout mice were intravenously injected with PGN (10 mg/kg) and serum was collected after 8 hours to assay TNFα and IL-6 levels by ELISA. (F) BMDMs were treated with PGN (10 μg/ml) for 8 hours and TNFα and IL-6 levels were assayed in the supernatant. The results are representative of at least two independent experiments. N.S., not significant.
TNFAIP8 regulates L. monocytogenes infection and the death of infected cells in vitro
Since differences in immune function did not adequately explain the resistance to lethal L. monocytogenes infection in TNFAIP8-knockout mice, we next investigated the roles of TNFAIP8 in non-immune cells involved in bacterial infection. Since hepatocytes are a major target of L. monocytogenes infection, we generated TNFAIP8 knockdown Hepa1-6 murine liver cells. Using a lentiviral vector expressing either a scrambled shRNA sequence or sequences targeting TNFAIP8, we created several knockdown cell lines and determined the protein levels of TNFAIP8 and AKT (Figure 3A). We found that shRNA-tnfaip8-1 (SH1) was a nearly complete knockdown, with increased AKT phosphorylation at Serine 473. AKT phosphorylation at Serine 473 is regulated by MTORC2 and PI3K and is important in regulating apoptotic cell death, and PI3K itself is important in L. monocytogenes cell invasion (14, 15).
Figure 3. TNFAIP8 regulates L. monocytogenes infection and the death of infected cells in vitro.

Hepa1-6 knockdown cells were generated using the pLKO.1 lentiviral vector to express either a scrambled shRNA (Scr) or one of the two antisense sequences targeting TNFAIP8 (SH1 and SH2). (A) Western blot was performed for the indicated proteins in the lysates of untransfected control (UT) and shRNA-treated Hepa1-6 cells. (B) L. monocytogenes infection of control and TNFAIP8 knockdown Hepa1-6 cells was measured in a colony formation assay. Cells were infected with 50 MOI of L. monocytogenes for 1 hour with or without 50 ng/ml of TNFα. NT, not treated with TNFα. Cells were washed and then treated with 150 μg/ml Gentamycin for 30 minutes to kill extracellular bacteria before being lysed to release the intracellular bacteria. (C) Hepa1-6 TNFAIP8 knockdown and control cells were challenged with L. monocytogenes (L) at 60 MOI with or without 50 ng/ml of TNFα (T) for 16 hours. Death was quantified by flow cytometry after Annexin V staining. (D) Hepa1-6 knockdown and control cell lines were treated with or without 5 ng/ml of TNFα and 20 ng/ml of cycloheximide (CHX) for 6 hours. Cell death was determined by flow cytometry after Annexin V and 7-AAD staining. The results are representative of at least two independent experiments. N.S., not significant; ** P < 0.01; *** P < 0.001.
Next we tested whether L. monocytogenes invasion of Hepa1-6 cells was affected by TNFAIP8 depletion. After allowing the bacteria to invade cells for 1 hour, we found that the knockdown cells had reduced bacterial burden, with or without TNFα treatment, and TNFα did not provide any protective effect against bacterial invasion (Figure 3B). TNFα is important in protecting mice against many bacterial pathogens. Hepatocytes produce TNFα upon exposure to L. monocytogenes, and its depletion sensitizes mice to death by L. monocytogenes (11, 16). Next we tested whether TNFAIP8 knockdown had an effect on L. monocytogenes -induced cell death. We incubated the cells with L. monocytogenes overnight with or without TNFα and found that the TNFAIP8 knockdown cells showed enhanced sensitivity towards cell death only when treated with both L. monocytogenes and TNFα (Figure 3C). Finally we assayed whether there was a difference in TNFα-induced cell death by treating cells with TNFα and cycloheximide and found increased sensitivity in the knockdown cell lines (Figure 3D). Our results suggest that the resistance to lethal L. monocytogenes infection in mice could be caused by the resistance of host cells to bacterial invasion and increased sensitivity to cell death following infection, thereby reducing bacterial load in the organism.
TNFAIP8 regulates RAC1 activation by TNFα
RAC1 is an important regulator of cell death following TNFα stimulation, and TNFAIP8 has been found to protect against TNFα-induced cell death (2, 3, 5, 17, 18). RAC1 has also been found to regulate L. monocytogenes invasion into endothelial cells (6, 19). To test whether there is a difference in RAC1 activation, PAK-GST beads were used to pull down RAC1. PAK only binds to active RAC1-GTP and is routinely used to assay for RAC1 activity. Knockdown and control cells were stimulated with TNFα for 5 minutes and the amount of RAC1 pulled down with PAK-GST beads was compared (Figure 4A). We found dysregulated RAC1 activation in TNFAIP8 knockdown cells, with higher basal levels of RAC1-GTP, but limited induction following TNFα stimulation. Next we checked whether there were any differences in RAC1 trafficking to the membrane and found impaired RAC1 translocation following TNFα stimulation (Figure 4B). In control cells, TNFAIP8 seemed to primarily localize to the cytosol, but surprisingly, there was more TNFAIP8 on the membrane than in the cytosol in the knockdown cells. To determine the consequence of increased basal RAC1 level, we assayed F-actin by flow cytometry and found increased F-actin in TNFAIP8 knockdown cells (Figure 4C).
