

Abstract
The uses of a method of coupling DNA is investigated for trapping and purifying transcription factors. Using the GFP-C/EBP fusion protein as a model, trapping gives higher purity and comparable yield to conventional affinity chromatography. The chemistry utilized is mild and was shown to have no detrimental effect on GFP fluorescence or GFP-C/EBP DNA-binding. The method involves introducing a ribose nucleotide to the 3′ end of a DNA sequence. Reaction with mM NaIO4 (sodium metaperiodate) produces a dialdehyde of ribose which couples to hydrazide-agarose. The DNA is combined at nM concentration with a nuclear extract or other protein mixture and DNA-protein complexes form. The complex is then coupled to hydrazide-agarose for trapping the DNA-protein complex and the protein eluted by increasing NaCl concentration. Using a different oligonucleotide with the proximal E-box sequence from the human telomerase promoter, USF-2 transcription factor was purified by trapping, again with higher purity than results from conventional affinity chromatography and similar yield. Other transcription factors binding E-boxes including E2A, c-myc, and myo-D were also purified but myogenenin and NFκB were not. Therfore, this approach proved valuable for both affinity chromatography and for the trapping approach.
1. INTRODUCTION
Recently, we reported a method [1] for coupling DNA to solid supports. The method involves introducing a ribose nucleotide at the 3′ end of a DNA sequence. Reaction with NaIO4 then produces a dialdehyde variant of ribose which then couples covalently to a hydrazide-agarose support for affinity chromatography. The coupling reaction was shown to be rapid, the linkage was shown to be stable over prolonged use, and coupling efficiencies in the range of 60–90% were obtained. As a model, the new supports were prepared using a DNA-sequence specifically bound by the CAAT –enhancer binding protein transcription factor (C/EBP). The columns produced allowed partial purification of a GFP-C/EBP chimeric fusion protein from a bacterial extract. Here we explore whether this new chemistry can be used for trapping affinity chromatography [2]. In this variant of affinity chromatography, a DNA sequence, is combined at low concentration with a protein mixture, typically nuclear extract. Proteins which bind the DNA sequence form a DNA-protein complex which is recovered on a column for subsequent elution. The trapping method [2] has been used to purify low abundance transcription factors, often to homogeneity, in a single operation. The method was later extended to intact DNA promoter sequences to purify active transcription complexes [3].
Affinity chromatography and trapping would not necessarily yield the same results. Transcription factors bind to their cognate DNA response element (RE) typically with nM-pM affinity. They also bind essentially any DNA sequence “non-specifically” with near micromolar affinity. This probably has a great deal to do with how they function in vivo. Von Hippel and colleagues originated the sliding model of TF-DNA binding [4–7]. This model predicts that TFs diffuse 3-dimensionally, binding euchromatin anywhere along its length (“non-specifically”), and then slide one-dimensionally along the DNA to locate their RE. This one-dimensional “diffusion” is much more rapid than the three-dimensional alternative and accounts for why some transcription factors bind RE DNA with on-rates more rapid than 3-dimensional diffusion would allow. Thus, this “non-specific binding” may be an essential component of their mechanism, for binding to DNA from solution, while their higher affinity RE-binding positions them correctly. This, however, has a profound effect on purification. Even columns containing as little as 1 nmol of DNA per ml of column bed contain μM DNA and as such can probably bind any TF “non-specifically”. For example, here we used a 0.1 ml column containing 500 pmole of EP18 oligonucleotide to purify GFP-C/EBP, an effective column concentration of 5 μM. To circumvent this problem, we developed the trapping method [2]. In this method, DNA is added to the protein sample at nM concentration, the DNA-protein complex forms and is then recovered on a column, circumventing high column DNA concentrations. For trapping of GFP-C/EBP, the formation of the DNA-protein complex was accomplished at 500 nM EP18. The effective DNA concentration alone may contribute to different results.
