

Abstract
Posttranscriptional regulation of RNA is an important mechanism for activating and resolving cellular stress responses. Poly(ADP-ribose) Polymerase-13 (PARP13), also known as ZC3HAV1 and Zinc-finger Antiviral Protein (ZAP), is an RNA-binding protein that regulates the stability, and translation of specific mRNAs, and modulates the miRNA silencing pathway to globally impact miRNA targets. These functions of PARP13 are important components of the cellular response to stress. In addition, the ability of PARP13 to restrict oncogenic viruses and to repress the pro-survival cytokine receptor TRAILR4 suggests that it can be protective against malignant transformation and cancer development. The relevance of PARP13 to human health and disease make it a promising therapeutic target.
Keywords: PARP13, ZAP, posttranscriptional RNA regulation, innate immune response, cancer
Stress responses, posttranscriptional RNA regulation and human health
The primary function of the poly(ADP-ribose) polymerase family of proteins (PARPs) is to modify target proteins with ADP-ribose using NAD+ as substrate. [1, 2]. ADP-ribosylation changes the function of protein targets, and can result in the recruitment of ADP-ribose binding proteins. PARPs regulate fundamental cellular processes and help initiate and maintain specific stress responses including the antiviral response, DNA damage, heat shock, cytoplasmic stress, and unfolded protein stress [3–7]. Cellular stress responses act as guardians against disease by allowing cells to rapidly respond to pathogens or unfavorable internal or external conditions that may otherwise lead to long-term pathologies. They are therefore important components of disease prevention including diseases such as viral infection and cancer [8–11].
Unfortunately, cellular stress responses can also have harmful effects on the cells and the organism as a whole. Activation of stress-response pathways in the absence of stressors, or their over activation in response to stressors can have detrimental consequences and can result in pathologies such as autoimmune or neurodegenerative diseases [12]. In addition, diseased tissues such as tumor cells can utilize stress responses to their advantage, allowing them to proliferate in the adverse conditions present in their microenvironment including hypoxia, nutrient starvation and inflammation [8, 9]. Gain of function mutations in stress response genes and co-opting stress response pathways to promote growth and survival are common adaptation mechanisms in cancers. For example, an important link has been established between inflammation and cancer progression (Box 1) [13]. Thus, tight control of the activation and inactivation of stress response programs is of critical importance.
Box 1. Viruses, inflammation and cancer.
Viral infection and inflammation play significant roles in the development of cancer. Many viruses promote or accelerate cancer progression. Although viruses primarily use host cells to replicate, this can inadvertently lead to cell transformation. Viruses can target a diverse range of host proteins to increase proliferation: for example the Human Papillomavirus (HPV) proteins E6 and E7 promote protein synthesis by activating mTOR [102]; numerous viral proteins including the Simian Virus 40 (SV40) large T protein or the Hepatitis B (HBV) X protein inactivate the tumor suppressors p53 [103, 104] and retinoblastoma (Rb) [105, 106], both of which prevent cancer, or the Epstein Barr Virus (EBV) protein LMP1 promotes survival and transformation by activating NFkB [107].
Viruses can also indirectly contribute to cancer by inducing inflammation at sites of infection. The release of cytokines by activated immune cells enables an efficient antiviral response by promoting the proliferation of immune cells in the infected area. However, chronic inflammation can also lead to increased proliferation of the surrounding epithelial cells leading to oncogenic transformation. Inflammatory conditions are also induced by non-viral stimuli, including bacterial infection, chemicals such as silica and asbestos, smoking, alcohol or UV induced DNA damage. Unresolved chronic inflammation can eventually give rise to cancer due to its highly proliferative microenvironment. Therefore the immune response must be efficiently shut off once the initial infection has been resolved.
The activation of cellular stress responses involves a coordinated switch in global gene expression both at the level of transcription, and at the posttranscriptional level [14]. The initial response is largely posttranscriptional [15]. Many stress response genes are actually constitutively transcribed, and their expression is suppressed through posttranscriptional mechanisms since expression in healthy cells would be harmful [16, 17]. In response to stress, the cell inhibits the active suppression of these stress response transcripts, resulting in a rapid increase in transcript levels and expression, and a rapid stress response without the need for de novo transcription [15]. This mechanism of regulation allows the cell to quickly establish an environment hostile to pathogens and favorable for cell repair. Once the stress is eliminated, suppression of the stress response transcripts is restored, facilitating the resolution of the stress response and return to steady state [14, 18].
The cell utilizes multiple mechanisms of posttranscriptional regulation during stress responses. Stress-dependent changes in the posttranscriptional regulation of RNAs include: (i) modulation of RNA decay, resulting in the stabilization of stress response transcripts, and destabilization of pathogenic RNAs if present; (ii) changes in translation rates that favor the expression of stress response genes; and (iii) alteration of microRNA silencing activity which results in the derepression of stress response transcripts, increasing their expression [18–22]. Together, these mechanisms result in the decreased expression of non-stress related transcripts and simultaneous increase in the expression of stress response transcripts. They also trigger the assembly of cytoplasmic and nuclear ribonucleoprotein particles such as stress granules and stress granule-like structures [23, 24]. Stress granules function as storage sites of stalled preinitiation complexes, RNA binding proteins, and mRNA and assemble as a result of stress-induced translational repression (see Glossary) [23, 24]. It is now thought that stress granules themselves also modulate signaling pathways in the cell during stress [24]. In total, stress dependent changes in posttranscriptional regulation of RNAs offer the potential for an immediate and global response to stress.
PARP13 is an RNA binding PARP that was initially identified in a screen for antiviral proteins [6]. It plays important roles in the posttranscriptional regulation of RNAs, particularly during stress conditions including viral infection and the general cytoplasmic stress response. PARP13 regulates each of the posttranscriptional pathways of RNA regulation that are altered during cellular stress responses: it targets specific cellular and viral RNAs for decay and translational repression [25–29], and alters the activity of the microRNA-silencing pathway [4, 22]. In addition, during cytoplasmic stress, PARP13 localizes to, and facilitates the assembly of, stress granules (along with PARP5a, PARP14, PARP15 and PARP12), recently recognized as a potent suppressors of general translation [4, 30, 31]. Thus, PARP13 acts as an important antiviral, pro-apoptotic and pro-inflammatory factor, and is a component of the TRAIL mediated immune response to cancer. As such PARP13 is a promising target for the treatment of multiple disease states including viral infections, autoimmune diseases and cancers. In this review we summarize the functions of PARP13 relevant to human health and disease, and discuss possible therapeutic approaches to regulate PARP13 function.
The ins and outs of PARP13
PARP13 (also known as Zinc-finger Antiviral Protein (ZAP)/ZC3HAV1) is a member of the CCCH-PARP subfamily; PARPs that contain one or more RNA-binding CCCH-type zinc-finger domains [2, 32]. Similar to the other CCCH PARPs, PARP12 and PARP7, PARP13 also contains a WWE domain, shown in other proteins to bind poly(ADP-ribose) (Figure 1) [33, 34]. Two major isoforms of PARP13, resulting from alternative splicing, are found in humans: the full length PARP13.1, and the truncated PARP13.2 [2, 35] (Figure 1). Both isoforms lack PARP activity and are unable to ADP-ribosylate target proteins; the catalytic domain of PARP13.1 lacks amino acid residues required for ADP-ribosylation activity, whereas PARP13.2 completely lacks a PARP domain. Three additional isoforms have been predicted based on sequence analyses but their expression in humans has not been experimentally verified (Uniprot) (Figure 1). PARP13 is unique among PARPs as it is the only catalytically inactive PARP shown to be targeted for ADP-ribosylation by other PARPs [2, 4, 33, 34, 36].
Figure 1. PARP13 domain structure.
