

Abstract
Migraine is the most common neurological disorder and one of the most common chronic pain conditions. Despite its prevalence, the pathophysiology leading to migraine is poorly understood and the identification of new therapeutic targets has been slow. Several processes are currently thought to contribute to migraine including altered activity in the hypothalamus, cortical-spreading depression (CSD), and afferent sensory input from the cranial meninges. Decreased extracellular pH and subsequent activation of acid-sensing ion channels (ASICs) may contribute to each of these processes and may thus play a role in migraine pathophysiology. Although few studies have directly examined a role of ASICs in migraine, studies directly examining a connection have generated promising results including efficacy of ASIC blockers in both preclinical migraine models and in human migraine patients. The purpose of this review is to discuss the pathophysiology thought to contribute to migraine and findings that implicate decreased pH and/or ASICs in these events, as well as propose issues to be resolved in future studies of ASICs and migraine.
Keywords: ASIC, migraine, headache, cortical spreading depression, meninges, dura
1. Introduction
Migraine is a complex disorder consisting of unilateral throbbing headache, aura, nausea, vomiting, photophobia, phonophobia, and cutaneous hypersensitivity. An attack typically follows a temporal pattern [1] that has four phases: premonitory, aura, headache, and postdrome. The premonitory phase consists of symptoms such as yawning or changes in mood, appetite, cognitive function, or energy levels [2]. In a minority of patients, the premonitory phase is followed hours to days by visual or other sensory disturbances commonly referred to as aura [3]. Approximately 1 hour after aura is the headache phase, which lasts between 4 and 72 hours (sometimes longer). The attack then resolves with a postdrome phase, lasting hours to days, and consisting of decreased energy levels, residual headache, and impaired cognitive function [4]. Together, the entire sequence of symptoms associated with migraine can last for many days. Patients suffering from migraine have a significant decrease in quality of life during attacks and in many cases migraine can be completely disabling.
Although classically considered to be a vascular disorder, it is now appreciated that maladaptive changes within the nervous system contribute to the pathology of migraine (described below). Given the likely contribution of the nervous system, migraine is now considered to be a neurological disorder. The recent Global Burden of Disease study analyzed 289 diseases worldwide between the years 1990 and 2010 [5]. This study placed migraine as the 3rd most prevalent disease on the planet (behind dental caries and tension-type headache). In terms of time spent disabled, migraine ranks 8th of all diseases. This prevalence easily makes migraine the most common neurological disorder and it is also clearly one of the most burdensome diseases on Earth. Despite this, the exact mechanisms that contribute to migraine are still poorly understood.
2. Proposed mechanisms contributing to migraine
2.1 Hypothalamic activity
Numerous sites within the central and peripheral nervous systems have been proposed to contribute to the various phases of migraine. The earliest obvious symptoms of an impending migraine occur in the premonitory phase where the collection of symptoms, e.g. the changes in energy levels, appetite, and food cravings, implicate the hypothalamus as the driver of migraine attacks. Indeed, human imaging studies show changes in hypothalamic activity during migraine attacks [6, 7], including during the premonitory phase [8], there are abnormal patterns in blood levels of hypothalamic hormones in chronic migraine patients [9], and increased functional connectivity between the hypothalamus and several other brain regions was recently reported in migraine patients [10]. Descending pathways originating in several subregions of the hypothalamus terminate bilaterally in the trigeminal nucleus caudalis (TNC) [11], the site of synapses of afferent trigeminal neurons. The hypothalamus is capable of extensive modulation of processes occurring in migraine, particularly pain transmission in the trigeminal system [12, 13], and can thus modulate noxious sensory input from the head. How and why the hypothalamus may ultimately contribute to a migraine attack is not clear, but it could be a response of this brain region to repeated episodes of stress [14]. Stress, or more appropriately the letdown from a stressful event, is commonly reported as a trigger of migraine [15] and the hypothalamus plays a critical role in the response to stress.
2.2 Cortical Spreading Depression
Another neuronal process linked to migraine is cortical spreading depression (CSD). CSD was first described in the cortex of rabbits by Aristides Leao in 1944 [16] and consists of brief neuronal excitation followed by a longer-lasting depression of activity. A CSD event propagates across the cortex in a wave-like pattern at a rate of 2–5 mm/min and is associated with numerous physiological changes within the cortex including alterations in intra-and extracellular ion concentrations, neurotransmitter release, and changes in both blood flow and oxygen levels [17, 18]. Similar to other phenomena of altered cortical excitability, e.g. seizures, the origin or triggering factors for a CSD are not known. Although CSD has not directly been measured in humans, imaging studies show patterns of changes in blood flow in the cortex of human migraine patients that are similar to CSD events [19]. The link between CSD and migraine has largely been with aura as the changes in vision that occur during aura, particularly the movement/expansion of geometric shapes or scintillating scotomas, can be mapped as electrical changes onto the visual cortex and they fit the pattern of CSD [20]. Whether CSD is linked to other phases of migraine is less clear.
One of the most intensely debated links is between CSD and the pain of migraine. Migraine pain most likely requires activation of peripheral nociceptors that innervate the cranial meninges [21, 22]. This innervation arises from neurons that have cell bodies either in the trigeminal ganglion and project into the TNC or in the upper cervical dorsal root ganglia and project into the cervical spinal cord. For many years, no direct evidence existed linking CSD to afferent activation outside of increased expression of c-fos in the TNC [23, 24], presumed due to afferent input, but also a finding that was not observed in all studies [25]. However, a series of papers showed that CSD could initiate prolonged activity in both meningeal afferents and in second-order TNC neurons that receive input from the meninges [26, 27]. More recently, it was found that spreading depression could lead to activity in meningeal afferents via the release of high-mobility group box 1 (HMGB1) protein from cortical neurons [28], providing one of the first demonstrations of a mechanism by which CSD could cause headache. And direct pathways from the cortex to the TNC can modulate neuronal activity in the TNC, providing anatomical mechanisms for CSD modulation of afferent trafficking in the trigeminal system [29]. But with these reports of CSD-induced afferent activation/modulation are other studies showing that induction of CSD events do not produce behavioral responses consistent with headache in rodents [30]. Along with the observation that the majority of migraine patients do not experience aura, these findings question whether CSD can be fully responsible for all of the symptoms during migraine attacks.
2.3 Meningeal afferents
As mentioned above, the pain of migraine likely requires activation of meningeal nociceptors. The spontaneous, throbbing pain is always specific to the head region and the most likely explanation for this finding is that peripheral afferents provide this specificity. It has been known for many decades that stimulation of much of the dura mater (particularly near blood vessels and sinuses) produces pain in humans and depending on the location of dural stimulation, pain can be produced in areas commonly reported by migraine patients [31]. What is not clear is how these neurons are activated during migraine attacks (CSD or other mechanisms). Prior work in preclinical models has found that dural afferents are mechanically sensitive [32–34], a finding that fits with the clinical observations that any change in intracranial pressure, such as that due to head movements, coughing, or changes in vertical position, can exacerbate existing headaches. However, the mechanisms mediating mechanical sensitivity remain unknown, as do the potential sources of mechanical stimulation within the skull during a migraine. Arterial pulsations have been traditionally thought to cause the throbbing pain of migraine. Although this may still be true, it appears to not be so simple due to a lack of correlation between throbbing pain and heart rate [35–37].
Dural afferents can also be activated and/or sensitized by application of chemical mediators such as capsaicin, mustard oil, hypotonic solutions, or an inflammatory soup (IS) [34, 38–40]. Various cytokines such as tumor necrosis factor-α (TNF- α), interleukin-6 (IL-6), and interleukin-1β (IL-1β) can also sensitize dural afferents increasing their excitability or mechanosensitivity [41–43], and TNF-α may actually mediate an interaction between blood vessels and sensory afferents by acting on both cell structures [42]. How these factors lead to sensitization of dural nociceptors is less well studied. Application of inflammatory mediators leads to sensitization of ionic currents (voltage-gated Na+ currents as well as depolarizing Cl− currents) that contribute to the increased excitability of dural afferents [44, 45]. IL-6 sensitizes dural afferents via increased interactions between extracellular signal-related kinase (ERK) and the sodium channel Nav1.7 [41]. In addition, there are likely other mechanisms that can lead to increased excitability of dural afferents and could mediate the increased sensitivity of these nerve endings to mechanical pressure from vessels.