Figure 4. TNFAIP8 and RAC1 in L. monocytogenes infection and cell death.

The levels of RAC1-GTP was determined by a PAK-GST pull down assay for control and TNFAIP8 knockdown Hepa1-6 cells stimulated with or without 50 ng/ml of TNFα for 5 minutes. Relative band intensities: 1, 1.5, 1.7, and 1.54 for Scr and SH1 with no treatment, and Scr and SH1 with TNFα treatment, respectively. (B) Control and TNFAIP8 knockdown Hepa1-6 cells were stimulated with 50 ng/ml of TNFα for 0, 2, 5, or 15 minutes. Lysates were fractionated into membrane [M] and cytoplasmic [C] portions, and TNFAIP8, RAC1, and actin were detected by Western blot. Relative band intensities: 1, 1.61, 2.86, 1.84, 1.72, 1.42, 1.91, and 2.04 for Scr with 0, 2, 5, and 15 minutes of TNFα treatment and SH1 with 0, 2, 5, and 15 minutes of TNFα treatment, respectively. Band intensities were determined by ImageJ software. (C) Control and TNFAIP8 knockdown Hepa1-6 cells were stained with phalloidin-FITC to quantify their total levels of F-actin by flow cytometry. (D) Control and TNFAIP8 knockdown Hepa1-6 cells were transiently transfected with pEGFP vectors containing either the EGFP cDNA alone [E], or EGFP plus RAC1-17N [R-] or RAC1-61L [R+] cDNAs. Cells were treated with 5 ng/ml of TNFα and 20 ng/ml of cycloheximide for 6 hours. EGFP positive cells were gated by flow cytometry and cell death was detected by Annexin V and 7-AAD staining. (E) Control and TNFAIP8 knowdown Hepa1-6 cells were infected with 50 MOI of L. monocytogenes for 1 hour with or without RAC1 inhibitor II [RI] Z62954982 at 100 μM. Cells were washed and then treated with 150 μg/ml Gentamycin for 30 minutes to kill extracellular bacteria before being lysed to release the intracellular bacteria. The results are representative of at least two independent experiments. * P < 0.05; ** P < 0.01; *** P < 0.001.
To test whether the differences in knockdown cell death are RAC1-dependent, we transfected cells with the constitutively active RAC1-61L or inactive RAC1-17N and treated them with TNFα and cycloheximide (CHX) (Figure 4D). We found that the dominant negative mutant partially rescued cell death while the constitutively active mutant greatly sensitized the control cells to death, but had a limited effect on the knockdown cells. We next tested the relevance of RAC1 signaling to L. monocytogenes invasion of the knockdown cells. We infected TNFAIP8 knockdown and control cells with L. monocytogenes, with or without RAC1 inhibitor treatment, and found that the inhibitor effectively protected cells from bacterial invasion, and negated any differences between control and TNFAIP8 knockdown cells (Figure 4E). Taken together, these results indicate that RAC1 activation is dysregulated in TNFAIP8 knockdown cells, which likely contributes to the altered response to TNFα-induced cell death and L. monocytogenes invasion.
TNFAIP8 associates with RAC1 with a higher affinity towards active RAC1-GTP
We first tested whether Flag-tagged TNFAIP8 associates with endogenous RAC1. We transfected 293T cells with a plasmid encoding Flag-tagged TNFAIP8 and immunoprecipitated cell lysates with anti-Flag antibody or control IGG and blotted for RAC1 and Flag-TNFAIP8 (Figure 5A). We found that endogenous RAC1 co-immunoprecipitated with TNFAIP8. We then tested whether this interaction was dependent on a particular region of RAC1. We created several HA-tagged Rac1 mutants and transfected them into 293T cells along with Flag-tagged TNFAIP8. We pulled down Flag and blotted for HA and found that RAC1 association was impaired with the Δ162-192 RAC1 mutant, which lacks the polybasic region (PBR) and CAAX motif (Figure 5B). The PBR is critical for RAC1 interaction with certain proteins and proper cellular localization (20, 21). The CAAX motif is responsible for post-translational modification and is crucial for the proper function of RAC1 (22, 23).
