

Abstract
Severely immunodeficient mice such as the NOD/SCID/IL2rγnull (NSG) strain can be engrafted with human hematopoietic stem cells (HSCs), resulting in chimeric mice containing many components of the human immune system (Human Immune System mice or HIS mice). HIS mice can both support the replication of and recapitulate much of the immunological response to a variety of pathogens, including ones with strict human tropism, such as HIV-1. In an effort to develop a better mouse model for human infectious pathogen infection and possible immune resolution, we compared the human immune system reconstitution of NSG mice following injection with human CD34+ HSCs purified from either fetal liver (FL) or umbilical cord blood (UCB). We analyzed reconstitution in standard NSG mice as well as a derivative of these mice containing an HLA.A2 encoding transgene (NSG.A2). HSCs from both sources effectively reconstituted hematopoietic lineages when injected into NSG mice. In marked contrast, total CD45+ human hematopoietic cells in NSG.A2 mice were well reconstituted by HSCs from UCB but very poorly by HSCs purified from FL. Moreover, the reconstitution of T cell lineages in NSG.A2 mice by HSCs from UCB was inferior to that obtained using NSG mice. We also found that FL CD34+ HSCs contain a much higher percentage of cells with a phenotype consistent with primitive progenitors than UCB HSCs. We discuss possible explanations for the influence of the HLA.A2 transgene on hematopoietic reconstitution using the two sources of HSCs.
Keywords: humanized mice, hematopoietic stem cells, HLA-A2 transgene
INTRODUCTION
The utility of rodent models for the elucidation of immunological mechanisms is unquestioned. However, particular aspects these mechanisms may be species specific. In addition, many human infectious pathogens demonstrate a strict host tropism and are unable to actively replicate in any current mouse model. These factors complicate the translation of knowledge gained from rodents to the development of better approaches for the prevention, diagnosis and treatment of human infectious diseases. Conversely, studying the factors responsible for immunity to infection in humans is fraught with caveats distinct from those that apply to rodent models. The human population is both genetically and environmentally diverse, and with the exception of vaccination, it is impossible to control the timing of infection, dose, and strain of pathogen received. Moreover, sources of responding lymphocytes and other cell types for analysis are usually restricted to peripheral blood, and these often cannot be obtained during active infection. Finally, treatment of patients with anti-viral and anti-inflammatory compounds and antibiotics alters the nature of the immune responses generated.
Human immune system (HIS) mice created by durable xeno engraftment of severely immunodeficient mice, such as the NOD-scid/γc−/− (NSG) strain, with human hematopoietic cells are now being widely used as an approach to avoid the limitations of rodent models discussed above [1–6]. Engrafting such immunodeficient mice with human hematopoietic stem cells (HSC) allows the generation of a “naive” human immune system in the resulting chimeric mice. Multiple human tropic pathogens that are reliant on human hematopoietic cells and their lineages for replication can be studied using this platform, as previously shown for HIV [7,8], Dengue [9] and EBV [10] viruses, and Salmonella typhi [11,12]. HIS mice can, in theory, be used to study human immune responses to any antigen or infectious pathogen.
However, limitation to the use of HIS mice created by the relatively straightforward technique of injection of human HSCs into severely immunodeficient mice have become apparent in the years following the initial development of this approach [13,14]. These include a variety of factors such as: only partial function and development of multiple innate immune cells, limited T dependent B cell response, and low levels of cytotoxic T cell mediated killing. Many of these stem from absent or insufficient regulatory interactions between the nonhematopoietic mouse cell types present in primary and secondary lymphoid organs, and the adoptively transferred human HSCs and their differentiating and differentiated derivatives [15–17]. Of all of the limitations applicable to the use of HIS mice, perhaps the most relevant to the study of human adaptive immune responses is that resulting from alterations in the selection of T cells during their development for their ability to bind to self major histocompatibility (MHC) molecules.
Since the thymus and its stromal elements in HIS mice are of mouse origin, selection of developing human T cells in this organ will predominantly take place via human T cell receptor-mouse MHC interactions. In the case of CD8 T cells this will bias responses towards antigenic peptides bound to mouse MHC class I molecules, and in the case of CD4 T cells towards such peptides bound to mouse MHC class II molecules. The former situation will result in complication of the use of HIS mice to study CD8 responses to antigenic peptide epitopes relevant to such responses in humans. The latter situation is likely to severely limit T cell dependent B cell immune responses in HIS mice. In such responses, antigen specific B cells receive critical co stimulatory signals from CD4 T cells that promote their proliferation and differentiation [18]. The receipt of these signals requires cognate CD4 T cell-B cell interaction in which the B cell presents antigenic peptides derived from antigen processing bound to MHC class II molecules to a CD4 T cell specific for this peptide-MHC complex. This interaction will not take place efficiently unless the T cell was selected during primary development in the thymus by the same MHC class II molecule expressed by the B cell. However, in conventional HIS mice, most CD4 T cells appear to be selected on mouse MHC II antigens and not on the human MHC II molecules expressed by the human B cells in these mice [19]. Indeed, we and others have found that TD B cells responses in HIS mice are limited, producing low levels of serum antibody deficient in IgG and no germinal center reactions [1,14,20].
For these reasons, alternative approaches to the generation of HIS mice have been developed. One of these is the Bone marrow, Liver, Thymus (BLT) approach, where immunodeficient mice are first transplanted with small pieces of liver (providing a human environment for human myeloid and B cell development) and thymus (providing a human environment for human T cell development), followed by injection of HSC, all from the same human fetal donor [10,21]. While this approach has been demonstrated to result in mice with more robust TD B cell and CD8 T cell responses than HIS mice [22–24], it is labor intensive and costly.
Another limitation to the use of HIS (and BLT) mice to study human immune responses and hematopoiesis may arise due the source of HSC used. Evidence for the unique origin of several mature murine hematopoietic subsets from fetal or adult HSCs has been obtained. Mouse B1a cells are predominantly derived from precursors present in fetal liver, but not adult BM [25,26]. Fetal liver, but not adult BM can reconstitute this subset when injected into lethally irradiated adult mice. Conversely, adult BM efficiently reconstitutes follicular (FO) but not B1a cells [27]. T cells expressing the TCR variable regions Vγ3 and Vγ4 are derived from fetal thymus and their progenitors are present in fetal liver [28]. Adult HSCs cannot give rise to these two subsets, even if injected into fetal thymus [29]. Human fetal HSCs give rise predominantly to regulatory T cells while adult HSCs do not [30]. Finally, erythrocytes produced in the yolk sac are larger than those found in adults, retain their nuclei and express fetal-type globins. Such erythrocytes are not produced during adult BM hematopoiesis [31].
