

Abstract
The role of ion channels is largely unknown in chemokine-induced migration in non-excitable cells such as dendritic cells. Here, we examined the role of KCa3.1 and chloride channels in lymphatic chemokines-induced migration of dendritic cells. The amplitude and kinetics of CCL19/21-induced Ca2+ influx were associated with CCR7 expression levels, extracellular free Ca2+ and Cl−, and independent of extracellular K+. Chemokines, CCL19 and CCL21, and KCa3.1 activator, 1-EBIO, induced plasma membrane hyperpolarization and K+ efflux, which was blocked by TRAM-34, suggesting that KCa3.1 carried larger conductance than the inward CRAC. Blockade of KCa3.1, low Cl− in the medium, and low dose of DIDS impaired CCL19/CCL21-induced Ca2+ influx, cell volume change, and DC migration. High doses of DIDS completely blocked DC migration possibly by significantly disrupting mitochondrial membrane potential. In conclusion, KCa3.1 and chloride channel are critical in human DC migration by synergistically regulating membrane potential, chemokine-induced Ca2+ influx, and cell volume.
Introduction
Dendritic cells (DCs) serve as principal antigen presenting cells and considered as initiators of the immune response in allergic asthma [1, 2]. Migration of DCs is meticulously governed by chemokines in a concentration, time and space-dependent manner [1]. Chemokine receptors expressed at different stages of maturating DCs drive the mobilization of selective DCs based on their maturation stage and type [1]. However, role of ion-channels in the chemokine-induced migration of DCs is largely unknown. DC migration to the secondary lymph organ is indispensible for the subsequent T helper cell-mediated adaptive immunity [3]. Upon antigen loading, activated DCs rapidly upregulate chemokine receptor 7 (CCR7) expression and acquire the ability to migrate to afferent lymphatics and draining lymph nodes [4] where lymphatic chemokine ligands, CCL19 and CCL21 are highly expressed [1, 5]. Based on current knowledge, entry of mature DCs to lymphatics and homing to draining lymph nodes are entirely dependent on CCR7-mediated directional migration [1, 4, 6] although semaphorins may also mediate DC entry to lymphatics [7]. Migrating DCs display high flexibility characterized by frequent shape/volume change and asymmetric cytoskeleton in order to adapt to the complex local anatomic structures [8, 9], which is putatively achieved by highly dynamic material exchange between intracellular compartment and extracellular matrix through aquaporin or ion-channels [10]. Unlike the ion-channels in excitable cells that normally generate large amplitude of rectified current, the electrophysiological properties of the ion-channels in immune cells are usually different and poorly characterized. Thus, the significance of ion-channels in the function of immune cells has long been neglected.
One of most extensively studied ionic activities is the chemokine-induced Ca2+ mobilization and subsequent Ca2+ influx. Activation of CCR7, a G protein-coupled receptor (GPCR), triggers entire migratory machinery in DC migration to draining lymph nodes [11]. Similar to most of the Gαi type GPCRs, CCR7 activates phospholipase C (PLCβ), which in turn, cleaves membrane lipids phosphatidylinositol 4,5-bisphosphate (PIP2) into membrane diacylglycerol (DAG) and cytosolic inositol 1, 4, 5 triphosphate (IP3). The latter triggers the Ca2+ release from endoplasmic reticulum (ER) lumen, a major intracellular Ca2+ stores, followed by a transient Ca2+ influx carried by calcium release-activated calcium channel (CRAC), as shown in mouse bone marrow-derived DCs [12, 13]. In human monocyte-derived DCs, however, no considerable Ca2+ influx was observed following the Ca2+ mobilization [14, 15]. The molecular identity and activation mechanisms of CRAC are unclear. Single transmembrane protein, STIM, has been identified as a putative sensor for ER Ca2+ depletion [16], while calcium release-activated calcium channel protein 1 (Orai1) is thought to be the candidate molecules for CRAC [17, 18].
Cell migration is dependent on a precisely integrated calcium signaling network which contains both spatial and temporal information [19]. A higher concentration of calcium has been observed in the trailing edge of migrating eosinophils in contrast to a lower concentration in the leading edge [20], while more recently, Wei and colleagues [21] have described active Ca2+ microdomains at the leading lamella of migrating cells. Although it is controversial as to whether or not calcium signal is indeed a part of the cell migration machinery due to lacking of identified pathways [22], most of the recent evidence has supported an indispensable role of Ca2+ in chemotaxis since the depletion of extracellular calcium or blockade of intracellular calcium mobilization always abolishes the cell migration [10, 23].
CCR7 activation-induced Ca2+ mobilization and influx exert substantial impact on overall calcium signal in two aspects: (i) it modulates the spatial and temporal dimensions of the Ca2+ signals necessary for cell migration, and (ii) asymmetrical exposure to chemokine gradient causes polarized CCR7 activation and leads to localized IP3-induced Ca2+ mobilization. Moreover, CCR7 undergoes desensitization or internalization to prevent constant activation of CCR7 [24], which sets the pace for intracellular calcium oscillation in a concerted manner. Secondly, the transient increase in intracellular Ca2+ leads to the activation of a number of calcium-activated ion channels, and possibly voltage-dependent channel if cell membrane potential is altered. These ion channels not only help to maintain a negative cell membrane potential through charge compensation mechanism to allow optimal calcium influx, they also potentially contribute to various cellular functions. Therefore, the simultaneous ion flows carried by multiple channels are far more complex than CRAC activity alone.
We have recently reported that intermediate conductance calcium-activated potassium channel, KCa3.1, is expressed in immunogenic and regulatory mouse lung DCs [25, 26] and is actively involved in DC migration to lymphatic chemokines by modulating CCL19/CCL21-induced Ca2+ influx [27]. In fact, KCa3.1 is expressed in almost all types of immune cells, including T cell [28], macrophage [29] and mast cells [30], and plays a key role in calcium-dependent cell activities such as migration [30-33], particularly under pathological conditions when calcium signaling is highly active. However, no in-depth investigation has been conducted to reveal the mechanisms by which KCa3.1 regulates human DC migration. The speculation for the mechanism of KCa3.1 in cell migration includes cell volume regulation, orchestrating membrane resting potential to maintain the Ca2+ fluctuation, and regulating rearrangement of the actin cytoskeleton [10].
We have also demonstrated that chloride channel is involved in monocyte migration [34], but no information is available as to if chloride flux occurs and serves as a regulating factor during chemokine-induced calcium influx. Volume sensitive chloride channel CLC3 is involved in neutrophil [35] migration, but the detailed underlying mechanism still has not yet been deciphered [36]. Moreover, chloride and potassium channels might work in concert with each other [37] in order to achieve the optimal volume change in cell migration [10]. In fact, the optimal regulation is usually accomplished by synergistic ion flow activity carried by multiple channels.
In the present study, we aimed to dissect the identity and direction of the ion flow during CCR7-induced Ca2+ influx and examined the role of these activities in the context of DC migration in response to lymphatic chemokines.
