

Abstract
Type 2 diabetes mellitus and Alzheimer’s disease are both associated with increasing age, and each increases the risk of development of the other. Epidemiological, clinical, biochemical and imaging studies have shown that elevated glucose levels and diabetes are associated with cognitive dysfunction, the most prevalent cause of which is Alzheimer’s disease. Cross sectional studies have clearly shown such an association, whereas longitudinal studies are equivocal, reflecting the many complex ways in which the two interact. Despite the dichotomy, common risk and etiological factors (obesity, dyslipidemia, insulin resistance, and sedentary habits) are recognized; correction of these by lifestyle changes and pharmacological agents can be expected to prevent or retard the progression of both diseases. Common pathogenic factors in both conditions span a broad sweep including chronic hyperglycemia per se, hyperinsulinemia, insulin resistance, acute hypoglycemic episodes, especially in the elderly, microvascular disease, fibrillar deposits (in brain in Alzheimer’s disease and in pancreas in type 2 diabetes), altered insulin processing, inflammation, obesity, dyslipidemia, altered levels of insulin like growth factor and occurrence of variant forms of the protein butyrylcholinesterase. Of interest not only do lifestyle measures have a protective effect against the development of cognitive impairment due to Alzheimer’s disease, but so do some of the pharmacological agents used in the treatment of diabetes such as insulin (especially when delivered intranasally), metformin, peroxisome proliferator-activated receptors γ agonists, glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors. Diabetes must be recognized as a risk for development of Alzheimer’s disease; clinicians must ensure preventive care be given to control and postpone both conditions, and to identify cognitive impairment early to manage it appropriately.
Keywords: Cognition, Insulin resistance, Insulin, Butyrylcholinesterase, Dementia
Core tip: Type 2 diabetes mellitus is a risk factor for future development of Alzheimer’s disease, the most prominent cause of cognitive failure in the elderly. Common pathogenic mechanisms underpin both conditions. Therapeutic strategies in prevention (lifestyle changes) and pharmacological agents (biguanides, intranasal insulin, thiazolidinediones, glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors could also be useful against Alzheimer’s disease.
INTRODUCTION
The prevalence of diabetes mellitus is inexorably rising as evidenced by data from local, national and international studies[1-3]. Improved care of diabetes as well as general increase of longevity are predictive of greater proportion of elderly in the general population. It is well known that the critical problems of the aged relate to impaired activities of daily living and of cognitive decline. It is also recognized that compared to persons without diabetes, those with diabetes have dementia two to three times more commonly[4]. The economic and social burden on the health-care system as well as on care-providers would be enormous. It is essential if methods are available, to prevent or postpone the onset of both conditions; lifestyle changes appear to be effective in preventing both conditions[5].
CAUSES OF DEMENTIA
Dementia may be broadly classed into Alzheimer’s disease, vascular dementia, dementia with Lewy bodies and frontotemporal dementia[4]. Of these, Alzheimer’s disease is the most common type[6].
Dementia in diabetes: Epidemiological studies
A number of epidemiological studies have shown an association of dementia with diabetes. In the Hisayama study (1995), the relative risk (RR) of Alzheimer’s disease was 2.18 (95%CI: 0.97-4.9) and of vascular dementia 2.77 (95%CI: 2.59-2.97)[7]. A later publication from the same study group (2011) showed an RR of 2.05 (95%CI: 1.18-3.57) for Alzheimer’s disease and 1.82 for vascular dementia (95%CI: 0.89-3.71)[8]. The Rochester Study (1997) reported differences in the risk of Alzheimer’s disease based on gender: RR was 2.27 for men (95%CI: 1.55-3.31) and 1.37 for women (95%CI: 0.94-2.01)[9]. The well cited Rotterdam Study (199) showed a relative risk of 1.9 for Alzheimer’s disease (95%CI: 1.2-3.1)[10] (Table 1).
Table 1.
Dementia and diabetes
| Relative risk of AD | Relative risk of vascular dementia | Ref. |
| 2.18 | 2.77 | [7] |
| 2.05 | 1.82 | [8] |
| 1.9 | - | [10] |
AD: Alzheimer’s disease.
Conflicting results: Cross sectional vs longitudinal studies
Although there is a consensus from cross-sectional studies that hyperglycemia causes cognitive impairment, results from longitudinal studies are conflicting[4]. Two recent longitudinal studies show up differences in the occurrence of Alzheimer’s disease and glucose tolerance.
The Baltimore Longitudinal Study of aging prospectively assessed a cohort of community-dwelling individuals. An investigation was made to relate serial glucose intolerance, insulin resistance with brain β amyloid burden measured in vivo using carbon 11-labelled Pittsburgh Compound B. The latter is utilized to image β-amyloid (Aβ) in vivo with PET scan; 11C-Pittsburgh compound-B (11C-PiB), a PET Aβ ligand, is widely employed for early diagnosis of Alzheimer disease. It allows quantitative analysis of Aβ burden. Derived as a carbon-11 labelled thioflavin-T amyloid dys, it binds to Aβ plaques with high specificity and affinity. 11C-Pittsburgh compound-B (11C-PiB), correlates with the rate of cerebral atrophy. Pathological process for Alzheimer’s disease was established at autopsy[11]. Analysis was carried out using grouped and continuous mixed-models analyses. The key result of the study was, there was no significant correlation of brain markers of Alzheimer with insulin resistance or glucose intolerance during the follow up period of 22.1 years (SD:8.0)[11].
