

Abstract
AIM
To analyze the CYP4V2 mutations in five unrelated Chinese patients with Bietti crystalline corneoretinal dystrophy (BCD) and to provide clinical features of these patients. BCD is a rare monogenic autosomal recessively inherited disorder characterized by the presence of crystals in the retina and retinal pigment epithelium atrophy. Mutations in the CYP4V2 gene have been found to be causative for BCD.
METHODS
Ophthalmic examinations were carried out in the affected individuals. Peripheral blood samples were collected and genomic DNA was extracted. All exons and flanking intronic regions of the CYP4V2 gene were amplified with polymerase chain reaction and screened for mutations by direct DNA sequencing. One hundred control chromosomes were also screened to exclude nonpathogenic polymorphisms.
RESULTS
Fundus examination revealed the presence of tiny yellowish-sparkling crystals at the posterior pole of the fundus and atrophy of the retinal pigment epithelium in all patients. Choroid neovascularization was noted in one patient. Five different CYP4V2 mutations were identified, including two missense mutations (p.F73L, p.R400H), two splice site mutations (c.802-8_810del17insGC, c.1091-2A>G), and one single base-pair deletion (p.T479TfsX7 or c.1437delC). The two splice site mutations were identified in three of the patients with BCD. Mutation p.T479TfsX7 was a novel mutation not observed in any of 100 ethnically matched control chromosomes.
CONCLUSION
Mutation c.802-8_810del17insGC and c.1091-2A>G are common mutations in Chinese patients with BCD. Our results expand the allelic heterogeneity of BCD.
Keywords: Bietti's dystrophy, corneoretinal dystrophy, CYP4V2 gene, mutation
INTRODUCTION
Bietti crystalline corneoretinal dystrophy [BCD, (MIM#210370)] is an autosomal recessive retinal dystrophy first described by Bietti in 1937 [1]. It is characterized by numerous tiny sparkling yellow-white spots at the posterior pole of the fundus and progressive atrophy of the retinal pigment epithelium (RPE) and choriocapillaris [1],[2]. The small glittering crystals can also occur in the corneal limbus and circulating lymphocytes [1],[3]. Clinically, BCD is progressive and usually manifests itself between the 2nd and 4th decades of life with nyctalopia, decreased visual acuity, and constriction of the visual fields. Affected patients usually progress to legal blindness by the 5th or 6th decade of life[3]. Although reported worldwide, it is relatively common in China and Japan. An epidemiological survey of genetic diseases in a general population in China estimated the gene frequency to be 0.005[4],[5].
In 2000, the locus of the gene responsible for BCD was mapped to 4q35-qter by genetic linkage analysis in several BCD families, and subsequently CYP4V2 was identified as the causative gene[6],[7]. The CYP4V2 gene consists of 11 exons that encode a widely expressed 525 amino acid cytochrome P450 family member[7]. The biochemical analysis of cultured lymphocytes from patients with BCD showed abnormally high levels of triglycerides and cholesterol with the absence of two fatty acid binding proteins[8]. It is assumed that the protein encoded by the CYP4V2 gene plays a role in fatty acid and corticosteroid metabolism[7]–[9]. Up to date, more than fifty different CYP4V2 mutations have been described, most of which are missense mutations (http://www.hgmd.cf.ac.uk/ac/index.php). Here we report our mutation screening of the CYP4V2 gene in five unrelated Chinese BCD families and the clinical findings in the patients.
SUBJECTS AND METHODS
This study was approved by the Institutional Review Board of Peking Union Medical College Hospital and confirmed to the tenets of the Declaration of Helsinki and the Guidance of Sample Collection of Human Genetic Diseases by the Ministry of Public Health of China. Informed consent was obtained from the participating individuals before entry into this study.
Clinical Studies
The pedigrees of the five unrelated patients with BCD who were examined at the Peking Union Medical College Hospital are shown in Figure 1. Ophthalmic examinations including best-corrected visual acuity (BCVA), slit lamp biomicroscopy, fundus examination and photography, fundus autofluorescence and optical coherence tomography (OCT) were performed.