Materials and Methods
Preparation of human dendritic cells
The research was in accordance with The Code of Ethics of the World Medical Association (Declaration of Helsinki). Informed consents, as approved by the Institutional Review Board of Creighton University, were obtained from healthy volunteers prior to blood draw. Dendritic cells were prepared from human peripheral mononuclear cells (PBMCs), as described previously [38]. Peripheral venous blood from healthy volunteers with EDTA was added into the equal volume of HBSS, and layered over Histopaque-1077 (Sigma, St Louis, MO). Mononuclear cells were separated by density gradient centrifugation at 1,500 rpm for 15 minutes without brake at room temperature. The interface layer between plasma and Histopaque layers, containing PBMCs, was collected with Pasteur pipette and transferred to another tube. PBMCs were then incubated with FcR blocking reagent (Miltenyi Biotec, Auburn CA) and anti-CD14 conjugated magnetic beads (Miltenyi Biotec, Auburn CA), and positively sorted using AutoMACS (Miltenyi Biotec, Auburn CA) under the program “Possel”. The CD14+ cell fraction (>93% purity) was cultured in 6-well suspension culture plate for 6 days at 1 x 106/ml in RPMI-1640 culture medium supplemented with 10% FBS in the presence of 50ng/ml GM-CSF (PeproTech, Rocky Hill, NJ) and 20ng/ml IL-4 (200-04, PeproTech, Rocky Hill, NJ) to achieve immature DCs. Maturation of the DCs was achieved with 1µg/ml LPS (L2755, from Escherichia coli 0128:B12), 100µg/ml ovalbumin (Sigma, St Louis, MO), and 25ng/ml TNF-α (PeproTech, Rocky Hill, NJ) into cell culture on day 7. The cells were harvested 48 hours after the addition of maturation stimuli cocktail.
RNA Isolation, Reverse Transcription and RT-PCR
The total RNA was isolated from mature DCs using the Trizol reagent (Sigma, St. Louis, MO) method, and quantified with Nanodrop (Thermo scientific, Rockford, IL). First-strand cDNA synthesis was done using 1µ g total RNA with oligo dT (1µ g), 5 X reaction buffer, MgCl2, dNTP mix, RNAse inhibitor and Improm II reverse transcriptase as per Improm II reverse transcription kit (Promega, Madison, WI). Following the first strand synthesis, Real-Time PCR was done using 8µ l cDNA, 10µ l SYBR green PCR master mix (BioRad Laboratories, Hercules, CA) and forward and reverse primers (10picomol/µ l) (Integrated DNA Technologies, San Diego, CA, USA) using a Real-Time PCR system (CFX96, BioRad Laboratories, Hercules, CA). The primers sequences used were GAPDH, 5’-GGGAAGGTGAAGGTCGGAGT-3’; Reverse-5’-TTGAGGTCAATGAAGGGGTCA-3’; KCNN4 (gene for KCa3.1), Forward-5’-AGCTGAAGAACTGGGTATGAGGCT-3’; Reverse-5’-AGCAGAG GCTGGTGAGTTACACTT-3’; 18S, Forward-5’-TCAACTTTCGATGGTAGTCGCCGT-3’; Reverse-5’-TCCTTGGATGTGGTAGCCGTTTCT-3’. The specificity of the primers was analyzed by running a melting curve. The PCR cycling conditions used were 5 min at 95°C for initial denaturation, 40 cycles of 30s at 95°C, 30s at 55°C (depending upon the primer annealing temperatures) and 30s at 72°C. Each real-time PCR was carried out using 4 individual samples in duplicates and the threshold cycle values were averaged. Analysis of data was done using the Bio-Rad CFX manager software. The results were normalized against housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or 18S.
Western blot
Mature DCs were lysed using RIPA buffer (Sigma St Louis MO) for 20 min supplemented with protease inhibitor cocktail (Sigma, St Louis MO) at 4°C. Homogenates were centrifuged for 10 min at 12,000×g at 4°C. Protein quantification was performed on the supernatant with bicinchoninic acid (BCA) assay kit (Sigma, St. Louis, MO). The colorimetric signal was read using EnSpire Multimode Plate Reader (PerkinElmer, Waltham MA). Protein was then denatured at 95°C for 5 min in Laemmli-SDS sample buffer containing β-mercaptoethanol. Equal amounts of protein (20µg) were loaded into each lane of acrylamide SDS-PAGE gel and resolved at 120V constant. Gels were transferred onto nitrocellulose membrane (Bio-Rad) at 400 mA constant for 1 hr at 4°C, and membranes were blocked in blocking buffer (PBS containing 0.5% Tween and 5% milk). Blots were incubated in primary polyclonal rabbit anti-KCa3.1 antibody (Abcam, MA) at 1:1000 dilution over night at 4°C, and rinsed 4 times followed by incubation with HRP-conjugated goat anti-rabbit IgG secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) at 1:1000 dilution for 120 min at room temperature. After being washed, blots were developed with SuperSignal West Dura Extended Duration Substrate Kit (Thermo Scientific, Rockford, IL). Densitometric analysis was performed directly from the blotted membrane using UVP BioImaging system (UVP Inc, Upland, CA).
Measurement of intracellular Ca2+ and K+
To measure intracellular Ca2+ levels, DCs were loaded with 5µM indo-1 in calcium-free PBS at 37°C incubator with 5% CO2 for 45 min and then suspended at 1 × 106/ml in L15 medium containing 2mM Ca2+, Ca2+-free PBS, low Cl−, Na+-free, or K+-free isotonic solutions. The indo-1 fluorescence was excited at 354 nm with a UV laser. The ratios of the fluorescence at wavelength of 390nm versus 530nm (bound indo-1/free indo-1) was recorded and calculated to indicate the intracellular Ca2+ levels. To measure intracellular K+ levels, DCs were loaded with 1µM PBFI in original cell culture at 37 °C incubator with 5% CO2 for 90 minutes. The PBFI fluorescence was excited at 354 nm with a UV laser and the fluorescence emission at 530 nm was recorded to indicate intracellular K+ levels. In all the assays, baseline fluorescence signal was recorded for about 90 seconds by a FACSAria flow cytometer (BD, San Jose, CA) prior to addition of CCL19, CCL21, or KCa3.1 opener 1-EBIO. In blockade assays, DCs were pre-treated with 2µM TRAM-34 for 6 hours or 1-100µM DIDS for 2 hours prior to dye loading. All controls were treated with same volume of the vehicle or saline.
Measurement of cell and mitochondrial membrane potential
Cell membrane potential was measured using a fluorescence dye DiBAC4(3) that can be detected by flow cytometry. DCs were loaded with 1 mM DiBAC4(3) in original cell culture at 37 °C incubator with 5% CO2 for 30 min. A blue laser (488 nm) was used for excitation and the signal acquisition for emission fluorescence was set at wavelength of 530 nm. A MitoProbe JC-1 assay kit was used to measure mitochondrial membrane potential as per manufacturer’s protocol. The cells were double-stained with JC-1 and annexin V-APC (eBioscience, San Diego, CA). Depolarization was indicated by a fluorescence emission shift from green (529 nm) to red (590 nm) or decrease in red fluorescence at emission wavelength of 575 nm.