The results presented in this manuscript stemmed from our desire to use HIS mice to study the human B and CD8 T cell responses to HIV infection. Given the caveats and limitations of HIS mice summarized above, we began by examining the nature of the human hematopoietic reconstitution obtained using two HSCs sources: umbilical cord blood (UCB) and fetal liver (FL); and two strains of severely immunodeficient mice: NSG mice, and a derivative of this strain in which the common allele of the human MHC class I molecule HLA-A2*0201 (HLA-2.1) is expressed from a transgene ([32], NOD.Cg-Prkdcscid Il2rgtm1Wjl Tg(HLA.A2.1)1Enge/SzJ (NSG.A2) mice, from Jackson Laboratories). Surprisingly, we found that NSG.A2 mice were very poorly reconstituted by FL-derived, but not UBC-derived HSCs. Moreover, while total hematopoietic reconstitution of NSG and NSG.A2 mice appeared similar using UCB HSCs, T cell reconstitution was poor using the latter type of mice. In total, and paradoxically, these results indicate severe limitations to the use of humanized NSG.A2 mice to study human TD B cell and CD8 T cell responses. We also report that HSCs purified from UCB and FL display phenotypic differences that may be relevant to the results we obtained.
MATERIALS AND METHODS
Mice
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) and NOD.Cg-Prkdcscid Il2rgtm1Wjl Tg(HLA.A2.1)1Enge/SzJ (NSG.A2) were purchased from The Jackson Laboratory (Bar Harbor, ME) and then bred in-house. NSG.A2 breeders were maintained in a hemizygous state and all offspring were screened by real time PCR for the presence of the transgene using primers and probes with sequences supplied by The Jackson Laboratory. All mice were maintained in specific pathogen free conditions, given autoclaved food and acidified water (pH 2.5–3.0), and housed in individually ventilated racks in the research animal facilities of Thomas Jefferson University. All animal experiments were conducted in accordance with guidelines of the Animal care and Use Committee of Thomas Jefferson University.
Isolation of CD34+ hematopoietic stem cells from human umbilical cord blood
Human umbilical cord blood was obtained from healthy full term newborns (Department of Obstetrics and Gynecology, Thomas Jefferson University) and with IRB approval. After Ficoll gradient centrifugation, CD34+ cells were enriched from umbilical cord blood mononuclear cells (PBMNs) using immune-magnetic beads according to the manufacturer’s instructions (CD34+ isolation kit: Miltenyi Biotec, Auburn, CA). Cells were frozen in 90% FBS 10% DMSO and stored in liquid nitrogen.
Isolation of CD34+ hematopoietic stem cells from human fetal liver
Human fetal livers obtained from aborted fetuses at 15–23 weeks of gestation were purchased (StemExpress, Placerville, CA). Fetuses were processed within 12 hours of the termination of pregnancy. Fetal liver tissue was initially cut into 1 mm slices followed by collagenase VI digestion (2 mg/ml in Dulbecco's modified Eagle's medium [DMEM]) for 1 hour at 37°C with periodic mixing. A single-cell suspension was prepared by passing the digested tissue through a 100 μm cell strainer (BD Biosciences, Franklin Lakes, NJ). PBMNs were isolated by Ficoll gradient centrifugation and CD34+ cells were purified with the use of a CD34-positive selection immune-magnetic bead kit (Miltenyi Biotec, Auburn, CA). Cell viability was examined by trypan blue staining. Isolated cells were frozen in liquid nitrogen in 90% FBS 10% DMSO.
Thawing of frozen CD34+ umbilical cord blood and fetal liver HSCs
Approximately 30 minutes prior to injection into mice, vials of frozen HSCs were removed from liquid nitrogen storage and rapidly warmed in a 37°C water bath until just prior to complete thawing (slurry of residual solids present). The contents of each vial were then aseptically transferred to a 15mL conical tube. PBS previously warmed to 37°C was then slowly added to the frozen cells while gently swirling the tube until the total volume reached 15 mL. The resulting cell suspension was centrifuged at 400x g at 4°C for 7 minutes. The supernatant was carefully aspirated off the cell pellet and the cells resuspended in an appropriate volume of fresh PBS for injection and transferred to a sterile 1.5mL Eppendorf tube. The tube was placed on wet ice until injections. Cell numbers and viability were determined by trypan blue dye-exclusion. For injections, a dose per mouse of 100,000 viable cells was calculated.
Xenotransplantation of hematopoietic stems cells into NSG and NSG.A2 mice
Forty-eight hours after birth, mouse pups were irradiated with a whole body dose of 2 Gy (200 Rads). Four hours post irradiation, frozen HSC were thawed as described above and injected at dose of 105 cells per mouse, intrahepatically in 25μl PBS using a Hamilton syringe with a 30 gauge needle (Hamilton Company USA, Reno, NV). Littermates were reconstituted with single donor derived cord blood or fetal liver stem cells. All stem cells were HLA.A2 typed by real time-PCR before implantation into NSG.A2 mice. In each individual experiment, the same preparation of CD34+ HSC from either UCB or FL was used to reconstitute both NSG and NSG.A2 mice. Reconstituted mice were weaned at three weeks of age and randomly distributed between different experimental groups. Blood was collected at the indicated time points by either retro-orbital bleeding, tail-vein collection or cardiac puncture (at sacrifice).
Flow cytometry
To evaluate human immune cell engraftment, 10 ul of peripheral blood was mixed with phosphate-buffered saline (PBS) containing 200 units/ml heparin. Red blood cells were lysed with ACK buffer before the sample was incubated with fluorochrome-coupled monoclonal antibodies and analyzed by flow cytometry. Monoclonal antibodies specific for the following human surface antigens were employed: CD45, CD19, CD20, and IgD FITC conjugates; CD34, CD3, CD56, and IgD PE conjugates; CD4, CD8, CD5, and CD27 Cy5.5 conjugates from BD Pharmingen, San Jose, CA. CD11b-APC, CD19 pacific blue conjugates were from Biolegend, San Diego, CA; CD3 APC-, and IgM-FITC conjugates were from Invitrogen, Grand Island, NY; CD45RA-PE-Cy7, CD38-APC, and CD90-PerCP5.5 conjugates from BD Pharmingen. Cells (0.1 to 1×106) were stained with indicated antibody mixtures in 100 μl of PBS containing 3% bovine serum albumin (BSA; Gemini Bio-Products, West Sacramento, CA USA), 0.02% sodium azide and 5 mM EDTA for 30 min on ice. Stained cells were analyzed on an LSRII Flow cytometer (BD Biosciences, San Jose, CA, USA). One hundred thousand events per sample were recorded and analyzed using the FlowJo X software (Tree-star, Ashland, OR). For absolute cell counting, AccuCount Rainbow Fluorescent Particles (Spherotech, Lake Forest, IL) were added at a known concentration after the final wash. The number of these particles counted by the flow cytometer during each analysis of was then used to calculate the volume of cell suspension that had been analyzed. This value was then used to calculate the number of cells analyzed per unit volume of original sample. In the case of spleens, each spleen was weighed before homogenization, allowing further calculation of the number of cells analyzed per unit weight of original sample.