Figure 1. Pedigrees of the Chinese families with BCD.

Squares represent males, and circles represent females. Solid symbols indicate affected subjects with BCD. Unfilled symbols represent unaffected family members. A diagonal line indicates a deceased family member. The arrow indicates the proband.
Molecular Genetic Studies
Genomic DNA was isolated from peripheral white blood cells using a QIA amp DNA Blood Mini Kit (Qiagen, Hilden, Germany), which was used as the template to amplify the CYP4V2 gene. All exons and the flanking introns of the CYP4V2 gene were amplified by polymerase chain reaction (PCR) using previously reported primers [7]. PCR products were purified and then sequenced on an automated sequencer ABI 3730 Genetic Analyzer (ABI, Foster City, CA, USA). The results were analyzed with Lasergene SeqMan software (DNASTAR, Madison, WI, USA) and compared with a CYP4V2 reference sequence (GeneBank accession number: NM_207352), 100 control chromosomes were also screened to exclude nonpathogenic polymorphisms.
RESULTS
Clinical Evaluation
A total of 5 Chinese BCD patients from 5 unrelated families were recruited into this study, including 4 females and one male. The age of patients ranges from 26 to 69y (mean age±standard deviation, 37.2±17.9y). The onset of disease in most patients was during their 2nd to 4th decades. The most common initial symptoms were decrease of visual acuity and nyctalopia. BCVA was highly variable, ranging from finger count to 20/25. All patients affected by BCD showed typical tiny glittering crystals at the posterior pole of the fundus with different extent of RPE atrophy (Table 1, Figure 2). Slit lamp examination revealed no crystals in the cornea and corneal limbus of all affected individuals. All patients underwent OCT examination, and OCT scans demonstrated RPE atrophy and intraretinal lesions with increased signal intensity consistent with intraretinal crystals. Disturbed organization of photoreceptor inner segment/outer segment layer was observed in all patients. OCT also confirmed the subretinal location of scarred choroid neovascularization (CNV) with the lesion being localized between the neural retina and the RPE in patient II: 2/family A (Figure 2). The patient had received intravitreous injection of ranibizumab for five times to treat this.
Table 1. Clinical features of patients with CYP4V2 mutations.
| Patients | Age (a)/Gender | Age at onset (a) | First symptoms | BCVA (R/L) | Fundus finding | Mutation |
| II:2/family A | 31/F | 29 | DVA distortion of vision | 20/50 | CD, CNV | c.802-8_810del17insGC |
| 20/100 | RPE atrophy | c.1091-2A>G | ||||
| II:2/family B | 69/F | 65 | DVA nyctalopia | 20/400 | CD | c.1091-2A>G |
| FC | RPE atrophy | p.F73L | ||||
| II:2/family C | 31/F | 27 | Nyctalopia | 20/25 | CD pigmentation | c.802-8_810del17insGC |
| 20/25 | RPE atrophy | p.R400H | ||||
| II:1/family D | 29/F | 28 | DVA | 20/25 | CD | Homozygous |
| 20/30 | RPE atrophy | c.1091-2A>G | ||||
| II:1/family E | 26/M | 14 | Nyctalopia | 20/30 | CD | c.802-8_810del17insGC |
| 20/50 | RPE atrophy | p.T479TfsX7 |
DVA: Decrease of visual acuity; CD: Crystalline deposits; CNV: Choroidal neavascularization; FC: Finger count.
Figure 2. Fundus photograph, fundus autofluorescence and OCT images of patients.