Chemotaxis assay
DCs were pre-treated with 2µM TRAM-34 for 6 hours or 10µM - 100µM DIDS for 2 hours before being loaded into the Transwell plates. The control DCs were treated with same amount of vehicle DMSO. The chemotaxis in response to 100ng/ml CCL19 and CCL21 (PeproTech, Rocky Hill, NJ) with the presence or absence of 2µM TRAM-34 or 10µM - 100µM DIDS were examined using a Transwell system as described previously [26]. The cells were counted with a Coulter Counter (Beckman Coulter, Brea CA). The chemotactic index, a measure of the specificity of migration, was calculated and normalized as follows: (number of cells migrating to chemokines) / (number of cells that migrated to medium alone). All the cell medium, low Cl−solution, and Ca2+-free PBS containing 2mM EGTA were supplemented with 4% FBS.
Data processing and statistical analysis
Data was processed and analyzed using Flowjo and GraphPad (GraphPad Software, La Jolla CA) and (Tree Star, Ashland OR). Values are expressed as means ± 95% confidence intervals unless otherwise indicated. The statistical analysis was performed using One-way ANOVA with Bonferroni’s corrections for multiple group comparisons. A value of p<0.05 was considered significant.
Results
CCR7 expression levels correlate with the amplitude of chemokine-induced Ca2+ increase
A combination of LPS, ovalbumin, and TNF-α induced a remarkable upregulation in CCR7 expression. As shown in Fig 1A, mature human DCs expressed significantly higher levels of CCR7 than immature DCs. Accordingly, lymphatic chemokine, CCL19, induced significantly greater chemotaxis in mature DCs than immature DCs (Fig 1B). Since CCR7 is a G-protein coupled receptor, its activation will lead to intracellular Ca2+ mobilization from endoplasmic reticulum (ER) via PLC-IP3 pathway, possibly followed by Ca2+ influx carried by store-operated calcium entry [14]. As expected, greater amplitude of Ca2+ influx was observed in mature DCs than in immature DCs within 15-20 seconds after the addition of 1µM CCL19 or CCL21 (Fig 1C). Thus, these findings demonstrate a correlation between CCR7 expression levels, the degree of chemokine-induced migration, and the amplitude of chemokine-induced Ca2+ influx. Additionally, the cell membrane texture, intracellular organelles, and cytoskeleton appeared to undergo re-organization (Fig 1D), as indicated by the simultaneous decrease in side scatter upon CCL19/CCL21-induced Ca2+ influx. The decrease in side scatter took place almost simultaneously as Ca2+ influx occurred (Fig 1D), while the decrease in forward scatter (average cell size) had a 100-150 seconds delay (Fig 1E). The values of the decrease in forward scatter indicate a decrease in the average cell size of the mature DCs following CCL19/CCL21 stimulation (Fig 1E).
Figure 1. CCR7 expression and CCL19/CCL21-induced cellular changes.

A, CCR7 expression in DCs matured with LPS, OVA and TNF-α cocktail as compared to immature DCs. B, Chemotaxis in response to CCL19 in immature and mature DCs. C, intracellular Ca2+ Kinetics: Ca2+ mobilization and influx in response to CCL19 and CCL21 in immature and mature DCs. Stimuli were added at 90 sec. D, CCL19/CCL21 decreased the side-scatter in mature DCs suggesting some intracellular reorganization. E, forward scatter also decreased in the matured DCs with CCL19/CCL21 stimulation suggesting a decrease in the cell size. Data are representatives of 3 independent experiments.
Ca2+ influx is the major component of chemokine-induced intracellular Ca2+ increase and not dependent on extracellular K+
Previously reported CCL19/CCL21-induced Ca2+ increase was mainly attributable to intracellular mobilization but not Ca2+ influx in monocyte-derived DCs [14]. In order to confirm that the Ca2+ influx is a major component of CCL19/CCL21-induced intracellular Ca2+ increase, calcium assay was performed with mature DCs suspended in Ca2+-free PBS. As shown in Fig 2A, CCL19/CCL21 did not induce large amplitude of intracellular Ca2+ increase in the absence of extracellular Ca2+, indicating that the increase in Ca2+ is primarily from extracellular medium. Additionally, depletion of extracellular K+ and blockade of inward K+ conductance with CsCl did not affect the amplitude and kinetics of CCL19/CCL21-induced Ca2+ influx (Fig 2B), suggesting that extracellular K+ and inward K+ conductance did not affect the properties of chemokine-induced Ca2+ influx.
Figure 2. Extracellular cations and CCL19/CCL21-induced intracellular Ca2+ increase.
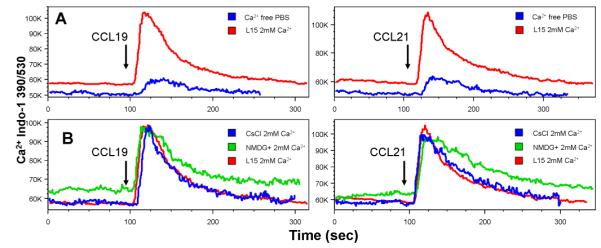
CCL21 (1µg/ml) was added at 90 sec. A, intracellular Ca2+ Kinetics: Ca2+ mobilization and influx in response to CCL19 (left panel) and CCL21 (right panel) in the presence of L15 medium or Ca2+-free PBS in mature DCs. B: intracellular Ca2+ Kinetics: Ca2+ mobilization and influx in response to CCL19 (left panel) and CCL21 (right panel) in the presence of L15 medium, or K+-free (CsCl2) solution. Data are the representatives of 3 independent experiments.
K+ efflux during Ca2+ influx is carried by KCa3.1
We further examined if an outward K+ conductance occurs concurrently with Ca2+ influx as a part of charge compensation mechanism because cationic Ca2+ influx tends to cause cell membrane depolarization by counteracting negative charge within intracellular compartment. Interestingly, CCL21-induced Ca2+ influx always led to cell membrane hyperpolarization (Fig 3A) but not depolarization in mature DCs, as measured by membrane potential dye DiBAC4(3). This strongly suggests that a greater outward cationic or inward anionic conductance was activated simultaneously to counteract the inward Ca2+ conductance leading to a net result of hyperpolarization. We then used K+-specific fluorescence dye, PBFI, to monitor the kinetics of intracellular K+ levels and observed that CCL21 also induced synchronized decreases in intracellular K+ levels (Fig 3B) concurrently with Ca2+ influx, suggesting that K+ efflux was triggered by CCR7 activation. This CCL21-induced K+ efflux was blocked by KCa3.1 blocker, TRAM-34 (Fig 3B). Additionally, addition of KCa3.1 opener 1-EBIO led to an instantaneous membrane hyperpolarization in DCs (Fig 3C), consistent with the manifestation of a cationic efflux. These findings further suggest that the K+ efflux is, at least in part, carried by KCa3.1. The quantitative real-time PCR and Western blot for the expression of KCa3.1 confirmed the KCa3.1 mRNA (Fig 3D) and protein (Fig 3E) expression, respectively, in mature DCs.