Figure 5. TNFAIP8 associates with RAC1 with a higher affinity towards active RAC1-GTP.

(A) 293T cells were transfected with the pRK5 expression plasmids containing Flag-tagged TNFAIP8. Anti-FLAG antibody or control mouse IGG was used to immunoprecipitate the Flag-tagged TNFAIP8 along with endogenous RAC1 and Western blotting was performed using the indicated antibodies. (B) HA-tagged RAC1 mutants were co-expressed with Flag-tagged TNFAIP8 in 293T cells, and IP was performed using anti-Flag. Western blots were developed using anti-HA antibody to identify RAC1 mutants co-immunoprecipitated with TNFAIP8. (C) 293T cells were cotransfected with Flag-tagged TNFAIP8 and Myc-tagged RAC1 or empty pRK5 vector. Total cell lysates were then treated with or without GTPγS or GDPβS, and anti-Myc antibody was used to immunoprecipitate RAC1. TNFAIP8 and RAC1 were determined by Western blotting. The results are representative of at least two independent experiments.
To test whether TNFAIP8-RAC1 interaction is dependent on the activation status of RAC1, we loaded 293T lysate expressing Myc-tagged RAC1 and Flag-tagged TNFAIP8 with non-hydrolyzable forms of GTP (GTPγS) and GDP (GDPβS). We found increased TNFAIP8 association with GTPγS loaded RAC1, suggesting that TNFAIP8 preferentially associates with the active form of RAC1 (Figure 5C). These results demonstrate that TNFAIP8 associates with active RAC1 and that this association may be responsible for the TNFAIP8 effect in L. monocytogenes infection.
Discussion
The role of TNFAIP8 in cell death appears to be condition-dependent. In most tumor cells, a protective effect of TNFAIP8 is reported; however in thymocytes, TNFAIP8 has been found to promote gluococorticoid-induced apoptosis (24). The molecular pathways of cell death that are regulated by TNFAIP8 are still largely unknown. Previous work suggests that TNFAIP8 protects against death through its ‘death effector domain (DED)’, but that hypothesis has been largely debunked by the structural analysis of TIPE2, which revealed a novel fold instead of a DED domain (3, 25). Others have suggested that TNFAIP8 works through Gα(i) proteins, and have shown that TNFAIP8 is important for the function of dopamine-D2short receptors in protecting against TNFα-induced cell death (5). Recently, TNFAIP8L1, another TNFAIP8 family member, has been implicated in activating mTOR to protect against oxidative stress in dopamine neurons by blocking autophagy (26). The mTOR protein itself plays an important role in promoting cancer, protecting against cell death, and increasing susceptibility to lethal L. monocytogenes infection, and has also been found to be regulated by the small GTPase RAC1 (27–29). We show here that TNFAIP8 associates with RAC1 and regulates TNFα-induced cell death and lethal L. monocytogenes infection in mice. We propose that it does so by regulating RAC1 signaling.
Listeria monocytogenes pathogenesis is dependent on a number of factors related to both immune and non-immune systems. Hepatocytes are quickly invaded by L. monocytogenes upon infection, and apoptotic cell death is important in releasing TNFα and other cytokines to trigger an immune response to the infected liver (9, 16, 30, 31). Neutrophils are attracted to the infected organs to phagocytose and kill bacteria. If the pathogen is not controlled, L. monocytogenes will escape to other organs and the organism can die of sepsis. Protecting immune cells from death has been found to be an important aspect in preventing lethal L. monocytogenes infection (32, 33). But promoting cell death in hepatocytes or blocking cellular invasion might be equally important for the survival of the organism (16, 19, 34).
Our results show that TNFAIP8 knockout animals are resistant to lethal L. monocytogenes infection with decreased bacterial load in the liver and spleen, similar to what has been previously observed with TIPE2 (6). However, unlike TIPE2 knockout mice that show heightened immunity to L. monocytogenes, there was little difference in phagocytosis, chemotaxis, or cytokine production between WT and TNFAIP8 KO innate immune cells. TNFα and IL-6 are important in the resistance to L. monocytogenes infection, and both were previously studied in TIPE2 knockout mice (2, 6, 11). In addition to these cytokines, IFNγ is also very important for immunity to L. monocytogenes (30, 35). However, due to limitations in serum quantities collected, we could not investigate IFNγ in this study. Despite reports suggesting a role for TNFAIP8 in Staphylococcus aureus infection, we found no difference in survival between WT and TNFAIP8 KO mice (4). This suggests that TNFAIP8 knockout mice are not resistant to extracellular pathogens, and that the immune system is no more capable of handling pathogens than that of WT mice. Rather the non-immune cells of the host may be more important in protecting themselves against intracellular pathogens.