In contrast, a group from the University of Washington, which evaluated whether higher glucose levels increase the risk of dementia in those without diabetes, found that they did[12]. Participants, without dementia were drawn from the Adult Changes in Thought study (839 men, 1228 women, mean baseline age 76 years). In all 35264 glucose levels and 10208 glycosylated hemoglobin levels were analyzed. They were followed up for a median of 6.8 years. 524 subjects developed dementia (74 of 243 with diabetes and 450 of 1228 without diabetes). Higher levels of average glucose levels were related to development of dementia in both groups, ie those with and without known diabetes. The authors conclude that “higher glucose levels may be a risk factor for dementia, even among persons without diabetes”[12].
Can the conflicting results of these two rigorous, well-designed studies be resolved? The Baltimore study used both neuroimaging as well as autopsy to identify Alzheimer’s pathological processes. The Adult Changes in Thought study performed a 6 year follow up in a large group of well-defined elderly. In the former, insulin resistance and glucose intolerance were not a risk factor for Alzheimer’s changes; in the latter, higher glucose levels even among those without diabetes may be a risk factor for dementia.
The apparent differences can be attributed to the variety of pathological changes in diabetes leading to dementia including Alzheimer’s disease: chronic hyperglycemia, hypoglycemia (acute and recurrent), glycosylated of proteins, vascular disease, endothelial dysfunction, inflammation, altered blood brain barrier, dyslipidemia, insulin resistance, genetic predisposition, amyloid deposition and depression, among others[13]. More sensitive methods of measuring brain volume as a surrogate of cognitive function may throw light[4]. In addition, there is a flaw in using brain markers such as plaques in diagnosis of Alzheimer’s disease: subjects may have amyloid plaques, yet display no symptoms of Alzheimer’s disease throughout their life. This discrepancy between the presence of plaques and Alzheimers could play a role for a lack of finding a link between diabetes and Alzheimer’s disease.
Hyperglycemia
Hyperglycemia is a recognized risk factor for cognitive impairment as shown by the ACCORD-MIND study and others[14,15]. Biological reasons for such changes were ascribed to neural damage following advanced glycosylated end products and oxidative stress, osmotic stress damaging the blood brain barrier and resultant leak of toxic substances leading to further damage of nervous structures[4]. In addition to chronic hyperglycemia as assessed by glycated hemoglobin, glycemic variability was also proposed to contribute to cognitive dysfunction. Measurements by continuous glucose monitoring revealed cognitive function was better correlated with diurnal variation in blood glucose[16]. Post prandial glucose levels could also be a contributing factor, acting via oxidative stress[4].
Hypoglycemia
Although severe hypoglycemia was shown to be associated with dementia in the elderly[17], when hypoglycemia is avoided by careful treatment as in the DCCT/EDIC study, there was no association between hypoglycemia and cognitive dysfunction[18]. In the elderly however, hypoglycemia, when coupled with atherosclerosis leads to organic brain damage which is often irreversible[4].
Role of insulin in brain
Cognition may be affected not only by alterations in the level of glucose, but also via the action of insulin. Upon transport through the blood brain barrier, insulin binds to its receptors, and is involved in modulating cognitive function. A large number of insulin receptors occur in brain areas related to memory such as the hippocampus and cerebral cortex. In addition it also aids the release of β-amyloid peptide extracellularly, and increases the expression of the enzyme which degrades insulin, insulin degrading enzyme (IDE)[4]. As the latter also degrades β-amyloid peptide, insulin deficiency results in accumulation of β-amyloid peptide. Both hyperinsulinemia and hyperglycemia were shown to increase neuritic plaque formation[19].
Information is being available about the origin of insulin in the brain and its role in cognition. Originally, brain was considered to be insulin insensitive because insulin did not influence the glucose uptake by the bulk brain. However insulin has been shown to be a neuroregulatory peptide playing a role in food intake and in monitoring the energy stores of the body[20]. Interestingly a role for insulin in modulation of memory and cognition is also emerging. Its action has been observed in regions associated with reward recognition such as hippocampus, and in global cognition and memory. Rather than passing across the blood brain barrier from the periphery, insulin appears to be produced locally for action as a neurotransmitter, regulated by glucose levels. In addition a para-arteriolar pathway for transport at the level of microvasculature has been proposed[20].
The role of insulin in the pathogenesis of Alzheimer disease has been described. Insulin can modulate Aβ peptide in vitro. The peptide is well known as a neuropathological hallmark of AD. Low levels of insulin in the brain can decrease the Aβ release into extracellular compartments. In addition hypoinsulinemia in the central nervous sytem can lower the levels of insulin-degrading enzyme, thereby impairing Aβ clearance. In all, chronic hyperinsulinemia in the peripheral circulation, along with decreased uptake of insulin into the brain can lead to dysregulation of Aβ and inflammation[21,22].