Figure 3. CCL21-induced K+ efflux is carried by KCa3.1.

A, kinetics of plasma membrane potential: in response to CCL21. CCL21 (1µg/ml) was added at 90 sec. (The figure is a representative of 6 independent experiments using CCL21 or CCL19 as stimulus) B, Kinetics of plasma membrane potential in response to KCa3.1 opener/activator, 1-EBIO. The 1-EBIO was added at 90 seconds with final concentration of 500mM and led to membrane hyperpolarization indicated by a decreased fluorescence intensity of DiBAC4(3). (The figure is a representative of 3 independent experiments) C, CCL21 (1µg/ml) was added at 90 sec to controls or 2µM TRAM-34-treated mature DCs and intracellular K+ kinetics was recorded as indicated by PBFI fluorescence intensity over the time. The figure is a representative of 3 independent experiments. D, mRNA expression KCa3.1 by quantitative real-time PCR (n=3) E, Western blot of KCa3.1 protein expression in mature DCs (n=3).
KCa3.1 is involved in DC migration and CCL19/CCL21-induced Ca2+ influx
Functional impact of KCa3.1 blockade in the context of CCL19/CCL21-induced DC migration was examined. As shown in Fig 4A, treatment with KCa3.1 specific blocker, TRAM-34 (2µM), for 6 hours partially blocked CCL19/CCL21-induced Ca2+ influx characterized by a significant decrease in the influx amplitude. There was no identifiable delay in intracellular Ca2+ recovery. Area under the curve (AUC), an indicator for overall Ca2+ loading upon chemokine-induced CCR7 activation, was also significantly decreased by TRAM-34 treatment (Fig 4B). Functionally, mature DCs treated with TRAM-34 (2µM) for 6 hours demonstrated impaired migration in response to 100ng/ml CCL19 or CCL21 (Fig 4C). Notably, TRAM-34 treatment did not depolarize mitochondrial membrane but slightly increased mitochondrial membrane potential as measured by JC-1 staining, indicating that the mitochondria-mediated cell metabolism was not negatively affected (Fig 4D). This is also consistent with the report showing low or no toxicity of TRAM-34 [31, 39].
Figure 4. KCa3.1 modulates CCL19/21-induced Ca2+ influx and migration in mature DCs.

A, CCL21 or CCL19 (1 µg/ml) was added at 90 sec to controls or 2µM TRAM-34-treated mature DCs. Intracellular Ca2+ kinetics were measured by indo-1 fluorescence over the time. B, The overall calcium loading indicated by the area under the curve (AUC) in control and 2µM TRAM-34-treated mature DCs in the context of CCL19/CCL21-induced Ca2+ mobilization and influx (n=5, *p<0.05, paired student t-test). C, DC migration to CCL19/CCL21 with the presence/absence of KCa3.1 blocker TRAM-34 as measured by Transwell (n=6, *p<0.05). Final concentration of chemokines and blocker: CCL19, 100ng/mL; CCL21, 100ng/mL; TRAM34, 2µM. D. Mitochondrial membrane potential as indicated by red fluorescence (575 nm) of JC-1 staining. Mitochondrial membrane potential disrupter, CCCP (carbonyl cyanide 3-chlorophenylhydrazone) serves as controls. The figure is a representative of three independent experiments.
Effect of TRAM-34 on KCa3.1 expression
To examine if the migration of mature DCs was due to change in the KCa3.1 expression following TRAM-34, mRNA transcripts of KCa3.1 were examined in mature DCs treated with TRAM-34. Amplification curve in log phase (Fig 5A) and sharp melt peak indicated the specific amplification of KCa3.1 and the housekeeping gene, 18S (Fig 5B). There was no statistically significant difference in the mRNA levels of KCa3.1 in mature DCs without TRAM-34 and mature DCs with TRAM-34 treatment (2µM) for 2 hours or 6 hours (Fig 5C), suggesting no effect of TRAM-34 on KCa3.1 gene regulation.
Figure 5. TRAM-34 does not change KCa3.1 expression in mature DCs.

Mature DCs were treated with 2µM TRAM-34 for 2 and 6 hours. A, Amplification curve (in log phase) showed the cycles at which KCa3.1 and 18S were amplified. B, Melt peak indicated the formation of specific products corresponding to KCa3.1 and 18S. C, No statistical change was observed in the mRNA levels of KCa3.1 in mature DCs without TRAM-34 and mature DCs with TRAM-34 treatment (2µM) for 2 hours or 6 hours (n=3, *p<0.05).
Chloride and KCa3.1 channel affect chemokine-induced cell volume change, Ca2+ influx, and DC migration
Apart from outward K+ conductance, CCL19/CCL21-induced Ca2+ influx might be accompanied by other ion-channel activity as a downstream event of CCR7 activation or increased intracellular calcium signaling. We next examined the role and activity of Cl− using CCL21 as a stimulus. Replacement of extracellular Cl− with Na+ gluconate did not affect the occurrence of Ca2+ influx but led to a decrease in flux amplitude (Fig 6A) and elevated baseline Ca2+ levels. Interestingly, low Cl− prevented cell volume decrease induced by CCL21, as indicated by forward scatter values (Fig 6B). The same effect was also observed in TRAM-34-treated mature DCs (Fig 6B). Furthermore, CCL21 still induced membrane hyperpolarization in the absence of extracellular Cl− (Fig 6C). Functionally, however, the DC migration was impaired (Fig 6D) in the absence of extracellular Cl− (Cl−/gluconate replacement) but is still more than that of Ca2+ depletion in the medium. Collectively, these data suggest that Cl− might be a part of charge compensation during Ca2+ influx and extracellular Cl− and KCa3.1-carried K+ efflux are important for CCL21-induced cell volume decrease.
Figure 6. Extracellular Cl-modulates CCL21-induced cell volume change, Ca2+ influx, and chemotaxis.

CCL21 was added at 90 sec. A, Intracellular Ca2+ kinetics in response to CCL21 (1 µg/ml) in the presence/absence of extracellular Cl− in mature DCs. B, Kinetics of average cell size of mature DCs in response to CCL21 (1µg/ml) in the presence/absence of extracellular Cl− and in the presence of TRAM-34 treatment for 6 hours, indicated by forward scatter values over the time. C, Kinetics of membrane potential in response to CCL21 (1µg/ml) in the absence of extracellular Cl− (Cl−/gluconate replacement) as indicated by DiBAC4(3) fluorescence over the time. D, Chemotaxis in response to 100 ng/ml CCL21 in medium containing 2mM EGTA, and medium in the presence and absence of Cl−.
