

Abstract
Signal peptide peptidase-like 3 (SPPL3) is a Golgi-resident intramembrane-cleaving protease that is highly conserved among multicellular eukaryotes pointing to pivotal physiological functions in the Golgi network which are only beginning to emerge. Recently, SPPL3 was shown to control protein N-glycosylation, when the key branching enzyme N-acetylglucosaminyltransferase V (GnT-V) and other medial/trans Golgi glycosyltransferases were identified as first physiological SPPL3 substrates. SPPL3-mediated endoproteolysis releases the catalytic ectodomains of these enzymes from their type II membrane anchors. Protein glycosylation is a multistep process involving numerous type II membrane-bound enzymes, but it remains unclear whether only few of them are SPPL3 substrates or whether SPPL3 cleaves many of them and thereby controls protein glycosylation at multiple levels. Therefore, to systematically identify SPPL3 substrates we used Sppl3-deficient and SPPL3-overexpression cell culture models and analyzed them for changes in secreted membrane protein ectodomains using the proteomics “secretome protein enrichment with click sugars (SPECS)” method. SPECS analysis identified numerous additional new SPPL3 candidate glycoprotein substrates, several of which were biochemically validated as SPPL3 substrates. All novel SPPL3 substrates adopt a type II topology. The majority localizes to the Golgi network and is implicated in Golgi functions. Importantly, most of the novel SPPL3 substrates catalyze the modification of N-linked glycans. Others contribute to O-glycan and in particular glycosaminoglycan biosynthesis, suggesting that SPPL3 function is not restricted to N-glycosylation, but also functions in other forms of protein glycosylation. Hence, SPPL3 emerges as a crucial player of Golgi function and the newly identified SPPL3 substrates will be instrumental to investigate the molecular mechanisms underlying the physiological function of SPPL3 in the Golgi network and in vivo. Data are available via ProteomeXchange with identifier PXD001672.
Signal peptide peptidase (SPP)1 and SPP-like (SPPL) proteases belong to the GxGD family of intramembrane-cleaving aspartyl proteases, which also comprises the presenilins, the catalytically active subunits of γ-secretase, implicated in Alzheimer's disease etiology (1). These proteases are multipass membrane proteins and their active site is buried within the membrane's hydrophobic interior. Similar to other intramembrane-cleaving proteases, SPP/SPPLs endoproteolyse membrane protein substrates within or adjacent to their membrane-spanning regions. In contrast to the well-documented selectivity of presenilins for type I membrane protein substrates, SPP/SPPLs appear to favor substrates adopting a type II topology in the membrane (1). In mammals, genes encoding five SPP/SPPL proteases (SPP, SPPL2a, SPPL2b, SPPL2c, and SPPL3) have been identified (2–4) and substrates have been described for all but SPPL2c (reviewed in (1)). Although SPP, SPPL2b, and γ-secretase only accept substrates with a rather short ectodomain (5–7) and thus usually require a preceding release of the substrates' ectodomain (shedding) SPPL3 was recently shown to directly endoproteolyse full-length membrane protein substrates close to their predicted transmembrane segments without prior ectodomain shedding (8, 9). Thus, SPPL3 unexpectedly acts as a type II membrane protein-selective sheddase catalyzing the release and secretion of the substrates' ectodomain.
SPPL3 orthologs are found in most multicellular eukaryotes and the degree of conservation is extraordinarily high pointing to a pivotal physiological function (1). Following up altered cellular N-glycosylation patterns associated with elevated or reduced SPPL3 expression in vitro and in vivo, Golgi-resident glycosyltransferases including the N-glycan branching enzyme N-acetylglucosaminyltransferase V (GnT-V) were recently uncovered as the first physiological type II membrane protein substrates of SPPL3 (9). Meanwhile, SPPL3 has additionally been shown to modulate cytosolic Ca2+ release and NFAT (nuclear factor of activated T cells) signaling following T-cell receptor engagement apparently in a nonproteolytic fashion (10).