Role of microvascular disease in cognitive decline
Studies have shown that retinopathy and nephropathy are associated with impairment of cognition[22,23]. Small vessels in both organs arise from a similar embryonic antecedent and share similar structures; it is conceivable therefore that insults (e.g., increased polyol pathway activity, myo-inositol dysmetabolism) result in similar adverse reactions in frontal lobe of the cerebral cortex leading to cognitive decline[4]. However, evidence is not unequivocal. Retinopathy is related to microvascular changes in diabetes. Because the retina shares many features with the brain, both developmental, anatomical (e.g., microvascular bed) and physiological (e.g., blood-tissue barrier), changes in retina were suggested to presage brain pathological processes. Alzheimer’s disease is known to involve the retina, such as the macula and the optic disc. It has been suggested that pathological changes in the retina such as macular deposits, reduced thickness of retinal nerve, cupping of optic disc and retinal microvascular changes may be related to cognitive dysfunction and Alzheimer disease[24].
Insulin resistance and Alzheimer’s disease
Downstream insulin signaling acts through a complex interplay involving phosphatidylinositol 3-kinase (P13K0 and mitogen activation protein kinase (MAPK). The latter is associated with most metabolic effects of insulin. Unlike its peripheral effects, the action of insulin in the central nervous system is dependent on its crossing the blood brain barrier via direct transfer[25] through an insulin receptor protein, which is selectively distributed. In addition there is also local synthesis of insulin in the CNS. Unlike peripheral insulin receptors, those in the CNS differ in terms of structure, function and size. They are highly populated in the olfactory bulb, hypothalamus, cortex, cerebellum, hippocampus and are expressed in both neurons and glia[26].
The physiological effects of insulin in the brain are unlike those in the periphery: in an animal model it suppressed food intake and increased the level of glucose[27], acting in a way as its own counterregulation[25]. Compared to its weight, the brain depends on a larger amount of glucose for its metabolic needs compared to other tissues. Glucose reaches it via facilitated diffusion transported by glucose transport proteins. In the brain, insulin does not have a major effect on either the transport of glucose to the brain or its basal metabolism[28,29]. While it is not a major regulator of glucose metabolism in the brain, insulin indirectly affects neurons by modulating neurotransmitter release, neural growth, tubulin activity, nerve survival and synaptic plasticity[26]. In humans, insulin improves cognition independent of its effects on peripheral glucose[30].
Whereas acute increases of insulin improve cognition, chronic hyperinsulinemia can adversely affect neuronal function in vitro by increasing susceptibility to toxin and stress-induced effects[31]. Glycated proteins and inflammatory mediators could also have a pathogenic role[25].
Protein aggregation, diabetes and Alzheimer’s disease
Protein aggregation has been suggested to be an underlying pathogenic factor between type 2 diabetes and Alzheimer’s disease[32]. A number of hypotheses were proposed to explain why a biological protein can be transformed into a pathological entity with the ability to self-assemble: aging, high concentrations of the protein, mutation of amino acids or abnormal post-translational modification, modulated by environmental factors[32].
Alzheimer’s disease is associated with accumulation of neurofibrillary tangles and amyloid fibers leading to neuronal cell loss. Amyloid β peptides form from cleavage of amyloid β-protein precursor seen in plaques. It is organized as amyloid fibrils, which are linear aggregates[32]. Diabetes is also characterized by localized and progressive amyloid deposition in the pancreatic β cell islets[33]. Common features of amyloid deposited in both Alzheimer’s disease and diabetes include: linear appearance, with a beta-sheet structure, which begin to form from spherical oligomers that can self-assemble[34]. The islet amyloid peptide is secreted by β cells of the pancreas and consists of 37 amino acids.
ApoE-ε4
Expression of ApoE-ε4, which is related to diabetes as well, increases the risk of early onset Alzheimer’s disease. It has increased ability to deposit Aβ, which is neurotoxic, and also impair its clearance[35]. ApoE-ε4 is less protective against oxidative stress and leads to cholinergic dysfunction seen in Alzheimer’s disease, besides modifying the cholesterol transporter protein ABCA1.
Other potential associations
In addition other associations are also being recognized as risk factors for both diabetes and Alzheimer’s disease: weight gain, acting perhaps through defective leptin signaling and increased formation of advanced glycation end products, which could have a pathogenic role in the amyloid plaques deposited in the brain[35]. Other emerging associations include disturbances in sleep and the circadian rhythm[36,37], and iron overload in brain among persons with obesity[38].
Insulin like growth factor
Animal studies have provided intriguing evidence that loss of insulin-like growth factors, along with insulin could lead to age-dependent brain atrophy with cognitive decline[39]. Insulin and its growth factors maintain brain protein content; their replacement can prevent brain protein loss, cell degeneration and demyelination. Lack of insulin and growth factors, which are common to type 2 diabetes and to Alzheimer’s disease could therefore play a role in their pathogenesis and provide therapeutic targets in their treatment[39].