One hypothesis for the source of many of these sensitizing agents is that sterile inflammation of the meninges occurs during migraine and initiates nociceptive signaling. This inflammation could be mediated by actions of meningeal macrophages [46] but more research has been devoted to studying a potential role of mast cells [47]. Mast cells are known to reside mostly within the dura mater compared to other meningeal layers [48, 49], and they have been found in direct contact with afferent endings within the dura [50]. Degranulation of these mast cells can activate and sensitize dural afferents via a variety of mechanisms [51–53]. Mast cell degranulation is known to occur following stress (via the release of corticotropin releasing factor or CRF), CGRP release, nitroglycerin infusion, and increased estrogen levels, all factors associated with migraine in humans [47], and there is an increased incidence of migraine in patients with mastocytosis [54]. Finally, CSD was shown to degranulate meningeal mast cells [28] providing a potential mechanistic link between CSD, mast cells, and pain. Degranulation of mast cells and the resulting release of pro-inflammatory factors could thus represent a significant factor in the excitation/sensitization of dural afferents.
2.4 Better mechanistic understanding of migraine is needed for new therapeutics
Ultimately, which of the above mechanisms play the greatest role in the pathophysiology of migraine, whether multiple mechanisms contribute, and whether there is an interrelationship between these mechanisms, remains to be determined. CSD remains a prominent hypothesis in the migraine literature but is largely a cortical phenomenon. However, in a mouse model of familial-hemiplegic migraine type 1 (FHM1), which in humans is due to a mutation in the voltage-gated calcium channel Cav2.1, CSD was found more likely to spread into subcortical structures [55, 56]. It is thus possible that CSDs may propagate into the hypothalamus under certain conditions. If so, animal studies have shown that CSDs can influence the activity of hypothalamic neurons [57–59]. These data provide a potential (but speculative) link between CSD and the hypothalamus. Regardless, both the cortex and the hypothalamus can modulate afferent trafficking in the TNC (see sections 2.1 and 2.2). Alterations in either brain region alone, as well as together, can influence afferent input from the meninges and contribute to the headache phase of migraine. Clearly, more work is necessary to better resolve whether and how these mechanisms contribute to migraine.
Despite the uncertainty on mechanisms contributing to migraine, what is clear is that there is a great need for the development of new therapeutics. Currently, migraine is most commonly treated using either prophylactic agents taken daily (e.g. topiramate) or abortive agents taken at the onset of headache (e.g. NSAIDs or triptans, a family of 5-HT1B/D agonists). Unfortunately, these therapies provide complete relief in less than 50% of migraine patients due to the lack of adequate pain control or the presence of intolerable side effects [60]. Many of these refractory patients are resistant to multiple therapies i.e. they do not respond to prophylactic or any of the abortive agents [61]. Consequently, a substantial percentage of migraine headache sufferers are not adequately treated. Ideally, among the mechanisms described above exist avenues for development of novel therapeutics for the treatment of migraine. Further investigation of these mechanisms, and identification of new drug targets, is of critical importance in the development of new therapies with greater efficacy for migraine.
3. ASICs as novel targets for migraine therapeutics
3.1 ASIC background
The acid-sensing ion channels (ASICs) are a family of ion channels, related to degenerin (DEG) and epithelial sodium (ENaC) channels, first cloned in 1997 [62] and consisting of 4 members, ASIC1-ASIC4, with several splice variants [63–66]. There is also a related family known as BASIC [67–69] about which little is known. ASICs exist as homomeric or heteromeric channels and are sensitive to different ranges of pH (ASIC4 is not pH sensitive) from a half-maximally activating pH (pH50) of between 4.0 and 5.0 (ASIC2a) to a pH50 range of between 5.8–6.8 (ASIC1 and ASIC3), although ASIC1 and ASIC3 can actually begin to open at pH values near 7.3 [64]. Interestingly, human ASIC3 is sensitive to alkaline pH up to 8.0 [70], a property not shared by any of the rodent ASICs. Thus, these channels are particularly well suited to detect changes in pH in both directions from 7.4, and in ranges that may not be considered pathological.
ASICs are expressed throughout the nervous system with the predominant ASIC in the CNS being ASIC1 [71]. They are expressed in the spinal cord and also in numerous brain regions including cortex, hippocampus, periaqueductal grey (PAG), striatum, and amygdala [72]. ASIC expression in this latter region may be important for fear circuitry as inhalation of CO2 decreases brain pH, produces fear responses in mice (humans as well), and these fear responses were reduced in mice deficient in ASIC1a [73]. Additionally, long-term potentiation (LTP) in the amygdala is dependent on decreased pH and is attenuated in ASIC1a knockout mice [74]. ASIC1a knockouts also have deficits in LTP in the hippocampus as well as in behavioral assays of learning and memory [63]. CNS ASIC1 is proposed to contribute to cell death following ischemia [75] since ASIC1 is a highly Ca++ permeable channel (potentially contributing to excitotoxicity) and the pH can fall below 6.5 during ischemic events in the brain [76]. In experimental models of stroke, ischemic injury was significantly decreased in ASIC1a knockout mice compared to wild-type, an effect that also occurred with pharmacological blockade of ASICs [77, 78]. In addition to fear processing and damage subsequent to ischemic events, ASIC1 has been speculated to contribute to epilepsy [63, 79] and a recent study linked single-nucleotide polymorphisms in ASIC1a to temporal-lobe epilepsy in humans [80]. What role ASICs may play in more normal CNS function is not entirely clear. They have been speculated to be an accessory sensor responding to high-frequency synaptic events as the contents of synaptic vesicles are acidic (pH 5.7) [81], and the release of vesicle contents can acidify the synapse leading to ASIC activation [74].
CNS ASICs have also been implicated in pain signaling. Increased expression of ASIC1a was found in the spinal cord after peripheral inflammation and either blockade or knockdown of this channel (both in the spinal cord) attenuated pain behaviors in inflamed rats [82, 83]. Intrathecal injection of an ASIC1a blocker (a toxin from the tarantula Psalmopoeus cambridgei named PcTx1) also attenuated acute pain responses as well as pain behaviors in more chronic inflammatory and neuropathic models [84]. And a recently identified blocker of ASIC1a from the venom of black mamba (Dendroapsis polylepis polylepis) named mambalgin-1, attenuated a variety of pain behaviors when given centrally [85]. Further, ASIC1a was found to be necessary for pain behavior following intrathecal injection of brain-derived neurotrophic factor (BDNF) [86]. These studies demonstrate that ASICs within CNS sites can contribute to pain signaling. Although the published data are specific to the lower spinal cord, these mechanisms may contribute to pain signaling at higher sites more relevant for pain in the trigeminal system.
In the peripheral nervous system, it has been known since 1980 that low pH generates currents in a subset of primary afferents [87]. Most ASIC subtypes are expressed on primary sensory neurons important for pain transduction [63, 88] and cutaneous pain from low pH solutions (at least above pH 6.0) is most likely due to ASIC activation [89, 90]. Unlike the CNS however, ASIC3 is highly expressed in the periphery and plays a major role, along with other subtypes, in processes contributing to nociception [91]. Peripheral sensory neurons expressing ASIC3 innervate visceral organs including the colon and heart as well as skeletal muscle and joints [92–96] and this channel has been proposed to participate in pain originating from these organs in conditions such as GI disorders, angina, intermittent claudication, and arthritis. Blockers of this channel have been shown to inhibit nociceptive behaviors in preclinical models of cutaneous inflammation as well as joint and muscle pain [97–99]. A toxin from sea anemone (Anthopleura elegantissima) that blocks ASIC3-containing channels, named APETx2, has been instrumental in discovering a role for peripheral ASIC3 in a variety of pain conditions including those described above and others including post-surgical pain due to incision of skin and muscle [100, 101]. Recently, a non-proton activating site has been identified on ASICs and ASIC3 can be activated selectively at normal pH through this site by compounds such as 2-guanidine-4-methylquinazoline (GMQ) and agmatine [102–105]. Peripheral cutaneous injection of these compounds causes pain in wild-type but not ASIC3 knockout mice, providing further evidence that activation of this channel in peripheral tissues contributes to pain signaling.