Affinity capture has also been accomplished using biotinylated oligonucleotides and (strept)avidin-coupled beads. However, as previously shown avidin and its various derivatives also retain other proteins which may interfere with some kinds of analysis [2]. Aldehyde-hydrazide coupling may provide an improved alternative.
The aldehyde coupling procedure used is so mild that we next investigate whether this coupling approach could be used for trapping. Here we used both the C/EBP binding oligonucleotide, and another which is derived from the human telomerase (hTERT) promoter. This latter sequence was shown previously to bind the USF-2 transcription factor [8]. The two sequences were then shown to purify GFP-C/EBP and USF-2, respectively, using the trapping approach and to give higher purity than is obtained by conventional affinity chromatography.
2. MATERIALS AND METHODS
2.1 Materials
Oligonucleotides used are given in Table 1. The prefix “r” denotes the ribonucleotide where appropriate. Antibodies against C/EBP (catalog SC-746, 1:1000), USF-2 (SC-862, 1:1000), E2A (N-649, 1:2000), MyoD (SC-760, 1:2000), and NFκB (SC-372, 1:1000) were from Santa Cruz Biotechnology (Dallas, TX, USA). Dilutions given are those used for Western blots. Antibody against c-Myc (catalog #9402, 1:1000) was from Cell Signaling Technology (Beverly, MA, USA). Ultralink© Hydrazide Agarose was from Thermo Scientific (Rockford, IL, USA). All chemicals were of the highest purity available commercially.
Table 1.
Oligonucleotides used
| Oligonucleotide | Sequence | Source |
|---|---|---|
| AP1 | 5′-CGCTTGATGACTCAGCCGGAA-3′ | IDT |
| SP1 | 5′-ACGGGCGGGCCCGCCCATGGGCGGGCCCGCCCGT-3′ | IDT |
| C/EBP | 5′-TGCAGATTGCGCAATCTGCA-3′ | IDT |
| EP18 | 5′-GCAGATTGCGCAATCTGrC-3′ | IDT |
| NFkB | 5′-AGTTGAGGGGACTTTCCCAGGC-3′ | Santa Cruz |
| SJ9 | 5′-GCTTCCCACGTGCGCArG-3′ | IDT |
| SJ11 | 5′-CTGCGCACGTGGGAAGC-3′ | IDT |
2.2 Aldehyde coupling chemistry
Oligonucleotides, a DNA sequences with a 3′-terminal ribonucleotide, were used for coupling (Table 1). Oligonucleotides (20 nM-1 μM) were placed in buffer KP (0.1 M potassium phosphate, pH 6.8) by desalting 50 μl on a 0.5 ml P-6 Biogel (BioRad Laboratories, Hercules, AC, USA) spin column in buffer KP. To the oligonucleotide was added 1/10 volume of 10 mM NaIO4 (sodium meta-periodate, Acros Organics, Fisher Chemical Co., Dallas, TX, USA) and incubated at room temperature (20°C) in the dark for 30 min. To quench the reaction 1/10 volume of 10 mM Na2S2O5 (sodium metabisulfite, Fisher) was added and incubation continued for 30 min. in the dark. Prior to use for trapping, the oligonucleotide was again desalted into buffer KP. For coupling, the oligonuclede was mixed with 0.01–0.1 ml of hydrazide-agarose which had been washed with buffer KP.
2.3 Expression and purification of GFP-C/EBP
BL21 bacteria containing the C/EBP-p22 plasmid, were induced and lysed as previously described [9]. The lysed bacteria were centrifuged at 42000 x g for 30 min. at 4°C and the supernatant saved. Some of the supernatant was saved as crude bacterial extract and stored as 0.1 ml aliquots at −85°C. The remainder was used to purify GFP-C/EBP fusion protein as described previously which was also stored at −85°C as aliquots [9].