Butyrylcholinesterase
Butyrylcholinesterase, belonging to the esterase family of enzymes that also contain acetylcholinesterase[40] has been evaluated in relation to insulin resistance, cardiovascular disease, obesity and dyslipidemia[36]. Variant forms of the enzyme with little or no activity exist in isolated geographic populations[41] with apparently no adverse health effects[42]. Butrylcholinesterase was studied in relation to both type 2 diabetes mellitus and to Alzheimer’s disease[43-45]. with studies suggesting a possible protective effect against Alzheimer’s disease and risk for fronto-temporal dementia[46].
A number of hypothesis were put forward for the association of butyrylcholinesterase with type 2 diabetes mellitus and Alzheimer’s disease[33,36]. Plaques in the brains of individuals with dementia had higher levels of butyrylcholinesterase, which is also localized in neurofibrillary tangles. It was also shown to attenuate amyloid formation[47].
Similarly in type 2 diabetes, butyrylcholinesterase may modify the expression of insulin resistance or by way of amyloid fibril deposition in β cells of the pancreas[33]. It was shown to interact with amylin and attenuate the formation of amylin fibril as well as its oligomer. When applied to cultured β cells, it was protective against amylin cytotoxicity[47]. Butyrylcholinesterase was shown to participate in the progression of metabolic syndrome to type 2 diabetes mellitus. A majority of subjects with type 2 diabetes show extracellular deposits formed by islet amyloid polypeptide (IAP), adjacent to the β cells. Elevated levels of IAP are found in conditions of insulin resistance. Disturbed balance of sympathetic and parasympathetic nervous system could participate in metabolic syndrome. Lower vagal activity could in part be caused by increased hydrolysis of acetylcholine mediated by increased butyrylcholinesterase[48]. Lowering of acetylcholinesterase results in reduced parasympathetic signals and increased ratio of sympathetic signals[48]. In addition BChE was reported to attenuate the formation of Aβ amyloid fibrils[47]. Essentially subjects with metabolic syndrome had elevated levels of BChE compared to those with type 2 diabetes and with controls. In vitro interaction of BChE was observed with amylin. It interacted with amylin, and attenuated the formation of both amylin fibril and oligomer formation, showing that it can protect cultured β cells from cytotoxicity due to amylin[47]. Thus increased BChE seen in metabolic syndrome could protect pancreatic β cells by reducing toxic amylin oligomer formation[47].
A bioinformatics study suggested the following sequences (E < e-5) were associated with both type 2 diabetes and Alzheimer’s disease: butyrylcholinesterase precursor K allele (NP_000046.1), acetylcholinesterase isoform E4-6 precorsor (NP_000656.1) and apoptosis-related acetylcholinesterase (1B41|A). In an animal study, streptozotocin-induced diabetes was associated with elevated butyrylcholinesterase activity, lowered superoxide dismutase and impaired cognitive function assessed by Morris water maze method[49]. Being low-grade inflammatory conditons, both type 2 diabetes mellitus and Alzheimer’s may be target conditions for utilization of butyrylcholinesterae as a biomarker[50], as well as a treatment target[33].
The principal drugs currently available to manage Alzheimer’s disease act via modifying butyrylcholinesterase levels. Renewed interest in the possible role of “missing genes” in people who are apparently healthy may aid in uncovering new treatment modalities[51]. Individuals with variant butyrylcholinesterae activity genes may be a potential group for such long-term follow up studies vis a vis their propensity or protection against diabetes and Alzheimer’s disease[40].
THERAPEUTIC IMPLICATIONS
The interest of linking type 2 diabetes mellitus and Alzheimer’s disease lies not in science, but more in translational science: how understanding the common pathogenesis can help in prevention and treatment of both conditions. Other than the scope for future therapies, evidence is now available for the currently available drugs used in diabetes[52] to have a modulatory effect on the cognitive decline due to Alzheimer’s disease.Along with its action on AMPK, metformin has been recently shown to influence the incretin system by increasing the secretion of glucagon-like peptide-1 (GLP-1)[53]. GLP-1 and GIP receptors are known to be expressed in the brain, and direct activation of these receptors may be a potential strategy to treat Allzheimer’s disease[54]. In addition, metformin also influences gut microbiome along with its other putative actions in the management of diabetes mellitus[55]. The use of drugs used in diabetes mellitus such as metformin, GLP-1 mimetics (exenatide and liraglutide) and peroxisome proliferator-activated receptors γ agonists may all be of potential benefit in the prevention and management of Alzheimer disease[56].
When a theoretical basis for insulin to affect brain responses was studied in clinical practice, a recent review of 8 published studies on effect of intranasal insulin on cognition, comprising 328 participants showed first of all, no significant adverse effects. Generally the authors concluded that “the limited clinical experience suggests potential beneficial cognitive effects of intranasal insulin”[57]. In a single study, use of 20 IU intranasal insulin showed improved immediate recall in Apoε4(-) subjects but not in Apoε4(+) subjects.
Other than correcting hyperglycemia, some of the conventional antidiabetic agents were shown to affect cognition. Metformin protected brain against oxidative stress in rats, and by preventing apoptosis[54]. However caution must be exercised because of metformin leading to increased biogenesis of Alzheimer’s amyloid peptides[58] and increased risk of cognitive impairment[59]. Thiazolidinediones, which act at the nuclear receptors of insulin-sensitive tissues, affect transcription of genes affecting lipid and glucose metabolism. Early studies suggested that this group of drugs may favourably affect cognition, before the clinical use tapered due to their adverse drug effect profile.