As mentioned above, ASIC3 does not contribute to pain signaling in peripheral tissues alone as pharmacological studies have shown that other ASICs also participate. A toxin isolated from the venom of the Texas coral snake (Micrurus tener tener), named MitTx, was recently found to activate ASIC1a and 1b homomers, it potentiates the proton sensitivity of ASIC2a, and at high concentrations, it activates ASIC3 [106]. Injection of MitTx into the skin of the mouse hindpaw produces nociceptive behavior that is lost in ASIC1a knockout mice and is also lost in ASIC3 knockout mice at high MitTx concentrations. Similarly, mambalgin-1 (the ASIC1/2 blocker) given via intraplantar injection in mouse, attenuates both acute thermal nociception as well as inflammatory hyperalgesia, an effect lost with ASIC1b knockdown [85]. These findings provide evidence that activation of numerous ASIC subtypes in peripheral tissues, as well as the CNS contribute to pain signaling. Ultimately, new pharmacological agents for pain based on an ASIC mechanism may need to be non-selective and also gain access to the CNS to produce their greatest effects.
3.2 pH changes and ASICs in the context of migraine
So far, this review has covered potential mechanisms contributing to migraine and has discussed how ASICs contribute to a variety of physiological and pathological processes. The remainder will now connect the two themes by focusing on studies that implicate ASICs in migraine pathophysiology. First, in addition to activation by decreased pH, ASICs are modulated by nerve growth factor (NGF) and serotonin [107], and serotonin was recently found to potentiate ASIC3 through the non-proton ligand-binding site [108]. Serotonin levels have long been linked to migraine [109–111] and NGF is increased in the CSF of chronic-daily headache patients with a history of migraine [112]. Nitric oxide (NO) donors, one of the most reliable migraine triggers in humans [113], also potentiate ASIC currents [114, 115]. Taken together, these data provide mechanistic links (albeit circumstantial) between ASIC expression/function and conditions contributing to migraine.
Regarding the hypothalamus and migraine, several in vitro studies have shown that decreased pH can produce currents in cultured hypothalamic neurons (pH 6.0 and below) [116], low pH can modulate GABAergic currents in hypothalamic slices (pH 6.4) [117, 118], and low pH can increase dopamine release (pH 6.5 and below) [119]. Although none of these studies directly tested whether ASICs mediate the effects of low pH, several other studies have examined ASICs in the hypothalamus. ASIC3 is expressed in nuclei throughout the hypothalamus [120] and both ASIC1- and ASIC3-like currents were demonstrated in cultured hypothalamic neurons [121]. Suprachiasmatic neurons generate currents at pH 7.0 and express mRNA for many ASICs [122]. ASIC-like currents are expressed in vasopressin hypothalamic neurons [123]. And pH 7.0 increased the excitability of neurons in the tuberomamillary nucleus (TMN) of the posterior hypothalamus, a nucleus that shows expression of multiple subtypes of ASIC [124]. Given that all of these studies were either in vitro or only examined ASIC expression, they do not show that ASIC activation can modulate hypothalamic output and physiological function. A recent study however, found that ASIC1a is expressed on orexinergic neurons in the lateral hypothalamus (LH) and pH 6.5 administration into the LH increased neuronal discharge in the phrenic nerve and increased respiratory drive, an effect blocked by ASIC antagonists [125]. This is the first direct evidence that activation of ASICs can modulate output from the hypothalamus and shows that these channels are capable of modulating hypothalamically-driven functions. Further, these data suggest that ASICs can play some role in hypothalamic contribution to migraine, especially given the proposed role of orexinergic outputs in primary headache disorders [12]. Future studies are required to provide a more conclusive link, as are studies examining processes that may lead to a decrease in hypothalamic pH.
Decreased pH and ASIC activation may also play a role in initiation or propagation of CSD (whether in the cortex or hypothalamus). As mentioned above, the events that initiate a CSD are unknown but one possibility is that they are due to mild ischemia or hypoxia. Microemboli traveling in the bloodstream can lead to ischemic events and CSDs without causing an infarction [126] and the ischemia may drop pH to a range detectible by ASICs. It is also known that there is a decrease in pH within the cortex during CSD as well as in related types of spreading depolarizations [17, 18], which may occur because CSD can increase metabolic demand without a corresponding increase in blood flow (due to neurovascular uncoupling) leading to hypoxia of cortical tissue [127–130]. Thus, the conditions may be appropriate for an ASIC contribution to CSD.
The most direct evidence of a role for ASICs in CSD comes from a study where needleprick-induced CSDs were inhibited by 2 ASIC blockers, the non-selective blocker amiloride and the tarantula toxin PcTx1 that blocks ASIC1a channels [131]. There was no effect of ASIC blockers against CSDs evoked by high K+ suggesting that ASICs may not contribute to all types of CSDs. Nonetheless, these data directly implicate ASICs in processes related to migraine and indicate that a decrease in pH may contribute to the initiating events of a CSD. More interestingly, and in order to connect these animal studies to human migraine patients, the authors gave amiloride (approved for use in humans as a diuretic) to 7 treatment-resistant migraine-with-aura patients. They found a decrease in both aura frequency and headache severity in 4 of 7 patients. Although amiloride is not selective for ASICs (addressed below), these findings provide exciting preliminary human data that not only suggest ASICs contribute to CSDs/aura but that they may be novel targets for migraine therapeutics.
ASICs may also contribute to afferent signaling from the meninges that mediates the pain of migraine. ASICs have been proposed as a sensor of decreased extracellular pH within the dura [132] but until recently, no data existed directly demonstrating their role. Studies from the late 1990s using in vivo electrophysiological recording techniques show that 75% of rat dural afferents respond to pH 4.7 [133], 37% respond to pH 5.0 in guinea pigs [38], and 64% respond to pH 6.1 in another rat study [134]. However, the mechanism responsible for this activation was not determined in these studies. More recently, electrical stimulation of the dura was found to produce vasodilation of the middle meningeal artery (MMA) as well as afferent activity into the TNC that was blocked by amiloride, implicating ASICs in the activation of dural nerve endings [131]. Furthermore, low pH (5.4 to 5.9) causes the release of the vasodilatory neuropeptide calcitonin gene-related peptide (CGRP) from isolated dura and trigeminal cell bodies, the latter in a mechanism blocked by APETx2 [135, 136].
The most direct evidence thus far for a role of ASICs in dural afferent activity are recent studies showing that 80% of dural afferents generate ASIC-like currents in response to pH 6.0 and over 50% generate currents at pH 7.0 [137]. Currents at pH 6.0 and 7.0 were blocked by amiloride, pH 6.0-responsive neurons generated currents in response to the ASIC3 activator GMQ, and 80% of dural afferents express ASIC3 immunoreactivity [138]. Additionally, these authors used a preclinical rat behavioral model of migraine where low pH stimuli is applied directly to the dura mater of awake animals, producing headache-related behaviors. Exposure of the dura to pH 5.0, 6.0, and 6.4 produced headache-like responses that were blocked by amiloride (pH 5.0 and 6.0) or the ASIC3 toxin APETx2 (pH 6.0). These latter studies directly demonstrate meningeal ASIC contributions to behavioral responses that are consistent with headache.
One mechanism that may lead to decreased meningeal pH and ASIC activation is sterile inflammation due to mast cell degranulation (discussed above). Mast cells are in direct contact with afferent endings within the dura [50] and the contents of mast cell granules are acidic (approximately pH 5.5) [139]. Thus, the degranulation of mast cells can decrease the pH surrounding afferent nerve endings. In relation to this, the percentage of dural afferents that fire action potentials at pH 7.0 is doubled in the presence of mast-cell mediators (40% vs. 20% in controls) [138]. Further, smaller drops in meningeal pH produce headache-related behavior in the presence of mast-cell mediators than under control conditions. So not only can mast cell degranulation provide the drop in pH necessary to activate ASICs, the mediators released from mast cells can also sensitize dural afferents leading to enhanced responses to low pH. Together with the studies described above, these data demonstrate that decreased meningeal pH can activate dural afferents via ASICs and produce behaviors consistent with headache. They also suggest that efficacy of amiloride for migraine in humans may also be due to actions on ASICs within the meninges.