Immunohistology
Sixteen weeks after CD34+ stem cell transplantation, reconstituted NSG mice were sacrificed and their spleens, small intestines and large intestines were harvested into ice cold RPMI containing 10% FBS. The intestines were flushed with ice cold PBS to remove fecal content and sliced into 1 cm pieces. Tissue samples were frozen in Tissue-Tek O.C.T compound (Sakura Finetek, Torrance, CA). Tissue sections of 6–8 μm were prepared on a cryotome and fixed on glass slides using cold 100% acetone for 10 minutes. After fixation, the sections were rehydrated in Tris-buffered saline (TBS) and blocked with TBS containing 20% w/v BSA and 0.1% v/v Tween-20. Sections were incubated with monoclonal antibodies targeting the following human antigens at room temperature for two hours: IgM-FITC (H15001), nucblue® fixed cell stain (both from Life Technologies, Grand Island, NY), CD3-PE (SK7), CD11c-APC (S-HCL-3), and CD45-FITC (2D1) all from (BD Pharmingen, San Diego, CA). Sections were subsequently washed with TBS containing 0.1% v/v Tween-20 and treated with Fluorogel (EMS, Hatfield, PA). The stained sections were analyzed using a fluorescence microscopy (Leica DM 5000B; Leica Microsystems, Wetzlar, Germany), and digital images were analyzed using LAS AF 1.8.1 software from Leica Microsystems.
Statistical analysis
A paired t-test was used to determine statistical significance. All graphs and statistics were performed using GraphPad Prism version 5.00 for Windows, GraphPad Software (San Diego, CA).
RESULTS
We began by comparing the reconstitution levels of NSG and NSG.A2 mice with HSCs from umbilical cord blood (UCB) purified using anti-CD34 MACS. We used NSG mice rather than the other commonly used Balb/c-Rag1−/−γc−/− mice as recipients, as the former have been demonstrated to display superior engraftment by human CD34+ HSCs [33]. This result appears to be due to a polymorphism in the sirpα gene in the Balb background that precludes recognition of the human CD47 molecule on HSCs by the Sirpα receptor on mouse macrophages, allowing the resident mouse macrophages cells to actively phagocytize human HSCs [33–35]. Mice were injected intra hepatically with HSCs 48 hours after birth and 4 to 6 hours after 2 Gy whole body irradiation. In all of these studies, only HSCs derived from HLA.A2 expressing donors were used. As shown in Figure 1A, while total human hematopoietic cell reconstitution (as measured by the percentage of human CD45+ cells in the blood) was similar in the resulting two types of mice at twelve weeks post HSC injection, the NSG.A2 recipients required a longer period to acquire these levels.
Figure 1. Human hematopoietic engraftment kinetics in NSG mice obtained from umbilical cord blood and fetal liver derived hematopoietic stem cells.
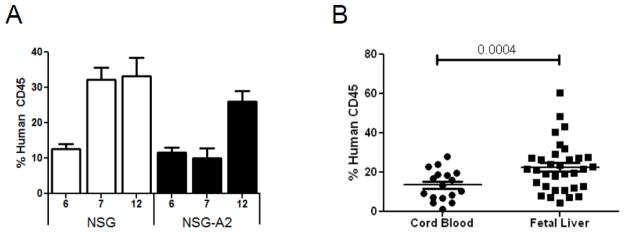
A) Engraftment of human CD45 cells in the blood of NSG and NSG-A2 mice reconstituted with umbilical cord blood derived stem cells at 6, 7, and 12 weeks. The data were compiled from 3 to 14 mice of each type at each time point. Standard deviations are show with error bars. B) Comparison of engraftment levels of human CD45+ cells in the blood of NSG mice at 12 weeks after reconstitution with either umbilical cord blood or fetal liver derived stem cells. Each symbol represents and individual mouse and each group contains mice generated with at least three different stem cell donors. In both panels mean values and standard deviations are indicated by bars. The significance of the difference between levels of reconstitution obtained with FL and UCB HSCs as measured by paired T test is shown in panel B.
We next compared reconstitution of NSG mice using HSCs purified from either UCB or fetal liver (FL). Figure 1B shows that, on average, total hematopoietic reconstitution percentage appeared to be significantly improved using HSCs from FL (p = 0.0004). These two findings were the first to indicate that both the HLA.A2 transgene and the source of HSCs might affect reconstitution and motivated more detailed studies on these issues.
HSCs from fetal liver do not effectively reconstitute human hematopoietic cells in the blood and spleen of NSG.A2 mice
We proceeded to do a pair-wise comparison of reconstitution of both types of mice with both types of HSCs, using HSCs purified from multiple donors. The resolution of these studies was also improved by measuring the absolute number of human CD34+ cells in the tissue being assayed. Figure 2 illustrates the surprising result that FL HSCs reconstituted total human hematopoietic cells in NSG.A2 mice extremely poorly, but their reconstitution of NSG mice was as good, if not better than that obtained using UBC HSCs. The data in both Figures 1 and 2 support the idea that there is a barrier to reconstitution of NSG.A2 mice not present in NSG mice.
Figure 2. Relative and absolute human hematopoietic engraftment of NSG and NSG-A2 mice.
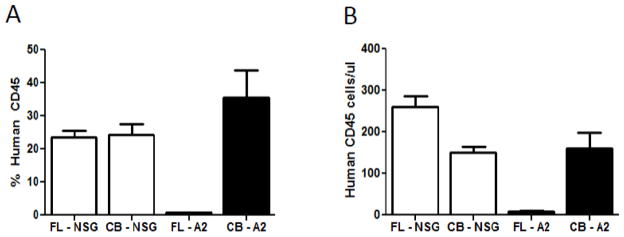
A) Relative engraftment of human CD45+ cells in the blood of NSG or NSG-A2 mice reconstituted with umbilical cord blood or fetal liver derived stem cells at 12 weeks. B) Absolute engraftment of human CD45+ cells in the blood of NSG or NSG-A2 mice reconstituted with umbilical cord blood or fetal liver derived stem cells at 12 weeks. The data were compiled from 8 to 20 mice of each type. In both panels standard deviations are shown with error bars.
Since the percentages of human CD45+ cells in blood we observed in our reconstituted mice differed somewhat from previous publications we conducted linear regression analyses comparing levels of reconstitution of HIS mice as measured by percent CD45+ cells in blood versus absolute number of CD45+ cells in blood. Surprisingly, we found in only one experimental condition and time point after reconstitution that these values were significantly related. These findings make it clear that measurement of absolute numbers of CD45+ cells in blood is superior to measurement of percentages of such cells when evaluating levels of human lymphocyte reconstitution in HIS mice. Unfortunately, the findings also indicate that comparison of our data with those of other studies in which percentages of human CD45+ cells in blood were measured will be inaccurate and perhaps misleading.