The cleavage mechanism of SPPL3 in contrast to that of γ-secretase, SPP and SPPL2b has not been investigated in more detail. γ-secretase, SPP and SPPL2b catalyze multiple cleavages within the center of the substrates' transmembrane domain to release the corresponding cleavage products into the cytosol or the extracellular space, respectively (reviewed in (1, 11)). In contrast to SPP and SPPL2b, SPPL3 seems to cleave its substrates within the C-terminal region of the substrates' transmembrane or possibly even within the juxtamembrane domain. This assumption, however, is based solely on the cleavage site of SPPL3 within GnT-V, which has been recently mapped (9). In order to fully understand how the individual members of the GxGD intramembrane protease family recognize and cleave their substrates it is crucial to map further cleavage sites.
Protease substrates can be identified with proteomic techniques that have already proven themselves as powerful tools for the identification of novel protease substrates and the in-depth characterization of protease degradomes (12). However, reliable detection of quantitative changes of rather small or very hydrophobic proteolytic cleavage products as a result of the successive proteolytic processing of membrane protein substrates by intramembrane proteases is challenging with proteomic approaches. Recent studies that aimed at identifying novel substrates of γ-secretase (13), SPP (14), and SPPL2a/SPPL2b (15) in cell culture observed a measurable accumulation of substrates in cellular membranes following pharmacological inhibition of the respective protease(s) or their genomic ablation. However, for SPP and SPPL2a/SPPL2b, these approaches only uncovered very few new candidate substrates (14, 15).
Here we identified novel SPPL3 substrates in a cell culture model over-expressing SPPL3 and in Sppl3-deficient cells. To circumvent previous difficulties with substrate identification for intramembrane proteases, we made use of the fact that SPPL3 cleavage results in the release of the substrates' ectodomain into the conditioned medium (also referred to as secretome) (9). There, proteomic identification and quantification of a membrane protein's ectodomain allows the protein to be scored as a SPPL3 substrate candidate, if the ectodomain levels are increased upon SPPL3 overxpression or reduced when SPPL3 is not expressed. To this aim we used the previously developed “secretome protein enrichment with click sugars” (SPECS) technology, which allows proteomic analysis of cellular secretomes at unprecedented depth enriching glycoproteins but renders nonglycosylated proteins invisible to detection. SPECS was previously used successfully to identify novel substrates of another membrane-bound protease, the β-site APP cleaving enzyme 1 (BACE1) (16).
We now employed this technology to define the spectrum of SPPL3 substrates in human and murine cell culture models. Besides the previously identified SPPL3 substrates (9), SPECS analyses of the same cell culture models revealed numerous additional candidate glycoprotein SPPL3 substrates. Interestingly, the majority of these novel substrates are type II-membrane Golgi-resident factors implicated in multiple cellular glycosylation pathways. This confirms SPPL3's crucial role in cellular N-glycosylation but also underscores a more general role of SPPL3 in Golgi homoeostasis and regulation of cellular glycosylation pathways. Hence, this study complements the substrate spectrum of SPPL3, confirms its role as a type II membrane protein-selective sheddase and underscores the importance of SPPL3 in Golgi biology.