4. The future of ASIC-based therapeutics for migraine
The studies described above provide a growing amount of evidence for a role of ASICs in processes believed to contribute to migraine (see Figure 1 for a summary). The ability of amiloride to inhibit CSD, the demonstration of ASICs on dural afferents, and the contribution of dural ASICs to headache-related behaviors provides solid preclinical support for a role of ASICs in migraine. The efficacy of amiloride against aura and headache in a small population of treatment-resistant migraine patients is the most convincing rationale for further investigation of ASICs as migraine targets in humans. Thus, the future of ASIC blockers as novel migraine therapeutics seems bright.
Figure 1. Mechanisms and ASIC subtypes that may play a role in the various phases of migraine.
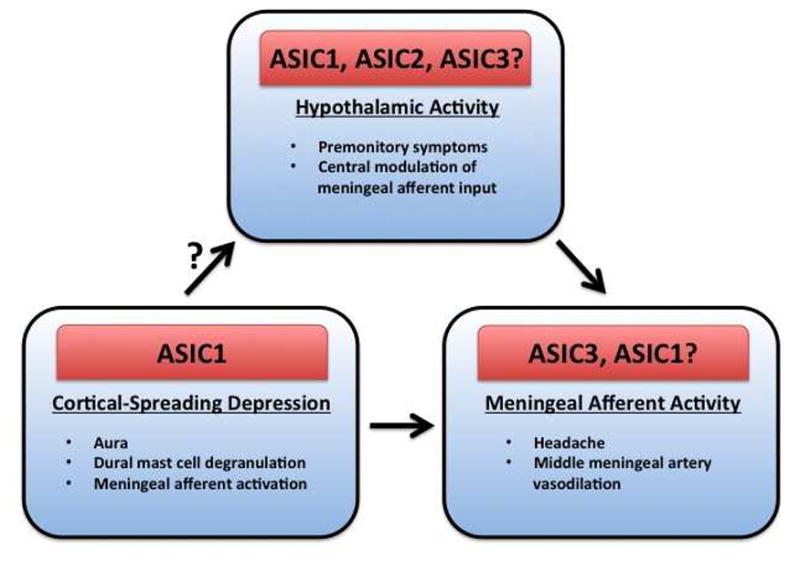
Hypothalamic activity is thought to drive both premonitory symptoms and to modulate incoming afferent activity from the meninges. Multiple ASIC subtypes and ASIC activity are present throughout the hypothalamus. A role for ASIC1 has been demonstrated in CSD, a process that may underlie aura, but can also lead to inflammatory events and afferent activity in the meninges. Whether CSD can propagate into the hypothalamus and contribute to migraine is not clear. ASIC3 can contribute to activation of meningeal afferents resulting in headache. ASIC1 participates in pain signaling from peripheral tissues but has not yet been linked to meningeal afferent activity.
Having said this, several important questions need to be resolved in future studies of ASICs and migraine. First, the efficacy of amiloride in human migraine patients is clearly exciting, but whether amiloride acts via ASICs or other targets (e.g. ENaCs, Na+/H+ exchangers) must be addressed. Unfortunately, this is not currently feasible, as no completely selective blockers of ASICs exist for use in humans. The pharmacological tools for preclinical studies, outside of natural toxins, are also lacking and even the toxins have limitations (for example see [140]). But this is a general issue across the field of ASIC research and has been a limitation in more fully documenting a role of these channels in many physiological/pathological conditions. Ideally, studies such as the ones described here will encourage the development of better tools (aided by recent crystal structures of ASICs [141]) so that a clear role for ASICs in migraine and other disorders can be examined. In the meantime, larger-scale studies of amiloride for migraine should be conducted. Whether or not amiloride works via an ASIC mechanism is important for future drug development. But amiloride may still provide relief to many migraine patients who are without adequate therapy. Knowing whether amiloride has efficacy for general migraine patients or only treatment-resistant patients, whether it should be a first-line therapy, and whether it can be given along with other agents (e.g. triptans) can still provide benefit without complete knowledge of mechanism.
The second question relates to the most important site of action for ASICs in migraine. Several nervous system locations have been discussed here, including the hypothalamus, cortex, spinal cord, and meninges, and whether one or more of these sites must be accessed for efficacy is important knowledge. For example, the dura is not protected by the blood-brain barrier and if blockade of dural ASICs alone is sufficient for efficacy, peripherally-restricted agents can be developed that may be devoid of CNS side effects. If CNS access of ASIC blockers is required, what are the adverse effects, particularly given the proposed role of ASICs in learning, memory, and fear circuitry?
Third, in which patient population does a pH change and subsequent ASIC activation contribute to migraine e.g. with or without aura? And where do pH changes occur e.g. hypothalamus, cortex, meninges? Assays focused on measuring changes in pH have traditionally been performed in humans by collecting plasma, CSF, or using magnetic-resonance spectroscopy or they could also be inferred from other markers e.g. lactate levels [142–144]. But these may not be sensitive enough to detect the small, and potentially local changes in pH that are necessary to activate ASICs. A modified MRI technique was used recently to image small changes in pH in the brain of humans and the pH of the visual cortex decreased in response to visual stimulation with a flashing checkerboard [145]. So in healthy subjects, drops in cortical pH occur with relatively normal activity. This technique could be instrumental in determining where and when pH changes occur in migraine patients in relation to progression of attacks and whether the magnitude of those pH changes are greater than in migraine-free patients. Addressing all of these questions may be difficult, but they may also lead to new ASIC-based therapies for the most common of neurological disorders.
Decreased extracellular pH and ASIC activation may play a role in migraine.
Preclinical studies implicate ASICs in many migraine-related processes.
Amiloride decreased aura and headache severity in human migraine patients.
Future questions including determining where, when, and how pH changes in migraine.
Acknowledgments
Grant support was provided by the National Institutes of Health (NS072204 and GM102575). The author wishes to acknowledge Drs. Eric Gonzales, Milena De Felice, and Theodore Price for critical reading of the manuscript.