As our initial intent was to study HIV-1 infection in HIS mice, so we went on to evaluate absolute levels of human CD3, CD4 and CD8 cells in the blood and spleens of these four populations of HIS mice. Figure 3A shows that the defect in human CD45+ cells in the blood of NSG.A2 mice reconstituted with FL HSCs extended to all T cell subsets. Moreover, while T cell reconstitution in the blood was comparable between NSG and NSG.A2 mice injected with UBC HSCs, splenic T cell reconstitution was sharply lower for NSG.A2 mice (Figure 3B). There appears to be yet another level of altered reconstitution intrinsic to NSG.A2 mice.
Figure 3.


Figure 3a. Absolute engraftment of human lymphoid cells in the blood of NSG and NSG-A2 mice twelve weeks after injection of fetal liver (FL) or cord blood (CB) derived hematopoietic stem cells A) Human CD45+ cell levels in the blood of NSG and NSG-A2 mice. B) Human CD3+ cell levels in the blood of NSG and NSG-A2 mice. C) Human CD4+ cell levels in the blood of NSG and NSG-A2 mice. D) Human CD8+ cell levels in the blood of NSG and NSG-A2 mice. The data were compiled from 3 to 4 mice of each type. In all panels standard deviations are shown with error bars.
Figure 3b. Absolute engraftment of human lymphoid cells in the spleens of NSG and NSG-A2 mice twelve weeks after injection of fetal liver and cord blood derived hematopoietic stem cells. A) Human CD45+ cell levels in the spleens of NSG and NSG-A2 mice. B) Human CD3+ cell levels in the spleens of NSG and NSG-A2 mice. C) Human CD4+ cell levels in the spleens of NSG and NSG-A2 mice. D) Human CD8+ cell levels in the spleens of NSG and NSG-A2 mice. The data were compiled from 3 to 4 mice of each type. In all panels standard deviations are shown with error bars.
Human lymphoid reconstitution in the intestine and spleen are poor in NSG.A2 mice injected with HSCs from fetal liver
Since the Gut Associated Lymphoid Tissue (GALT) is particularly relevant to the establishment of natural HIV infection [36], we histologically examined levels of human hematopoietic reconstitution in the small and large intestine of the HIS mice. Figure 4 illustrates that the intestines of all HSC->NSG mice contained abundant human CD45+ cells and human CD3+ cells, all localized to the villi and lamina propria regions. In contrast, the FL and UCB HSC->NSG.A2 mice revealed GALT structures containing reduced frequencies of human CD45+ cells and human CD3+ cells relative to the other types of HIS mice. These data are presented in a semi-quantitative form in Table I.
Figure 4. Gut-associated lymphoid tissue (GALT) lymphoid micro architecture in NSG and NSG-A2 mice reconstituted with umbilical cord blood or fetal liver hematopoietic stem cells.
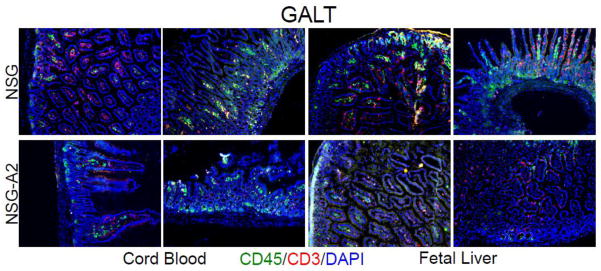
Intestines were isolated from either umbilical cord blood (left panels) or fetal liver (right panels) stem cell reconstituted mice of the indicated types, processed for histologic analysis, and sections stained with fluorescence antibodies specific for the indicated markers as well as DAPI to delineate all cell nuclei. Stained sections were analyzed by fluorescence microscopy. Note the rather low human immune cell reconstitution in the lamina propria and villi regions in umbilical cord blood or fetal liver stem cell reconstituted NSG-A2 intestines (lower panels) as compared to the well reconstituted areas in the intestines of NSG mice. Data are representative of those obtained from at least two reconstituted mice of each type.
Table 1. Semi quantitative analysis of spleen and intestine immuno histology data obtained from HIS mice.
Spleen and intestine sections stained with fluorescence antibodies specific for the indicated markers were analyzed by fluorescence microscopy. For the spleen sections, the number and size of cell clusters containing human cells were quantified, as was the number of such clusters with clear segregation of B cells and T cells. Cell segregation and various numbers of human immune cell populations in individual clusters were quantified as high(+++), medium(++), and low(+). For GALT sections, the number of villi and areas of lamina propria that contained human cells were quantified and data are presented as: most of the villi with numerous human cells (+++), and few villi with numerous human cells (+).
| Group | Spleen | GALT | ||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Number of Clusters | Size of Clusters | Cell Segregation | IgM+ | CD3+ | CD11c+ | CD45+ | CD3+ | |
| NSG-CB (n=3) | 15 | Large=9 Medium=4 Small=2 | +++ | +++ | +++ | +++ | ||
| +++ | +++ | +++ | + | +++ | +++ | |||
| ++ | ++ | + | + | |||||
| NSG.A2 - CB (n=3) | 8 | Large=2 Medium=2 Small=4 | ++ | +++ | ++ | + | ||
| + | + | + | + | +++ | + | |||
| + | + | + | + | |||||
| NSG - FL (n=3) | 12 | Large=6 Medium=4 Small=2 | +++ | +++ | +++ | + | ||
| ++ | +++ | ++ | + | +++ | +++ | |||
| + | ++ | + | + | |||||
| NSG.A2 - FL (n=3) | 7 | Large=0 Medium=2 Small=5 | --- | --- | --- | --- | ||
| + | + | +++ | + | + | + | |||
| + | + | + | + | |||||
In the spleen, NSG mice reconstituted with both sources of HSCs displayed frequent and often large clusters of human hematopoietic cells containing abundant B cells, T cells and DCs (Figure 5). In addition, these clusters usually displayed segregation of B cells, T cells and often DCs into discrete zones, as is characteristic of natural white pulp areas in mice and humans [37]. The formation of such clusters of lymphocytes as well as their segregation into discrete regions is clearly an important component of functional immune system reconstitution. Previous studies have shown that in the absence of such well-organized lymphoid microenvironments immune response are severely compromised [37–39]. In the spleens of NSG.A2 mice reconstituted with UCB HSCs, fewer such clusters were observed and they were, in general, smaller than those in reconstituted NSG mice. Finally, in the spleens of NSG.A2 mice reconstituted with FL HSCs, human hematopoietic cells were rarely observed in discrete clusters, and even in the infrequent small clusters the segregation of different subsets was minimal. Table I presents the results of semi-quantitative analysis of these histology data.
Figure 5. Splenic lymphoid micro architecture in reconstituted NSG and NSG-A2 mice.