Blockade of chloride channel abolishes Ca2+ influx and DC migration
The treatment of the cells with a chloride channel blocker, DIDS (1-100µM) completely abolished CCL19/CCL21-induced Ca2+ influx (Fig 7A-upper panel) and chemotaxis (Fig 7B) at all doses (1-100µM). DIDS at 1µM blocked the CCL19-induced cell size decrease (Fig 7A-lower panel) but did not alter mitochondrial membrane potential (Fig 7C). However, higher dose of DIDS caused permanent cell shrinkage and CCL19 was not able to induce cell size change (Fig 7A-lower panel). This remarkable blocking effect appeared to be due to severe disruption of mitochondrial membrane potential towards depolarization, as indicated by JC-1 staining (Fig 7C). Interestingly, the cells with depolarized mitochondrial membrane did not bind to annexin V, a specific cell surface marker binding to phosphatidylserine during early apoptosis (Fig 7D). This suggests that these cells experience metabolic depression but not apoptosis at the time of measurement.
Figure 7. DIDS abolishes CCL19/CCL21-induced Ca2+ influx and migration in mature DCs.

A, mature DCs were treated with various concentrations of DIDS (0, 1, 10, 100µM) were used to measure CCL19/CCL21-induced intracellular Ca2+ mobilization and influx indicated by indo-1 fluorescence (upper panel) and cell size change indicated by forward scatter values (lower panel) over the time. CCL19/CCL21 was added at 90 seconds. B, mature DCs were treated with various concentrations of DIDS (0, 10, 100µM) for 2 hours and the chemotaxis were examined with Transwell (n=3, ***<0.001, error bars indicate SEM). Final chemokine concentration: CCL19, 100ng/mL; CCL21, 100ng/mL. C. Mitochondrial membrane potential as indicated by red fluorescence (575 nm) of JC-1 staining. Valinomycin, a potassium channel ionophore, was used as a positive control for depolarization. D, Expression of annexin-V in DIDS-treated mature DCs. The figures are a representative of three independent experiments.
Discussion
We have demonstrated the expression of KCa3.1 channel and its positive role in modulating lymphatic chemokine-induced Ca2+ influx, cell volume change, and migration in human DCs. Most importantly, we for the first time, report that the calcium mobilization simultaneously triggers multiple ion flows including Ca2+, K+, and possibly Cl− in human DCs. The initial transient rise in Ca2+ signal temporally coupled the Ca2+-activated channels into one synergistic complex leading to favorable changes for cell migration: change in cell size/volume, cytoskeleton re-organization and polarization, and optimized temporal and spatial dynamics of calcium loading. The desensitization and internalization of CCR7 prevent constant activation of its downstream signaling [24], which sets up the pace for the rhythmic firing of chemokine-induced ion channel activity in regard to DC migration to chemokine gradient. Thus, these ion-channel activities are indispensable for DC migration. Furthermore, the fluorescent ion indicators make it possible to isolate a directional ion flow without having to alter the membrane integrity and internal ionic composition as normally patch clamping does, which renders a big advantage over the conventional electrophysiological approach.
We also confirmed that Ca2+ influx is the major component of CCL19/CCL21-induced intracellular Ca2+ increase relative to Ca2+ release from ER in human monocyte-derived DCs, but the small amplitude of intracellular Ca2+ mobilization triggers the subsequent Ca2+ influx. Additionally, we did not observe that CCL19/CCL21-induced Ca2+ influx was dependent on PGE2, as reported by Scandella and colleagues [14], possibly due to the different maturation stimuli applied and the lack of evidence for CCR7 expression levels in PGE2-treated monocyte-derived DCs in their studies. Nevertheless, since Ca2+ mobilization is the downstream signaling event of CCR7 activation, it is likely that CCR7 expression levels could be the major determinant for the Ca2+ mobilization from intracellular stores and the subsequent Ca2+ influx, as shown by our data.
Several lines of evidence supported the notion that KCa3.1 was involved in DC migration to lymphatic chemokines in this study: the co-expression of CCR7 and KCa3.1, CCR7 activation-induced Ca2+ influx carried by CRAC and K+ efflux carried by KCa3.1, Ca2+ dependency of KCa3.1 activation, and 1-EBIO-induced membrane hyperpolarization in human DCs. The blockade of KCa3.1 by TRAM-34 not only largely disrupted the temporal coupling between KCa3.1 and Ca2+ influx; it also prevented the chemokine-induced cell shrinkage. All these subsequently impaired CCR7-induced chemotaxis. However, sustained opening of KCa3.1 will disrupt the coupling between Ca2+ influx and calcium-activated potassium channel, and thus impairs temporal dynamics of calcium signal necessary for cell migration. This is evident by the fact that 1-EBIO reduces migration rate in transformed renal epithelial cells [33]. Expression data showed that TRAM-34 does not affect KCa3.1 expression in mature DCs, and is only involved with the functional aspects of the channel.
Indeed, only a small increase in intracellular Ca2+ is required to activate KCa3.1 and allow for K+ efflux and higher intracellular Ca2+ levels ease the KCa3.1 activation [39, 40], which counteracts the depolarizing effect of Ca2+ influx. The net result of a CCR7 activation-induced Ca2+ influx is membrane depolarization provided that no other ion channel is involved. However, if K+ efflux couples with Ca2+ influx temporally, the overall consequence could be either depolarization or hyperpolarization, depending on the kinetics and conductance of the involved calcium and potassium channels. Our findings are supported by a recent report stating that store-operated Ca2+ influx leads to cell membrane hyperpolarization in human monocyte-derived macrophages, and the outward cationic current is carried by KCa3.1 [41]. Although CRAC is highly Ca2+ selective, it has an extremely small unitary conductance for Ca2+ [42]. The estimated conductance of CRAC is 0.2pS in Jurkat E6-1 human leukemic T cells [43] versus a conductance of 12pS for KCa3.1 in Chinese hamster ovary cells [40]. Therefore, it is reasonable to speculate that KCa3.1 carries a significantly larger conductance than CRAC so that an “over charge compensation” leads to membrane hyperpolarization in DCs. Nonetheless, we still cannot rule out the involvement of voltage-dependent potassium channel (Kv) and potentially other non-selective cation channels in this process. However, the activation of Kv channel relies on cell membrane depolarization. If Kv channels were expressed in DCs, it might serve as a backup mechanism in the absence of sufficient KCa3.1 conductance when sustained Ca2+ influx causes cell membrane depolarization. Since KCa3.1 channels are not sensitive to voltage stimulation, they seem to be perfectly designed to maintain the resting membrane potential in non-excitable cells, such as DCs.
TRAM-34 is a small molecule and highly specific blocker of KCa3.1 that does not affect cytochrome p450 [39]. However, since the binding site is located on the cytosolic side of KCa3.1 channel, the optimal blocking effect requires relatively long incubation in cell culture to allow for its transmembrane diffusion. The TRAM-34 has low toxicity and only causes minimal or no apoptosis or necrosis [31, 44], which is also demonstrated in this study. In our preliminary studies, intratracheal administration of TRAM-34 (up to 5 µM) did not induce any apparent toxicity. None-the-less, additional studies on the potential toxicity or adverse effects of TRAM-34 on KCa3.1 blocking would strengthen the therapeutic potential of TRAM-34. If the in vitro findings on the role of KCa3.1 in DC migration from this study are confirmed in an in vivo study under inflammatory condition, TRAM-34 could be a potential drug to target KCa3.1 in the treatment of allergic airway inflammation.