MATERIALS AND METHODS
Cell Lines, Antibodies, and Reagents
HEK293 T-Rex™ cells stably expressing ectopic, HA epitope-tagged SPPL3 under control of a doxycycline-inducible CMV promoter are described elsewhere (17) and were kept under standard conditions. Immortalized Sppl3+/+ and Sppl3−/− mouse embryonic fibroblast (MEF) lines had been originally obtained from litter-mate embryos and were described recently (9). siRNA pools targeting SPPL3 (#M-006042-02), EXTL3 (#M-012578-00), XYLT2 (#M-013040-01), B3GALT6 (#M-021340-01), HS6ST1 (#M-011944-01), HS6ST2 (#M-015558-01), and SGK196 (#M-005321-00) as well as a nontargeting control siRNA pool were purchased from Thermo Fisher Scientific (Waltham, MA). Antibodies used for substrate validation by immunoblotting were purchased from the following commercial sources: anti-ASPH (pAb, H-300, Santa Cruz Biotechnologies, Dallas, TX), anti-β4GalT1 (pAb, AF3609, R&D Systems, Minneapolis, MN), anti-β3GalT6 (pAb, ab103375, Abcam, Cambridge, UK), anti-Calnexin (pAb, Enzo Life Sciences, Farmingdale, NY), anti-Cant1 (mAb, 861206, R&D Systems), anti-EXTL3 (mAb, clone G-5, Santa Cruz Biotechnologies), anti-GalNAcT10 (pAb, AF7575, R&D Systems), anti-GnT-V (mAb, clone 706824, R&D Systems), anti-HS6ST1 (pAb, AF5057, R&D Systems), anti-HS6ST2 (pAb, HPA034625, Sigma, St. Louis, MO), anti-Integrin α5 (pAb, 4705, Cell Signaling Technology, Cambridge, UK), anti-OGFOD3 (mAb, clone F-19, Santa Cruz Biotechnologies), anti-Sgk196 (mAb, clone S-23, Santa Cruz Biotechnologies), anti-TOR1AIP1 (pAb, 60–1001, EMD Millipore, Billerica, MA), anti-TOR1AIP1 (pAb, NBP1–19122, Novus Biologicals, Littleton, CO), and anti-XylT2 (mAb, clone G-1, Santa Cruz Biotechnologies). The anti-Sppl3 mAb (clone 7F9) was described earlier (8). ManNAz was synthesized as previously described (16, 18). DBCO-PEG12-Biotin was obtained from Click Chemistry Tools (Scottsdale, AZ).
SPECS Workflow
Secretome enrichment and subsequent proteomic analyses were essentially performed as detailed earlier (16). In brief, 40 million cells were plated and incubated for 48 h in 20 ml DMEM supplemented with 10% (v/v) FCS and 200 nm tetraacetyl-N-azidoacetyl mannosamine (ManNAz). Cells ectopically expressing SPPL3 were pre-incubated with media supplemented with 1 μg/ml doxycycline and labeling was likewise performed in the presence of doxycycline. Conditioned media were collected and filtered through 0.45 mm PVDF filter (Millex) into a VivaSpin 20 ultrafiltration unit (cutoff, 30 kDa) at 4 °C. VivaSpin 20 columns were centrifuged at 4600 r.p.m. at 4 °C to remove nonmetabolized ManNAz. The retentate was filled with 20 ml H2O. This procedure was repeated three times. In the last step, the H2O refill step was omitted. Instead, 100 nm of DBCO-PEG12-biotin (Click-chemistry tools) diluted in 1 ml ddH2O was added to biotinylate metabolically azide-labeled glycoproteins. Columns were incubated overnight at 4 °C. For removal of nonreacted DBCO-PEG12-Biotin, VivaSpin20 columns were subject to three times of centrifugation with subsequent PBS buffer refill. After the last centrifugation step, the retentate was diluted in 5 ml PBS with 2% SDS (v/v) and 2 mm Tris(2-carboxyethyl)phosphine (TCEP). For purification of biotinylated proteins, the sample was loaded on a 10-ml polyprep column with a streptavidin bed of 300 μl streptavidin slurry. After binding of proteins, streptavidin beads were washed 3 times with 10 ml PBS supplemented with 1% SDS. To elute the biotinylated and azide-labeled glycoproteins, streptavidin beads were boiled with urea sample buffer containing 3 mm biotin to compete for the binding of biotinylated proteins.