2.4 Affinity chromatography and trapping
All steps were performed at 4°C unless otherwise noted. For affinity chromatography, 0.1 ml columns were prepared containing either 500 pmol (EP18) or 20 pmole (SJ9/SJ11) DNA. Other details are provided in the figure legends. Typically, protein (GFP-C/EBP or HEK293 nuclear extract) as diluted to 1 ml in TE0.1 buffer (10 mM Tris, pH 8, 1 mM EDTA, 0.1 M NaCl) and either applied to the column or mixed with the resin before pouring into the column. For trapping, protein was desalted into buffer KP using a P-6 spin column, mixed with either 500 pmole (EP18) or 20 pmole (SJ9/SJ11) in a total volume of 1 ml containing 0.5 mg protein. This was mixed with hydrazide-agarose (0.1 ml) which had been washed with buffer KP and pelleted in a centrifuge. The mixture was either allowed to couple for 2 h at room temperature (bacterial extract) or 4 h at 4°C (nuclear extract). The resin was then packed into a column, retaining the flow through fraction. For both trapping and affinity chromatography, the column was washed 5–10 times with 0.5 ml portions of TE0.1 buffer and eluted with 0.5 ml of buffer TE0.5, TE1.0, or TE1.5 (10 mM Tris, pH 8, 1 mM EDTA, with either 0.5, 1.0 or 1.5 M NaCl) as specified in the figure legends.
2.5 Electrophoresis
For electrophoretic gel shift assay, protein samples were incubated with 10 nM 32P-duplex oligonucleotide on ice for 30 min. Then 4 volumes of buffer GS (50 mM Tris, pH8, 5% glycerol, 1 mM EDTA, 1 mM 2-mercaptoethanol) and 0.5 volume BPE-Glyc (0.015% bromophenol blue in 50% glycerol) was added and the sample (20 μl) applied to a 5% acrylamide gel that had been pre-electrophoresed. The gel and running buffer was in 0.5X TBE buffer (45 mM H3BO3, 45 mM Tris, 1 mM EDTA), the voltage 130 v, and when the tracking dye was 0.5 cm from the gel’s bottom, it was removed. The gel was dried and detected by autoradiography.
For SDS-PAGE, samples in Laemmli [10] sample buffer was applied to a 10–12% acrylamide gel and electrophoresed at 200 v for 45 min. The gel was stained with silver using the kit provided by BioRad Laboratories or electroblotted to polyvinylidene difluoride (PVDF). The electroblot was blocked with 5% non-fat dry milk in TTBS (20 mM Tris, pH 7.5, 100 mM NaCl, 0.1% Tween-20) for 1 h and detected with antibodies, followed by horseradish peroxidase-conjugate second antibody, and enhanced chemiluminescence (ECL).
3. RESULTS and DISCUSSION
3.1 Does the chemistry used affect protein activity?
One significant question when combining a coupling chemistry with trapping affinity chromatography is whether any of the chemicals used during coupling will adversely affect proteins. If the chemistry is mild enough, reaction in the presence of proteins may be possible, and even if not, it is important to know how critical it will be to remove all reagents before introduction of proteins. The dialdehyde of DNA could conceivably react with proteins. Aldehydes and amines, such as the N-terminus or the lysine side chains, can combine to form imidates (Schiff base) adducts, however, such adducts are only transiently stable and reaction of the aldehyde with the hydrazide support should be favored and provide a stable linkage. We begin by testing whether or not the coupling chemistry affects protein activity.
We determined if reaction has any effect on the purified GFP-C/EBP fusion protein. As shown in Fig. 1, the fluorescence intensity of GFP is unaffected. Other experiments (data not shown) demonstrate that neither the excitation nor emission spectra of the GFP moiety are affected, and the protein still binds DNA as shown in a gel shift assay. Thus, the chemistry appears to be quite mild, even when performed in the presence of protein.
Figure 1. The chemistry does not alter the fluorescence of GFP-C/EBP.