Footnotes
The author declares no conflict of interest in relation to this work.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Charles A. The evolution of a migraine attack - a review of recent evidence. Headache. 2013;53(2):413–9. doi: 10.1111/head.12026. [DOI] [PubMed] [Google Scholar]
- 2.Schoonman GG, et al. The prevalence of premonitory symptoms in migraine: a questionnaire study in 461 patients. Cephalalgia. 2006;26(10):1209–13. doi: 10.1111/j.1468-2982.2006.01195.x. [DOI] [PubMed] [Google Scholar]
- 3.Lipton RB, et al. Classification of primary headaches. Neurology. 2004;63(3):427–35. doi: 10.1212/01.wnl.0000133301.66364.9b. [DOI] [PubMed] [Google Scholar]
- 4.Kelman L. The postdrome of the acute migraine attack. Cephalalgia. 2006;26(2):214–20. doi: 10.1111/j.1468-2982.2005.01026.x. [DOI] [PubMed] [Google Scholar]
- 5.Vos T, et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990–2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2163–96. doi: 10.1016/S0140-6736(12)61729-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alstadhaug KB. Migraine and the hypothalamus. Cephalalgia : an international journal of headache. 2009;29(8):809–17. doi: 10.1111/j.1468-2982.2008.01814.x. [DOI] [PubMed] [Google Scholar]
- 7.Denuelle M, et al. Hypothalamic activation in spontaneous migraine attacks. Headache. 2007;47 (10):1418–26. doi: 10.1111/j.1526-4610.2007.00776.x. [DOI] [PubMed] [Google Scholar]
- 8.Maniyar FH, et al. Brain activations in the premonitory phase of nitroglycerin-triggered migraine attacks. Brain : a journal of neurology. 2013 doi: 10.1093/brain/awt320. [DOI] [PubMed] [Google Scholar]
- 9.Peres MF, et al. Hypothalamic involvement in chronic migraine. J Neurol Neurosurg Psychiatry. 2001;71(6):747–51. doi: 10.1136/jnnp.71.6.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moulton EA, et al. Altered hypothalamic functional connectivity with autonomic circuits and the locus coeruleus in migraine. PLoS One. 2014;9(4):e95508. doi: 10.1371/journal.pone.0095508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abdallah K, et al. Bilateral descending hypothalamic projections to the spinal trigeminal nucleus caudalis in rats. PLoS One. 2013;8(8):e73022. doi: 10.1371/journal.pone.0073022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holland P, Goadsby PJ. The hypothalamic orexinergic system: pain and primary headaches. Headache. 2007;47(6):951–62. doi: 10.1111/j.1526-4610.2007.00842.x. [DOI] [PubMed] [Google Scholar]
- 13.Noseda R, Burstein R. Migraine pathophysiology: anatomy of the trigeminovascular pathway and associated neurological symptoms, CSD, sensitization and modulation of pain. Pain. 2013;154(Suppl 1) doi: 10.1016/j.pain.2013.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Borsook D, et al. Understanding migraine through the lens of maladaptive stress responses: a model disease of allostatic load. Neuron. 2012;73(2):219–34. doi: 10.1016/j.neuron.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 15.Lipton RB, et al. Reduction in perceived stress as a migraine trigger: testing the “let-down headache” hypothesis. Neurology. 2014;82(16):1395–401. doi: 10.1212/WNL.0000000000000332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leao AAP. Spreading depression of activity in the cerebral cortex. Journal of Neurophysiology. 1944;7(6):359–390. doi: 10.1152/jn.1947.10.6.409. [DOI] [PubMed] [Google Scholar]
- 17.Pietrobon D, Moskowitz MA. Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nat Rev Neurosci. 2014;15(6):379–93. doi: 10.1038/nrn3770. [DOI] [PubMed] [Google Scholar]
- 18.Somjen GG. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiological Reviews. 2001;81(3):1065–1096. doi: 10.1152/physrev.2001.81.3.1065. [DOI] [PubMed] [Google Scholar]
- 19.Charles AC, Baca SM. Cortical spreading depression and migraine. Nature Reviews Neurology. 2013;9(11):637–644. doi: 10.1038/nrneurol.2013.192. [DOI] [PubMed] [Google Scholar]
- 20.Hansen JM, et al. Distinctive anatomical and physiological features of migraine aura revealed by 18 years of recording. Brain. 2013;136:3589–3595. doi: 10.1093/brain/awt309. [DOI] [PubMed] [Google Scholar]
- 21.Levy D. Migraine pain and nociceptor activation--where do we stand? Headache. 2010;50(5):909–16. doi: 10.1111/j.1526-4610.2010.01670.x. [DOI] [PubMed] [Google Scholar]
- 22.Bernstein C, Burstein R. Sensitization of the trigeminovascular pathway: perspective and implications to migraine pathophysiology. Journal of clinical neurology. 2012;8(2):89–99. doi: 10.3988/jcn.2012.8.2.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moskowitz MA, Nozaki K, Kraig RP. Neocortical Spreading Depression Provokes the Expression of C-Fos Protein-Like Immunoreactivity within Trigeminal Nucleus Caudalis Via Trigeminovascular Mechanisms. Journal of Neuroscience. 1993;13(3):1167–1177. doi: 10.1523/JNEUROSCI.13-03-01167.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maneesri S, et al. Cortical spreading depression, meningeal inflammation and trigeminal nociception. Neuroreport. 2004;15(10):1623–1627. doi: 10.1097/01.wnr.0000134989.89428.3b. [DOI] [PubMed] [Google Scholar]
- 25.Ingvardsen BK, et al. Possible mechanism of c-fos expression in trigeminal nucleus caudalis following cortical spreading depression. Pain. 1997;72(3):407–415. doi: 10.1016/s0304-3959(97)00069-9. [DOI] [PubMed] [Google Scholar]
- 26.Zhang XC, et al. Activation of Central Trigeminovascular Neurons by Cortical Spreading Depression. Annals of Neurology. 2011;69(5):855–865. doi: 10.1002/ana.22329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang XC, et al. Activation of Meningeal Nociceptors by Cortical Spreading Depression: Implications for Migraine with Aura (June, pg 8807, 2010) Journal of Neuroscience. 2010;30(30):10259–10259. doi: 10.1523/JNEUROSCI.0511-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Karatas H, et al. Spreading Depression Triggers Headache by Activating Neuronal Panx1 Channels. Science. 2013;339(6123):1092–1095. doi: 10.1126/science.1231897. [DOI] [PubMed] [Google Scholar]
- 29.Noseda R, et al. Changes of meningeal excitability mediated by corticotrigeminal networks: a link for the endogenous modulation of migraine pain. J Neurosci. 2010;30(43):14420–9. doi: 10.1523/JNEUROSCI.3025-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fioravanti B, et al. Evaluation of cutaneous allodynia following induction of cortical spreading depression in freely moving rats. Cephalalgia. 2011;31(10):1090–1100. doi: 10.1177/0333102411410609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ray BS, Wolff HG. Experimental studies on headache - Pain-sensitive structures of the head and their significance in headache. Archives of Surgery. 1940;41(4):813–856. [Google Scholar]
- 32.Kaube H, Hoskin KL, Goadsby PJ. Activation of the trigeminovascular system by mechanical distension of the superior sagittal sinus in the cat. Cephalalgia. 1992;12(3):133–6. doi: 10.1046/j.1468-2982.1992.1203133.x. [DOI] [PubMed] [Google Scholar]
- 33.Levy D, Strassman AM. Mechanical response properties of A and C primary afferent neurons innervating the rat intracranial dura. J Neurophysiol. 2002;88(6):3021–31. doi: 10.1152/jn.00029.2002. [DOI] [PubMed] [Google Scholar]
- 34.Strassman AM, Raymond SA, Burstein R. Sensitization of meningeal sensory neurons and the origin of headaches. Nature. 1996;384(6609):560–4. doi: 10.1038/384560a0. [DOI] [PubMed] [Google Scholar]
- 35.Ahn AH. On the Temporal Relationship Between Throbbing Migraine Pain and Arterial Pulse. Headache. 2010;50(9):1507–1510. doi: 10.1111/j.1526-4610.2010.01765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mirza AF, et al. Is There a Relationship between Throbbing Pain and Arterial Pulsations? Journal of Neuroscience. 2012;32(22):7572–7576. doi: 10.1523/JNEUROSCI.0193-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mo J, et al. Does throbbing pain have a brain signature? Pain. 2013;154(7):1150–1155. doi: 10.1016/j.pain.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bove GM, Moskowitz MA. Primary afferent neurons innervating guinea pig dura. J Neurophysiol. 1997;77(1):299–308. doi: 10.1152/jn.1997.77.1.299. [DOI] [PubMed] [Google Scholar]