A: Fundus photograph of the patient with mutation p.F73L showed many small yellowish-white sparkling crystalline deposits scattered throughout the fundus and RPE atrophy (II: 2/family B); B: Fundus image of the patient with mutation p.T479TfsX7 revealed typical crystalline deposits distributed in the posterior pole with most of them located at the macular (II: 1/family E); C: OCT image demonstrated the presence of RPE atrophy and irregularity of the tri-laminar structure of the high reflectance band (II: 1/family E); D: Fundus photograph revealed diffuse RPE atrophy with numerous crystalline deposits (II: 2/family A); E: OCT images showed foveal contour with scarred subfoveal CNV and intraretinal hyperintense lesions (the red arrow) confirming the presence of intraretinal crystals (II: 2/family A); F: Fundus autofluorescence picture depicted widespread hypo-autofluorescent atrophic areas (II: 2/family A).
Genetic Evaluation
Four patients from families A, B, C and E were found to harbor compound heterozygous CYP4V2 mutations, and one patient from family D carried homozygous mutations in the CYP4V2 gene (Table 1). In all, five different CYP4V2 mutations were identified among them, including two missense mutations (p.F73L, p.R400H), two splice site mutations (c.802-8_810del17insGC, c.1091-2A>G), and one single base-pair deletion (p.T479TfsX7 or c.1437delC). The two splice site mutations have been identified in three of the patients. To the best of our knowledge, mutation p.T479TfsX7 has not been described previously (Figure 3A, 3B). Furthermore, it was not observed in the 100 control chromosomes or present in Single Nucleotide Polymorphism database (dbSNP) or in the 1000 Genomes Project data set.
Figure 3. Direct sequencing of the CYP4V2 gene revealed one novel mutation p.T479TfsX7 (c.1437delC).
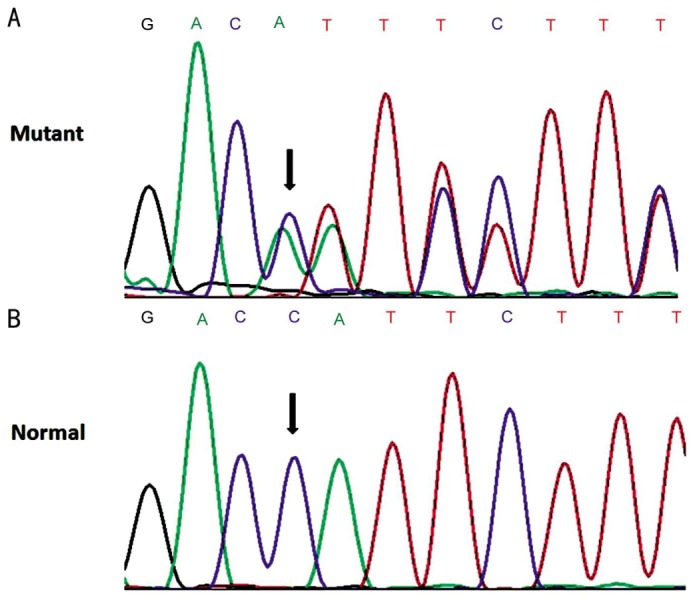
A: Heterozygous one-base-pair deletion of C at c.1437; B: The corresponding normal sequence.
DISCUSSION
In the present study, we collected five Chinese patients with clinically diagnosed BCD and screened for gene mutations in the CYP4V2 gene. By directly sequencing we identified four reported mutations (c.802-8_810del17insGC, c.1091-2A>G, p.F73L, p.R400H) and one novel mutation (p.T479TfsX7).
Clinically, BCD patients usually experience decreased vision and nyctalopia between the 2nd to 4th decades of life. In our study, the onset of disease in most patients falls into this time span, while patient II:2/family B had a late onset age of 65y and patient II:1/family E had an early onset age of 14y. A review of the literature shows patients with onset age older than 60y or earlier than 20y were frequently reported[10]–[14]. This could be related to the wide variation of the disease phenotype of BCD. The features and the combination of different CYP4V2 mutations, as well as other genetic modifiers and/or environmental factors may influence phenotype expression.