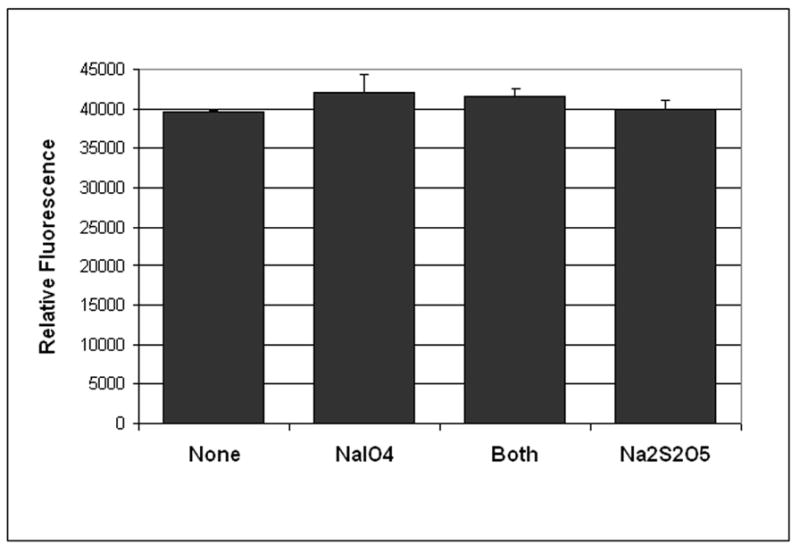
When 100 μl of 8.5 μg/ml (~200 nM) purified GFP-C/EBP was reacted with 20 μl of 50 mM NaIO4 (or water) and 20 μl Na2S2O5 (or water) and then diluted with 400 μl of buffer GS (50 mM Tris, pH 8, 5% glycerol, 1mM EDTA, 1 mM 2-mercaptoethanol), the fluorescence (λex = 398 nm, λem = 512 nm) is not altered. Shown are the average and standard deviation for duplicates. In other experiments (data not shown), the excitation and emission spectra were also unaltered.
The reacted GFP-C/EBP also has the same affinity for DNA as the unreacted protein, as shown in Fig. 2. For this experiment, different concentrations of radiolabeled EP18 DNA and the same amount of purified GFP-C/EBP and the amount of bound complex and free DNA determined using densitometry of the autoradiogram. DNA-binding affinity is unaffected by reaction.
Figure 2. The chemistry also has minimal effect on the DNA-binding or affinity of purified GFP-C/EBP.

To 45 μl of 0.31 mg/ml (~8.2 μM) purified GFP-C/EBP in buffer KP (0.1 M potassium phosphate, pH 6.8) was added 5 μl 10 mM NaIO4. The mixture was wrapped in foil and incubated in the dark for 30 min. on a rocking platform. 5 μl of 10 mM Na2S2O5 was added and incubation continued an additional 30 min and then 36 μl of buffer KP was added (0.155 mg/ml final). Similarly, 25 μl 2 μM radiolabeled rEP18 in buffer KP was treated with 2.5 μl of each reagent and diluted with 61 μl of buffer KP (0.82 μM final). The 90 μl of reacted GFP-C/EBP was mixed with 54 μl buffer GS, 18 μl of 0.5 mg/ml poly dI:dC, and 18 μl H2O and 20 μl of this mixture was incubated with 10 μl of the rEP18 or serial 1:2 dilutions of it in buffer KP for 30 min at room temperature, 5 μl of BPB-Glyc (0.015% bromophenol blue, 50% glycerol) was added and 20 μl was loaded onto 5% native PAGE for gel shift assay. During incubation, the concentration of GFP-C/EBP was 77 μg/ml and the DNA concentrations are shown in the graph. Without chemicals (squares, dashed line), the line is generated using the equation [Bound] = 0.004 x Ln [Free] + 0.0162, R2 = 0.91; with chemicals (triangles, solid line), the line is generated using [Bound] = 0.0035 x Ln [Free] + 0.0145, R2 = 0.96.