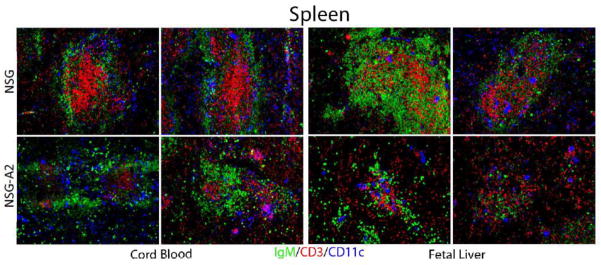
Spleens were isolated from either umbilical cord blood (left panels) or fetal liver (right panels) stem cell injected adult naive HIS mice. Spleens were processed for histologic analysis, and sections stained with fluorescence antibodies specific for the indicated markers. Stained sections were analyzed by fluorescence microscopy. Note the rather disorganized structure of the white pulp regions in cord blood or fetal liver stem cell injected naive NSG-A2 spleens (lower panels) as compared to the well segregated B, T, and DC areas in the spleens of NSG HIS mice. Data are representative of those obtained from at least two mice of each type.
HSCs purified from fetal liver contain more primitive hematopoietic progenitors that those purified from UCB
Our experiments demonstrated that reconstitution of NSG.A2 mice with FL HSCs was reproducibly poor. Moreover, inoculation of these mice with UCB HSCs led to good human hematopoietic reconstitution in peripheral blood but relatively poor reconstitution in splenic and intestinal tissues compared to NSG HIS mice. Together, our data suggest that differences in both HLA.A2 transgene expression and HSC source influenced the outcome of reconstitution. To investigate how the HSC source might account for these results, we conducted flow-cytometric phenotypic analysis of HSCs isolated from UCB and FL immediately post anti-CD34 MACS purification and after frozen storage.
Figure 6A illustrates that immediately prior to CD34-MACS purification the vast majority of cells in the HSC preparations were CD34+, with CD3+ cell contamination at levels of 2–5%. However, after cryopreservation and thawing the percentage of CD34+ cells in the preparations were substantially reduced, perhaps reflective relatively poor viability of these cells (Figure 6B).
Figure 6. Flow cytometric analysis of the multiple populations of CD34+ stem cells isolated from either cord blood or fetal liver.
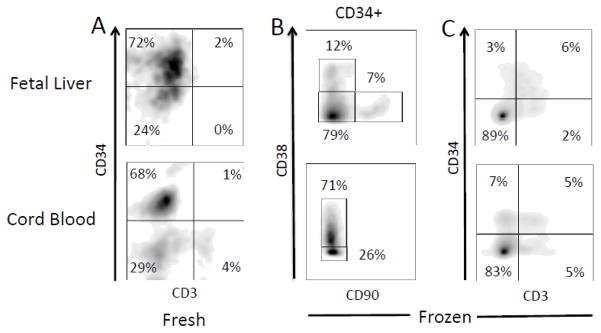
A) Cells analyzed immediately after CD34-MACs purification for percentages of CD34+ and CD3+ cells; B) Cells analyzed after freezing and thawing for percentages of cells in the CD34+ gate expressing CD38 and CD90; C) Cells analyzed after freezing and thawing for expression of CD34 and CD3. The data are representative of those obtained from three cord blood and three fetal liver samples.
Different populations of human CD34+ hematopoietic precursors can be distinguished through the expression pattern of cell surface antigens CD90, CD38, and CD45RA [40]. The least differentiated, or primitive, CD34+ HSCs capable of long-term renewal are thought to express CD90 but lack the other two markers. Maturation into multi potent progenitors results in cells that lack expression of all three of these antigens. As these cells differentiate into common lymphoid progenitors, they express CD38 and CD45RA but continue to lack CD90.
Analysis of the cryo-preserved and thawed HSC preparations revealed that the primitive HSC pool (CD90+CD38−CD45RA−) could be detected at low frequency in FL samples, but was undetectable in UCB samples (Figure 6, Table II). The majority of CD34+ cells from UCB expressed CD38 and lacked CD90, as did a smaller, but still significant, fraction of cells from FL. Of these CD38+CD90− cells from both tissues, 10–17% also expressed CD45RA. These data suggest that CD34+ cells from FL are enriched for primitive HSCs and multipotent progenitor cells compared to UCB cells, while the UCB samples contain more lymphoid progenitor cells.
Table 2. Flow cytometric analysis of the multiple populations of CD34+ stem cells isolated from either cord blood or fetal liver.
Three separate samples of either isolated fetal liver stem cells or cord blood stem cells were analyzed by flow cytometry to quantify the overall breakdown of the various stages of stem cells present in each isolated sample. The data were compiled from three samples of each type. The various functional subpopulations of HSC were defined as: Long Term Self Renewal - CD34+CD38−CD90+CD45RA−; Multipotent Progenitor - CD34+CD38−CD90−CD45RA−; Common Lymphoid Progenitor - CD34+CD38+CD45RA+. Standard deviations are shown for all values.
| Stem Cells: Cord Blood | % CD38+CD90− of CD34+ | % CD45RA− of CD38+CD90− | % CD45RA+ of CD38+CD90− |
| 55.40±8.15 | 75.33±2.06 | 16.50±2.31 | |
| % CD38−CD90− of CD34+ | %CD45RA− of CD38−CD90− | % CD45RA+ of CD38−CD90− | |
| 41.97±8.09 | 90.70±0.40 | 6.83±0.76 | |
| Stem Cells: Fetal Liver | % CD38+CD90− of CD34+ | % CD45RA− of CD38+CD90− | % CD45RA+ of CD38+CD90− |
| 17.20±2.65 | 72.33±1.82 | 24.57±1.83 | |
| % CD38−CD90− of CD34+ | % CD45RA− of CD38−CD90− | % CD45RA+ of CD38−CD90− | |
| 78.60±0.79 | 85.50±1.08 | 10.01±2.34 | |
| % CD38−CD90+ of CD34+ | % CD45RA− of CD38−CD90+ | ||
| 2.71±2.23 | 96.40±1.97 |
Long Term Self Renewal: CD34+CD38−CD90+CD45RA−
Multipotent Progenitor: CD34+CD38−CD90−CD45RA−
Common Lymphoid Progenitor: CD34+CD38+CD45RA+
We also analyzed the CD34+ HSCs for expression of mature markers for NK cells (CD56), monocytes (CD14), B-cells (CD27 and CD19) and T-cells (CD3) (Figure 6C and Table II). Of all of these markers, only significant staining with CD3 was observed. Unexpectedly, most of this staining was detected on cells that were also CD34+ (Figure 6C).
DISCUSSION
In an attempt to develop an improved HIS mouse model for human pathogen studies, we compared the ability of human HSCs purified from HLA.A2.1-expressing UCB or FL to reconstitute NSG mice that did or did not express a human HLA-A2.1 transgene. The most striking, and unexpected result of our studies was the inability of HSCs from either source to effectively reconstitute NSG.A2 mice. Overall reconstitution of these mice by FL HSCs was extremely poor (Figures 2 and 3). Moreover, while reconstitution by UCB HSCs in peripheral blood was comparable between NSG and NSG.A2 mice, lymphoid reconstitution in spleen and intestine was very limited in the HLA.A2 expressing mice (Figures 3b, 4 and Table I).