Apoptosis of host cells is an important defense mechanism against intracellular pathogens like L monocytogenes (10). Hepatocyte apoptosis may inhibit L. monocytogenes spread within the liver and allow access of the pathogen to professional phagocytes (31). Previous work has focused on leukocyte-mediated cell death of hepatocytes, and it has been discovered that depletion of Fas or the downstream Caspase-8 enzyme in hepatocytes reduced CD8+ T-cell-mediated cell death and resulted in increased bacterial burden in the liver (36, 37). The leukocytes responsible for lysis of hepatocytes during the early stages of infection are not clear, with both neutrophils and macrophages likely involved (38, 39). We therefore decided to investigate L. monocytogenes infection in the non-immune hepatoma cell line Hepa1-6. We found that TNFAIP8 knockdown Hepa1-6 cells were more sensitive to death by L. monocytogenes when treated with TNFα, and were resistant to L. monocytogenes invasion. TNFα is critical for protecting against lethal L. monocytogenes infection, which can block certain aspects of TNFα signaling to protect itself (11, 40). Therefore, it stands to reason that TNFAIP8-knockout mice are protected against lethal L. monocytogenes infection by first reducing hepatocyte invasion and second by increasing apoptosis of infected hepatocytes in response to TNFα, thus blocking a productive infection of the pathogen.
We observed an increase in pAKT at serine 473 in TNFAIP8 knockdown cells, suggesting that mTOR may also be regulated by TNFAIP8. RAC1 has been implicated in regulating mTOR complexes by its interaction with mTORC1 and mTORC2 proteins (29). In addition, TNFAIP8L2 has been found to associate with RAC1 (6). This led us to investigate whether TNFAIP8 effector functions are dependent on RAC1 signaling because both RAC1 and mTOR are implicated in L. monocytogenes invasion of cells and apoptosis (15, 19, 28).
After testing several conditions using TNFAIP8 knockdown cells, we concluded that TNFAIP8’s regulation of cell death is RAC1 dependent. By introducing the dominant negative mutant of RAC1, TNFα-induced cell death in TNFAIP8 knockdown cells is partially rescued. Furthermore, the constitutively active mutant had a stronger effect on the control cells than they did on the knockdown cells, presumably because the RAC1 signaling was already heightened in knockdown cells. We observed increased RAC1-GTP at basal levels in knockdown cells and enrichment of TNFAIP8 in the membrane fraction when compared to the controls. The localization of TNFAIP8 in knockdown cells suggests that the membrane association is crucial for proper cell function and that it may be related to the increased RAC1 activation. In addition, we found increased levels of F-actin in knockdown cells at the basal state, supporting the heightened level of RAC1-GTP in these cells. This led us to the conclusion that TNFAIP8 knockdown cells are more susceptible to TNFα-induced cell death because there is insufficient TNFAIP8 present to inhibit RAC1.
Bacterial entry into knockdown cells is inhibited in spite of increased RAC1 activation and reports that RAC1 activation is necessary for bacterial entry (19, 41). The RAC1 inhibitor did inhibit bacterial invasion of both knockdown and control cells and eliminated the differences in bacterial counts, suggesting that RAC1 is responsible for the differences in bacterial invasion. A possible explanation for reduced bacterial invasion of knockdown cells in the face of elevated RAC1 activation is that RAC1 is activated nonspecifically throughout the cellular membrane in TNFAIP8 deficient cells, increasing ROS production and activating molecular pathways that do not require localized activation. Functions that require localized activation, like the internalization of a L. monocytogenes cell on a host membrane, may be inhibited because there is too much background RAC1 activation to efficiently recruit the required proteins to a specific location and internalize the bacterium. An alternative explanation is that infected TNFAIP8 knockdown cells are dying rapidly, reducing the total number of bacteria protected from the gentamycin treatment. This explanation, however, seems unlikely due to the short incubation time with L. monocytogenes for the infection experiments, and that no difference in death induced by L. monocytogenes was observed after 24 hours compared to control cells.
There are several different forms of endocytosis, and L. monocytogenes has been shown to take advantage of Clathrin dependent MET receptor-mediated endocytosis to gain entry into the cell (42). Contrary to the differences observed with L. monocytogenes invasion of the Hepa 1-6 cell lines, phagocytosis by TNFAIP8 knockout bone marrow-derived macrophages showed no abnormalities. One possible explanation is that the lack of TNFAIP8 in the knockout macrophages is compensated by the presence of TIPE2. TIPE2 protein is undetectable in Hepa 1-6 cells (data not shown), but is expressed in macrophages where it regulates phagocytosis (6). A second potential explanation is that the molecular mechanism for phagocytosis of L. monocytogenes in macrophages is sufficiently different from the endocytosis of L. monocytogenes in Hepa 1-6 cells that TNFAIP8 deficiency can affect one but not the other.