SDS-PAGE Separation, Trypsinization
Proteins were separated on a 10% Tris/glycine SDS gel. Afterward, qualitatively equal gel slices were cut out from the gel with the exception of the remaining albumin band at around 60 kDa. Proteins in the gel slices were subject to trypsinization according to standard protocols (19).
Mass Spectrometric Analysis
Mass spectrometry experiments were performed on an Easy nLCnanoflow HPLC system II (Proxeon) connected to an LTQ-Velos Orbitrap Pro (Thermo Fisher Scientific). Peptides were separated by reverse phase chromatography using in-house made 30 cm columns (New Objective, FS360-75-8-N-S-C30) packed with C18-AQ 2.4 mm resin (Dr Maisch GmbH, Part No. r124.aq). A 90-min gradient (5–40%) at a flow rate of 200 nl/min was used. The measurement method consisted of an initial FTMS scan recorded in profile mode with 30 000 m/z resolution, a mass range from 300 to 2000 m/z and a target value of 1,000,000. Subsequently, collision-induced dissociation (CID) fragmentation was performed for the 15 most intense ions with an isolation width of 2 Da in the ion trap. A target value of 10,000, enabled charge state screening, a monoisotopic precursor selection, 35% normalized collision energy, an activation time of 10 ms, wide band activation and a dynamic exclusion list with 30 s exclusion time were applied.
Analysis of Mass Spectrometry Data
For data analysis five biological replicates of the HEK293T secretome were analyzed with the freely available MaxQuant suite (version 1.4.1.2) and five biological replicates with two technical replicates of the MEF secretome were analyzed with the freely available MaxQuant suite (version 1.5.0.124) (20). Protein identification was performed using the integrated Andromeda search algorithm (21). Mass recalibration was done by a first search in a reduced murine protein database. First search, mass recalibration and Mmain search was of tryptic peptides were performed using a human Uniprot database downloaded 08/21/2012 (86749 entries) for the HEK293T secretome and a murine Uniprot database downloaded on 05/16/2014 (version 3.68) (51389 entries) for the MEF secretome allowing for N-terminal acetylation and oxidation of methionine as variable modifications and carbamidomethylation of cysteine as fixed modifications. Two missed cleavages were allowed. Peptide as well as protein false discovery rate was set to 1%. Mass accuracy was set to 20 ppm for the first search and 5 ppm for the main search. Label-free quantification (LFQ) was performed between the respective control and Sppl3 knockout or SPPL3 overexpression control and SPPL3 overexpression or knockout conditions on the basis of unique and razore peptides. Missing LFQLFQ values were imputed in Perseus 1.5.16 following a standard distribution. p values were calculated with a heteroscedastic, two-sided student's t test for all proteins based on the log2 transformed absolute LFQ values in case of the HEK293 secretome and on log2 transformed relative intensity ratio values for the MEF secretome where two technical replicates allowed calculation of the variance within one biological replicate. Proteins with a p value of p ≤ 0.05 were considered as hits. MaxQuant output files (protein groups and peptides) (supplemental Table S2–S3 and S5–S6) and calculated values (supplemental Table S1 and S4) for both data sets are attached as supplementary files. The mass spectrometry proteomics raw data including MaxQuant outputfiles have been deposited to at the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the PRIDE partner repository with the data set identifier PXD001672. GO term analysis was performed on the SPPL3 overexpression data set considering elevated type II membrane proteins as hits and the remaining secretome as background using GOrilla (http://cbl-gorilla.cs.technion.ac.il/) (22).
Substrate Validation by Immunoblotting
HEK293 cells were transfected with siRNA pools at a final concentration of 20 nm using Lipofectamine® RNAiMax (Life Technologies, Carlsbad, CA) according to the manufacturer's instructions. Ectopic expression of catalytically active SPPL3 was induced by supplementing cell culture media with 1 μg/ml doxycycline. 48 h after transfection or 48 h after doxycycline mediated induction of SPPL3 overexpression cells were washed twice with prewarmed Opti-MEM™ (Life Technologies) and then cultivated in Opti-MEM™ for additional 48 h. Conditioned supernatants of MEFs were collected for 8 h. All supernatants were cleared from detached cells by centrifugation and secreted proteins were isolated by TCA precipitation (9). Supernatant samples were normalized to lysate protein content prior to loading. Cell lysis, SDS-PAGE and immunoblotting were conducted as described earlier (9).