- 39.Edelmayer RM, et al. Activation of TRPA1 on dural afferents: A potential mechanism of headache pain. Pain. 2012;153(9):1949–58. doi: 10.1016/j.pain.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei X, et al. Activation of TRPV4 on dural afferents produces headache-related behavior in a preclinical rat model. Cephalalgia : an international journal of headache. 2011;31(16):1595–600. doi: 10.1177/0333102411427600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yan J, et al. Sensitization of dural afferents underlies migraine-related behavior following meningeal application of interleukin-6 (IL-6) Molecular pain. 2012;8:6. doi: 10.1186/1744-8069-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang XC, et al. Tumor necrosis factor-alpha induces sensitization of meningeal nociceptors mediated via local COX and p38 MAP kinase actions. Pain. 2011;152(1):140–149. doi: 10.1016/j.pain.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang XC, Burstein R, Levy D. Local action of the proinflammatory cytokines IL-1 beta and IL-6 on intracranial meningeal nociceptors. Cephalalgia. 2012;32(1):66–U109. doi: 10.1177/0333102411430848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vaughn AH, Gold MS. Ionic mechanisms underlying inflammatory mediator-induced sensitization of dural afferents. J Neurosci. 2010;30(23):7878–88. doi: 10.1523/JNEUROSCI.6053-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Harriott AM, Gold MS. Electrophysiological properties of dural afferents in the absence and presence of inflammatory mediators. J Neurophysiol. 2009;101(6):3126–34. doi: 10.1152/jn.91339.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reuter U, et al. Delayed inflammation in rat meninges: implications for migraine pathophysiology. Brain : a journal of neurology. 2001;124(Pt 12):2490–502. doi: 10.1093/brain/124.12.2490. [DOI] [PubMed] [Google Scholar]
- 47.Levy D. Migraine pain, meningeal inflammation, and mast cells. Curr Pain Headache Rep. 2009;13 (3):237–40. doi: 10.1007/s11916-009-0040-y. [DOI] [PubMed] [Google Scholar]
- 48.Dimlich RV, et al. Linear arrays of homogeneous mast cells in the dura mater of the rat. J Neurocytol. 1991;20(6):485–503. doi: 10.1007/BF01252276. [DOI] [PubMed] [Google Scholar]
- 49.Strassman AM, et al. Axon diameters and intradural trajectories of the dural innervation in the rat. J Comp Neurol. 2004;473(3):364–76. doi: 10.1002/cne.20106. [DOI] [PubMed] [Google Scholar]
- 50.Rozniecki JJ, et al. Morphological and functional demonstration of rat dura mater mast cell-neuron interactions in vitro and in vivo. Brain Res. 1999;849(1–2):1–15. doi: 10.1016/s0006-8993(99)01855-7. [DOI] [PubMed] [Google Scholar]
- 51.Zhang XC, Levy D. Modulation of meningeal nociceptors mechanosensitivity by peripheral proteinase-activated receptor-2: the role of mast cells. Cephalalgia. 2008;28(3):276–84. doi: 10.1111/j.1468-2982.2007.01523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang XC, et al. Sensitization and activation of intracranial meningeal nociceptors by mast cell mediators. J Pharmacol Exp Ther. 2007;322(2):806–12. doi: 10.1124/jpet.107.123745. [DOI] [PubMed] [Google Scholar]
- 53.Levy D, et al. Mast cell degranulation activates a pain pathway underlying migraine headache. Pain. 2007;130(1–2):166–76. doi: 10.1016/j.pain.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith JH, Butterfield JH, Cutrer FM. Primary headache syndromes in systemic mastocytosis. Cephalalgia. 2011;31(15):1522–1531. doi: 10.1177/0333102411421683. [DOI] [PubMed] [Google Scholar]
- 55.Eikermann-Haerter K, et al. Enhanced Subcortical Spreading Depression in Familial Hemiplegic Migraine Type 1 Mutant Mice. Journal of Neuroscience. 2011;31(15):5755–5763. doi: 10.1523/JNEUROSCI.5346-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eikermann-Haerter K, et al. Genetic and hormonal factors modulate spreading depression and transient hemiparesis in mouse models of familial hemiplegic migraine type 1. J Clin Invest. 2009;119(1):99–109. doi: 10.1172/JCI36059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chowdhury GMI, et al. Cortical spreading depression affects Fos expression in the hypothalamic paraventricular nucleus and the cerebral cortex of both hemispheres. Neuroscience Research. 2003;45 (2):149–155. doi: 10.1016/s0168-0102(02)00207-9. [DOI] [PubMed] [Google Scholar]
- 58.Monda M, et al. Cortical spreading depression reduces paraventricular activation induced by hippocampal neostigmine injection. Brain Research. 1999;824(1):119–124. doi: 10.1016/s0006-8993(99)01227-5. [DOI] [PubMed] [Google Scholar]
- 59.Shibata M, et al. Activity of Hypothalamic Thermosensitive Neurons during Cortical Spreading Depression in the Rat. Brain Research. 1984;308(2):255–262. doi: 10.1016/0006-8993(84)91064-3. [DOI] [PubMed] [Google Scholar]
- 60.Stovner LJ, Tronvik E, Hagen K. New drugs for migraine. J Headache Pain. 2009 doi: 10.1007/s10194-009-0156-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Goadsby PJ, et al. Towards a definition of intractable headache for use in clinical practice and trials. Cephalalgia. 2006;26(9):1168–70. doi: 10.1111/j.1468-2982.2006.01173.x. [DOI] [PubMed] [Google Scholar]
- 62.Waldmann R, et al. A proton-gated cation channel involved in acid-sensing. Nature. 1997;386(6621):173–7. doi: 10.1038/386173a0. [DOI] [PubMed] [Google Scholar]
- 63.Wemmie JA, Taugher RJ, Kreple CJ. Acid-sensing ion channels in pain and disease. Nature Reviews Neuroscience. 2013;14(7):461–471. doi: 10.1038/nrn3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Deval E, et al. Acid-Sensing Ion Channels (ASICs): Pharmacology and implication in pain. Pharmacology & Therapeutics. 2010;128(3):549–558. doi: 10.1016/j.pharmthera.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 65.Sherwood TW, Frey EN, Askwith CC. Structure and activity of the acid-sensing ion channels. American Journal of Physiology-Cell Physiology. 2012;303(7):C699–C710. doi: 10.1152/ajpcell.00188.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zha XM. Acid-sensing ion channels: trafficking and synaptic function. Molecular Brain. 2013;6 doi: 10.1186/1756-6606-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sakai H, et al. Cloning and functional expression of a novel degenerin-like Na+ channel gene in mammals. Journal of Physiology-London. 1999;519(2):323–333. doi: 10.1111/j.1469-7793.1999.0323m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schaefer L, et al. Molecular cloning, functional expression and chromosomal localization of an amiloride-sensitive Na+ channel from human small intestine. Febs Letters. 2000;471(2–3):205–210. doi: 10.1016/s0014-5793(00)01403-4. [DOI] [PubMed] [Google Scholar]
- 69.Wiemuth D, et al. BASIC-a bile acid-sensitive ion channel highly expressed in bile ducts. Faseb Journal. 2012;26(10):4122–4130. doi: 10.1096/fj.12-207043. [DOI] [PubMed] [Google Scholar]
- 70.Delaunay A, et al. Human ASIC3 channel dynamically adapts its activity to sense the extracellular pH in both acidic and alkaline directions. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(32):13124–13129. doi: 10.1073/pnas.1120350109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Grunder S, Chen X. Structure, function, and pharmacology of acid-sensing ion channels (ASICs): focus on ASIC1a. Int J Physiol Pathophysiol Pharmacol. 2010;2(2):73–94. [PMC free article] [PubMed] [Google Scholar]
- 72.Wemmie JA, et al. Acid-sensing ion channel 1 is localized in brain regions with high synaptic density and contributes to fear conditioning. Journal of Neuroscience. 2003;23(13):5496–5502. doi: 10.1523/JNEUROSCI.23-13-05496.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ziemann AE, et al. The amygdala is a chemosensor that detects carbon dioxide and acidosis to elicit fear behavior. Cell. 2009;139(5):1012–21. doi: 10.1016/j.cell.2009.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Du J, et al. Protons are a neurotransmitter that regulates synaptic plasticity in the lateral amygdala. Proc Natl Acad Sci U S A. 2014 doi: 10.1073/pnas.1407018111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang YZ, Xu TL. Acidosis, acid-sensing ion channels, and neuronal cell death. Mol Neurobiol. 2011;44(3):350–8. doi: 10.1007/s12035-011-8204-2. [DOI] [PubMed] [Google Scholar]