All affected individuals had obvious fundus characteristics of BCD, while crystals in the cornea or corneal limbus were detected in none of them. Although corneal crystals were described in the original cases[1], it has been reported that cornea crystalline deposits were not present in half of the reported cases of BCD[3]. A recent study by Yin et al[15] which investigated the genotypes and phenotypes of 17 BCD patients from 14 unrelated Chinese families did not find crystals in the cornea or corneal limbal in patients either. It appears that the pure retinal form of BCD is more common in Asians, in particular Chinese and Japanese, compared with Caucasians[3],[5],[15]–[18]. The other possibility is that the corneal crystals in some patients are too subtle to be detected by slit lamp biomicroscopy. The corneal rotating Scheimpflug imaging or the contact specular microscopy may be used to detect corneal deposits that are not apparent by other methods[19],[20].
Interestingly, molecular genetic analysis of the CYP4V2 gene demonstrated three of the five patients had the splice site mutation c.802-8_810del17insGC or c.1091-2A>G, indicating that they are common mutations in Chinese patients with BCD. This agrees with previous studies[7],[15],[16],[18]. The two mutations could result in skipping of exon 7 or exon 9 respectively. Unlike missense mutations, these two exon-skipping mutations are expected to cause a gross conformational change in the CYP4V2 protein structure and thus result in a more severe disease phenotype than those with homozygous or heterozygous amino acid substitution mutations. CNV is usually associated with defects of the RPE and Bruch's membrane and is a rare event in BCD. To date there are only six documented cases of BCD associated with CNV[13],[15],[21]–[23] while molecular genetic analysis was performed in three of them. Mamatha et al[23] reported two BCD patients of Asian Indian origin with parafoveal CNV. However, screening of the CYP4V2 gene and exon 5 of tissue inhibitor of metalloproteinase 3 (TIMP3) gene did not reveal any pathogenic variation[23]. Yin et al[15] recently reported one Chinese BCD patient associated with unilateral subfoveal CNV. Compound heterozygous mutations, c.1091-2A>G, and a newly detected single base-pair duplication mutation c.1062dupA, were identified in this patient, while no pathogenic variant was observed in the TIMP3 gene. Mutation c.1062dupA could result in a defective amino acid sequence (p.V355SfsX4) and cause gross conformational change in the CYP4V2 protein. In the present study, patient II: 2/family A had typical clinical feature of BCD and CNV at a relatively early age, and had received intravitreous injection of ranibizumab for five times. Genetic analysis revealed compound heterozygous mutations c.802-8_810del17insGC and c.1091-2A>G in the patient. This partly confirmed that compound heterozygous mutations c.802-8_810del17insGC and c.1091-2A>G could cause clinically severe BCD and agreed with a previous study[14]. However, electroretinogram examination and visual field examination were needed to further assess the phenotype. The underlying mechanism of CNV formation in this case is unclear while RPE and choroidal atrophy as well as the chronic irritation of Bruch's membrane by the crystals might have played a role[23].
Although more than fifty disease-causing mutations in the CYP4V2 gene have been identified, there are only three reported small deletions, all of which occurred in the intron-exon boundary of the gene (http://www.hgmd.cf.ac.uk/ac/index.php). The p.T479TfsX7 (c.1437delC) mutation in the coding region of the CYP4V2 gene was firstly described in the present study. It could cause a frame shift effect and give rise to a premature stop after eighteen nucleotides. This may result in nonsense-mediated decay of the mutant transcript and loss of the protein and thus cause BCD. Patient II: 1/family E with this mutation did not show any special fundus appearance when compared with other BCD patients. Recently mutation p.F73L was reported for the first time by Yin et al[15]. It was frequently detected in seven out of 34 alleles in their study, suggesting it may be another recurrent mutation in Chinese patients with BCD[15]. The detection of mutation p.F73L in the present study has further supported this point.
In conclusion, we described the genetic and phenotypic characteristics of five Chinese patients affected by BCD. Our study confirmed that c.802-8_810del17insGC and c.1091-2A>G are common mutations in Chinese patients with BCD and reported one novel CYP4V2 mutation. Understanding the genetics of BCD will allow for the development of potential new diagnostic modalities and gene therapy regimens.