RESULTS
Identification of Novel SPPL3 Substrates in Cells Ectopically Expressing SPPL3
Because SPPL3 was previously shown to endoproteolyse individual full-length type II membrane proteins triggering their secretion (8, 9), we applied a mass spectrometry-based method to identify novel SPPL3 substrates in a more systematic fashion. Our first experimental approach followed the rational that ectopic SPPL3 overexpression should lead to a quantitative increase of SPPL3 substrate ectodomains in the secretome compared with control cells with endogenous SPPL3 expression. Using the SPECS method, we determined and quantitatively compared the glycoprotein secretome of HEK293 cells inducibly overexpressing SPPL3 (Fig. 1). The same cell line without doxycycline induction served as a control, ensuring that essentially identical cell populations were compared and ruling out the possibility that cell line- or clone-specific effects result in false positive hits. Under these experimental conditions, we reproducibly detected secretion of 428 proteins with a protein and peptide false discovery rate of ≤1%, of which the majority (48.5%) were annotated as secreted proteins and 29.4% and 18.6% as type I and type II transmembrane proteins, respectively, suggesting that the latter had undergone proteolytic cleavage prior to secretion (Fig. 1A and Table I). The volcano plot depicted in Fig. 1B summarizes changes in protein secretion from cells ectopically expressing SPPL3 compared with control cells. Statistically significant changes (p value cutoff: 0.05) are shown above the dashed line. A substantial number of proteins were more abundantly secreted from cells overexpressing SPPL3 (Fig. 1B, top right quadrant). Type II membrane proteins were strongly enriched constituting the majority (83%) of all proteins with increased secretion (Fig. 1B, yellow circles and Fig. 1C). Furthermore, mapping of the identified peptides against the protein topology of these hits via QARIP analysis (23) revealed that, with the exception of one single peptide, all detected peptides exclusively originated from extracellular/luminal protein moieties and did not cover the N-terminal cytosolic or membrane-spanning regions (supplemental Fig. S1). This suggests that these proteins had undergone proteolytic processing prior to secretion (supplemental Fig. S1). Table I lists all type II membrane proteins that displayed a significantly increased or decreased secretion upon SPPL3 overexpression, including, among others, the previously identified SPPL3 substrate ER α-mannosidase I (gene name: MAN1B1) (9). Secretion of a number of proteins (Fig. 1B, red dots, top left quadrant and Table I) including a few type II membrane proteins appeared to be reduced following ectopic SPPL3 expression, suggesting that SPPL3 overexpression may cause a reduced secretion of these proteins. However, it was shown that in cells ectopically expressing SPPL3, the extent of complex N-glycosylation is diminished as overexpressed SPPL3 prematurely liberates Golgi glycosyltransferases and thus impairs their intracellular activity on nascent glycoproteins (9). In line with this, the extent of terminal glycan sialylation may also be reduced, resulting in a less efficient metabolic labeling of de novo synthesized glycoproteins proteins with ManNAz and subsequent biotin labeling with bioorthogonal click chemistry. Thus, it is more likely that the apparent reduction in secretion may simply be because of less efficient metabolic labeling of glycoproteins in cells overexpressing SPPL3.
Fig. 1.