The Cl− could be an element in charge compensation mechanism during Ca2+ influx because chloride influx will lead to cell membrane hyperpolarization. It is commonly believed that Cl− efflux facilitates cell shrinkage [45]. In our study, replacement of extracellular Cl− affected the amplitude of Ca2+ influx and baseline Ca2+ levels. However, the Cl− replacement did not abolish membrane hyperpolarization, suggesting that it is not dispensable for charge compensation. The potassium efflux per se is able to counteract depolarizing Ca2+ influx. However, Cl− replacement prevented the CCL21-induced cell shrinkage, indicating the critical role of chloride in cell volume regulation. It is reasonable to speculate that a poor shape/size change led to impaired chemotaxis in response to CCL21 in the absence of extracellular Cl−. In fact, because the difference between intracellular and extracellular Cl− concentrations is not very prominent, the actual Cl− flow might be highly dynamic to cope with the cell volume dynamics. Of note, the KCa3.1 blocker, TRAM-34, also prevented the CCL21-induced cell shrinkage, suggesting a synergistic effect carried out by both Cl− and K+ on cell volume change.
The findings by the blocking of chloride channel provided information as to the general effect of chloride channel deficiency since the blockers for a specific subtype of chloride channel is not available. DIDS completely abolished the cell migration and Ca2+ influx. We observed that lower dose (1µM) seemed to only impair cell volume change and Ca2+ influx without affecting mitochondrial function. Higher doses (10-100µM) caused remarkable cellular changes including permanent cell shrinkage, irresponsiveness to chemokine stimulation, and cease of chemotaxis, which can be explained by highly depolarized mitochondrial membrane potential in DIDS-treated DCs. These effects are probably mediated by different anion channel expressed on plasma and mitochondrial membrane. Mitochondrial membrane potential is normally maintained at very negative levels by proton pump [46, 47], and serves as the driving force of proton movement for ATP production in the respiratory chain. Since DIDS has inhibitory effect on a broad range of anion channels, the identity of the ion channels that accounted for mitochondrial potential disruption is unclear but likely relate to anion/proton antiporter activity. Previously reported DIDS-sensitive Cl−/H+ antiporter in plasma membrane and lysosome [48-50], if expressed on DC mitochondria, could be one of the putative mechanisms by which cells maintain its negative mitochondrial membrane potential. More in-depth studies are needed to clarify the molecular identity of the responsible ion channels and to delineate the underlying mechanisms. Metabolic depression is the direct consequence of mitochondrial membrane depolarization. As a result, all ATP-dependent cell activities were ceased including the GPCR downstream pathways.
In conclusion, our data suggest that Ca2+ influx is the major source of CCL19/CCL21-induced intracellular Ca2+ increase. Potassium channel, KCa3.1, positively regulates the amplitude of CLL19/CCL21-induced Ca2+ influx, cell volume, and chemotaxis. Plasma membrane and mitochondrial chloride channel activity is required for chemokine-induced cell volume change and maintaining regular mitochondrial membrane potential. Our findings decipher the mechanisms underlying in the migration of DCs and may help to tailor the therapy for allergic airway inflammation in asthma.
Brief Commentary.
Background
The precise molecular mechanism on the migration of dendritic cells from the lung to lymph nodes during allergic airway inflammation is not known. Specifically, no information is available as to if chloride flux alone or in conjunction with another channel regulate chemokine-induced calcium influx. In this study, we characterized the ion channels and the direction of the ion flow during CCR7-induced Ca2+ influx and their role in DC migration in response to lymphatic chemokines.
Translational Significance
The KCa3.1 channel could be a target to develop better therapeutic approaches in the treatment of allergic airway inflammation in asthma.
Acknowledgements
This work was supported by the National Institutes of Health Grants R01HL085680 and R01AI075315 to Devendra K Agrawal, and LB506 DHHS State of Nebraska Cancer and Smoking-Related Disease Program Grant to Zhifei Shao. We thank Dr. Gregory A Perry and Creighton University Flow Cytometry Core Facility for the assistance with the flow cytometry experiments.
Abbreviations
- ANOVA
Analysis of variance
- APC
Antigen presenting cell
- ATP
Adenosine triphosphate
- AUC
Area under the curve
- BCA
Bicinchoninic acid
- CD
Cluster of differentiation
- CCCP
Carbonyl cyanide 3-chlorophenylhydrazone
- CCL
Chemokine ligand
- CCR
Chemokine receptor
- CLC3
Chloride channel
- CRAC
Calcium release-activated calcium channel
- CsCl
Cesium chloride
- DAG
Diacylglycerol
- DC
Dendritic cell
- DiBAC4
Bis-(1,3-Dibutylbarbituric Acid)Trimethine Oxonol
- DIDS
4,4'-Diisothiocyano-2,2'-stilbenedisulfonic acid
- 1-EBIO
1-Ethyl-1,3-dihydro-2H-benzimidazol-2-one; 1-Ethylbenzimidazolinone
- EDTA
Ethylenediaminetetraacetic acid
- EGTA
Ethylene glycol tetraacetic acid
- ER
Endoplasmic Reticulum
- FACS
Fluorescence-activated cell sorting
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GM-CSF
Granulocyte macrophage colony-stimulating factor
- GPCR
G-Protein Coupled Receptor
- HBSS
Hanks Balance Salt Solution
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HRP
Horseradish peroxidase
- IgG
Immunoglobulin G
- IL
Interleukin
- IP3
Inositol 1,4,5-trisphosphate
- KCa3.1
Intermediate-conductance Calcium-activated Potassium Channel
- Kv
Voltage-dependent potassium channel
- LPS
Lipopolysaccharide
- MQAE
N-(ethoxycarbonylmethyl)-6-methoxyquinolinium bromide
- ORAI 1
Calcium release-activated calcium channel protein 1
- PBFI
Potassium-binding benzofuran isophthalate
- PBMCs
Peripheral blood mononuclear cells
- PBS
Phosphate buffered saline
- PCR
Polymerase chain reaction
- PGE2
Prostaglandin E2
- PIP2
Phosphatidylinositol 4,5-bisphosphate
- PLC
Phospholipase C
- RIPA
Radioimmunoprecipitation assay buffer
- RPMI
Roswell Park Memorial Institute
- SDS
Sodium dodecyl sulfate
- STIM
Stromal interaction molecule
- TNF
Tumor necrosis factor
- TRAM-34
1-[(2-Chlorophenyl)diphenylmethyl]-1H-pyrazole
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure
All authors have read the journal's authorship agreement and policy on disclosure of potential conflicts of interest. The authors declared no conflict of interest.