Acknowledgments
The authors thank all study participants.
Conflicts of Interest: Tian R, None; Wang SR, None; Wang J, None; Chen YX, None.
REFERENCES
- 1.Bietti G. Ueber familiaeres Vorkommen von ‘Retinitis punctata albescens’ (verbunden mit ‘Dystrophia marginalis cristallinea corneae’), Glitzern des Glaskoerpers und anderen degenerativen Augenveraenderungen. Klin Mbl Augenheilk. 1937;99:737–757. [Google Scholar]
- 2.Bagolini B, Ioli-Spada G. Bietti's tapetoretinal degeneration with marginal corneal dystrophy. Am J Ophthalmol. 1968;65(1):53–60. doi: 10.1016/0002-9394(68)91028-3. [DOI] [PubMed] [Google Scholar]
- 3.Kaiser-Kupfer MI, Chan CC, Markello TC, Crawford MA, Caruso RC, Csaky KG, Guo J, Gahl WA. Clinical biochemical and pathologic correlations in Bietti's crystalline dystrophy. Am J Ophthalmol. 1994;118(5):569–582. doi: 10.1016/s0002-9394(14)76572-9. [DOI] [PubMed] [Google Scholar]
- 4.Hu DN. Genetic aspects of retinitis pigmentosa in China. Am J Med Genet. 1982;12(1):51–56. doi: 10.1002/ajmg.1320120107. [DOI] [PubMed] [Google Scholar]
- 5.Hu DN. Ophthalmic genetics in China. Ophthal Paediat Genet. 1983;2:39–45. [Google Scholar]
- 6.Jiao X, Munier FL, Iwata F, Hayakawa M, Kanai A, Lee J, Schorderet DF, Chen MS, Kaiser-Kupfer M, Hejtmancik JF. Genetic linkage of Bietti crystallin corneoretinal dystrophy to chromosome 4q35. Am J Hum Genet. 2000;67(5):1309–1313. doi: 10.1016/s0002-9297(07)62960-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li A, Jiao X, Munier FL, Schorderet DF, Yao W, Iwata F, Hayakawa M, Kanai A, Shy Chen M, Alan Lewis R, Heckenlively J, Weleber RG, Traboulsi EI, Zhang Q, Xiao X, Kaiser-Kupfer M, Sergeev YV, Hejtmancik JF. Bietti crystalline corneoretinal dystrophy is caused by mutations in the novel gene CYP4V2. Am J Hum Genet. 2004;74(5):817–826. doi: 10.1086/383228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee J, Jiao X, Hejtmancik JF, Kaiser-Kupfer M, Chader GJ. Identification, isolation, and characterization of a 32-kDa fatty acid-binding protein missing from lymphocytes in humans with Bietti crystalline dystrophy (BCD) Mol Genet Metab. 1998;65(2):143–154. doi: 10.1006/mgme.1998.2723. [DOI] [PubMed] [Google Scholar]
- 9.Lee J, Jiao X, Hejtmancik JF, Kaiser-Kupfer M, Gahl WA, Markello TC, Guo J, Chader GJ. The metabolism of fatty acids in human Bietti crystalline dystrophy. Invest Ophthalmol Vis Sci. 2001;42(8):1707–1714. [PubMed] [Google Scholar]
- 10.Yokoi Y, Sato K, Aoyagi H, Takahashi Y, Yamagami M, Nakazawa M. A novel compound heterozygous mutation in the CYP4V2 gene in a Japanese patient with Bietti's crystalline corneoretinal dystrophy. Case Rep Ophthalmol. 2011;2(3):296–301. doi: 10.1159/000331885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiao X, Mai G, Li S, Guo X, Zhang Q. Identification of CYP4V2 mutation in 21 families and overview of mutation spectrum in Bietti crystalline corneoretinal dystrophy. Biochem Biophys Res Commun. 2011;409(2):181–186. doi: 10.1016/j.bbrc.2011.04.112. [DOI] [PubMed] [Google Scholar]