It is paradoxical that the presence of the HLA-A2 transgene should so dramatically negatively affect reconstitution levels. In theory, the expression of HLA-A2 should have resulted in positive selection of human CD8+ T cells during their primary development, thus promoting overall reconstitution of CD3+, CD8+ cells. Indeed, a previous publication reports evidence for such selection when UCB HSCs were used to reconstitute neonatal NSG mice expressing a transgene encoding a single chain human HLA-A2/beta-2 microglobulin molecule [41]. However, we observed poor reconstitution of human CD3 and CD8 expressing cells in NSG.A2 mice as compared to NSG mice.
In contrast to our findings, two previous publications report the successful reconstitution of human hematopoietic lineages in neonatal NSG.A2 mice, and a derivative of these mice also expressing an HLA-DR1 transgene via intra hepatic injection of purified CD34+ FL HSCs isolated from HLA-A2 positive donors [42,43]. In these studies, as in ours, HSCs were purified using anti-human CD34 MACS approaches. However, we found using multiple FL samples that reconstitution of total human CD45+ cells as well as the CD3+, CD4+ and CD8+ subsets was extremely poor in FL -> NSG.A2 mice, and reconstitution of CD3+, CD4+ and CD8+ cells limited in UCB ->NSG.A2 mice, indicating a reproducible and global defect in hematopoietic reconstitution in NSG.A2 mice. The reason for these discordant results is not clear, but might have been influenced by differences in the composition of the CD34+ HSC samples used for reconstitution (see below) or more poorly defined factors such as environmental differences in the mouse colonies in which these experiments were performed. Also, as mentioned above, comparison of our data with those of others is complicated by the fact that previous reports have used percentages of CD45+ cells in blood as a measure of reconstitution. Our linear regression analyses demonstrated that such percentages are not accurately reflective of the absolute number of human CD45+ cells in HIS mice.
Since all of the HSCs used in our studies were derived from HLA-A2 positive donors, one possible explanation for the poor reconstitution of NSG.A2 mice is that the transgenic expression of HLA-A2 elicits a population of mouse cells that targets developing human HLA-A2 expressing hematopoietic cells for destruction. It seems unlikely that either mouse T cells or NK cells could comprise such a hypothetical rejecting population, as both the scid mutation and the common cytokine receptor gamma chain mutations in the NSG strain should block the development of such mouse cells. Further studies to test these ideas appear warranted.
Since most types of humanized mice with the NOD background develop graft versus host disease (GVHD) at some point in their lives [44,45], such a process could also have contributed to the poor engraftment and post engraftment viability observed in NSG.A2 mice. The reason for that GVHD develops in humanized mice with the NOD background is not clear, but is reduced by an MHC class I or II deficiency in the host mice, suggesting that human T cells xenoreactive with mouse MHC antigens are in part responsible [45]. In the case of NSG.A2 mice a more likely explanation is that human T cells selected on HLA-A2 bound to human peptide antigens react with host cells expressing HLA-A2 bound to mouse peptide antigens (i.e. a form of alloreactivity). Perhaps consistent with this idea, we observed that reconstituting NSG.A2 mice, but not reconstituting NSG mice, became ill beginning at two to three months of age and many did not survive beyond four months of age. However, we found that FL CD34-MACS preparations contained a lower frequency of cells expressing CD3 than UCB CD34-MACS preparations, despite the fact that FL HSCs preparations were inferior to UCB HSC preparations in reconstituting NSG.A2 mice. Nonetheless, after cryopreservation and thawing, both type of preparations contained significant fractions of viable CD34+, CD3+ and CD34−,CD3+ cells. The former population might be explained by selective viability to freezing and thawing of contaminating T cells in the original HSC preparations. Indeed, the total cell and HSC viability we obtained using our cell freezing and thawing protocol was poor, perhaps exacerbating effects due to differential viability of contaminating subpopulations of cells. Such differential viability issues have not been previously reported, but could also complicate comparison of our data with those of others.
Detection of CD34+, CD3+ cells in the thawed HSC preparations was surprising given that a stringent “singlet” gate was used in the flow cytometric analysis. However, a previous report suggests that even with such stringent gating that doublets of CD34+ and CD3+ cells can appear as single flow cytometric events [46]. Taken together, these data suggest that the possibility that contaminating HLA-A2 restricted T cells are responsible for the poor engraftment and viability of NSG.A2 mice must be seriously considered.
We, and others (personal communication with Jackson Laboratories) have also found that presence of the HLA-A2 transgene in the NOD.Cg-Prkdcscid Il2rgtm1Wjl Tg(HLA.A2.1)1Enge/SzJ line affects the fecundity of these mice substantially. If either the male or the female (or both) are homozygous for this transgene, no litters are obtained. The simplest explanation for this finding is that the integration of the transgene has inactivated an endogenous gene important for mating behavior or fertility. It is possible that this same genetic defect influences human hematopoietic reconstitution levels, but how these phenotypes would be linked is far from obvious. In this context, we should point out that we used both HLA-A2 homozygous and hemizygous mice as recipients throughout our studies and observed no systematic differences in hematopoietic reconstitution between mice of these two genotypes.
We also found differences in the ability of HSCs derived from UCB versus FL to reconstitute both NSG mice, with the latter being superior. A previous study also reported differences in these two types of HSCs to reconstitute NSG mice [47], but not to the extent that we report here. Our data indicate that anti-CD34 MACs purification from both FL and UCB results in isolation of a very heterogeneous mixture of hematopoietic precursors. However, CD34+, CD90+, CD38−, CD45RA− cells, the phenotype associated with the most primitive HSC pool, were only detected in FL HSC preparations, perhaps accounting for the enhanced levels of NSG reconstitution observed with these preparations.
In total, our data illustrate major limitations to the use of HIS mice constructed using HSCs isolated from FL and the currently available line of NSG HLA-A2.1 transgenic mice. Moreover, even when UCB HSCs are used for reconstitution, the formation of lymphoid microenvironments in these mice is limited compared to when NSG mice are used as recipients. It appears that generation of new lines of such mice will be necessary to determine whether an integration site effect intrinsic to this particular transgene is responsible for the engraftment barrier we observed, or whether an immune rejection-based process is responsible. Since we found that the CD34+ cells isolated by conventional approaches from both FL and UCB were rather heterogeneous, it will be important to determine if additional purification steps designed to enrich specific hematopoietic precursors from these tissue sources will improve human hematopoietic reconstitution levels in NSG.A2 mice from such precursors.
Highlights.
Hematopoietic stem cells (HSCs) purified from human fetal liver appear superior to HSCs purified from human umbilical cord blood for reconstitution of a human hematopoietic compartment in NSG mice.