References
- [1].Gaurav R, Agrawal DK. Clinical view on the importance of dendritic cells in asthma. Expert review of clinical immunology. 2013;9:899–919. doi: 10.1586/1744666X.2013.837260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bharadwaj AS, Agrawal DK. Transcription factors in the control of dendritic cell life cycle. Immunologic research. 2007;37:79–96. doi: 10.1007/BF02686091. [DOI] [PubMed] [Google Scholar]
- [3].Agrawal DK, Shao Z. Pathogenesis of allergic airway inflammation. Current allergy and asthma reports. 2010;10:39–48. doi: 10.1007/s11882-009-0081-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Braun A, Worbs T, Moschovakis GL, Halle S, Hoffmann K, Bolter J, et al. Afferent lymph-derived T cells and DCs use different chemokine receptor CCR7-dependent routes for entry into the lymph node and intranodal migration. Nature immunology. 2011;12:879–87. doi: 10.1038/ni.2085. [DOI] [PubMed] [Google Scholar]
- [5].Ricart BG, John B, Lee D, Hunter CA, Hammer DA. Dendritic cells distinguish individual chemokine signals through CCR7 and CXCR4. Journal of immunology. 2011;186:53–61. doi: 10.4049/jimmunol.1002358. [DOI] [PubMed] [Google Scholar]
- [6].Yanagihara S, Komura E, Nagafune J, Watarai H, Yamaguchi Y. EBI1/CCR7 is a new member of dendritic cell chemokine receptor that is up-regulated upon maturation. Journal of immunology. 1998;161:3096–102. [PubMed] [Google Scholar]
- [7].Takamatsu H, Takegahara N, Nakagawa Y, Tomura M, Taniguchi M, Friedel RH, et al. Semaphorins guide the entry of dendritic cells into the lymphatics by activating myosin II. Nature immunology. 2010;11:594–600. doi: 10.1038/ni.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Van Haastert PJ, Devreotes PN. Chemotaxis: signalling the way forward. Nature reviews Molecular cell biology. 2004;5:626–34. doi: 10.1038/nrm1435. [DOI] [PubMed] [Google Scholar]
- [9].Friedl P, Weigelin B. Interstitial leukocyte migration and immune function. Nature immunology. 2008;9:960–9. doi: 10.1038/ni.f.212. [DOI] [PubMed] [Google Scholar]
- [10].Schwab A, Nechyporuk-Zloy V, Fabian A, Stock C. Cells move when ions and water flow. Pflugers Archiv : European journal of physiology. 2007;453:421–32. doi: 10.1007/s00424-006-0138-6. [DOI] [PubMed] [Google Scholar]
- [11].Riol-Blanco L, Sanchez-Sanchez N, Torres A, Tejedor A, Narumiya S, Corbi AL, et al. The chemokine receptor CCR7 activates in dendritic cells two signaling modules that independently regulate chemotaxis and migratory speed. Journal of immunology. 2005;174:4070–80. doi: 10.4049/jimmunol.174.7.4070. [DOI] [PubMed] [Google Scholar]
- [12].Hsu S, O'Connell PJ, Klyachko VA, Badminton MN, Thomson AW, Jackson MB, et al. Fundamental Ca2+ signaling mechanisms in mouse dendritic cells: CRAC is the major Ca2+ entry pathway. Journal of immunology. 2001;166:6126–33. doi: 10.4049/jimmunol.166.10.6126. [DOI] [PubMed] [Google Scholar]
- [13].Matzner N, Zemtsova IM, Nguyen TX, Duszenko M, Shumilina E, Lang F. Ion channels modulating mouse dendritic cell functions. Journal of immunology. 2008;181:6803–9. doi: 10.4049/jimmunol.181.10.6803. [DOI] [PubMed] [Google Scholar]
- [14].Scandella E, Men Y, Legler DF, Gillessen S, Prikler L, Ludewig B, et al. CCL19/CCL21-triggered signal transduction and migration of dendritic cells requires prostaglandin E2. Blood. 2004;103:1595–601. doi: 10.1182/blood-2003-05-1643. [DOI] [PubMed] [Google Scholar]
- [15].Chan VW, Kothakota S, Rohan MC, Panganiban-Lustan L, Gardner JP, Wachowicz MS, et al. Secondary lymphoid-tissue chemokine (SLC) is chemotactic for mature dendritic cells. Blood. 1999;93:3610–6. [PubMed] [Google Scholar]
- [16].Zhang SL, Yu Y, Roos J, Kozak JA, Deerinck TJ, Ellisman MH, et al. STIM1 is a Ca2+ sensor that activates CRAC channels and migrates from the Ca2+ store to the plasma membrane. Nature. 2005;437:902–5. doi: 10.1038/nature04147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Prakriya M, Feske S, Gwack Y, Srikanth S, Rao A, Hogan PG. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443:230–3. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- [18].Feske S. ORAI1 and STIM1 deficiency in human and mice: roles of store-operated Ca2+ entry in the immune system and beyond. Immunological reviews. 2009;231:189–209. doi: 10.1111/j.1600-065X.2009.00818.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, et al. Cell migration: integrating signals from front to back. Science. 2003;302:1704–9. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- [20].Brundage RA, Fogarty KE, Tuft RA, Fay FS. Calcium gradients underlying polarization and chemotaxis of eosinophils. Science. 1991;254:703–6. doi: 10.1126/science.1948048. [DOI] [PubMed] [Google Scholar]
- [21].Wei C, Wang X, Chen M, Ouyang K, Song LS, Cheng H. Calcium flickers steer cell migration. Nature. 2009;457:901–5. doi: 10.1038/nature07577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Marasco WA, Becker EL, Oliver JM. The ionic basis of chemotaxis. Separate cation requirements for neutrophil orientation and locomotion in a gradient of chemotactic peptide. The American journal of pathology. 1980;98:749–68. [PMC free article] [PubMed] [Google Scholar]
- [23].Evans JH, Falke JJ. Ca2+ influx is an essential component of the positive-feedback loop that maintains leading-edge structure and activity in macrophages. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:16176–81. doi: 10.1073/pnas.0707719104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kohout TA, Nicholas SL, Perry SJ, Reinhart G, Junger S, Struthers RS. Differential desensitization, receptor phosphorylation, beta-arrestin recruitment, and ERK1/2 activation by the two endogenous ligands for the CC chemokine receptor 7. The Journal of biological chemistry. 2004;279:23214–22. doi: 10.1074/jbc.M402125200. [DOI] [PubMed] [Google Scholar]
- [25].Shao Z, Bharadwaj AS, McGee HS, Makinde TO, Agrawal DK. Fms-like tyrosine kinase 3 ligand increases a lung DC subset with regulatory properties in allergic airway inflammation. The Journal of allergy and clinical immunology. 2009;123:917–24. doi: 10.1016/j.jaci.2009.01.052. e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Shao Z, Makinde TO, McGee HS, Wang X, Agrawal DK. Fms-like tyrosine kinase 3 ligand regulates migratory pattern and antigen uptake of lung dendritic cell subsets in a murine model of allergic airway inflammation. Journal of immunology. 2009;183:7531–8. doi: 10.4049/jimmunol.0901341. [DOI] [PubMed] [Google Scholar]