Secretome analysis of HEK293 cells ectopically expressing active SPPL3. A, Pie chart of all glycoproteins which have been identified in at least 4 out of 5 biological replicates of the HEK293 secretome subdivided according to their topology and the presence of a transmembrane domain. B, Volcano plot: Plotted is the negative lg of the p value (y axis) of log2 LFQ values versus log2 LFQ ratio of SPPL3 overexpressing and control cells (x axis) of a given identified protein. The interspaced light gray line defines the significance level of p = 0.05 (Student's t test, multiple hypothesis testing was not applied). Almost exclusively type II membrane proteins clustered in the upper right quadrant upon SPPL3 overexpression. The previously identified substrate MAN1B1 (9) is given in bold. C, Pie chart of all proteins that were significantly increased in secretomes of HEK293 cells overexpressing SPPL3.
Table I. Type II membrane proteins that are significantly altered ectopic expression of catalytically active SPPL3 in HEK293 cells. The table contains all membrane proteins with type II orientation which are significantly (p value ≤ 0.05) increased or decreased upon SPPL3 overexpression. Those type II proteins that were more abundantly secreted following SPPL3 overexpression are shown above the dotted line, those secreted less below. Indicated are the names of the proteins, the gene name, number of unique peptides, the mean of the ratio between SPPL3 overexpression and control (con) of 5 biological replicates and the p value calculated with a two-sided, heteroscedastic t-test based on the log2 transformed LFQ values for the control and the SPPL3 overexpression condition. Previously published SPPL3 substrates are highlighted in bold, those identified also in the MEF screen (Fig. 3 and Table II) in italics.
| Protein names | Gene name | Unique peptides | Mean ratio SPPL3/Con | p value |
|---|---|---|---|---|
| Aspartyl/asparaginyl beta-hydroxylase | ASPH | 35 | 287.58 | 1.53E-04 |
| Gamma-glutamyltransferase 7 | GGT7 | 13 | 122.60 | 1.00E-06 |
| Torsin-1A-interacting protein 2 | TOR1AIP2 | 2 | 77.17 | 7.11E-05 |
| Cytoskeleton-associated protein 4 | CKAP4 | 17 | 21.11 | 6.05E-03 |
| Exostosin-like 3 | EXTL3 | 13 | 17.98 | 1.51E-02 |
| Adipocyte plasma membrane-associated protein | APMAP | 6 | 17.93 | 8.08E-04 |
| Glutaminyl-peptide cyclotransferase-like protein | QPCTL | 3 | 11.88 | 2.49E-03 |
| Sodium/potassium-transporting ATPase subunit beta-1 | ATP1B1 | 4 | 10.37 | 4.79E-05 |
| Probable inactive protein kinase-like protein SgK196 | SGK196 | 12 | 7.35 | 3.32E-05 |
| Endoplasmic reticulum mannosyl-oligosaccharide 1,2-alpha-mannosidase | MAN1B1 | 14 | 6.99 | 9.64E-05 |
| 2-oxoglutarate and iron-dependent oxygenase domain-containing protein 3 | OGFOD3 | 7 | 6.63 | 4.13E-03 |
| Beta-1,4-galactosyltransferase 7 | B4GALT7 | 3 | 5.96 | 8.16E-05 |
| Heparan-sulfate 6-O-sulfotransferase 2 | HS6ST2 | 15 | 5.05 | 1.28E-04 |
| Beta-1,3-galactosyltransferase 6 | B3GALT6 | 9 | 3.92 | 5.54E-04 |
| Xylosyltransferase 2 | XYLT2 | 15 | 3.27 | 9.39E-03 |
| Alpha-mannosidase 2x | MAN2A2 | 33 | 3.18 | 3.42E-03 |
| Type 2 lactosamine alpha-2,3-sialyltransferase | ST3GAL6 | 5 | 3.08 | 1.11E-02 |
| Bifunctional heparan sulfate N-deacetylase/N-sulfotransferase 1 | NDST1 | 21 | 3.05 | 1.48E-06 |
| Protein ITFG3 | ITFG3 | 4 | 2.78 | 3.34E-02 |