Pharmacological agents modulating the incretin system have now become mainstream in many markets world-wide. Pathological studies showed that sitagliptin lowered APP and Aβ deposition in the hippocampus of transgenic Alzheimer’s disease mice[53]. However whether it does so by lowering glucose levels or independently remains to be clarified. Interest arises because liraglutide, another drug in the same group had similar protective effects (Figure 1).
Figure 1.
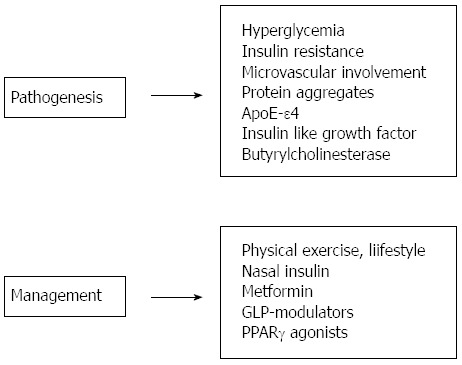
Links between Alzheimer’s disease and type 2 diabetes mellitus.
CONCLUSION
In conclusion, diabetes mellitus and Alzheimer’s disease are both common and increasing in incidence in the aging population. Recent evidence has shown common pathogenic factors operating in both conditions. Thereby common preventive and therapeutic agents may be used in their prevention and treatment. Physicians caring for the elderly must be aware of the increased risk of the other when one condition is present. Common pathogenesis and therapeutic agents make it possible to manage both using similar lifestyle changes and pharmacological agents.
Footnotes
Conflict-of-interest: None.
Open-Access: This article is an open-access article which was selected by an in-house editor and fully peer-reviewed by external reviewers. It is distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Peer-review started: August 30, 2014
First decision: November 27, 2014
Article in press: March 18, 2015
P- Reviewer: Hssan M, Hwu CM, Laher I S- Editor: Tian YL L- Editor: A E- Editor: Zhang DN
References
- 1.Sridhar GR, Putcha V, Lakshmi G. Time trends in the prevalence of diabetes mellitus: ten year analysis from southern India (1994-2004) on 19,072 subjects with diabetes. J Assoc Physicians India. 2010;58:290–294. [PubMed] [Google Scholar]
- 2.Anjana RM, Pradeepa R, Deepa M, Datta M, Sudha V, Unnikrishnan R, Bhansali A, Joshi SR, Joshi PP, Yajnik CS, et al. Prevalence of diabetes and prediabetes (impaired fasting glucose and/or impaired glucose tolerance) in urban and rural India: phase I results of the Indian Council of Medical Research-INdia DIABetes (ICMR-INDIAB) study. Diabetologia. 2011;54:3022–3027. doi: 10.1007/s00125-011-2291-5. [DOI] [PubMed] [Google Scholar]
- 3.Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 4.Kawamura T, Umemura T, Hotta N. Cognitive impairment in diabetic patients: Can diabetic control prevent cognitive decline? J Diabetes Investig. 2012;3:413–423. doi: 10.1111/j.2040-1124.2012.00234.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodríguez-Gómez O, Palacio-Lacambra ME, Palasí A, Ruiz-Laza A, Boada-Rovira M. Prevention of Alzheimer’s disease: a global challenge for next generation neuroscientists. J Alzheimers Dis. 2014;42 Suppl 4:S515–S523. doi: 10.3233/JAD-141479. [DOI] [PubMed] [Google Scholar]
- 6.Inestrosa NC, Alvarez A, Pérez CA, Moreno RD, Vicente M, Linker C, Casanueva OI, Soto C, Garrido J. Acetylcholinesterase accelerates assembly of amyloid-beta-peptides into Alzheimer’s fibrils: possible role of the peripheral site of the enzyme. Neuron. 1996;16:881–891. doi: 10.1016/s0896-6273(00)80108-7. [DOI] [PubMed] [Google Scholar]
- 7.Yoshitake T, Kiyohara Y, Kato I, Ohmura T, Iwamoto H, Nakayama K, Ohmori S, Nomiyama K, Kawano H, Ueda K. Incidence and risk factors of vascular dementia and Alzheimer’s disease in a defined elderly Japanese population: the Hisayama Study. Neurology. 1995;45:1161–1168. doi: 10.1212/wnl.45.6.1161. [DOI] [PubMed] [Google Scholar]
- 8.Ohara T, Doi Y, Ninomiya T, Hirakawa Y, Hata J, Iwaki T, Kanba S, Kiyohara Y. Glucose tolerance status and risk of dementia in the community: the Hisayama study. Neurology. 2011;77:1126–1134. doi: 10.1212/WNL.0b013e31822f0435. [DOI] [PubMed] [Google Scholar]
- 9.Leibson CL, Rocca WA, Hanson VA, Cha R, Kokmen E, O’Brien PC, Palumbo PJ. Risk of dementia among persons with diabetes mellitus: a population-based cohort study. Am J Epidemiol. 1997;145:301–308. doi: 10.1093/oxfordjournals.aje.a009106. [DOI] [PubMed] [Google Scholar]
- 10.Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology. 1999;53:1937–1942. doi: 10.1212/wnl.53.9.1937. [DOI] [PubMed] [Google Scholar]