We also investigated whether the gel shifts for HEK293 nuclear extract with C/EBP, NFκB, AP1, or SP1 were adversely affected by reaction with NaIO4 alone or in combination with Na2S2O5. None of these gel shifts were adversely affected by the chemicals as shown in Supplemental Data Fig. 1. The presence of protein is also not detrimental to the coupling of the modified oligonucleotide to the support. Coupling for 2 h with 5′-32P- rEP18, either in the presence of the same amount of nuclear extract used for trapping or its absence, allowed 72.5 ± 1.6% or 64.2 ±1.5% (n=2), respectively, of the DNA to couple to the support. We conclude that neither protein activity or oligonucleotide coupling is substantially affected by the presence of either the chemicals or of proteins.
These results show that the DNA-aldehyde or the chemicals used to produce it are not affecting transcription factors. This means that using the same chemistry for trapping could be approached in more than one way: The DNA can be converted to the aldehyde, and the chemicals removed prior to trapping or the DNA could be mixed with protein prior to chemical modification, followed by chemistry and coupling. We chose the former for the experiments presented below although we have used both approaches successfully (data not shown). Therefore, the DNA is reacted with NaIO4, the reaction quenched with Na2S2O5, and then desalted by gel filtration on a P-6 spin column before trapping is performed.
3.2 Does trapping or affinity chromatography yield higher purity?
To test whether trapping may yield high enrichment, we used the GFP-C/EBP model system, this time using a crude bacterial extract containing the fusion protein. In Fig. 3A is shown the result of using the rEP18 for trapping after reaction, DNA-protein complex formation, and finally coupling for 2 h to the hydrazide agarose. Fig. 3B shows affinity purification using the rEP18 pre-coupled to the hydrazide-agarose. In either case, the same amount of bacterial extract was used, the columns prepared were the same size, washed the same, and the protein was eluted with the same TE0.5 buffer. The position of the band which comigrates with our purified GFP-C/EBP and is stained with a C/EBP antibody (data not shown), is indicated with an arrow. Although neither method purifies the protein to homogeneity, clearly the trapping approach gave less contaminant proteins, especially those of higher molecular mass.
Figure 3. Comparing GFP-C/EBP purity from affinity chromatography and trapping methods.

Bacterial (BL21) crude extract containing fusion protein GFP-C/EBP was used as a model to compare purity by affinity chromatography or trapping methods. A, For affinity chromatography, UltraLink Hydrazide resin was first coupled with rEP18. Briefly, 5 μL of 100 μM rEp18 oligonucleotide and 85 μL of buffer KP was reacted with 10 μL of 10 mM sodium metaperiodate in the dark for 30 minutes at room temperature. The reaction was quenched by addition of 10 μL of 10 mM sodium metabisulfite for 10 minutes at room temperature. The mixture was desalted on a 1 ml P-6 spin column to remove any unreacted chemicals. This aldehyde form of rEP18 DNA was mixed with a 200 μL UltraLink Hydrazide resin slurry (1:1), which was pre-washed thrice with buffer KP. After two-hour coupling at room temperature on a rotating wheel, the resin was washed with TE0.1 thrice, and incubated with 100 μL of bacterial extract (C) in a final 1.0 mL volume at 4°C for 30 minutes with gentle mixing. And then, the mixture was packed in a chromatographic column for collecting flow through (F). Following ten wash (0.5 mL each) with TE0.1, the last wash was collected (W10), and the packed resin was eluted by TE0.5 buffer (E). B, For trapping the aldehyde form of rEP18 was prepared as in panel A was mixed with bacterial extract in a final volume of 1 ml for 30 min. on a rocker at room temperature. The mixture was then combined with the hydrazide resin, and incubated for two-hour at room temperature before packing in a column. Column washing and elution were the same as in panel A. For both experiments, each fraction (20 μL) was loaded on a 12% SDS PAGE, and the proteins were stained by silver. The arrow to the right shows the position of the GFP-C/EBP purified by Ni2+-NTA-agarose.