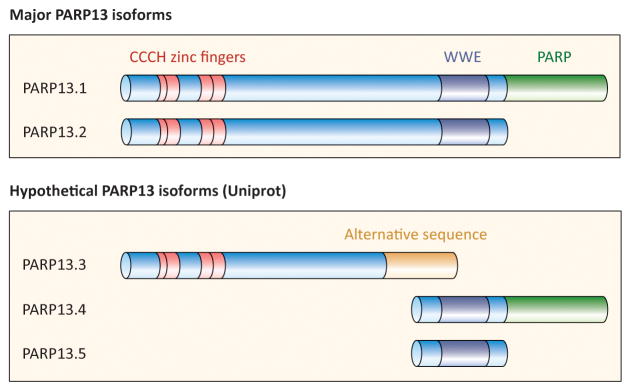
PARP13.1 and PARP13.2 are the two major isoforms of PARP13 in humans. Both contain four CCCH-type RNA-binding zinc fingers (red) and a WWE domain predicted to bind poly(ADP-ribose) (dark blue). PARP13.1 also contains a catalytically inactive PARP domain (green). Three additional predicted isoforms of PARP13 are shown below.
The four CCCH zinc finger domains found in the N-terminus of PARP13 bind target RNAs with high specificity [37]. The zinc fingers form two RNA-interacting clefts that provide specificity and selectivity of binding, and allow recognition of and interaction with looped RNA [38]. Therefore PARP13 is thought to bind to tridimensional structures within its cognate RNAs, rather than to a linear motif [38]. In addition, PARP13 can form a dimer, potentially increasing the complexity of the RNA-interaction surface and the specificity of RNA binding [38, 39]. While PARP13 can bind certain transcripts, it lacks nuclease or helicase activity, and cannot directly affect the integrity or structure of its RNA targets. Instead, PARP13 functions as a recruiting factor, bringing destabilizing factors, and likely other effector proteins, to the transcripts it binds, leading to their destabilization, and in certain cases decreasing the rate of their translation [25–28].
Posttranscriptional RNA regulation and disease
Changes in posttranscriptional RNA regulation occur during the initial stage of cellular stress responses. Increases in the half-lives of stress response transcripts and global inhibition of translation, accompanied by specific translation of specialized proteins helps to establish an antiviral state [20, 40–42]. Resolution of the stress response and return to steady state also relies on posttranscriptional regulation of RNA [19, 41]. Given the important role posttranscriptional RNA regulation plays in both initiating and abolishing the stress response, misregulation of any of the posttranscriptional mechanisms that regulate the expression of stress response genes can result in disease. Below we discuss the three major mechanisms of posttranscriptional RNA regulation in detail and describe how misregulation of any of these processes is linked to disease.
RNA decay
RNA decay has two major functions: (i) quality control and elimination of defective transcripts; and (ii) regulating the abundance of specific transcripts in order to control their expression [43]. This review focuses on the second of these functions. For more information onRNA quality control, or aspects of nonsense mediated decay, refer to [43, 44].
RNA decay is initiated by interactions of specialized RNA-binding proteins with specific target mRNAs. It involves removal of the 3′ poly-A tail by deadenylases such as the CCR4-NOT complex, poly(A) nuclease (PAN) and the poly(A)-specific ribonuclease (PARN), and removal of the 5′ 7-methyl guanine cap structure by the decapping enzymes DCP1 and DCP2 (Figure 2a) [43, 45]. The 5′-3′ exoribonuclease XRN1 or the 3′-5′ exonuclease complex, the RNA exosome, degrade the remaining transcript. RNA binding proteins bound to cis-acting elements found in the 3′UTRs of mRNA transcripts facilitate or prevent the binding of RNA decay factors, resulting in degradation or stabilization of the RNA, respectively (Figure 2a). Both endogenous and viral transcripts are targeted by this mechanism. RNA decay is often compartmentalized in non-membrane-bound structures called P-bodies, and recent evidence suggests that RNA decay components can also be enriched in specialized RNA granules during viral infection [46–49].
Figure 2. Mechanisms of RNA regulation.

a) RNA decay. Cis-regulatory elements found within an RNA sequence (usually within the 3′UTR) mediate binding of a destabilizing RNA-binding protein (RBP). The RBP can recruit specific RNA decay factors (arrows), but RNA decay usually initiates with removal of the poly(A) tail by deadenylases PAN, PARN or the CCR4-NOT complex. Removal of the poly(A) tail recruits decapping enzymes, including DCP1 and DCP2, which remove the 5′ methyl-guanine cap structure. The now unprotected RNA is degraded by the processive 5′-3′ exonuclease, XRN1, or 3′-5′ exonuclease, the exosome complex. Decay factors recruited specifically by PARP13 are boxed in red. An indent shows the structure of the exosome complex, with EXOSC1-EXOSC9 subunits identified. Arrow points to EXOSC7, the subunit that binds PARP13 in human.. b) Translation initiation and translational repression. Translation initiation begins by activating the mRNA via binding of the cap-binding factor eIF4E, the scaffold protein eIF4G and the helicase eIF4A. Interactions between eIF4G and PABP are required to stabilize the complex which then recruits the 43S complex, consisting of the small ribosomal subunit, loaded with the ternary complex (Met-tRNA and GTP bound eIF2), and additional initiation factors. The 43S-RNA complex is then competent for translation initiation, scanning and 60S subunit joining. T-bars show steps of translation initiation known to be inhibited by RNA-binding proteins. PARP13 represses translation by binding eIF4A and preventing it from interacting with eIF4G, a step required for cap-dependent translation initiation. c) miRNA silencing. Left, RISC complex loaded with miRNA silences its target by repressing translation and recruiting deadenylation factors, thus initiating decay. Right, PARP13 targets Argonaute (Ago), a component of the RISC complex, for poly(ADP-ribos)ylation by another PARP protein. Modification of Ago inhibits its function, likely by decreasing the binding affinity of the Ago-miRNA complex for the target mRNA. This results in global repression of miRNA silencing and increased expression of miRNA targets.
RNA decay plays important roles during both physiological conditions and during stress and disease. In healthy cells RNA decay helps regulate the levels of stress-response genes, cytokines, growth factors and pro-apoptotic factors [16]. This mechanism ensures that the expression of these transcripts does not exceed tolerable levels, and that their expression can be rapidly induced by inhibiting their decay [15, 41, 50, 51].
During cell stress, RNA decay is modulated through multiple mechanisms. For example, the RNA-destabilizing activity of Tristetraprolin (TTP), a key regulatory protein for mRNA decay, is inhibited by posttranslational modification during the initial stages of inflammation, leading to an accumulation of cellular transcripts encoding inflammatory cytokines, and their increased expression [19]. When inhibition of TTP is reversed, cytokine mRNA decay is upregulated resulting in the resolution phase of the response [19]. This so called “peaked” response allows for a rapid increase of cytokine expression followed by an equally rapid decrease in their expression facilitating the quick resolution of inflammatory signaling [18].
Misregulation of RNA decay can have important disease consequences. For example, loss of TTP function creates an imbalance in inflammatory signaling and leads to uncontrolled expression of inflammatory cytokines, resulting in chronic inflammation and conditions such as cachexia and arthritis [40]. Loss of TTP is also common in breast cancer and correlates with infiltration of monocytes and macrophages into the tumor; a poor prognostic indicative of increased local inflammatory signaling [52, 53]. Another RNA-regulatory protein, AUF1, is upregulated in human hepatocellular carcinomas [54], and increased levels of the RNA-stabilizing factor HuR have been found in multiple human cancer samples, including breast, lung and ovarian [55].
Translational repression
The upregulation of stress-response gene expression is often facilitated by a switch from cap-dependent to cap-independent translation initiation [15, 20, 56]. Cap-dependent initiation of translation is a multistep process (Figure 2b) that requires the recognition of the 5′ cap of mRNAs by a complex consisting of the cap-binding factor eIF4E, the scaffold protein eIF4G, and the helicase eIF4A [42]. Interactions between eIF4G and the poly(A)-binding protein (PABP) stabilize the complex and allow it to recruit the 43S subunit, which includes the 40s ribosomal subunit, the ternary complex, composed of Met-tRNA, and the eIF2 complex bound to GTP, and additional initiation factors (Figure 2b). Once bound, the 43S subunit begins scanning the mRNA and upon recognition of a start codon allows the joining of the 60S ribosomal subunit to initiate elongation. During physiological conditions, RNA-binding proteins that repress the translation of specific mRNAs often target eIF4E function by either inhibiting its binding to the 5′cap or by interfering with its binding to eIF4G [42, 57]. More rarely, later steps in the initiation process can also be targeted.