Finally, we showed that TNFAIP8 associates with RAC1 by co-immunoprecipitation. We were forced to use transiently expressed TNFAIP8 due to the very weak binding affinity of the commercial TNFAIP8 antibodies. We further found that the TNFAIP8 association is dependent on the C-terminus of RAC1, which contains the PBR and CAAX motif. These two regions are responsible for protein interactions, membrane localization signaling, and post-translational modifications. Our finding that the association is dependent on whether RAC1 is primed with GTP or GDP, suggests that RAC1 interaction with TNFAIP8 is enhanced with RAC1 activation. Understanding the molecular interaction of TNFAIP8 with RAC1 is important in identifying therapeutic targets for treating a wide variety of diseases, including bacterial infections and cancer.
Here, we propose that TNFAP8 regulates TNFα-induced cell death by its inhibition of RAC1 (Figure 6). The TNFα signaling cascade activates pro-death pathways through caspase cleavage and ROS production by the NAPDH oxidase complex, and pro-survival pathways through the activation of the NF-κB transcription factor. Inhibition of RAC1 and the associated NADPH oxidase activation has been documented to protect cells against both apoptotic and necrotic cell death following TNFα stimulation (17, 18). TNFAIP8 expression is reported to be up-regulated by TNFα signaling through NF-κB (2, 43). Our findings suggest that NF-κB can protect against cell death by preventing ROS production through RAC1 inhibition by the expression of TNFAIP8.
Figure 6. Model of TNFAIP8 molecular function in cell death.
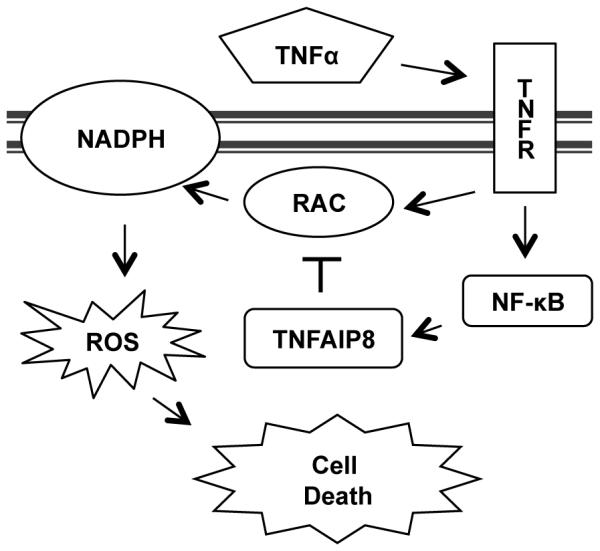
TNFα activates the tumor necrosis factor receptor (TNFR), which results in RAC1 and NF-κB activation. RAC1 then activates the NAPDH oxidase complex to generate reactive oxygen species (ROS) that promote cell death. The NF-κB transcription factor promotes TNFAIP8 expression to inhibit RAC1 activation and protect against cell death.
We also propose that RAC1 regulation by TNFAIP8 is important for L. monocytogenes invasion of host cells. L. monocytogenes Internalin A and B surface proteins bind to MET (also known as the hepatocyte growth factor receptor), β-integrin and E-cadherin receptors to activate RAC1 and induce F-actin cytoskeletal remodeling that allow for bacterial entry into the cells (41). We hypothesize that TNFAIP8 functions to reduce background RAC1 activation, allowing for precise control of RAC1 signaling. Insufficient TNFAIP8 likely interferes with this aspect of bacterial infection by preventing localized RAC1 signaling at the site of bacterial entry thus preventing the recruitment of F-actin and inhibiting bacterial internalization. In conclusion, our results indicate that TNFAIP8 regulates RAC1 signaling during TNFα receptor activation and L. monocytogenes invasion of cells, and that this regulation is crucial for controlling cell death and lethal L. monocytogenes infection of mice.
Acknowledgements
The authors thank Ji Qi, Qingguo Ruan, and George Luo for reagents and/or valuable advice. The authors additionally acknowledge Drs. Margaret M. Chou, Nicola J. Mason, Robert H. Vonderheide and Kenneth S. Zaret for helpful comments and discussions.
This work was funded by National Institutes of Health, USA (AI-077533, AI-050059, and GM-085112 to YHC).