In Fig. 4, we investigate a different DNA sequence. The hTERT promoter contains two E-box transcription factor binding sites. These, proximal and distal, were shown to bind the USF-2 transcription factor earlier [8]. This promoter, however, has no binding site for NFκB. We coupled the proximal E-box sequence DNA (a duplex containing the hybrid of SJ9 and SJ11 oligonucleotides) to hydrazide-agarose and used the resulting support for affinity chromatography of HEK 293 nuclear extract. The fractions were then further separated on a 12% SDS-PAGE gel and the Western blot is shown in the figure. The nuclear extract (C) clearly contains both USF-2 and NFκB. USF-2 has two bands stained by its antibody, which is consistent with the three isoforms of USF2 previously reported, one of which is 44 kDa. [11]. Not all isoforms are expressed in all cells; we observe two in HEK293. The upper band flows through (F) the column while the lower band, at 43 kDa., binds the column and elutes (E) in NaCl. NFκB does not bind the column. We conclude the column specifically binds one of the USF-2 isoforms but not the other or NFκB.
Figure 4. Western blot of USF-2 and a negative control on column fraction.

HEK293 nuclear extract was diluted 5-fold in TE0.1 and affinity chromatographic purification performed using SJ9 oligonucleotide (5′-GCTTCCCACGTGCGCArG-3′) annealed with the SJ11 complementary strand. The sequence is from the human telomerase (hTERT) promoter proximal E-box. Previously, we had shown that this sequence binds USF-2 [13]. The initial mixture (C), flow through (F), last wash through (W10) and eluate (E) by high salt were resolved by 12% SDS PAGE gel. Gels were electroblotted onto 0.2 μm PVDF membranes as previously described [14]. The dilution of USF2 (N-18) antibody was 1:1,000. Immunoreactive proteins were visualized using 1:10,000 goat anti-rabbit secondary antibody-HRP conjugate (Santa Cruz Biotechnology, CA) and detected by enhanced chemiluminescence (ECL). The Western blot of NFκB was done on the same blot after stripping, and the primary and secondary antibodies were diluted as 1:1,000 and 1:10,000, respectively. The arrows indicate where USF2 (~43kDa) or NFκB (~65 kDa) are located.
In Supplemental Data Fig. 2, is shown a Western blot of USF-2 used to determine the yield of USF-2 obtained in Fig. 4. Shown are dilutions of both the nuclear extract starting material and of the purified 43 kDa. band. The density was measured and fit to the equation of a straight line well (for nuclear extract, R2 = 0.97, for the eluate R2 = 0.93). From these results we determine that 31.7 ± 3.4% of the USF-2 present in nuclear extract was purified by affinity chromatography.
In Fig. 5 is shown the results of a silver stained SDS-PAGE gel. The same USF-2 binding E-box oligonucleotide was used for both affinity chromatography or for trapping, using aldehyde coupling for both. Affinity chromatography clearly binds the 43 kDa. band (arrow) shown in the Western blot (Figs. 4 and 6), however, using the trapping approach, a similar amount of this band is purified with far fewer contaminants apparent. While the results for trapping show predominantly a single band in Fig. 5, closer inspection of the stained gel do reveal several minor bands elsewhere on the gel, predominantly in the 60 and 70 kDa. range.
Figure 5. Comparing purity by affinity chromatography and trapping methods using the USF-2 oligonucleotide.

HEK293 nuclear extract (500 μg for each method) was diluted 5-fold in buffer KP and affinity chromatographic or trapping purification using annealed SJ9/11 oligonucleotide was performed. The initial mixture (C), flow through (F), last wash through (W10) and elute (E) by TE1.0 were resolved by 12% SDS PAGE gel. Proteins on each gel were stained by silver. The arrow denotes where USF-2 is located, based upon Western blotting.
Figure 6. Other E-box binding transcription factors are also purified by the trapping method.