- 11.Thambisetty M, Jeffrey Metter E, Yang A, Dolan H, Marano C, Zonderman AB, Troncoso JC, Zhou Y, Wong DF, Ferrucci L, et al. Glucose intolerance, insulin resistance, and pathological features of Alzheimer disease in the Baltimore Longitudinal Study of Aging. JAMA Neurol. 2013;70:1167–1172. doi: 10.1001/jamaneurol.2013.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crane PK, Walker R, Hubbard RA, Li G, Nathan DM, Zheng H, Haneuse S, Craft S, Montine TJ, Kahn SE, et al. Glucose levels and risk of dementia. N Engl J Med. 2013;369:540–548. doi: 10.1056/NEJMoa1215740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McCrimmon RJ, Ryan CM, Frier BM. Diabetes and cognitive dysfunction. Lancet. 2012;379:2291–2299. doi: 10.1016/S0140-6736(12)60360-2. [DOI] [PubMed] [Google Scholar]
- 14.Cukierman-Yaffe T, Gerstein HC, Williamson JD, Lazar RM, Lovato L, Miller ME, Coker LH, Murray A, Sullivan MD, Marcovina SM, et al. Relationship between baseline glycemic control and cognitive function in individuals with type 2 diabetes and other cardiovascular risk factors: the action to control cardiovascular risk in diabetes-memory in diabetes (ACCORD-MIND) trial. Diabetes Care. 2009;32:221–226. doi: 10.2337/dc08-1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Umegaki H, Kawamura T, Mogi N, Umemura T, Kanai A, Sano T. Glucose control levels, ischaemic brain lesions, and hyperinsulinaemia were associated with cognitive dysfunction in diabetic elderly. Age Ageing. 2008;37:458–461. doi: 10.1093/ageing/afn051. [DOI] [PubMed] [Google Scholar]
- 16.Rizzo MR, Marfella R, Barbieri M, Boccardi V, Vestini F, Lettieri B, Canonico S, Paolisso G. Relationships between daily acute glucose fluctuations and cognitive performance among aged type 2 diabetic patients. Diabetes Care. 2010;33:2169–2174. doi: 10.2337/dc10-0389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whitmer RA, Karter AJ, Yaffe K, Quesenberry CP, Selby JV. Hypoglycemic episodes and risk of dementia in older patients with type 2 diabetes mellitus. JAMA. 2009;301:1565–1572. doi: 10.1001/jama.2009.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jacobson AM, Musen G, Ryan CM, Silvers N, Cleary P, Waberski B, Burwood A, Weinger K, Bayless M, Dahms W, et al. Long-term effect of diabetes and its treatment on cognitive function. N Engl J Med. 2007;356:1842–1852. doi: 10.1056/NEJMoa066397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsuzaki T, Sasaki K, Tanizaki Y, Hata J, Fujimi K, Matsui Y, Sekita A, Suzuki SO, Kanba S, Kiyohara Y, et al. Insulin resistance is associated with the pathology of Alzheimer disease: the Hisayama study. Neurology. 2010;75:764–770. doi: 10.1212/WNL.0b013e3181eee25f. [DOI] [PubMed] [Google Scholar]
- 20.Gray SM, Meijer RI, Barrett EJ. Insulin regulates brain function, but how does it get there? Diabetes. 2014;63:3992–3997. doi: 10.2337/db14-0340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Craft S. The role of insulin dysregulation in aging and Alzheimer’s disease. In: Craft S, Christen Y, editors. Diabetes, Insulin and Alzheimer’s Disease. Berlin: Springer-Verlag Berlin Heidelberg; 2010. pp. 109–127. [Google Scholar]
- 22.Ding J, Strachan MW, Reynolds RM, Frier BM, Deary IJ, Fowkes FG, Lee AJ, McKnight J, Halpin P, Swa K, et al. Diabetic retinopathy and cognitive decline in older people with type 2 diabetes: the Edinburgh Type 2 Diabetes Study. Diabetes. 2010;59:2883–2889. doi: 10.2337/db10-0752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Bresser J, Reijmer YD, van den Berg E, Breedijk MA, Kappelle LJ, Viergever MA, Biessels GJ. Microvascular determinants of cognitive decline and brain volume change in elderly patients with type 2 diabetes. Dement Geriatr Cogn Disord. 2010;30:381–386. doi: 10.1159/000321354. [DOI] [PubMed] [Google Scholar]
- 24.Ikram MK, Cheung CY, Wong TY, Chen CP. Retinal pathology as biomarker for cognitive impairment and Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2012;83:917–922. doi: 10.1136/jnnp-2011-301628. [DOI] [PubMed] [Google Scholar]
- 25.Neumann KF, Rojo L, Navarrete LP, Farías G, Reyes P, Maccioni RB. Insulin resistance and Alzheimer’s disease: molecular links & amp; clinical implications. Curr Alzheimer Res. 2008;5:438–447. doi: 10.2174/156720508785908919. [DOI] [PubMed] [Google Scholar]
- 26.Banks WA. The source of cerebral insulin. Eur J Pharmacol. 2004;490:5–12. doi: 10.1016/j.ejphar.2004.02.040. [DOI] [PubMed] [Google Scholar]