The regulation of translation initiation is an important component of stress responses. During stress, phosphorylation of eIF2α, a subunit of eIF2, globally inhibits cap-dependent translation, blocking general protein synthesis [20, 56]. However, specific stress-response factors continue to be translated through cap-independent internal ribosome entry site (IRES) initiation. The expression of several stress response transcripts is regulated in this manner. IRES-containing RNAs can for instance promote proliferation (c-Myc), inhibit apoptosis (XIAP) or stimulate angiogenesis (HIF1, VEGF) [58]. Global translational repression also induces stress granules, which can themselves alter cell signaling by sequestering signaling factors, and have been shown to be protective against cell death [23, 24]. Translational repression is usually transient and is abolished upon resolution of the stress [59].
Escape from translational repression is one mechanism by which pathogens evade inhibition through the cellular stress response. Multiple viruses employ IRES elements to avoid the general translational repression triggered during infection while others have evolved mechanism to inhibit eIF2α phosphorylation, allowing for the production of viral proteins [60, 61]. Prolonged inhibition of protein synthesis and sustained IRES-dependent translation of cellular transcripts can be detrimental as it can facilitate the ability of tumors to survive and proliferate in the stress-inducing tumor microenvironment [56, 62].
microRNA silencing
MicroRNAs (miRNAs) are small non-coding RNAs that bind to specific mRNAs through base-pairing with sequences found in the 3′UTR of the transcripts and, by doing so, suppress the expression and abundance of their targets [51, 63, 64]. Gene silencing by miRNAs is therefore an important component of posttranscriptional RNA regulation and is essential for optimizing gene expression programs [65]. In addition, misregulation of miRNAs can result in pathologies and correlates with many diseases, including cancer.
miRNAs repress target mRNAs through the activity of a ribonucleoprotein complex called the RNA-induced Silencing Complex (RISC) [51, 63, 64]. Two proteins, Argonaute (AGO) and Glycine-Tryptophan Protein 182 (GW182), constitute the core components of the complex. In humans there are four AGO proteins (AGO1-4), all of which confer silencing upon base-pairing between miRNAs and their targets. AGO, loaded with a miRNA, mediates target mRNA recognition and binding, while GW182 initiates target decay [66]. In addition RISC induces translational repression of its targets (Figure 2c).
Changes in miRNA silencing activity can facilitate rapid and in some cases global changes in gene expression programs. For example, under physiological conditions, miRNA silencing represses the expression of antiviral, but potentially cytotoxic genes [67]. During stress and infection, the activity of the miRNA pathway is itself repressed, allowing for the rapid upregulation of these targets which helps to establish an antiviral stress state in the cell [22].
Not surprisingly, misregulation of miRNA silencing can have widespread physiological effects and has the potential to result in disease. General loss of miRNA silencing is associated with cancers, while specific miRNAs can act as tumor suppressors (e.g. let7g in lung cancer models) or as oncogenes (e.g. miR-17-92 in lymphoma models; reviewed in [68]). One miRNA from the mir-17-92 cluster, miR-19, induces inflammation and NF-kB signaling [69], which is consistent with the link between chronic inflammation and cancer.
PARP13 and posttranscriptional regulation of RNAs
PARP13 is an important regulator of each of the three mechanisms of posttranscriptional RNA regulation described above. By regulating the miRNA pathway, PARP13 promotes the rapid initiation of the cellular antiviral stress responses. It also directly targets specific pathogenic and cellular RNAs for decay, and can repress translation of some viral RNAs. In addition, PARP13 facilitates the assembly of stress granules and viral RNA granules, with potential global effects on posttranscriptional gene expression. Proper regulation of PARP13 function is therefore important for human health and disease.
PARP13 destabilizes target RNAs by binding to defined regions within the transcripts, and recruiting RNA decay factors to initiate their degradation (Figure 2a). In humans, it recruits the exosome complex by interacting with the exosome component EXOSC7 and binds to and engages PARN [27], promoting 3′-5′ decay of target RNAs (Figure 2a) [25, 27]. PARP13 can also indirectly recruit 5′-3′ decay factors XRN1, DCP1 and DCP2 through the DEAD-box RNA helicase DDX17, and initiate 5′-3′ mRNAs decay [27].
PARP13 also regulates the translation efficiency of specific RNAs and plays a role in general translation inhibition during the cellular stress response. It represses the translation of specific targets by binding the translation-initiation factor eIF4A, and preventing it from interacting with the scaffold protein eIF4G (Figure 2b) [28]. This blocks an early step in translation initiation and inhibits expression of the target mRNA. High levels of PARP13 expression can also induce global translational repression by triggering de-novo stress granule assembly [4]. Similarly, during infection PARP13 facilitates the formation of RNA granules enriched in mRNA decay factors that promote degradation of the viral RNA [48]. These observations suggest that RNA granule assembly could be an important general function of PARPs in RNA regulation [4, 48].
Finally, PARP13 represses miRNA silencing by targeting AGO proteins for poly(ADP-ribosyl)ation during physiological and stress conditions, including viral infection (Figure 2c) [4, 22]. Modification of AGO results in decreased silencing activity, possibly by reducing the affinity of the miRNA-AGO complex for target mRNA [4, 22, 70].
PARP13 and disease: innate immunity
PARP13 was initially discovered in a screen for host factors that restrict proliferation of the retroviral murine leukemia virus (MLV), and was therefore named Zinc Finger Antiviral Protein (ZAP) [6]. Its antiviral function has since been expanded to other retroviruses (HIV) [27] as well as different viral families such as alpha viruses (Sindbis Virus, SINV; Semliki Forest Virus, SFV; Ross River Virus, RRV; Venezuelan Equine Encephalitus Virus, VEEV) [29], filoviruses (Ebola Virus, EBOV; Marburg Virus, MARV) [71], and hepadna viruses (Hepatitis B Virus, HBV) [72] (Table 1). In addition, PARP13 promotes latency upon infection with herpes viruses (murine gamma herpes virus 68, MHV-68) [73]. In many of these cases the activity of PARP13 was demonstrated to be dependent on direct recognition and binding to specific regions of the viral RNAs and the initiation of their decay via 3′-5′ and 5′-3′ cytoplasmic RNA decay (Figure 2a, 3a). However, whether or not this is the dominant mechanism of PARP13 antiviral activity is unknown. Additional PARP13 functions that have been recently identified, including translational repression and regulation of miRNA silencing, could also contribute to its antiviral function significantly (see below).
Table 1.
| Virus | Viral family | PARP13- sensitive region | Citation |
|---|---|---|---|
| Sindbis Virus (SINV) | Alphavirus | Multiple fragments | [29, 37] |
| Semliki Forest Virus (SFV) | Alphavirus | NA | [29] |
| Ross River Virus (RRV) | Alphavirus | NA | [29] |
| Venezuelan Equine Encephalitis Virus (VEEV) | Alphavirus | NA | [29] |
| Moloney Murine Leukemia Virus (MMLV) | Retrovirus | 3′ Long Terminal Repeat | [6, 37] |
| Xenotropic Murine Leukemia Virus- related Virus (XMRV) | γ-retrovirus | 3′ UTR | Wang, X., Tu, F., Zhu, Y., & Gao, G. (2012). Zinc-finger antiviral protein inhibits XMRV infection. PloS one, 7(6), e39159. |
| Human Immunodeficiency Virus (HIV) | Retrovirus | nef 5′ UTR | [27] |
| Ebola Virus (EBOV) | Filovirus | L gene sequences | [71] |
| Marburg virus (MARV) | Filovirus | L gene sequences | [71] |
| Hepatitus B Virus (HBV) | Hepadna- virus | Terminal redundancy sequences | [72] |
| Murine gammaherpesvirus 68 (MHV-68) | Gamma herpesvirus | M2 gene, ORF64 gene | [73] Xuan, Y., Gong, D., Qi, J., Han, C., Deng, H., & Gao, G. (2013). ZAP Inhibits Murine Gammaherpesvirus 68 ORF64 Expression and Is Antagonized by RTA. Journal of virology, 87(5), 2735–2743. |
Figure 3. PARP13 functions in human health.