500 μg of freshly prepared HEK293 nuclear extract was prepared for trapping using 200 nM annealed SJ9/SJ11 duplex oligonucleotide in buffer KP. After incubation for 30 min on ice, the mixture was mixed with 100 μl of hydrazide agarose and allowed to couple at 4°C for 4 h on a rotating wheel mixer. The mixture was then packed into a column and the unretained (FT, flow-through) fraction saved. The column was washed with 0.5 ml of TE0.1 buffer five times and then eluted with 0.5 ml of TE1.5. The eluted fraction (E) was collected, concentrated in a centrifugal concentrator to 40 μl. 20 μl was loaded onto 12% SDS-PAGE along with 20 μl of 10 μg HEK293 nuclear extract (C) and 20 μl of the flow-through fraction (FT) for electrophoresis and electroblotting. The blots were then probed with an antibody against USF-2, E2A, c-Myc or MyoD as indicated in the figure. A duplicate gel was also stained with silver in a separate experiment.
3.3 Is USF-2 the only E-box binding transcription factor purified by either method?
We next investigated whether USF-2 is the only E-box binding transcription factor purified by trapping or affinity chromatography. These results are shown in Figs. 6 and 7, respectively. For these experiments, we used a freshly prepared nuclear extract and eluted the column with TE1.5 instead of a stored, frozen nuclear extract and TE1.0 elution used in Fig. 5 to investigate these more minor components. We expanded the experiments to investigate four transcription factors known to bind the E-box motif. For trapping, E2A is retained to only a small extent by trapping and most is in the unretained, flow-through fraction. For MyoD and USF-2, much more is in the eluate and less is in the flow through. The c-Myc is well retained and elutes, with little in the flow-through. The silver stained gel shows band at the position of all of these proteins and little else.
Figure 7. Other E-box binding transcriptions are also purified by affinity chromatography with some differences.

500 μg HEK293 nuclear extract was prepared for affinity chromatography by dilution into buffer TE0.1. This was applied to a 0.1 ml column prepared from 20 pmoles annealed SJ9/SJ11 duplex oligonucleotide by aldehyde coupling. The flow-through (FT) was collected and the column was washed with 0.5 ml of TE0.1 buffer and then eluted with 0.5 ml of TE1.5. The conditions otherwise are the same as in Fig. 6.
For affinity chromatography (Fig. 7), the results were similar but different in some regards. c-Myc and E2A are both well retained by the column and elute, while MyoD is much less well retained and little elutes. The results for USF-2 were presented in Fig. 4 and are most similar to c-Myc and E2A. From these results, we conclude that all four of these E-box binding transcription factors are present in nuclear extract, can bind to the proximal E-box of the hTERT promoter, and are purified. However, the amount purified depends upon the method used. Trapping which gives the highest enrichment effectively purifies USF-2, MyoD and c-Myc while affinity chromatography would give the highest yield for E2A. Thus, both approaches of using aldehyde coupling of DNA are valuable tools, though the results may differ somewhat. Myogenin is also an E-box binding transcription factor, present in HEK293 nuclear extract, but it was purified by neither trapping nor affinity chromatography (data not shown).
The results show that for the model GFP-C/EBP and for USF-2, the trapping approach yields higher purity. The recovery from either affinity chromatography or trapping appears to be quite similar, though somewhat lower than anticipated. Clearly, the aldehyde based coupling procedure under development is mild chemistry which appears to have no detrimental effects to proteins activity for those proteins investigated. However, the method has some limitations. Intracellular proteins are typically not glycoproteins, which are normally found on secreted and membrane proteins or proteins targeted to specific organelles. However, the NaIO4 chemistry would also produce aldehydes in other glycans such as those present in glycoproteins. The chemistry has been widely used by others as a means of coupling glycoproteins such as antibodies and some glycoprotein enzymes such as horseradish peroxidase [12]. This more extensive literature also agrees that the activity of these proteins such as antigen-binding by antibodies and catalysis by enzymes is unaffected, though careful investigation of the yield of activity is seldom found. Here, we showed that transcription factors belonging to a variety of homology families are also unaffected. Clearly, the aldehyde based coupling of DNA that we previously reported is an important tool for transcription factor purification, whether used for affinity chromatography or the trapping approach, though the latter is likely to yield higher enrichment.
We are currently investigating the characterization of the purified proteins by mass spectrometry.