- 27.Florant GL, Singer L, Scheurink AJ, Park CR, Richardson RD, Woods SC. Intraventricular insulin reduces food intake and body weight of marmots during the summer feeding period. Physiol Behav. 1991;49:335–338. doi: 10.1016/0031-9384(91)90053-q. [DOI] [PubMed] [Google Scholar]
- 28.Marfaing P, Penicaud L, Broer Y, Mraovitch S, Calando Y, Picon L. Effects of hyperinsulinemia on local cerebral insulin binding and glucose utilization in normoglycemic awake rats. Neurosci Lett. 1990;115:279–285. doi: 10.1016/0304-3940(90)90469-p. [DOI] [PubMed] [Google Scholar]
- 29.Hasselbalch SG, Knudsen GM, Videbaek C, Pinborg LH, Schmidt JF, Holm S, Paulson OB. No effect of insulin on glucose blood-brain barrier transport and cerebral metabolism in humans. Diabetes. 1999;48:1915–1921. doi: 10.2337/diabetes.48.10.1915. [DOI] [PubMed] [Google Scholar]
- 30.Kern W, Born J, Schreiber H, Fehm HL. Central nervous system effects of intranasally administered insulin during euglycemia in men. Diabetes. 1999;48:557–563. doi: 10.2337/diabetes.48.3.557. [DOI] [PubMed] [Google Scholar]
- 31.Schäfer M, Erdö SL. Development of glutamate neurotoxicity in cortical cultures: induction of vulnerability by insulin. Brain Res Dev Brain Res. 1991;62:293–296. doi: 10.1016/0165-3806(91)90179-m. [DOI] [PubMed] [Google Scholar]
- 32.Di Carlo M, Pasquale Picone P, Carrotta R, Giacomazza D, Biagio S. Alzheimer’s disease and type 2 diabetes: different pathologies and same features. In: Zimering M, editor. Topics in the prevention, treatment and complications of type 2 diabetes. Croatia: INTECH Open Access Publisher; 2011. [Google Scholar]
- 33.Sridhar GR, Thota H, Allam AR, Suresh Babu C, Siva Prasad A, Divakar Ch. Alzheimer’s disease and type 2 diabetes mellitus: the cholinesterase connection? Lipids Health Dis. 2006;5:28. doi: 10.1186/1476-511X-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu S. Aggregation drives “misfolding” in protein amyloid fiber formation. Amyloid. 2007;14:119–131. doi: 10.1080/13506120701260059. [DOI] [PubMed] [Google Scholar]
- 35.Akter K, Lanza EA, Martin SA, Myronyuk N, Rua M, Raffa RB. Diabetes mellitus and Alzheimer’s disease: shared pathology and treatment? Br J Clin Pharmacol. 2011;71:365–376. doi: 10.1111/j.1365-2125.2010.03830.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sridhar GR, Rao AA, Srinivas K, Nirmala G, Lakshmi G, Suryanarayna D, Rao PV, Kaladhar DG, Kumar SV, Devi TU, et al. Butyrylcholinesterase in metabolic syndrome. Med Hypotheses. 2010;75:648–651. doi: 10.1016/j.mehy.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 37.Kang JE, Lim MM, Bateman RJ, Lee JJ, Smyth LP, Cirrito JR, Fujiki N, Nishino S, Holtzman DM. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326:1005–1007. doi: 10.1126/science.1180962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blasco G, Puig J, Daunis-I-Estadella J, Molina X, Xifra G, Fernández-Aranda F, Pedraza S, Ricart W, Portero-Otín M, Fernández-Real JM. Brain iron overload, insulin resistance, and cognitive performance in obese subjects: a preliminary MRI case-control study. Diabetes Care. 2014;37:3076–3083. doi: 10.2337/dc14-0664. [DOI] [PubMed] [Google Scholar]
- 39.Serbedžija P, Ishii DN. Insulin and insulin-like growth factor prevent brain atrophy and cognitive impairment in diabetic rats. Indian J Endocrinol Metab. 2012;16:S601–S610. doi: 10.4103/2230-8210.105578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sridhar GR, Nirmala G, Apparao A, Madhavi AS, Sreelatha S, Rani JS, Vijayalakshmi P. Serum butyrylcholinesterase in type 2 diabetes mellitus: a biochemical and bioinformatics approach. Lipids Health Dis. 2005;4:18. doi: 10.1186/1476-511X-4-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sridhar GR, Nirmala G. Inborn errors in lipid metabolism. In: Lipid disorders., editor. Tripathy BB, Das S, editors. Association of Physicians of India. Mumbai: API College of Physicians; 2002. pp. 59–80. [Google Scholar]
- 42.Manoharan I, Boopathy R, Darvesh S, Lockridge O. A medical health report on individuals with silent butyrylcholinesterase in the Vysya community of India. Clin Chim Acta. 2007;378:128–135. doi: 10.1016/j.cca.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 43.Lehmann DJ, Johnston C, Smith AD. Synergy between the genes for butyrylcholinesterase K variant and apolipoprotein E4 in late-onset confirmed Alzheimer’s disease. Hum Mol Genet. 1997;6:1933–1936. doi: 10.1093/hmg/6.11.1933. [DOI] [PubMed] [Google Scholar]