a) PARP13 antiviral functions. Left, PARP13 mediates translational repression of target viral RNA transcripts and initiates their degradation by recruiting RNA decay factors as described in Fig. 2a. In the case of MLV, this process occurs in the context of viral RNA granules, and granule compartmentalization may be a common element of PARP13 function. Right, in parallel, PARP13 targets Ago for ADP-ribosylation, inhibiting its function. The global repression of miRNA silencing results in upregulation of pro-inflammatory cytokines, common miRNA targets, and helps mount the antiviral response. b) PARP13 functions in cancer. Top, chronic infection with an oncogenic virus can result in cancer onset. Restriction of oncogenic viruses by PARP13 prevents chronic infection and may be protective against cancer. Bottom, PARP13-mediated destabilization of TRAILR4 mRNA promotes TRAIL binding to the apoptotic receptors TRAILR1 and TRAILR2 and favors cell death. Loss of PARP13 function leads to stabilization of TRAILR4 mRNA and the increased expression of the protein. This inhibits TRAIL signaling through TRAILR1 and TRAILR2, favoring cell survival.
During MLV infection, PARP13 and MLV transcripts localize to RNA granules that share characteristics of stress granules and P-bodies (Figure 3a) [48]. PARP13 recruits both MLV RNA and the exosome complex to these structures, likely facilitating the compartmentalization of viral RNA decay and promoting its efficiency. Whether or not this is related to PARP13 function in stress granule assembly is unknown, but given the biochemical similarities between the structures, it is a likely possibility [4, 48].
PARP13 can also repress the translation of specific viral RNAs, inhibiting the production of viral proteins (Figure 2b, 3a). PARP13 blocks translation initiation of HIV and SINV RNA reporters without affecting global translation [28]. Translation inhibition appears independent of RNA decay, and may precede recruitment of RNA decay factors by PARP13 [28]. Whether or not this mechanism of translation inhibition is specific for HIV and SINV or is relevant to the restriction for other viruses remains unknown Figure 2b, 3a).
Finally, upon viral infection, PARP13 suppresses miRNA silencing by downregulating AGO activity, resulting in the rapid upregulation and expression of miRNA targets, particularly those repressed by mir-17 and mir-93 [22]. Targets of these miRNA families are enriched for antiviral and cytotoxic transcripts and miR-93 was recently shown to silence inflammatory cytokines [22, 74]. PARP13-mediated repression of the miRNA pathway, therefore, facilitates the expression of antiviral transcripts, and promotes the switch to an antiviral state [22].
Thus, PARP13 employs multiple mechanisms of posttranscriptional RNA regulation to contribute to the innate immune response (Figure 3a). In addition, PARP13 synergizes with more than 30 interferon-stimulated genes, and interferon signaling appears to be required for its antiviral activity through unknown mechanisms [75, 76]. PARP13 was recently reported to facilitate the antiviral signaling of the retinoic acid-inducible gene 1(RIG-I), an important sensor of viral RNA in the cytosol that triggers the type I interferon response [35]. However, this interaction, initially observed in human cells, was absent in mouse cells, therefore its relevance to the antiviral activity of PARP13 remains controversial [48]. Nevertheless it is clear that PARP13 forms an important node in the signaling pathways that detect and restrict foreign RNA and establish the cellular antiviral state. As a result, the PARP13 gene, and specifically the PARP domain, has experienced positive diversifying selection pointing to rapid evolution in an arm’s race with viral genomes [77].
PARP13 and disease: cancer
PARP13 plays direct and indirect roles in cancer and could be a useful prognostic biomarker as several cancer types show lower levels of PARP13 expression [78]. PARP13 restricts the proliferation of oncogenic viruses [6, 72] and regulates the cellular response to the TNF-related apoptosis-inducing ligand (TRAIL), a pro-apoptotic cytokine secreted primarily by immune cells that selectively targets cancer cells for killing [13].
Viral infections pose dangers beyond the immediate impact of acute infection. Some human viruses can cause cancer: for example specific human papillomavirus (HPV) types encode oncogenes that can initiate malignant transformation, and HPV is the primary cause of cervical cancer [79]. Chronic infection with Hepatitis B Virus (HBV) is tightly linked to the increased risk of liver cancer and appears to contribute to oncogenesis through a combination of genetic alterations of host cells and the induction of chronic inflammation [80]. PARP13 restricts HBV infection in human hepatocytes during the acute phase [72], thereby preventing the development of chronic infection and could be protective against HBV associated cancers (Figure 3b). In mice PARP13 restricts the murine leukemia virus (MLV) and can thus be protective against leukemia in those models [6].
In addition to targeting viral RNA for degradation, PARP13 also represses the expression of cellular RNAs through RNA decay. One important example is the regulation of the TNF-Related Apoptosis Inducing Ligand (TRAIL) receptor, TRAILR4 [26]. Repression of TRAILR4 by PARP13 has direct implications for cell survival, apoptosis, and cancer therapy (Figure 3b). Four TRAIL receptors are found in humans: TRAILR1-R4. TRAILR3 and R4 are pro-survival receptors that protect cells from TRAIL-induced apoptosis, whereas TRAILR1 and TRAILR2 are proapoptotic, and induce programmed cell death upon binding to TRAIL [81, 82]. TRAILR4 is incapable of signaling apoptosis upon TRAIL binding and sequesters TRAIL from binding to TRAILR1 and R2, inhibiting apoptotic signaling. Thus, TRAILR4 expression is a critical component of the TRAIL response, and high TRAILR4 expression can result in resistance to TRAIL-mediated apoptosis [83–86]. Interestingly, TRAIL treatment increases PARP13 levels consistent with positive feedback between TRAIL signaling and PARP13 expression [26]. Thus, by targeting TRAILR4 mRNA for decay, PARP13 acts as a pro-apoptotic factor and may increase the cellular response to TRAIL in transformed cells (Figure 3b).
It is therefore becoming clear that the antiviral and pro-apoptotic activity of PARP13 is protective against cancer development and survival, and consequently loss of PARP13 may be detrimental for human health. Indeed, decreased PARP13 levels were recently identified in liver, colon and bladder cancer samples from human patients compared to normal tissue, suggesting that loss of PARP13 may indeed be favorable for the progression of certain cancers [78].
Regulation of PARP13 function and its therapeutic potential
Given its importance in the cellular stress response and the posttranscriptional regulation of RNA, PARP13 is an attractive target for the treatment of diseases including cancer, autoimmunity and viral infection, although the effects of targeting PARP13 on an organismal level is unknown. Based on its complex function, both increasing and decreasing PARP13 activity could have therapeutic benefits for different disease indications, even though inhibiting PARP13 should be balanced against the potential risk of increased susceptibility to certain viruses. But how does one target a protein that lacks enzymatic activity? Several attractive approaches exist. PARP13 activity is regulated posttranslationally [4, 22, 87, 88] and inhibiting the enzymes that modify it could be an effective approach to decrease its activity. On the other hand, PARP13 levels can be transcriptionally upregulated by interferon treatment [35, 89], an approach with potential clinical relevance.
Several posttranslational modifications of PARP13 have been identified that modulate its function, localization and interactions with other proteins (Figure 4). Therapeutically targeting the enzymes that posttranslationally modify PARP13 can therefore suppress its activity. Due to its function in the innate immune response, PARP13 inhibition can be beneficial in conditions characterized by chronic inflammation, such as autoimmunity, which often exhibit elevated interferon signaling [12, 90]. In addition PARP13 was recently identified as a host factor protective against the oncolytic alphavirus M1 [78]. Consequently, the oncolytic therapeutic potential of infection with M1 was restricted to cancers defective in PARP13 expression [78]. Therapeutic inhibition of PARP13 can therefore be used in combination with oncolytic virotherapy to sensitize a more diverse set of cancers to the effects of the viral infection.
Figure 4. Mechanisms of PARP13 regulation.

Bullet points indicate how the indicated regulation affects PARP13 levels and activity; boxed text indicates how inhibition (red) or activation (green) of these regulatory mechanisms can be used therapeutically. Clockwise: 1) PARP13 expression is upregulated at the level of transcription by interferons. 2) PARP13 is poly(ADP-ribos)ylated by another member of the PARP family; ADP-ribosylation of PARP13 increases during stress and infection, and likely increases PARP13 activity. 3) PARP13 is phosphorylated at multiple residues by GSK3β; phosphorylation promotes the translational repression function of PARP13. 4) PARP13.1 is farnesylated at Cys899, a modification that results in its anchoring to cellular membranes.