- 76.Nedergaard M, et al. Dynamics of Interstitial and Intracellular Ph in Evolving Brain Infarct. American Journal of Physiology. 1991;260(3):R581–R588. doi: 10.1152/ajpregu.1991.260.3.R581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Xiong ZG, et al. Neuroprotection in ischemia: Blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118(6):687–698. doi: 10.1016/j.cell.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 78.Pignataro G, Simon RP, Xiong ZG. Prolonged activation of ASIC1a and the time window for neuroprotection in cerebral ischaemia. Brain. 2007;130:151–158. doi: 10.1093/brain/awl325. [DOI] [PubMed] [Google Scholar]
- 79.Xiong ZG, et al. Acid-sensing ion channels (ASICs) as pharmacological targets for neurodegenerative diseases. Current Opinion in Pharmacology. 2008;8(1):25–32. doi: 10.1016/j.coph.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lv RJ, et al. ASIC1a polymorphism is associated with temporal lobe epilepsy. Epilepsy Research. 2011;96(1–2):74–80. doi: 10.1016/j.eplepsyres.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 81.Miesenbock G, De Angelis DA, Rothman JE. Visualizing secretion and synaptic transmission with pH-sensitive green fluorescent proteins. Nature. 1998;394(6689):192–195. doi: 10.1038/28190. [DOI] [PubMed] [Google Scholar]
- 82.Duan B, et al. Upregulation of acid-sensing ion channel ASIC1a in spinal dorsal horn neurons contributes to inflammatory pain hypersensitivity. Journal of Neuroscience. 2007;27(41):11139–11148. doi: 10.1523/JNEUROSCI.3364-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wu LJ, et al. Characterization of acid-sensing ion channels in dorsal horn neurons of rat spinal cord. Journal of Biological Chemistry. 2004;279(42):43716–43724. doi: 10.1074/jbc.M403557200. [DOI] [PubMed] [Google Scholar]
- 84.Mazzuca M, et al. A tarantula peptide against pain via ASIC1a channels and opioid mechanisms. Nature Neuroscience. 2007;10(8):943–945. doi: 10.1038/nn1940. [DOI] [PubMed] [Google Scholar]
- 85.Diochot S, et al. Black mamba venom peptides target acid-sensing ion channels to abolish pain. Nature. 2012;490(7421):552–+. doi: 10.1038/nature11494. [DOI] [PubMed] [Google Scholar]
- 86.Duan B, et al. PI3-kinase/Akt Pathway-Regulated Membrane Insertion of Acid-Sensing Ion Channel 1a Underlies BDNF-Induced Pain Hypersensitivity. Journal of Neuroscience. 2012;32(18):6351–6363. doi: 10.1523/JNEUROSCI.4479-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Krishtal OA, Pidoplichko VI. A Receptor for Protons in the Nerve-Cell Membrane. Neuroscience. 1980;5(12):2325–2327. doi: 10.1016/0306-4522(80)90149-9. [DOI] [PubMed] [Google Scholar]
- 88.Alvarez de la Rosa D, et al. Functional implications of the localization and activity of acid- sensitive channels in rat peripheral nervous system. Proc Natl Acad Sci U S A. 2002;99(4):2326–31. doi: 10.1073/pnas.042688199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jones NG, et al. Acid-induced pain and its modulation in humans. J Neurosci. 2004;24(48):10974–9. doi: 10.1523/JNEUROSCI.2619-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ugawa S, et al. Amiloride-blockable acid-sensing ion channels are leading acid sensors expressed in human nociceptors. J Clin Invest. 2002;110(8):1185–90. doi: 10.1172/JCI15709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wu WL, et al. Targeting ASIC3 for pain, anxiety, and insulin resistance. Pharmacology & Therapeutics. 2012;134(2):127–138. doi: 10.1016/j.pharmthera.2011.12.009. [DOI] [PubMed] [Google Scholar]
- 92.Jones RC, 3rd, Xu L, Gebhart GF. The mechanosensitivity of mouse colon afferent fibers and their sensitization by inflammatory mediators require transient receptor potential vanilloid 1 and acid-sensing ion channel 3. J Neurosci. 2005;25(47):10981–9. doi: 10.1523/JNEUROSCI.0703-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Page AJ, et al. Different contributions of ASIC channels 1a, 2, and 3 in gastrointestinal mechanosensory function. Gut. 2005;54(10):1408–15. doi: 10.1136/gut.2005.071084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Molliver DC, et al. ASIC3, an acid-sensing ion channel, is expressed in metaboreceptive sensory neurons. Mol Pain. 2005;1:35. doi: 10.1186/1744-8069-1-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sutherland SP, et al. Acid-sensing ion channel 3 matches the acid-gated current in cardiac ischemia-sensing neurons. Proc Natl Acad Sci U S A. 2001;98(2):711–6. doi: 10.1073/pnas.011404498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Ikeuchi M, Kolker SJ, Sluka KA. Acid-Sensing Ion Channel 3 Expression in Mouse Knee Joint Afferents and Effects of Carrageenan-Induced Arthritis. Journal of Pain. 2009;10(3):336–342. doi: 10.1016/j.jpain.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ikeuchi M, et al. Role of ASIC3 in the primary and secondary hyperalgesia produced by joint inflammation in mice. Pain. 2008;137(3):662–669. doi: 10.1016/j.pain.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sluka KA, et al. Chronic hyperalgesia induced by repeated acid injections in muscle is abolished by the loss of ASIC3, but not ASIC1. Pain. 2003;106(3):229–239. doi: 10.1016/S0304-3959(03)00269-0. [DOI] [PubMed] [Google Scholar]
- 99.Price MP, et al. The DRASIC cation channel contributes to the detection of cutaneous touch and acid stimuli in mice. Neuron. 2001;32(6):1071–1083. doi: 10.1016/s0896-6273(01)00547-5. [DOI] [PubMed] [Google Scholar]
- 100.Baron A, et al. Venom toxins in the exploration of molecular, physiological and pathophysiological functions of acid-sensing ion channels. Toxicon. 2013;75:187–204. doi: 10.1016/j.toxicon.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 101.Deval E, et al. Acid-Sensing Ion Channels in Postoperative Pain. Journal of Neuroscience. 2011;31 (16):6059–6066. doi: 10.1523/JNEUROSCI.5266-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yu Y, et al. A nonproton ligand sensor in the acid-sensing ion channel. Neuron. 2010;68(1):61–72. doi: 10.1016/j.neuron.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 103.Li WG, et al. ASIC3 Channels Integrate Agmatine and Multiple Inflammatory Signals through the Nonproton Ligand Sensing Domain. Molecular Pain. 2010;6 doi: 10.1186/1744-8069-6-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Alijevic O, Kellenberger S. Subtype-specific Modulation of Acid-sensing Ion Channel (ASIC) Function by 2-Guanidine-4-methylquinazoline. Journal of Biological Chemistry. 2012;287(43):36059–36070. doi: 10.1074/jbc.M112.360487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Smith RN, Gonzales EB. Protons and Psalmotoxin-1 reveal nonproton ligand stimulatory sites in chicken acid-sensing ion channel: Implication for simultaneous modulation in ASICs. Channels (Austin) 2014;8(1):49–61. doi: 10.4161/chan.26978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bohlen CJ, et al. A heteromeric Texas coral snake toxin targets acid-sensing ion channels to produce pain. Nature. 2011;479(7373):410–U167. doi: 10.1038/nature10607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mamet J, et al. Proinflammatory mediators, stimulators of sensory neuron excitability via the expression of acid-sensing ion channels. J Neurosci. 2002;22(24):10662–70. doi: 10.1523/JNEUROSCI.22-24-10662.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang X, et al. Serotonin facilitates peripheral pain sensitivity in a manner that depends on the nonproton ligand sensing domain of ASIC3 channel. J Neurosci. 2013;33(10):4265–79. doi: 10.1523/JNEUROSCI.3376-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hamel E. Serotonin and migraine: biology and clinical implications. Cephalalgia : an international journal of headache. 2007;27(11):1293–300. doi: 10.1111/j.1468-2982.2007.01476.x. [DOI] [PubMed] [Google Scholar]