4. Conclusions
The aldehyde coupling chemistry can be used for either affinity chromatography or trapping. With one model protein (GFP-C/EBP) and the E-box binding transcription factors, trapping yielded somewhat higher purity.
Supplementary Material
Acknowledgments
This work was supported by NIH grant R01GM043609.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jia Y, Larionov O, Jarrett HW. Coupling of DNA to solid supports using 3′ terminal ribose incorporation. J Chromatogr A. 2014;1339:73–79. doi: 10.1016/j.chroma.2014.02.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gadgil H, Jarrett HW. Oligonucleotide trapping method for purification of transcription factors. J Chromatogr A. 2002;966:99–110. doi: 10.1016/s0021-9673(02)00738-0. [DOI] [PubMed] [Google Scholar]
- 3.Jiang D, Moxley RA, Jarrett HW. Promoter trapping of c-jun promoter-binding transcription factors. J Chromatogr A. 2006;1133:83–94. doi: 10.1016/j.chroma.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 4.Revzin A, von Hippel PH. Direct measurement of association constants for the binding of Escherichia coli lac repressor to non-operator DNA. Biochemistry. 1977;16:4769–4776. doi: 10.1021/bi00641a002. [DOI] [PubMed] [Google Scholar]
- 5.Berg OG, Winter RB, von Hippel PH. Diffusion-driven mechanisms of protein translocation on nucleic acids. 1. Models and theory. Biochemistry. 1981;20:6929–6948. doi: 10.1021/bi00527a028. [DOI] [PubMed] [Google Scholar]
- 6.Winter RB, Berg OG, von Hippel PH. Diffusion-driven mechanisms of protein translocation on nucleic acids. 3. The Escherichia coli lac repressor--operator interaction: kinetic measurements and conclusions. Biochemistry. 1981;20:6961–6977. doi: 10.1021/bi00527a030. [DOI] [PubMed] [Google Scholar]
- 7.Winter RB, von Hippel PH. Diffusion-driven mechanisms of protein translocation on nucleic acids. 2. The Escherichia coli repressor-operator interactions: Equilibrium measurements. Biochemistry. 1981;20:6961–6977. doi: 10.1021/bi00527a029. [DOI] [PubMed] [Google Scholar]
- 8.Jiang S, Galindo MR, Jarrett HW. Purification and identification of a transcription factor, USF-2, binding to E-box element in the promoter of human telomerase reverse transcriptase (hTERT) Proteomics. 2010;10:203–211. doi: 10.1002/pmic.200800693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jarrett HW, Taylor WL. Transcription factor-green fluorescent protein chimeric fusion proteins and their use in studies of DNA affinity chromatography. J Chromatogr A. 1998;803:131–139. doi: 10.1016/s0021-9673(97)01257-0. [DOI] [PubMed] [Google Scholar]
- 10.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 11.Viollet B, Lefrancois-Martinez AM, Henrion A, Kahn A, Raymondjean M, Martinez A. Immunochemical Characterization and Transacting Properties of Upstream Stimulatory Factor Isoforms. J Biol Chem. 1996;271:1405–1415. doi: 10.1074/jbc.271.3.1405. [DOI] [PubMed] [Google Scholar]
- 12.Nakane PK, Kawaoi A. Peroxidase-labeled antibody. A new method of conjugation. J Histochem Cytochem. 1974;22:1084–1091. doi: 10.1177/22.12.1084. [DOI] [PubMed] [Google Scholar]
- 13.Jiang S, Galindo M, Jarrett HW. Purification and identification of a transcription factor, USF-2, binding to E-box element in the promoter of human telomerase reverse transcriptase (hTERT) Proteomics. 2010;10:203–211. doi: 10.1002/pmic.200800693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jiang D, Jia Y, Zhou Y, Jarrett HW. Two–Dimensional Southwestern Blotting and Characterization of Transcription Factors On-blot. J Proteome Res. 2009;8:3693–3701. doi: 10.1021/pr900214p. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.