PARP13 is phosphorylated at four serine residues in the N-terminal RNA binding domain by Glycogen Synthase Kinase 3b (GSK3β) (Figure 4) [87]. Phosphorylation by GSK3β facilitates the ability of PARP13 to repress the translation of HIV mRNA, likely by promoting its interaction with factors involved in the regulation of translation initiation. Additional regulatory functions in the absence of viral infection have not been examined. If phosphorylation contributes to the role of PARP13 in promoting inflammation and an antiviral cell state, specific kinase inhibitors may be useful in suppressing this function of the protein, especially in pathologies characterized by chronic inflammation such as autoimmunity and cancer. GSK3 inhibitors are already used for the treatment of pathologies such as diabetes, Alzheimer’s, neurological diseases and cancer [91].
PARP13 is also activated by poly(ADP-ribos)ylation by other PARPs (Figure 4). It is heavily ADP-ribosylated during stress and infection: cellular contexts when its activity in repressing the miRNA pathway increases [4, 22]. This observation suggests that ADP-ribosylation of the protein helps it target AGO for modification and thus suppress miRNA silencing. Whether ADP-ribosylation also activates the RNA decay and translational repression functions of PARP13 is currently unknown, but is a likely possibility. Inhibiting the poly(ADP-ribos)ylation of PARP13 with specific PARP inhibitors could therefore decrease its activity for therapeutic purposes. To this end, it is essential to determine which PARP family member(s) is responsible for modifying PARP13 and to target that PARP selectively. Highly specific and active inhibitors of PARPs 1, 2 and 5a are already in clinical trials for the treatment of cancer, and there is a growing interest in developing specific inhibitors for the remaining members of the family [3, 92, 93].
PARP13.1 is specifically targeted to cellular membranes through farnesylation at its C-terminal CaaX-motif, which is not present in PARP13.2 (Figure 4) [88]. The addition of this hydrophobic lipid group onto target proteins facilitates interaction with membranes [94]. Farnesylation is required for optimal antiviral activity of PARP13.1 against SINV [88]. The importance of this modification for the antiviral activity against other viruses, or for other non-antiviral functions for PARP13 has not been explored. Farnesyl transferase inhibitors therefore have the potential to modulate PARP13 function. Farnesyl transferase inhibitors are already in clinical trials and are promising agents for the treatment of cancers driven by Rho GTPases, as they prevent their proper localization and function [95]. It would be interesting to examine their effects on PARP13 function.
Upregulation of PARP13 function can be induced therapeutically via interferon treatment [35, 89]. PARP13.2 expression is transcriptionally activated during viral infection by interferons and the Interferon-regulatory factor 3 (IRF3) [89]. PARP13.2 is therefore classified as an Interferon-stimulated gene (ISG) and is a key component of the cellular transcriptional response to infection [35]. Consequently, treatment with interferons can be employed to increase PARP13.2 levels and promote PARP13.2-mediated RNA decay (Figure 4).
Therapeutic increase of PARP13 levels can be beneficial for the treatment of viral infections and cancers that are TRAIL sensitive. For cancer therapy, interferon treatment in combination with TRAIL therapy can be used to decrease the mRNA levels of the pro-survival TRAILR4 receptor, maximizing the efficacy of TRAIL therapy. TRAIL-induced apoptosis has been already shown to contribute to the antitumor effects of Interferon-α in hepatocellular carcinoma cell lines [96]. Interferon therapy is a common approach to treat viral infections and autoimmune diseases including multiple sclerosis, and the increase in PARP13.2 expression could be partially responsible for some of the beneficial effects [97].
An important consideration, however, is that interferon treatment fails to increase PARP13.1 expression [35], and the question of which PARP13 isoform has more potent antiviral activity remains controversial. Several reports suggest that PARP13.1 is more efficient at restricting viruses [77, 88, 98]. Positive selection, indicative of antiviral function, has only affected PARP13.1 and not PARP13.2, suggesting a more important role for the longer isoform [77]. This question is further complicated by the fact that many of the early studies of PARP13 were performed with the rat gene, and rats only express a single PARP13 isoform that lacks the PARP domain, a PARP13.2 homologue (Uniprot). Therefore much of the founding work on PARP13 provides no information about possible functional differences between the two human isoforms. Additional analysis comparing the function of the two isoforms is therefore needed in order to examine the effectiveness of increasing PARP13 function through interferon treatment.
Concluding remarks
Posttranscriptional regulation of RNA, including decay, translational repression and miRNA silencing, is an important component of gene expression that facilitates the fine-tuning of transcript levels during physiological conditions, and the rapid switch in global gene expression programs that occur during cellular stress responses. Long-term misregulation of stress-response pathways dependent on posttranscriptional RNA regulation can often result in disease states including cancer. PARP13 has emerged as an important regulator of multiple facets of posttranscriptional RNA regulation, with promising therapeutic potential.
During the innate immune response PARP13 helps eliminate viral RNA through RNA decay, inhibits the translation of specific viral proteins, and helps establish an antiviral cellular state by repressing the activity of the miRNA pathway. PARP13 activity is also likely protective against cancer, as its antiviral activity can help prevent the development of chronic infections correlated with cancer onset; in addition, by inhibiting the expression of the pro-survival receptor TRAILR4, it helps sensitize cells to TRAIL mediated apoptosis and thus may restrict cancer survival. Indeed multiple cancers show decreased PARP13 expression compared to normal tissue. PARP13, therefore, emerges as an important pro-inflammatory and pro-apoptotic factor with effects on the cellular response to stress and the onset and resolution of disease.
The direct involvement of PARP13 in multiple stress response pathways relevant to human health and disease suggests that activating or inhibiting PARP13 may show therapeutic potential. Transcriptional upregulation of PARP13 levels by interferon treatment can be useful during infection or for the therapy of cancers in combination with TRAIL treatment. On the other hand, PARP13-deficient cancers are susceptible to oncolytic viral therapies, and PARP13 inhibition may be an important mechanism to broaden the impact of this approach to more cancer types. In addition, PARP13 inhibition may be useful during chronic inflammation or stress pathway activation. Importantly, all approaches to inhibit or upregulate PARP13 activity, including GSK3 inhibitors, PARP inhibitors, farnesyltransferase inhibitors and interferon treatment are already in use in the clinic or are undergoing clinical trials. Data suggests that these treatments are well tolerated and could therefore be useful approaches to modulate PARP13 function in patients [97, 99–101]. Elucidating how PARP13 is regulated and exploiting this knowledge for the therapeutic repression of PARP13 is the next step in understanding this multifaceted protein (Box 2).
Box 2. Outstanding questions.
How do the known posttranslational modifications of PARP13 affect its activity and function?
Is PARP13 regulated differently during different stress conditions, or is it generally activated during stress?
Is PARP13 function misregulated during chronic inflammation or in cancer? Is PARP13 downregulated in multiple cancer types? What is the activity of the remaining PARP13 protein?
What other cellular transcripts and signaling pathways are regulated by PARP13 and do they contribute to immunity and cancer?
Which enzymatically active PARPs modify PARP13 and how does modification with poly(ADP-ribose) affect PARP13 function, RNA binding affinity and target specificity?
What role does PARP13 play in the compartmentalization of viral RNAs to RNA granules? Is such compartmentalization required for efficient destabilization?
PARP13 regulates RNA in stress and disease
RNA regulation is important for health
PARP13 is a target for treatment of disease
Acknowledgments
This work was partially supported by Cancer Center Support (core; grant P30-CA14051) and RO1GM087465 from the National Institutes of Health to PC. FB was funded by a Ludwig Postdoctoral Fellowship and TT was funded by an MIT School of Science Fellowship in Cancer Research.