- 110.Panconesi A. Serotonin and migraine: a reconsideration of the central theory. The journal of headache and pain. 2008;9(5):267–76. doi: 10.1007/s10194-008-0058-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dussor G. Serotonin, 5HT1 agonists, and migraine: new data, but old questions still not answered. Curr Opin Support Palliat Care. 2014;8(2):137–42. doi: 10.1097/SPC.0000000000000044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sarchielli P, et al. Levels of nerve growth factor in cerebrospinal fluid of chronic daily headache patients. Neurology. 2001;57(1):132–4. doi: 10.1212/wnl.57.1.132. [DOI] [PubMed] [Google Scholar]
- 113.Olesen J. The role of nitric oxide (NO) in migraine, tension-type headache and cluster headache. Pharmacol Ther. 2008;120(2):157–71. doi: 10.1016/j.pharmthera.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 114.Cadiou H, et al. Modulation of acid-sensing ion channel activity by nitric oxide. J Neurosci. 2007;27 (48):13251–60. doi: 10.1523/JNEUROSCI.2135-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wang JQ, et al. Modulation of ionotropic glutamate receptors and Acid-sensing ion channels by nitric oxide. Front Physiol. 2012;3:164. doi: 10.3389/fphys.2012.00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ueno S, Nakaye T, Akaike N. Proton-Induced Sodium Current in Freshly Dissociated Hypothalamic Neurons of the Rat. Journal of Physiology-London. 1992;447:309–327. doi: 10.1113/jphysiol.1992.sp019004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Huang RQ, Dillon GH. Effect of extracellular pH on GABA-activated current in rat recombinant receptors and thin hypothalamic slices. Journal of Neurophysiology. 1999;82(3):1233–1243. doi: 10.1152/jn.1999.82.3.1233. [DOI] [PubMed] [Google Scholar]
- 118.Chen ZL, Huang RQ. Extracellular pH modulates GABAergic neurotransmission in rat hypothalamus. Neuroscience. 2014;271:64–76. doi: 10.1016/j.neuroscience.2014.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cannizzaro C, et al. [H-3]-DA release evoked by low pH medium and internal H+ accumulation in rat hypothalamic synaptosomes: involvement of calcium ions. Neurochemistry International. 2003;43 (1):9–17. doi: 10.1016/s0197-0186(02)00211-5. [DOI] [PubMed] [Google Scholar]
- 120.Meng QY, et al. Distribution of acid-sensing ion channel 3 in the rat hypothalamus. Neuroscience. 2009;159(3):1126–34. doi: 10.1016/j.neuroscience.2009.01.069. [DOI] [PubMed] [Google Scholar]
- 121.Wang W, Yu Y, Xu TL. Modulation of acid-sensing ion channels by Cu(2+) in cultured hypothalamic neurons of the rat. Neuroscience. 2007;145(2):631–41. doi: 10.1016/j.neuroscience.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 122.Chen CH, et al. Acid-sensing ion channels in neurones of the rat suprachiasmatic nucleus. J Physiol. 2009;587(Pt 8):1727–37. doi: 10.1113/jphysiol.2008.166918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Ohbuchi T, et al. Acid-sensing ion channels in rat hypothalamic vasopressin neurons of the supraoptic nucleus. J Physiol. 2010;588(Pt 12):2147–62. doi: 10.1113/jphysiol.2010.187625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yanovsky Y, et al. Proton- and ammonium-sensing by histaminergic neurons controlling wakefulness. Front Syst Neurosci. 2012;6:23. doi: 10.3389/fnsys.2012.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Song N, et al. Acid sensing ion channel 1 in lateral hypothalamus contributes to breathing control. PLoS One. 2012;7(7):e39982. doi: 10.1371/journal.pone.0039982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nozari A, et al. Microemboli May Link Spreading Depression, Migraine Aura, and Patent Foramen Ovale. Annals of Neurology. 2010;67(2):221–229. doi: 10.1002/ana.21871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Takano T, et al. Cortical spreading depression causes and coincides with tissue hypoxia. Nat Neurosci. 2007;10(6):754–62. doi: 10.1038/nn1902. [DOI] [PubMed] [Google Scholar]
- 128.Chang JC, et al. A mathematical model of the metabolic and perfusion effects on cortical spreading depression. PLoS One. 2013;8(8):e70469. doi: 10.1371/journal.pone.0070469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Chang JC, et al. Biphasic direct current shift, haemoglobin desaturation and neurovascular uncoupling in cortical spreading depression. Brain. 2010;133(Pt 4):996–1012. doi: 10.1093/brain/awp338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Piilgaard H, Lauritzen M. Persistent increase in oxygen consumption and impaired neurovascular coupling after spreading depression in rat neocortex. J Cereb Blood Flow Metab. 2009;29 (9):1517–27. doi: 10.1038/jcbfm.2009.73. [DOI] [PubMed] [Google Scholar]
- 131.Holland PR, et al. Acid-sensing ion channel 1: a novel therapeutic target for migraine with aura. Ann Neurol. 2012;72(4):559–63. doi: 10.1002/ana.23653. [DOI] [PubMed] [Google Scholar]
- 132.Burstein R. Deconstructing migraine headache into peripheral and central sensitization. Pain. 2001;89 (2–3):107–10. doi: 10.1016/s0304-3959(00)00478-4. [DOI] [PubMed] [Google Scholar]
- 133.Burstein R, et al. Chemical stimulation of the intracranial dura induces enhanced responses to facial stimulation in brain stem trigeminal neurons. J Neurophysiol. 1998;79(2):964–82. doi: 10.1152/jn.1998.79.2.964. [DOI] [PubMed] [Google Scholar]
- 134.Schepelmann K, et al. Response properties of trigeminal brain stem neurons with input from dura mater encephali in the rat. Neuroscience. 1999;90(2):543–54. doi: 10.1016/s0306-4522(98)00423-0. [DOI] [PubMed] [Google Scholar]
- 135.Durham PL, Masterson CG. Two mechanisms involved in trigeminal CGRP release: implications for migraine treatment. Headache. 2013;53(1):67–80. doi: 10.1111/j.1526-4610.2012.02262.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zimmermann K, Reeh PW, Averbeck B. ATP can enhance the proton-induced CGRP release through P2Y receptors and secondary PGE(2) release in isolated rat dura mater. Pain. 2002;97(3):259–65. doi: 10.1016/S0304-3959(02)00027-1. [DOI] [PubMed] [Google Scholar]
- 137.Yan J, et al. Dural afferents express acid-sensing ion channels: a role for decreased meningeal pH in migraine headache. Pain. 2011;152(1):106–13. doi: 10.1016/j.pain.2010.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Yan J, et al. pH-Evoked Dural Afferent Signaling Is Mediated by ASIC3 and Is Sensitized by Mast Cell Mediators. Headache. 2013 doi: 10.1111/head.12152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.De Young MB, Nemeth EF, Scarpa A. Measurement of the internal pH of mast cell granules using microvolumetric fluorescence and isotopic techniques. Arch Biochem Biophys. 1987;254(1):222–33. doi: 10.1016/0003-9861(87)90098-1. [DOI] [PubMed] [Google Scholar]
- 140.Blanchard MG, Rash LD, Kellenberger S. Inhibition of voltage-gated Na(+) currents in sensory neurones by the sea anemone toxin APETx2. Br J Pharmacol. 2012;165(7):2167–77. doi: 10.1111/j.1476-5381.2011.01674.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Baconguis I, Hattori M, Gouaux E. Unanticipated parallels in architecture and mechanism between ATP-gated P2X receptors and acid sensing ion channels. Current Opinion in Structural Biology. 2013;23(2):277–284. doi: 10.1016/j.sbi.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Durham P, Papapetropoulos S. Biomarkers associated with migraine and their potential role in migraine management. Headache. 2013;53(8):1262–77. doi: 10.1111/head.12174. [DOI] [PubMed] [Google Scholar]
- 143.Harrington MG. Cerebrospinal fluid biomarkers in primary headache disorders. Headache. 2006;46 (7):1075–87. doi: 10.1111/j.1526-4610.2006.00501.x. [DOI] [PubMed] [Google Scholar]
- 144.Reyngoudt H, Achten E, Paemeleire K. Magnetic resonance spectroscopy in migraine: what have we learned so far? Cephalalgia. 2012;32(11):845–59. doi: 10.1177/0333102412452048. [DOI] [PubMed] [Google Scholar]
- 145.Magnotta VA, et al. Detecting activity-evoked pH changes in human brain. Proc Natl Acad Sci U S A. 2012;109(21):8270–3. doi: 10.1073/pnas.1205902109. [DOI] [PMC free article] [PubMed] [Google Scholar]