References
- 1.Patel S, Wang FH, Whiteside TL, Kasid U. Identification of seven differentially displayed transcripts in human primary and matched metastatic head and neck squamous cell carcinoma cell lines: implications in metastasis and/or radiation response. Oral Oncol. 1997;33:197–203. doi: 10.1016/s0964-1955(96)00065-6. [DOI] [PubMed] [Google Scholar]
- 2.Kumar D, Whiteside TL, Kasid U. Identification of a novel tumor necrosis factor-alpha-inducible gene, SCC-S2, containing the consensus sequence of a death effector domain of fas-associated death domain-like interleukin-1beta-converting enzyme-inhibitory protein. J. Biol. Chem. 2000;275:2973–8. doi: 10.1074/jbc.275.4.2973. [DOI] [PubMed] [Google Scholar]
- 3.Kumar D, Gokhale P, Broustas C, Chakravarty D, Ahmad I, Kasid U. Expression of SCC-S2, an antiapoptotic molecule, correlates with enhanced proliferation and tumorigenicity of MDA-MB 435 cells. Oncogene. 2004;23:612–616. doi: 10.1038/sj.onc.1207123. [DOI] [PubMed] [Google Scholar]
- 4.Ahn S-H, Deshmukh H, Johnson N, Cowell LG, Rude TH, Scott WK, Nelson CL, Zaas AK, Marchuk DA, Keum S, Lamlertthon S, Sharma-Kuinkel BK, Sempowski GD, Fowler VG. Two genes on A/J chromosome 18 are associated with susceptibility to Staphylococcus aureus infection by combined microarray and QTL analyses. PLoS Pathog. 2010;6:e1001088. doi: 10.1371/journal.ppat.1001088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Laliberté B, Wilson AM, Nafisi H, Mao H, Zhou YY, Daigle M, Albert PR. TNFAIP8: a new effector for Galpha(i) coupling to reduce cell death and induce cell transformation. J. Cell. Physiol. 2010;225:865–74. doi: 10.1002/jcp.22297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Z, Fayngerts S, Wang P, Sun H, Johnson DS, Ruan Q, Guo W, Chen YH. TIPE2 protein serves as a negative regulator of phagocytosis and oxidative burst during infection. Proc. Natl. Acad. Sci. U. S. A. 2012;109:15413–8. doi: 10.1073/pnas.1204525109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mateus T, Silva J, Maia RL, Teixeira P. Listeriosis during Pregnancy: A Public Health Concern. ISRN Obstet. Gynecol. 20132013:851712. doi: 10.1155/2013/851712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hernandez-Milian A, Payeras-Cifre A. What Is New in Listeriosis? Biomed Res. Int. 20142014:358051. doi: 10.1155/2014/358051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vázquez-Boland JA, Kuhn M, Berche P, Chakraborty T, Domínguez-Bernal G, Goebel W, González-Zorn B, Wehland J, Kreft J. Listeria pathogenesis and molecular virulence determinants. Clin. Microbiol. Rev. 2001;14:584–640. doi: 10.1128/CMR.14.3.584-640.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaufmann SH. Immunity to intracellular bacteria. Annu. Rev. Immunol. 1993;11:129–63. doi: 10.1146/annurev.iy.11.040193.001021. [DOI] [PubMed] [Google Scholar]
- 11.Miura T, Nishikawa S, Sasaki S, Yamada K, Hasegawa S, Mizuki D, Mizuki M, Hatayama I, Sekikawa K, Tagawa Y, Iwakura Y. Roles of endogenous cytokines in liver apoptosis of mice in lethal Listeria monocytogenes infection. FEMS Immunol. Med. Microbiol. 2000;28:335–41. doi: 10.1111/j.1574-695X.2000.tb01495.x. a Nakane. [DOI] [PubMed] [Google Scholar]
- 12.Lee J, Cacalano G, Camerato T, Toy K, Moore MW, Wood WI. Chemokine binding and activities mediated by the mouse IL-8 receptor. J. Immunol. 1995;155:2158–64. [PubMed] [Google Scholar]
- 13.Das ST, Rajagopalan L, Guerrero-Plata A, Sai J, Richmond A, Garofalo RP, Rajarathnam K. Monomeric and dimeric CXCL8 are both essential for in vivo neutrophil recruitment. PLoS One. 2010;5:e11754. doi: 10.1371/journal.pone.0011754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hatano E, a Brenner D. Akt protects mouse hepatocytes from TNF-alpha- and Fas-mediated apoptosis through NK-kappa B activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2001;281:G1357–68. doi: 10.1152/ajpgi.2001.281.6.G1357. [DOI] [PubMed] [Google Scholar]