Glossary
- Apoptosis
The process of programmed cell death used to eliminate cells damaged beyond repair in a controlled fashion without eliciting inflammation. Apoptosis is also utilized by the developing organism to remove cells that are no longer needed
- Cellular stress response
Cellular pathways that are activated in response to specific stressors, including pathogens, hypoxia, heat shock, unfolded proteins, reactive oxygen species and others. In general, cellular stress responses are used to eliminate the stress and to prevent or minimize possible cellular damage. If a cell is unable to resolve the stress, most cellular stress responses activate separate apoptotic pathways to prevent damage to the whole organism
- Cytokines
Secreted proteins that act as messengers and mediate the local crosstalk between immune and non-immune cells during an immune response
- Farnesylation
A posttranslational modification constituting the addition of a farnesyl lipid group onto a target protein by a farnesyl transferase. The presence of a farnesyl group facilitates interactions of the modified protein with cellular membranes and can affect protein localization, activity and interactions with other polypeptides
- Inflammation
A protective response by the body that consists of a complex set of signaling pathways aimed at removing harmful agents. At the tissue level, inflammation involves the increased permeability of the vascular endothelium that allows immune cells to invade the damaged space and repair the damage. Inflammation can be triggered by wounding and microbial or viral infection and is tightly linked to the innate immune response. In the case of infection it often helps to recruit and activate cells of the adaptive immune system
- Innate immune response
A set of defined cellular responses stimulated by the detection of foreign molecules of pathogenic origin. The innate immune response triggers the upregulation of pro-inflammatory cytokines, which initiate the process of inflammation and help to recruit adaptive immune cells
- P-bodies
Sites of active RNA decay. These structures are enriched in RNA-decay factors, such as the decapping enzymes DCP1/2, the 5′-3′ exonuclease XRN1, the components of RISC, GW182 and AGO, and others
- PARPs
Poly(ADP-ribose) polymerases are a family of proteins that use NAD+ as substrate to modify target proteins with a posttranslational protein modification called ADP-ribose. PARPs modify proteins with two types of ADP-ribose modifications: mono(ADP-ribose), the addition of a single ADP-ribose unit; and poly(ADP-ribose),the addition of multiple ADP-ribose subunits in linear or branched polymers. 17 PARPs are found in humans that have multiple cellular functions
- Stress granules
Non-membrane-bound structures that consist of stalled preinitiation complexes, mRNAs and RNA-binding proteins. Their formation is induced by global translational repression in response to different stresses, including hypoxia, heat shock, cytoplasmic stress, and others
Footnotes
Conflict of interest statement: The authors declare no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ame JC, et al. The PARP superfamily. BioEssays : news and reviews in molecular, cellular and developmental biology. 2004;26:882–893. doi: 10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]
- 2.Vyas S, et al. A systematic analysis of the PARP protein family identifies new functions critical for cell physiology. Nature communications. 2013;4:2240. doi: 10.1038/ncomms3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vyas S, Chang P. New PARP targets for cancer therapy. Nature reviews. Cancer. 2014;14:502–509. doi: 10.1038/nrc3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leung AK, et al. Poly(ADP-ribose) regulates stress responses and microRNA activity in the cytoplasm. Molecular cell. 2011;42:489–499. doi: 10.1016/j.molcel.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jwa M, Chang P. PARP16 is a tail-anchored endoplasmic reticulum protein required for the PERK- and IRE1alpha-mediated unfolded protein response. Nature cell biology. 2012;14:1223–1230. doi: 10.1038/ncb2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao G, et al. Inhibition of retroviral RNA production by ZAP, a CCCH-type zinc finger protein. Science. 2002;297:1703–1706. doi: 10.1126/science.1074276. [DOI] [PubMed] [Google Scholar]
- 7.Di Giammartino DC, et al. PARP1 represses PAP and inhibits polyadenylation during heat shock. Molecular cell. 2013;49:7–17. doi: 10.1016/j.molcel.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li X, et al. Unfolded protein response in cancer: the physician’s perspective. Journal of hematology & oncology. 2011;4:8. doi: 10.1186/1756-8722-4-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cairns RA, et al. Regulation of cancer cell metabolism. Nature reviews. Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 10.Majmundar AJ, et al. Hypoxia-inducible factors and the response to hypoxic stress. Molecular cell. 2010;40:294–309. doi: 10.1016/j.molcel.2010.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reineke LC, Lloyd RE. Diversion of stress granules and P-bodies during viral infection. Virology. 2013;436:255–267. doi: 10.1016/j.virol.2012.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Park H, et al. Lighting the fires within: the cell biology of autoinflammatory diseases. Nature reviews. Immunology. 2012;12:570–580. doi: 10.1038/nri3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zamarron BF, Chen W. Dual roles of immune cells and their factors in cancer development and progression. International journal of biological sciences. 2011;7:651–658. doi: 10.7150/ijbs.7.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Nadal E, et al. Controlling gene expression in response to stress. Nature reviews. Genetics. 2011;12:833–845. doi: 10.1038/nrg3055. [DOI] [PubMed] [Google Scholar]
- 15.Thomas MP, Lieberman J. Live or let die: posttranscriptional gene regulation in cell stress and cell death. Immunological reviews. 2013;253:237–252. doi: 10.1111/imr.12052. [DOI] [PubMed] [Google Scholar]
- 16.Sharova LV, et al. Database for mRNA half-life of 19 977 genes obtained by DNA microarray analysis of pluripotent and differentiating mouse embryonic stem cells. DNA research : an international journal for rapid publication of reports on genes and genomes. 2009;16:45–58. doi: 10.1093/dnares/dsn030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu X, Brewer G. The regulation of mRNA stability in mammalian cells: 2.0. Gene. 2012;500:10–21. doi: 10.1016/j.gene.2012.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rabani M, et al. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nature biotechnology. 2011;29:436–442. doi: 10.1038/nbt.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kratochvill F, et al. Tristetraprolin-driven regulatory circuit controls quality and timing of mRNA decay in inflammation. Molecular systems biology. 2011;7:560. doi: 10.1038/msb.2011.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holcik M, Sonenberg N. Translational control in stress and apoptosis. Nature reviews. Molecular cell biology. 2005;6:318–327. doi: 10.1038/nrm1618. [DOI] [PubMed] [Google Scholar]
- 21.Leung AK, Sharp PA. MicroRNA functions in stress responses. Molecular cell. 2010;40:205–215. doi: 10.1016/j.molcel.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seo GJ, et al. Reciprocal inhibition between intracellular antiviral signaling and the RNAi machinery in mammalian cells. Cell host & microbe. 2013;14:435–445. doi: 10.1016/j.chom.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anderson P, Kedersha N. Stress granules: the Tao of RNA triage. Trends in biochemical sciences. 2008;33:141–150. doi: 10.1016/j.tibs.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 24.Kedersha N, et al. Stress granules and cell signaling: more than just a passing phase? Trends in biochemical sciences. 2013;38:494–506. doi: 10.1016/j.tibs.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo X, et al. The zinc-finger antiviral protein recruits the RNA processing exosome to degrade the target mRNA. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:151–156. doi: 10.1073/pnas.0607063104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Todorova T, et al. PARP13 regulates cellular mRNA post-transcriptionally and functions as a pro-apoptotic factor by destabilizing TRAILR4 transcript. Nature communications. 2014;5:5362. doi: 10.1038/ncomms6362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu Y, et al. Zinc-finger antiviral protein inhibits HIV-1 infection by selectively targeting multiply spliced viral mRNAs for degradation. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:15834–15839. doi: 10.1073/pnas.1101676108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhu Y, et al. Translational repression precedes and is required for ZAP-mediated mRNA decay. The EMBO journal. 2012;31:4236–4246. doi: 10.1038/emboj.2012.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bick MJ, et al. Expression of the zinc-finger antiviral protein inhibits alphavirus replication. Journal of virology. 2003;77:11555–11562. doi: 10.1128/JVI.77.21.11555-11562.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Welsby I, et al. PARP12, an Interferon Stimulated Gene Involved in the Control of Protein Translation and Inflammation. The Journal of biological chemistry. 2014 doi: 10.1074/jbc.M114.589515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Atasheva S, et al. Interferon-stimulated poly(ADP-Ribose) polymerases are potent inhibitors of cellular translation and virus replication. Journal of virology. 2014;88:2116–2130. doi: 10.1128/JVI.03443-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nature reviews. Molecular cell biology. 2012;13:411–424. doi: 10.1038/nrm3376. [DOI] [PubMed] [Google Scholar]