Human hematopoietic cells are very poorly reconstituted in NSG mice containing a human HLA-A2 transgene using HSCs purified from human fetal liver.
Human T cells and lymphoid microenvironments are not well reconstituted in NSG mice containing a human HLA-A2 transgene using HSCs purified from human umbilical cord blood.
HSCs preparations from human fetal liver contain more cells with a phenotype consistent with primitive and multipotent progenitors than HSC preparations from human umbilical cord blood.
Acknowledgments
We thank Scot Fenn for assistance with mouse breeding and the Laboratory Animal Services and Kimmel Cancer Center Flow Cytometry Facilities for technical support. These studies were supported by NIH grants to T.M. (R21AI097677) and MR (RO1GM66682)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Manz MG. Human-hemato-lymphoid-system mice: opportunities and challenges. Immunity. 2007;26:537–541. doi: 10.1016/j.immuni.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 2.Traggiai E, Chicha L, Mazzucchelli L, Bronz L, Piffaretti JC, et al. Development of a human adaptive immune system in cord blood cell-transplanted mice. Science. 2004;304:104–107. doi: 10.1126/science.1093933. [DOI] [PubMed] [Google Scholar]
- 3.Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat Rev Immunol. 2007;7:118–130. doi: 10.1038/nri2017. [DOI] [PubMed] [Google Scholar]
- 4.Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, et al. Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol. 2005;174:6477–6489. doi: 10.4049/jimmunol.174.10.6477. [DOI] [PubMed] [Google Scholar]
- 5.Ito M, Hiramatsu H, Kobayashi K, Suzue K, Kawahata M, et al. NOD/SCID/gamma(c)(null) mouse: an excellent recipient mouse model for engraftment of human cells. Blood. 2002;100:3175–3182. doi: 10.1182/blood-2001-12-0207. [DOI] [PubMed] [Google Scholar]
- 6.Ishikawa F, Yasukawa M, Lyons B, Yoshida S, Miyamoto T, et al. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor {gamma} chain(null) mice. Blood. 2005;106:1565–1573. doi: 10.1182/blood-2005-02-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang L, Kovalev GI, Su L. HIV-1 infection and pathogenesis in a novel humanized mouse model. Blood. 2007;109:2978–2981. doi: 10.1182/blood-2006-07-033159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baenziger S, Tussiwand R, Schlaepfer E, Mazzucchelli L, Heikenwalder M, et al. Disseminated and sustained HIV infection in CD34+ cord blood cell-transplanted Rag2−/−gamma c−/− mice. Proc Natl Acad Sci U S A. 2006;103:15951–15956. doi: 10.1073/pnas.0604493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jaiswal S, Pearson T, Friberg H, Shultz LD, Greiner DL, et al. Dengue virus infection and virus-specific HLA-A2 restricted immune responses in humanized NOD-scid IL2rgammanull mice. PLoS One. 2009;4:e7251. doi: 10.1371/journal.pone.0007251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Melkus MW, Estes JD, Padgett-Thomas A, Gatlin J, Denton PW, et al. Humanized mice mount specific adaptive and innate immune responses to EBV and TSST-1. Nat Med. 2006;12:1316–1322. doi: 10.1038/nm1431. [DOI] [PubMed] [Google Scholar]
- 11.Libby SJ, Brehm MA, Greiner DL, Shultz LD, McClelland M, et al. Humanized nonobese diabetic-scid IL2rgammanull mice are susceptible to lethal Salmonella Typhi infection. Proc Natl Acad Sci U S A. 2010;107:15589–15594. doi: 10.1073/pnas.1005566107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mian MF, Pek EA, Chenoweth MJ, Coombes BK, Ashkar AA. Humanized mice for Salmonella typhi infection: new tools for an old problem. Virulence. 2011;2:248–252. doi: 10.4161/viru.2.3.16133. [DOI] [PubMed] [Google Scholar]
- 13.Shultz LD, Brehm MA, Garcia-Martinez JV, Greiner DL. Humanized mice for immune system investigation: progress, promise and challenges. Nat Rev Immunol. 2012;12:786–798. doi: 10.1038/nri3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rongvaux A, Takizawa H, Strowig T, Willinger T, Eynon EE, et al. Human hemato-lymphoid system mice: current use and future potential for medicine. Annu Rev Immunol. 2013;31:635–674. doi: 10.1146/annurev-immunol-032712-095921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brehm MA, Wiles MV, Greiner DL, Shultz LD. Generation of improved humanized mouse models for human infectious diseases. J Immunol Methods. 2014 doi: 10.1016/j.jim.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Q, Khoury M, Chen J. Expression of human cytokines dramatically improves reconstitution of specific human-blood lineage cells in humanized mice. Proc Natl Acad Sci U S A. 2009;106:21783–21788. doi: 10.1073/pnas.0912274106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Willinger T, Rongvaux A, Strowig T, Manz MG, Flavell RA. Improving human hemato-lymphoid-system mice by cytokine knock-in gene replacement. Trends Immunol. 2011;32:321–327. doi: 10.1016/j.it.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 18.McHeyzer-Williams LJ, Malherbe LP, McHeyzer-Williams MG. Helper T cell-regulated B cell immunity. Curr Top Microbiol Immunol. 2006;311:59–83. doi: 10.1007/3-540-32636-7_3. [DOI] [PubMed] [Google Scholar]
- 19.Watanabe Y, Takahashi T, Okajima A, Shiokawa M, Ishii N, et al. The analysis of the functions of human B and T cells in humanized NOD/shi-scid/gammac(null) (NOG) mice (hu-HSC NOG mice) Int Immunol. 2009;21:843–858. doi: 10.1093/intimm/dxp050. [DOI] [PubMed] [Google Scholar]
- 20.Vuyyuru R, Patton J, Manser T. Human immune system mice: current potential and limitations for translational research on human antibody responses. Immunol Res. 2011;51:257–266. doi: 10.1007/s12026-011-8243-9. [DOI] [PubMed] [Google Scholar]
- 21.Lan P, Tonomura N, Shimizu A, Wang S, Yang YG. Reconstitution of a functional human immune system in immunodeficient mice through combined human fetal thymus/liver and CD34+ cell transplantation. Blood. 2006;108:487–492. doi: 10.1182/blood-2005-11-4388. [DOI] [PubMed] [Google Scholar]