- 15.Jiwani S, Wang Y, Dowd GC, Gianfelice A, Pichestapong P, Gavicherla B, Vanbennekom N, Ireton K. Identification of components of the host type IA phosphoinositide 3-kinase pathway that promote internalization of Listeria monocytogenes. Infect. Immun. 2012;80:1252–66. doi: 10.1128/IAI.06082-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santos S, Dos A, De Andrade DR, De Andrade Júnior DR. Rat hepatocyte invasion by Listeria monocytogenes and analysis of TNF-alpha role in apoptosis. Rev. Inst. Med. Trop. Sao Paulo. 2005;47:73–80. doi: 10.1590/s0036-46652005000200003. [DOI] [PubMed] [Google Scholar]
- 17.Jin S, Ray RM, Johnson LR. TNF-alpha/cycloheximide-induced apoptosis in intestinal epithelial cells requires Rac1-regulated reactive oxygen species. Am. J. Physiol. Gastrointest. Liver Physiol. 2008;294:G928–37. doi: 10.1152/ajpgi.00219.2007. [DOI] [PubMed] [Google Scholar]
- 18.Kim Y-S, Morgan MJ, Choksi S, Liu Z-G. TNF-induced activation of the Nox1 NADPH oxidase and its role in the induction of necrotic cell death. Mol. Cell. 2007;26:675–87. doi: 10.1016/j.molcel.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 19.Schmeck B, Beermann W, van Laak V, Opitz B, Hocke AC, Meixenberger K, Eitel J, Chakraborty T, Schmidt G, Barth H, Suttorp N, Hippenstiel S. Listeria monocytogenes induced Rac1-dependent signal transduction in endothelial cells. Biochem. Pharmacol. 2006;72:1367–74. doi: 10.1016/j.bcp.2006.06.033. [DOI] [PubMed] [Google Scholar]
- 20.Modha R, Campbell LJ, Nietlispach D, Buhecha HR, Owen D, Mott HR. The Rac1 polybasic region is required for interaction with its effector PRK1. J. Biol. Chem. 2008;283:1492–500. doi: 10.1074/jbc.M706760200. [DOI] [PubMed] [Google Scholar]
- 21.Williams CL. The polybasic region of Ras and Rho family small GTPases: a regulator of protein interactions and membrane association and a site of nuclear localization signal sequences. Cell. Signal. 2003;15:1071–1080. doi: 10.1016/s0898-6568(03)00098-6. [DOI] [PubMed] [Google Scholar]
- 22.Casey PJ, Solski PA, Der CJ, Buss JE. p21ras is modified by a farnesyl isoprenoid. Proc. Natl. Acad. Sci. U. S. A. 1989;86:8323–7. doi: 10.1073/pnas.86.21.8323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kreck ML, Uhlinger DJ, Tyagi SR, Inge KL, Lambeth JD. Participation of the small molecular weight GTP-binding protein Rac1 in cell-free activation and assembly of the respiratory burst oxidase. Inhibition by a carboxyl-terminal Rac peptide. J. Biol. Chem. 1994;269:4161–8. [PubMed] [Google Scholar]
- 24.Woodward MJ, de Boer J, Heidorn S, Hubank M, Kioussis D, Williams O, Brady HJM. Tnfaip8 is an essential gene for the regulation of glucocorticoid-mediated apoptosis of thymocytes. Cell Death Differ. 2010;17:316–23. doi: 10.1038/cdd.2009.125. [DOI] [PubMed] [Google Scholar]
- 25.Zhang X, Wang J, Fan C, Li H, Sun H, Gong S, Chen YH, Shi Y. Crystal structure of TIPE2 provides insights into immune homeostasis. Nat. Struct. Mol. Biol. 2009;16:89–90. doi: 10.1038/nsmb.1522. [DOI] [PubMed] [Google Scholar]
- 26.Choi K-C, Kim S-H, Ha J-Y, Kim S-T, Son JH. A novel mTOR activating protein protects dopamine neurons against oxidative stress by repressing autophagy related cell death. J. Neurochem. 2010;112:366–76. doi: 10.1111/j.1471-4159.2009.06463.x. [DOI] [PubMed] [Google Scholar]
- 27.Strimpakos AS, Karapanagiotou EM, Saif MW, Syrigos KN. The role of mTOR in the management of solid tumors: an overview. Cancer Treat. Rev. 2009;35:148–59. doi: 10.1016/j.ctrv.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 28.Weichhart T, Costantino G, Poglitsch M, Rosner M, Zeyda M, Stuhlmeier KM, Kolbe T, Stulnig TM, Hörl WH, Hengstschläger M, Müller M, Säemann MD. The TSC-mTOR signaling pathway regulates the innate inflammatory response. Immunity. 2008;29:565–77. doi: 10.1016/j.immuni.2008.08.012. [DOI] [PubMed] [Google Scholar]