- 33.Wang Z, et al. Recognition of the iso-ADP-ribose moiety in poly(ADP-ribose) by WWE domains suggests a general mechanism for poly(ADP-ribosyl)ation-dependent ubiquitination. Genes & development. 2012;26:235–240. doi: 10.1101/gad.182618.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.He F, et al. Structural insight into the interaction of ADP-ribose with the PARP WWE domains. FEBS letters. 2012;586:3858–3864. doi: 10.1016/j.febslet.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 35.Hayakawa S, et al. ZAPS is a potent stimulator of signaling mediated by the RNA helicase RIG-I during antiviral responses. Nature immunology. 2011;12:37–44. doi: 10.1038/ni.1963. [DOI] [PubMed] [Google Scholar]
- 36.Vyas S, et al. Family-wide analysis of poly(ADP-ribose) polymerase activity. Nature communications. 2014;5:4426. doi: 10.1038/ncomms5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo X, et al. The zinc finger antiviral protein directly binds to specific viral mRNAs through the CCCH zinc finger motifs. Journal of virology. 2004;78:12781–12787. doi: 10.1128/JVI.78.23.12781-12787.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen S, et al. Structure of N-terminal domain of ZAP indicates how a zinc-finger protein recognizes complex RNA. Nature structural & molecular biology. 2012;19:430–435. doi: 10.1038/nsmb.2243. [DOI] [PubMed] [Google Scholar]
- 39.Law LM, et al. Identification of a dominant negative inhibitor of human zinc finger antiviral protein reveals a functional endogenous pool and critical homotypic interactions. Journal of virology. 2010;84:4504–4512. doi: 10.1128/JVI.02018-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sanduja S, et al. The role of tristetraprolin in cancer and inflammation. Frontiers in bioscience. 2012;17:174–188. doi: 10.2741/3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carpenter S, et al. Post-transcriptional regulation of gene expression in innate immunity. Nature reviews. Immunology. 2014;14:361–376. doi: 10.1038/nri3682. [DOI] [PubMed] [Google Scholar]
- 42.Jackson RJ, et al. The mechanism of eukaryotic translation initiation and principles of its regulation. Nature reviews. Molecular cell biology. 2010;11:113–127. doi: 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schoenberg DR, Maquat LE. Regulation of cytoplasmic mRNA decay. Nature reviews. Genetics. 2012;13:246–259. doi: 10.1038/nrg3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kervestin S, Jacobson A. NMD: a multifaceted response to premature translational termination. Nature reviews. Molecular cell biology. 2012;13:700–712. doi: 10.1038/nrm3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garneau NL, et al. The highways and byways of mRNA decay. Nature reviews. Molecular cell biology. 2007;8:113–126. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- 46.Onomoto K, et al. Critical role of an antiviral stress granule containing RIG-I and PKR in viral detection and innate immunity. PloS one. 2012;7:e43031. doi: 10.1371/journal.pone.0043031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kedersha N, Anderson P. Mammalian stress granules and processing bodies. Methods in enzymology. 2007;431:61–81. doi: 10.1016/S0076-6879(07)31005-7. [DOI] [PubMed] [Google Scholar]
- 48.Lee H, et al. Zinc-finger antiviral protein mediates retinoic acid inducible gene I-like receptor-independent antiviral response to murine leukemia virus. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:12379–12384. doi: 10.1073/pnas.1310604110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Onomoto K, et al. Antiviral innate immunity and stress granule responses. Trends in immunology. 2014;35:420–428. doi: 10.1016/j.it.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Griseri P, Pages G. Control of pro-angiogenic cytokine mRNA half-life in cancer: the role of AU-rich elements and associated proteins. Journal of interferon & cytokine research : the official journal of the International Society for Interferon and Cytokine Research. 2014;34:242–254. doi: 10.1089/jir.2013.0140. [DOI] [PubMed] [Google Scholar]
- 51.Meister G. Argonaute proteins: functional insights and emerging roles. Nature reviews. Genetics. 2013;14:447–459. doi: 10.1038/nrg3462. [DOI] [PubMed] [Google Scholar]
- 52.Griseri P, Pages G. Regulation of the mRNA half-life in breast cancer. World journal of clinical oncology. 2014;5:323–334. doi: 10.5306/wjco.v5.i3.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Milke L, et al. Depletion of tristetraprolin in breast cancer cells increases interleukin-16 expression and promotes tumor infiltration with monocytes/macrophages. Carcinogenesis. 2013;34:850–857. doi: 10.1093/carcin/bgs387. [DOI] [PubMed] [Google Scholar]
- 54.Zucconi BE, Wilson GM. Modulation of neoplastic gene regulatory pathways by the RNA-binding factor AUF1. Frontiers in bioscience. 2011;16:2307–2325. doi: 10.2741/3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang J, et al. Multiple Functions of the RNA-Binding Protein HuR in Cancer Progression, Treatment Responses and Prognosis. International journal of molecular sciences. 2013;14:10015–10041. doi: 10.3390/ijms140510015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Spriggs KA, et al. Translational regulation of gene expression during conditions of cell stress. Molecular cell. 2010;40:228–237. doi: 10.1016/j.molcel.2010.09.028. [DOI] [PubMed] [Google Scholar]
- 57.Abaza I, Gebauer F. Trading translation with RNA-binding proteins. Rna. 2008;14:404–409. doi: 10.1261/rna.848208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mokrejs M, et al. IRESite: the database of experimentally verified IRES structures (www.iresite.org) Nucleic acids research. 2006;34:D125–130. doi: 10.1093/nar/gkj081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Novoa I, et al. Stress-induced gene expression requires programmed recovery from translational repression. The EMBO journal. 2003;22:1180–1187. doi: 10.1093/emboj/cdg112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Walsh D, Mohr I. Viral subversion of the host protein synthesis machinery. Nature reviews. Microbiology. 2011;9:860–875. doi: 10.1038/nrmicro2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hanson PJ, et al. IRES-Dependent Translational Control during Virus-Induced Endoplasmic Reticulum Stress and Apoptosis. Frontiers in microbiology. 2012;3:92. doi: 10.3389/fmicb.2012.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Blagden SP, Willis AE. The biological and therapeutic relevance of mRNA translation in cancer. Nature reviews. Clinical oncology. 2011;8:280–291. doi: 10.1038/nrclinonc.2011.16. [DOI] [PubMed] [Google Scholar]
- 63.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wilson RC, Doudna JA. Molecular mechanisms of RNA interference. Annual review of biophysics. 2013;42:217–239. doi: 10.1146/annurev-biophys-083012-130404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang Y, et al. Biological functions of microRNAs: a review. Journal of physiology and biochemistry. 2011;67:129–139. doi: 10.1007/s13105-010-0050-6. [DOI] [PubMed] [Google Scholar]
- 66.Fabian MR, Sonenberg N. The mechanics of miRNA-mediated gene silencing: a look under the hood of miRISC. Nature structural & molecular biology. 2012;19:586–593. doi: 10.1038/nsmb.2296. [DOI] [PubMed] [Google Scholar]
- 67.O’Neill LA, et al. MicroRNAs: the fine-tuners of Toll-like receptor signalling. Nature reviews. Immunology. 2011;11:163–175. doi: 10.1038/nri2957. [DOI] [PubMed] [Google Scholar]