- [27].Shao Z, Makinde TO, Agrawal DK. Calcium-activated potassium channel KCa3.1 in lung dendritic cell migration. American journal of respiratory cell and molecular biology. 2011;45:962–8. doi: 10.1165/rcmb.2010-0514OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Ghanshani S, Wulff H, Miller MJ, Rohm H, Neben A, Gutman GA, et al. Up-regulation of the IKCa1 potassium channel during T-cell activation. Molecular mechanism and functional consequences. The Journal of biological chemistry. 2000;275:37137–49. doi: 10.1074/jbc.M003941200. [DOI] [PubMed] [Google Scholar]
- [29].Schmid-Antomarchi H, Schmid-Alliana A, Romey G, Ventura MA, Breittmayer V, Millet MA, et al. Extracellular ATP and UTP control the generation of reactive oxygen intermediates in human macrophages through the opening of a charybdotoxin-sensitive Ca2+-dependent K+ channel. Journal of immunology. 1997;159:6209–15. [PubMed] [Google Scholar]
- [30].Cruse G, Duffy SM, Brightling CE, Bradding P. Functional KCa3.1 K+ channels are required for human lung mast cell migration. Thorax. 2006;61:880–5. doi: 10.1136/thx.2006.060319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Toyama K, Wulff H, Chandy KG, Azam P, Raman G, Saito T, et al. The intermediate-conductance calcium-activated potassium channel KCa3.1 contributes to atherogenesis in mice and humans. The Journal of clinical investigation. 2008;118:3025–37. doi: 10.1172/JCI30836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Duffy SM, Cruse G, Cockerill SL, Brightling CE, Bradding P. Engagement of the EP2 prostanoid receptor closes the K+ channel KCa3.1 in human lung mast cells and attenuates their migration. European journal of immunology. 2008;38:2548–56. doi: 10.1002/eji.200738106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Schwab A, Schuricht B, Seeger P, Reinhardt J, Dartsch PC. Migration of transformed renal epithelial cells is regulated by K+ channel modulation of actin cytoskeleton and cell volume. Pflugers Archiv : European journal of physiology. 1999;438:330–7. doi: 10.1007/s004240050917. [DOI] [PubMed] [Google Scholar]
- [34].Kim MJ, Cheng G, Agrawal DK. Cl-channels are expressed in human normal monocytes: a functional role in migration, adhesion and volume change. Clinical and experimental immunology. 2004;138:453–9. doi: 10.1111/j.1365-2249.2004.02635.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Moreland JG, Davis AP, Bailey G, Nauseef WM, Lamb FS. Anion channels, including ClC-3, are required for normal neutrophil oxidative function, phagocytosis, and transendothelial migration. The Journal of biological chemistry. 2006;281:12277–88. doi: 10.1074/jbc.M511030200. [DOI] [PubMed] [Google Scholar]
- [36].Cheng G, Ramanathan A, Shao Z, Agrawal DK. Chloride channel expression and functional diversity in the immune cells of allergic diseases. Current molecular medicine. 2008;8:401–7. doi: 10.2174/156652408785160934. [DOI] [PubMed] [Google Scholar]
- [37].Schwab A, Wojnowski L, Gabriel K, Oberleithner H. Oscillating activity of a Ca(2+)-sensitive K+ channel. A prerequisite for migration of transformed Madin-Darby canine kidney focus cells. The Journal of clinical investigation. 1994;93:1631–6. doi: 10.1172/JCI117144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Tedder TF, Jansen PJ. Coligan John E, et al., editors. Isolation and generation of human dendritic cells. Current protocols in immunology. 2001 doi: 10.1002/0471142735.im0732s23. Chapter 7:Unit 7 32. [DOI] [PubMed] [Google Scholar]
- [39].Bradding P, Wulff H. The K+ channels K(Ca)3.1 and K(v)1.3 as novel targets for asthma therapy. British journal of pharmacology. 2009;157:1330–9. doi: 10.1111/j.1476-5381.2009.00362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Joiner WJ, Wang LY, Tang MD, Kaczmarek LK. hSK4, a member of a novel subfamily of calcium-activated potassium channels. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:11013–8. doi: 10.1073/pnas.94.20.11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gao YD, Hanley PJ, Rinne S, Zuzarte M, Daut J. Calcium-activated K(+) channel (K(Ca)3.1) activity during Ca(2+) store depletion and store-operated Ca(2+) entry in human macrophages. Cell calcium. 2010;48:19–27. doi: 10.1016/j.ceca.2010.06.002. [DOI] [PubMed] [Google Scholar]
- [42].Prakriya M, Lewis RS. CRAC channels: activation, permeation, and the search for a molecular identity. Cell calcium. 2003;33:311–21. doi: 10.1016/s0143-4160(03)00045-9. [DOI] [PubMed] [Google Scholar]
- [43].Prakriya M, Lewis RS. Separation and characterization of currents through store-operated CRAC channels and Mg2+-inhibited cation (MIC) channels. The Journal of general physiology. 2002;119:487–507. doi: 10.1085/jgp.20028551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Shepherd MC, Duffy SM, Harris T, Cruse G, Schuliga M, Brightling CE, et al. KCa3.1 Ca2+ activated K+ channels regulate human airway smooth muscle proliferation. American journal of respiratory cell and molecular biology. 2007;37:525–31. doi: 10.1165/rcmb.2006-0358OC. [DOI] [PubMed] [Google Scholar]
- [45].Hoffmann EK, Lambert IH, Pedersen SF. Physiology of cell volume regulation in vertebrates. Physiological reviews. 2009;89:193–277. doi: 10.1152/physrev.00037.2007. [DOI] [PubMed] [Google Scholar]
- [46].Ramzan R, Staniek K, Kadenbach B, Vogt S. Mitochondrial respiration and membrane potential are regulated by the allosteric ATP-inhibition of cytochrome c oxidase. Biochimica et biophysica acta. 2010;1797:1672–80. doi: 10.1016/j.bbabio.2010.06.005. [DOI] [PubMed] [Google Scholar]
- [47].Zhang H, Huang HM, Carson RC, Mahmood J, Thomas HM, Gibson GE. Assessment of membrane potentials of mitochondrial populations in living cells. Analytical biochemistry. 2001;298:170–80. doi: 10.1006/abio.2001.5348. [DOI] [PubMed] [Google Scholar]
- [48].Graves AR, Curran PK, Smith CL, Mindell JA. The Cl-/H+ antiporter ClC-7 is the primary chloride permeation pathway in lysosomes. Nature. 2008;453:788–92. doi: 10.1038/nature06907. [DOI] [PubMed] [Google Scholar]
- [49].Picollo A, Pusch M. Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature. 2005;436:420–3. doi: 10.1038/nature03720. [DOI] [PubMed] [Google Scholar]
- [50].Scheel O, Zdebik AA, Lourdel S, Jentsch TJ. Voltage-dependent electrogenic chloride/proton exchange by endosomal CLC proteins. Nature. 2005;436:424–7. doi: 10.1038/nature03860. [DOI] [PubMed] [Google Scholar]