- 12.Haddad NM, Waked N, Bejjani R, Khoueir Z, Chouery E, Corbani S, Mégarbané A. Clinical and molecular findings in three Lebanese families with Bietti crystalline dystrophy: report on a novel mutation. Mol Vis. 2012;18:1182–1188. [PMC free article] [PubMed] [Google Scholar]
- 13.Gupta B, Parvizi S, Mohamed MD. Bietti crystalline dystrophy and choroidal neovascularisation. Int Ophthalmol. 2011;31(1):59–61. doi: 10.1007/s10792-010-9406-8. [DOI] [PubMed] [Google Scholar]
- 14.Lai TY, Ng TK, Tam PO, Yam GH, Ngai JW, Chan WM, Liu DT, Lam DS, Pang CP. Genotype phenotype analysis of Bietti's crystalline dystrophy in patients with CYP4V2 mutations. Invest Ophthalmol Vis Sci. 2007;48(11):5212–5220. doi: 10.1167/iovs.07-0660. [DOI] [PubMed] [Google Scholar]
- 15.Yin H, Jin C, Fang X, Miao Q, Zhao Y, Chen Z, Su Z, Ye P, Wang Y, Yin J. Molecular analysis and phenotypic study in 14 Chinese families with Bietti crystalline dystrophy. PLoS One. 2014;9(4):e94960. doi: 10.1371/journal.pone.0094960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin J, Nishiguchi KM, Nakamura M, Dryja TP, Berson EL, Miyake Y. Recessive mutations in the CYP4V2 gene in East Asian and Middle Eastern patients with Bietti crystalline corneoretinal dystrophy. J Med Genet. 2005;42(6):e38. doi: 10.1136/jmg.2004.029066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chan WM, Pang CP, Leung AT, Fan DS, Cheng AC, Lam DS. Bietti crystalline retinopathy affecting all 3 male siblings in a family. Arch Ophthalmol. 2000;118(1):129–131. [PubMed] [Google Scholar]
- 18.Shan M, Dong B, Zhao X, Wang J, Li G, Yang Y, Li Y. Novel mutations in the CYP4V2 gene associated with Bietti crystalline corneoretinal dystrophy. Mol Vis. 2005;11:738–743. [PubMed] [Google Scholar]
- 19.Zenteno JC, Ayala-Ramirez R, Graue-Wiechers F. Novel CYP4V2 gene mutation in a Mexican patient with Bietti's crystalline corneoretinal dystrophy. Curr Eye Res. 2008;33(4):313–318. doi: 10.1080/02713680801983217. [DOI] [PubMed] [Google Scholar]
- 20.Wada Y, Abe T, Shiono T, Tamai M. Specular microscopic findings of corneal deposits in patients with Bietti's crystalline corneal retinal dystrophy. Br J Ophthalmol. 1999;83(9):1095. doi: 10.1136/bjo.83.9.1088k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Atmaca LS, Muftuoglu O, Atmaca-Sonmez P. Peripapillary choroidal neovascularization in Bietti crystalline retinopathy. Eye (Lond) 2007;21(6):839–842. doi: 10.1038/sj.eye.6702673. [DOI] [PubMed] [Google Scholar]
- 22.Le Tien V, Atmani K, Querques G, Massamba N, Souied EH. Ranibizumab for subfoveal choroidal neovascularization in Bietti crystalline retinopathy. Eye (Lond) 2010;24(11):1728–1729. doi: 10.1038/eye.2010.116. [DOI] [PubMed] [Google Scholar]
- 23.Mamatha G, Umashankar V, Kasinathan N, Krishnan T, Sathyabaarathi R, Karthiyayini T. Molecular screening of the CYP4V2 gene in Bietti crystalline dystrophy that is associated with choroidal neovascularization. Mol Vis. 2011;17:1970–1977. [PMC free article] [PubMed] [Google Scholar]