- 68.Farazi TA, et al. miRNAs in human cancer. The Journal of pathology. 2011;223:102–115. doi: 10.1002/path.2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gantier MP, et al. A miR-19 regulon that controls NF-kappaB signaling. Nucleic acids research. 2012;40:8048–8058. doi: 10.1093/nar/gks521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Leung A, et al. Poly(ADP-ribose) regulates post-transcriptional gene regulation in the cytoplasm. RNA biology. 2012;9:542–548. doi: 10.4161/rna.19899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Muller S, et al. Inhibition of filovirus replication by the zinc finger antiviral protein. Journal of virology. 2007;81:2391–2400. doi: 10.1128/JVI.01601-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mao R, et al. Inhibition of hepatitis B virus replication by the host zinc finger antiviral protein. PLoS pathogens. 2013;9:e1003494. doi: 10.1371/journal.ppat.1003494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Xuan Y, et al. Zinc finger antiviral protein inhibits murine gammaherpesvirus 68 M2 expression and regulates viral latency in cultured cells. Journal of virology. 2012;86:12431–12434. doi: 10.1128/JVI.01514-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu Y, et al. MicroRNA-93 inhibits inflammatory cytokine production in LPS-stimulated murine macrophages by targeting IRAK4. FEBS letters. 2014;588:1692–1698. doi: 10.1016/j.febslet.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 75.Karki S, et al. Multiple interferon stimulated genes synergize with the zinc finger antiviral protein to mediate anti-alphavirus activity. PloS one. 2012;7:e37398. doi: 10.1371/journal.pone.0037398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.MacDonald MR, et al. The zinc finger antiviral protein acts synergistically with an interferon-induced factor for maximal activity against alphaviruses. Journal of virology. 2007;81:13509–13518. doi: 10.1128/JVI.00402-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kerns JA, et al. Positive selection and increased antiviral activity associated with the PARP-containing isoform of human zinc-finger antiviral protein. PLoS genetics. 2008;4:e21. doi: 10.1371/journal.pgen.0040021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lin Y, et al. Identification and characterization of alphavirus M1 as a selective oncolytic virus targeting ZAP-defective human cancers. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E4504–4512. doi: 10.1073/pnas.1408759111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Munoz N, et al. Chapter 1: HPV in the etiology of human cancer. Vaccine. 2006;24(Suppl 3):S3/1–10. doi: 10.1016/j.vaccine.2006.05.115. [DOI] [PubMed] [Google Scholar]
- 80.Fallot G, et al. Diverse roles of hepatitis B virus in liver cancer. Current opinion in virology. 2012;2:467–473. doi: 10.1016/j.coviro.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 81.LeBlanc HN, Ashkenazi A. Apo2L/TRAIL and its death and decoy receptors. Cell death and differentiation. 2003;10:66–75. doi: 10.1038/sj.cdd.4401187. [DOI] [PubMed] [Google Scholar]
- 82.Marsters SA, et al. A novel receptor for Apo2L/TRAIL contains a truncated death domain. Current biology : CB. 1997;7:1003–1006. doi: 10.1016/s0960-9822(06)00422-2. [DOI] [PubMed] [Google Scholar]
- 83.Morizot A, et al. Chemotherapy overcomes TRAIL-R4-mediated TRAIL resistance at the DISC level. Cell death and differentiation. 2011;18:700–711. doi: 10.1038/cdd.2010.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Degli-Esposti MA, et al. The novel receptor TRAIL-R4 induces NF-kappaB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity. 1997;7:813–820. doi: 10.1016/s1074-7613(00)80399-4. [DOI] [PubMed] [Google Scholar]
- 85.Vindrieux D, et al. Down-regulation of DcR2 sensitizes androgen-dependent prostate cancer LNCaP cells to TRAIL-induced apoptosis. Cancer cell international. 2011;11:42. doi: 10.1186/1475-2867-11-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bouralexis S, et al. Progressive resistance of BTK-143 osteosarcoma cells to Apo2L/TRAIL-induced apoptosis is mediated by acquisition of DcR2/TRAIL-R4 expression: resensitisation with chemotherapy. British journal of cancer. 2003;89:206–214. doi: 10.1038/sj.bjc.6601021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sun L, et al. Glycogen synthase kinase 3beta (GSK3beta) modulates antiviral activity of zinc-finger antiviral protein (ZAP) The Journal of biological chemistry. 2012;287:22882–22888. doi: 10.1074/jbc.M111.306373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Charron G, et al. Prenylome profiling reveals S-farnesylation is crucial for membrane targeting and antiviral activity of ZAP long-isoform. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:11085–11090. doi: 10.1073/pnas.1302564110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang N, et al. Viral induction of the zinc finger antiviral protein is IRF3-dependent but NF-kappaB-independent. The Journal of biological chemistry. 2010;285:6080–6090. doi: 10.1074/jbc.M109.054486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity. 2006;25:383–392. doi: 10.1016/j.immuni.2006.08.010. [DOI] [PubMed] [Google Scholar]
- 91.Cohen P, Goedert M. GSK3 inhibitors: development and therapeutic potential. Nature reviews. Drug discovery. 2004;3:479–487. doi: 10.1038/nrd1415. [DOI] [PubMed] [Google Scholar]
- 92.Wahlberg E, et al. Family-wide chemical profiling and structural analysis of PARP and tankyrase inhibitors. Nature biotechnology. 2012;30:283–288. doi: 10.1038/nbt.2121. [DOI] [PubMed] [Google Scholar]
- 93.Steffen JD, et al. Structural Implications for Selective Targeting of PARPs. Frontiers in oncology. 2013;3:301. doi: 10.3389/fonc.2013.00301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sebti SM. Protein farnesylation: implications for normal physiology, malignant transformation, and cancer therapy. Cancer cell. 2005;7:297–300. doi: 10.1016/j.ccr.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 95.Berndt N, et al. Targeting protein prenylation for cancer therapy. Nature reviews. Cancer. 2011;11:775–791. doi: 10.1038/nrc3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yamamoto T, et al. Partial contribution of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)/TRAIL receptor pathway to antitumor effects of interferon-alpha/5-fluorouracil against Hepatocellular Carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2004;10:7884–7895. doi: 10.1158/1078-0432.CCR-04-0794. [DOI] [PubMed] [Google Scholar]
- 97.Killestein J, Polman CH. Determinants of interferon beta efficacy in patients with multiple sclerosis. Nature reviews. Neurology. 2011;7:221–228. doi: 10.1038/nrneurol.2011.22. [DOI] [PubMed] [Google Scholar]
- 98.Glasker S, et al. The alternate triad motif of the PARP-like domain of the human zinc finger antiviral protein is essential for its antiviral activity. The Journal of general virology. 2014 doi: 10.1099/vir.0.060988-0. [DOI] [PubMed] [Google Scholar]
- 99.Hong DS, et al. Inhibition of the Ras/Raf/MEK/ERK and RET kinase pathways with the combination of the multikinase inhibitor sorafenib and the farnesyltransferase inhibitor tipifarnib in medullary and differentiated thyroid malignancies. The Journal of clinical endocrinology and metabolism. 2011;96:997–1005. doi: 10.1210/jc.2010-1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kozikowski AP, et al. Highly potent and specific GSK-3beta inhibitors that block tau phosphorylation and decrease alpha-synuclein protein expression in a cellular model of Parkinson’s disease. ChemMedChem. 2006;1:256–266. doi: 10.1002/cmdc.200500039. [DOI] [PubMed] [Google Scholar]
- 101.Javle M, Curtin NJ. The role of PARP in DNA repair and its therapeutic exploitation. British journal of cancer. 2011;105:1114–1122. doi: 10.1038/bjc.2011.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Spangle JM, Munger K. The human papillomavirus type 16 E6 oncoprotein activates mTORC1 signaling and increases protein synthesis. Journal of virology. 2010;84:9398–9407. doi: 10.1128/JVI.00974-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Scheffner M, et al. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 104.Bargonetti J, et al. Wild-type but not mutant p53 immunopurified proteins bind to sequences adjacent to the SV40 origin of replication. Cell. 1991;65:1083–1091. doi: 10.1016/0092-8674(91)90560-l. [DOI] [PubMed] [Google Scholar]
- 105.DeCaprio JA, et al. SV40 large tumor antigen forms a specific complex with the product of the retinoblastoma susceptibility gene. Cell. 1988;54:275–283. doi: 10.1016/0092-8674(88)90559-4. [DOI] [PubMed] [Google Scholar]
- 106.Munger K, et al. Complex formation of human papillomavirus E7 proteins with the retinoblastoma tumor suppressor gene product. The EMBO journal. 1989;8:4099–4105. doi: 10.1002/j.1460-2075.1989.tb08594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mosialos G, et al. The Epstein-Barr virus transforming protein LMP1 engages signaling proteins for the tumor necrosis factor receptor family. Cell. 1995;80:389–399. doi: 10.1016/0092-8674(95)90489-1. [DOI] [PubMed] [Google Scholar]