- 44.Johansen A, Nielsen EM, Andersen G, Hamid YH, Jensen DP, Glümer C, Drivsholm T, Borch-Johnsen K, Jørgensen T, Hansen T, et al. Large-scale studies of the functional K variant of the butyrylcholinesterase gene in relation to Type 2 diabetes and insulin secretion. Diabetologia. 2004;47:1437–1441. doi: 10.1007/s00125-004-1459-7. [DOI] [PubMed] [Google Scholar]
- 45.Pohanka M. Butyrylcholinesterase as a biochemical marker. Bratisl Lek Listy. 2013;114:726–734. doi: 10.4149/bll_2013_153. [DOI] [PubMed] [Google Scholar]
- 46.Diamant S, Podoly E, Friedler A, Ligumsky H, Livnah O, Soreq H. Butyrylcholinesterase attenuates amyloid fibril formation in vitro. Proc Natl Acad Sci USA. 2006;103:8628–8633. doi: 10.1073/pnas.0602922103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shenhar-Tsarfaty S, Bruck T, Bennett ER, Bravman T, Aassayag EB, Waiskopf N, Rogowski O, Bornstein N, Berliner S, Soreq H. Butyrylcholinesterase interactions with amylin may protect pancreatic cells in metabolic syndrome. J Cell Mol Med. 2011;15:1747–1756. doi: 10.1111/j.1582-4934.2010.01165.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rao AA, Siva Reddy C, Sridhar GR, Annapurna A, Hanuman T, Prameela M, Suresh K, Prasannalaxmi S, Das UN. Enhanced butyrylcholinesterase activity may be the common link in triggering low-grade systemic inflammation and decrease in cognitive function in diabetes mellitus and Alzheimer’s disease. CurrNutr Food Sci. 2008;4:213–216. [Google Scholar]
- 49.Rao AA, Sridhar GR, Das UN. Elevated butyrylcholinesterase and acetylcholinesterase may predict the development of type 2 diabetes mellitus and Alzheimer’s disease. Med Hypotheses. 2007;69:1272–1276. doi: 10.1016/j.mehy.2007.03.032. [DOI] [PubMed] [Google Scholar]
- 50.Sridhar GR. Proteins of the esterase family: patents for some proteins in search of metabolic functions. Recent Patents on Biomarkers. 2011;1:205–212. [Google Scholar]
- 51.Kaiser J. The hunt for missing genes. Science. 2014;344:687–689. doi: 10.1126/science.344.6185.687. [DOI] [PubMed] [Google Scholar]
- 52.Cummings JL. Use of cholinesterase inhibitors in clinical practice: evidence-based recommendations. Am J Geriatr Psychiatry. 2003;11:131–145. [PubMed] [Google Scholar]
- 53.Shemesh E, Rudich A, Harman-Boehm I, Cukierman-Yaffe T. Effect of intranasal insulin on cognitive function: a systematic review. J Clin Endocrinol Metab. 2012;97:366–376. doi: 10.1210/jc.2011-1802. [DOI] [PubMed] [Google Scholar]
- 54.Vardarli I, Arndt E, Deacon CF, Holst JJ, Nauck MA. Effects of sitagliptin and metformin treatment on incretin hormone and insulin secretory responses to oral and “isoglycemic” intravenous glucose. Diabetes. 2014;63:663–674. doi: 10.2337/db13-0805. [DOI] [PubMed] [Google Scholar]
- 55.Napolitano A, Miller S, Nicholls AW, Baker D, Van Horn S, Thomas E, Rajpal D, Spivak A, Brown JR, Nunez DJ. Novel gut-based pharmacology of metformin in patients with type 2 diabetes mellitus. PLoS One. 2014;9:e100778. doi: 10.1371/journal.pone.0100778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Strittmatter WJ. Alzheimer’s disease: the new promise. J Clin Invest. 2012;122:1191. doi: 10.1172/JCI62745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sebastião I, Candeias E, Santos MS, de Oliveira CR, Moreira PI, Duarte AI. Insulin as a Bridge between Type 2 Diabetes and Alzheimer Disease - How Anti-Diabetics Could be a Solution for Dementia. Front Endocrinol (Lausanne) 2014;5:110. doi: 10.3389/fendo.2014.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen Y, Zhou K, Wang R, Liu Y, Kwak YD, Ma T, Thompson RC, Zhao Y, Smith L, Gasparini L, et al. Antidiabetic drug metformin (GlucophageR) increases biogenesis of Alzheimer’s amyloid peptides via up-regulating BACE1 transcription. Proc Natl Acad Sci USA. 2009;106:3907–3912. doi: 10.1073/pnas.0807991106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moore EM, Mander AG, Ames D, Kotowicz MA, Carne RP, Brodaty H, Woodward M, Boundy K, Ellis KA, Bush AI, et al. Increased risk of cognitive impairment in patients with diabetes is associated with metformin. Diabetes Care. 2013;36:2981–2987. doi: 10.2337/dc13-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]