- 22.Dudek TE, No DC, Seung E, Vrbanac VD, Fadda L, et al. Rapid evolution of HIV-1 to functional CD8(+) T cell responses in humanized BLT mice. Sci Transl Med. 2012;4:143ra198. doi: 10.1126/scitranslmed.3003984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jaiswal S, Pazoles P, Woda M, Shultz LD, Greiner DL, et al. Enhanced humoral and HLA-A2-restricted dengue virus-specific T-cell responses in humanized BLT NSG mice. Immunology. 2012;136:334–343. doi: 10.1111/j.1365-2567.2012.03585.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Biswas S, Chang H, Sarkis PT, Fikrig E, Zhu Q, et al. Humoral immune responses in humanized BLT mice immunized with West Nile virus and HIV- 1 envelope proteins are largely mediated via human CD5+ B cells. Immunology. 2011;134:419–433. doi: 10.1111/j.1365-2567.2011.03501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hardy RR, Hayakawa K. A developmental switch in B lymphopoiesis. Proc Natl Acad Sci U S A. 1991;88:11550–11554. doi: 10.1073/pnas.88.24.11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dorshkind K, Montecino-Rodriguez E. Fetal B-cell lymphopoiesis and the emergence of B-1-cell potential. Nat Rev Immunol. 2007;7:213–219. doi: 10.1038/nri2019. [DOI] [PubMed] [Google Scholar]
- 27.Hayakawa K, Hardy RR, Herzenberg LA. Progenitors for Ly-1 B cells are distinct from progenitors for other B cells. J Exp Med. 1985;161:1554–1568. doi: 10.1084/jem.161.6.1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Havran WL, Allison JP. Developmentally ordered appearance of thymocytes expressing different T-cell antigen receptors. Nature. 1988;335:443–445. doi: 10.1038/335443a0. [DOI] [PubMed] [Google Scholar]
- 29.Ikuta K, Kina T, MacNeil I, Uchida N, Peault B, et al. A developmental switch in thymic lymphocyte maturation potential occurs at the level of hematopoietic stem cells. Cell. 1990;62:863–874. doi: 10.1016/0092-8674(90)90262-d. [DOI] [PubMed] [Google Scholar]
- 30.Mold JE, Venkatasubrahmanyam S, Burt TD, Michaelsson J, Rivera JM, et al. Fetal and adult hematopoietic stem cells give rise to distinct T cell lineages in humans. Science. 2010;330:1695–1699. doi: 10.1126/science.1196509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Metcalf DaM, MAS Embryonic aspects of haemopoiesis. Frontiers in Biology. 1971;24 [Google Scholar]
- 32.Le AX, Bernhard EJ, Holterman MJ, Strub S, Parham P, et al. Cytotoxic T cell responses in HLA-A2.1 transgenic mice. Recognition of HLA alloantigens and utilization of HLA-A2.1 as a restriction element. J Immunol. 1989;142:1366–1371. [PubMed] [Google Scholar]
- 33.Takenaka K, Prasolava TK, Wang JC, Mortin-Toth SM, Khalouei S, et al. Polymorphism in Sirpa modulates engraftment of human hematopoietic stem cells. Nat Immunol. 2007;8:1313–1323. doi: 10.1038/ni1527. [DOI] [PubMed] [Google Scholar]
- 34.Legrand N, Huntington ND, Nagasawa M, Bakker AQ, Schotte R, et al. Functional CD47/signal regulatory protein alpha (SIRP(alpha)) interaction is required for optimal human T- and natural killer- (NK) cell homeostasis in vivo. Proc Natl Acad Sci U S A. 2011;108:13224–13229. doi: 10.1073/pnas.1101398108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Strowig T, Rongvaux A, Rathinam C, Takizawa H, Borsotti C, et al. Transgenic expression of human signal regulatory protein alpha in Rag2−/−gamma(c)−/− mice improves engraftment of human hematopoietic cells in humanized mice. Proc Natl Acad Sci U S A. 2011;108:13218–13223. doi: 10.1073/pnas.1109769108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pope M, Haase AT. Transmission, acute HIV-1 infection and the quest for strategies to prevent infection. Nat Med. 2003;9:847–852. doi: 10.1038/nm0703-847. [DOI] [PubMed] [Google Scholar]
- 37.Mebius RE, Kraal G. Structure and function of the spleen. Nat Rev Immunol. 2005;5:606–616. doi: 10.1038/nri1669. [DOI] [PubMed] [Google Scholar]
- 38.Kroemer G, Cuende E, Martinez C. Compartmentalization of the peripheral immune system. Adv Immunol. 1993;53:157–216. doi: 10.1016/s0065-2776(08)60500-3. [DOI] [PubMed] [Google Scholar]
- 39.Phan TG, Gray EE, Cyster JG. The microanatomy of B cell activation. Curr Opin Immunol. 2009;21:258–265. doi: 10.1016/j.coi.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kondo M, Wagers AJ, Manz MG, Prohaska SS, Scherer DC, et al. Biology of hematopoietic stem cells and progenitors: implications for clinical application. Annu Rev Immunol. 2003;21:759–806. doi: 10.1146/annurev.immunol.21.120601.141007. [DOI] [PubMed] [Google Scholar]
- 41.Shultz LD, Saito Y, Najima Y, Tanaka S, Ochi T, et al. Generation of functional human T-cell subsets with HLA-restricted immune responses in HLA class I expressing NOD/SCID/IL2r gamma(null) humanized mice. Proc Natl Acad Sci U S A. 2010;107:13022–13027. doi: 10.1073/pnas.1000475107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Strowig T, Gurer C, Ploss A, Liu YF, Arrey F, et al. Priming of protective T cell responses against virus-induced tumors in mice with human immune system components. J Exp Med. 2009;206:1423–1434. doi: 10.1084/jem.20081720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Billerbeck E, Horwitz JA, Labitt RN, Donovan BM, Vega K, et al. Characterization of human antiviral adaptive immune responses during hepatotropic virus infection in HLA-transgenic human immune system mice. J Immunol. 2013;191:1753–1764. doi: 10.4049/jimmunol.1201518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Greenblatt MB, Vrbanac V, Tivey T, Tsang K, Tager AM, et al. Graft versus host disease in the bone marrow, liver and thymus humanized mouse model. PLoS One. 2012;7:e44664. doi: 10.1371/journal.pone.0044664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.King MA, Covassin L, Brehm MA, Racki W, Pearson T, et al. Human peripheral blood leucocyte non-obese diabetic-severe combined immunodeficiency interleukin-2 receptor gamma chain gene mouse model of xenogeneic graft-versus-host-like disease and the role of host major histocompatibility complex. Clin Exp Immunol. 2009;157:104–118. doi: 10.1111/j.1365-2249.2009.03933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kudernatsch RF, Letsch A, Stachelscheid H, Volk HD, Scheibenbogen C. Doublets pretending to be CD34+ T cells despite doublet exclusion. Cytometry A. 2013;83:173–176. doi: 10.1002/cyto.a.22247. [DOI] [PubMed] [Google Scholar]
- 47.Lepus CM, Gibson TF, Gerber SA, Kawikova I, Szczepanik M, et al. Comparison of human fetal liver, umbilical cord blood, and adult blood hematopoietic stem cell engraftment in NOD-scid/gammac−/−, Balb/c-Rag1−/−gammac−/−, and C.B-17-scid/bg immunodeficient mice. Hum Immunol. 2009;70:790–802. doi: 10.1016/j.humimm.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]