- 29.Saci A, Cantley LC, Carpenter CL. Rac1 regulates the activity of mTORC1 and mTORC2 and controls cellular size. Mol. Cell. 2011;42:50–61. doi: 10.1016/j.molcel.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pamer EG. Immune responses to Listeria monocytogenes. Nat. Rev. Immunol. 2004;4:812–23. doi: 10.1038/nri1461. [DOI] [PubMed] [Google Scholar]
- 31.Rogers HW, Callery MP, Deck B, Unanue ER. Listeria monocytogenes induces apoptosis of infected hepatocytes. J. Immunol. 1996;156:679–84. [PubMed] [Google Scholar]
- 32.Zheng S-J, Jiang J, Shen H, Chen YH. Reduced apoptosis and ameliorated listeriosis in TRAIL-null mice. J. Immunol. 2004;173:5652–8. doi: 10.4049/jimmunol.173.9.5652. [DOI] [PubMed] [Google Scholar]
- 33.Gurung P, Rai D, Condotta SA, Babcock JC, Badovinac VP, Griffith TS. Immune unresponsiveness to secondary heterologous bacterial infection after sepsis induction is TRAIL dependent. J. Immunol. 2011;187:2148–54. doi: 10.4049/jimmunol.1101180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dos Santos SA, de Andrade Júnior DR, de Andrade DR. TNF-α production and apoptosis in hepatocytes after Listeria monocytogenes and Salmonella Typhimurium invasion. Rev. Inst. Med. Trop. Sao Paulo. 2011;53:107–12. doi: 10.1590/s0036-46652011000200009. [DOI] [PubMed] [Google Scholar]
- 35.Harty JT, Bevan MJ. Specific immunity to Listeria monocytogenes in the absence of IFN gamma. Immunity. 1995;3:109–17. doi: 10.1016/1074-7613(95)90163-9. [DOI] [PubMed] [Google Scholar]
- 36.Jensen ER, Glass AA, Clark WR, Wing EJ, Miller JF, Gregory SH. Fas (CD95)-dependent cell-mediated immunity to Listeria monocytogenes. Infect. Immun. 1998;66:4143–50. doi: 10.1128/iai.66.9.4143-4150.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ben Moshe T, Barash H, Kang T-B, Kim J-C, Kovalenko A, Gross E, Schuchmann M, Abramovitch R, Galun E, Wallach D. Role of caspase-8 in hepatocyte response to infection and injury in mice. Hepatology. 2007;45:1014–24. doi: 10.1002/hep.21495. [DOI] [PubMed] [Google Scholar]
- 38.Conlan JW, Dunn PL, North RJ. Leukocyte-mediated lysis of infected hepatocytes during listeriosis occurs in mice depleted of NK cells or CD4+ CD8+ Thy1.2+ T cells. Infect. Immun. 1993;61:2703–7. doi: 10.1128/iai.61.6.2703-2707.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Haschtmann D, Gerber HJ, a Mielke ME. Cytotoxic activity of murine resident peritoneal cells against Listeria monocytogenes-infected hepatocytes in vitro. Microbes Infect. 2005;7:1177–83. doi: 10.1016/j.micinf.2005.03.031. [DOI] [PubMed] [Google Scholar]
- 40.Gouin E, Adib-Conquy M, Balestrino D, Nahori M-A, Villiers V, Colland F, Dramsi S, Dussurget O, Cossart P. The Listeria monocytogenes InlC protein interferes with innate immune responses by targeting the I{kappa}B kinase subunit IKK{alpha} Proc. Natl. Acad. Sci. U. S. A. 2010;107:17333–8. doi: 10.1073/pnas.1007765107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Da Silva CV, Cruz L, Araújo NDS, Angeloni MB, Fonseca BB, Gomes ADO, Carvalho FDR, Gonçalves ALR, Barbosa BDF. A glance at Listeria and Salmonella cell invasion: different strategies to promote host actin polymerization. Int. J. Med. Microbiol. 2012;302:19–32. doi: 10.1016/j.ijmm.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 42.Veiga E, Cossart P. Listeria hijacks the clathrin-dependent endocytic machinery to invade mammalian cells. Nat. Cell Biol. 2005;7:894–900. doi: 10.1038/ncb1292. [DOI] [PubMed] [Google Scholar]
- 43.You Z, Ouyang H, Lopatin D, Polver PJ, Wang CY. Nuclear factor-kappa B-inducible death effector domain-containing protein suppresses tumor necrosis factor-mediated apoptosis by inhibiting caspase-8 activity. J. Biol. Chem. 2001;276:26398–404. doi: 10.1074/jbc.M102464200. [DOI] [PubMed] [Google Scholar]