| Endothelin-converting enzyme 1 | ECE1 | 15 | 2.76 | 6.11E-04 |
| Ectonucleoside triphosphate diphosphohydrolase 6 | ENTPD6 | 10 | 2.71 | 1.49E-03 |
| Fukutin-related protein | FKRP | 2 | 2.46 | 2.59E-02 |
| Alpha-1,3-mannosyl-glycoprotein 2-beta-N-acetylglucosaminyltransferase | MGAT1 | 15 | 2.38 | 4.44E-02 |
| Heparan-sulfate 6-O-sulfotransferase 1 | HS6ST1 | 10 | 2.13 | 3.14E-02 |
| Bifunctional heparan sulfate N-deacetylase/N-sulfotransferase 2 | NDST2 | 14 | 1.90 | 3.52E-02 |
| Heparan sulfate glucosamine 3-O-sulfotransferase 3B1 | HS3ST3B1 | 9 | 0.58 | 4.27E-02 |
| Cyclic AMP-dependent transcription factor ATF-6 alpha | ATF6 | 3 | 0.36 | 3.06E-02 |
| N-acetyllactosaminide beta-1,3-N-acetylglucosaminyltransferase | B3GNT1 | 19 | 0.30 | 1.95E-02 |
A number of the candidate SPPL3 substrates identified in this first proteomic approach localize to the Golgi compartment, for instance, the bifunctional heparin sulfate N-deacetylase/N-sulfotransferase 1 (NDST1) (24), β1,3-galactosyltransferase 6 (β3GalT6) (25), the ER α-mannosidase I (gene name: MAN1B1) (26) and others. Individual other candidate substrates were reported to localize to the ER (e.g. the O-mannose kinase Sgk196 (27)) or even to the nuclear lamina (e.g. TOR1AIP1 (28)). From a functional perspective (supplemental Table S7), the type II membrane proteins identified are implicated in diverse cellular processes, but the majority of candidate substrates has been linked to cellular Golgi glycan anabolism, including N-glycosylation (e.g. α-mannosidase 2x, gene name: MAN2A2), glycosaminoglycan biosynthesis (e.g. exostosin-like 3 (EXTL3)) and O-mannosylation (Sgk196).
In order to validate selected type II substrate candidates, we next analyzed protein levels of selected candidate substrates in TCA-precipitated conditioned supernatants and whole-cell lysates of HEK293 cells. Transfection with candidate substrate-specific siRNA pools strongly reduced signals for secreted and cellular proteins demonstrating that the antibodies used are specific. In control cells (Fig. 2 and supplemental Fig. S2, − Dox) we observed constitutive secretion of endogenous XylT2 (Fig. 2A), HS6ST2 (Fig. 2B), HS6ST1 (Fig. 2C), β3GalT6 (Fig. 2D), Sgk196 (Fig. 2E), EXTL3 (Fig. 2F) and OGFOD3 (supplemental Fig. S2). A robust boost in secretion of all candidate substrates was observed upon ectopic expression of catalytically active SPPL3 (Fig. 2 and supplemental Fig. S2, + Dox), fully confirming the SPECS data (Fig. 1B and Table I). In addition, SPPL3 overexpression reduced intracellular levels of some candidate substrates, confirming that the increased secretion is facilitated by SPPL3 rather than by elevated intracellular protein levels. In contrast, we detected no constitutive secretion of the candidate substrates ASPH and TOR1AIP1, likely because secreted levels were below the detection limit (supplemental Fig. S3). This likewise explains the substantial boost of their secretion in cells overexpressing SPPL3 (supplemental Fig. S3, + Dox), which is well in line with the stark increase (> 130-fold) in secretion observed in the proteomics screen (Fig. 1 and Table I). Compared with all other candidate substrates the cytosolic domain of TOR1AIP1 is relatively long (supplemental Fig. S1), therefore the secreted cleavage product, detected with a polyclonal antibody directed against TOR1AIP1's luminal part, is substantially smaller than full-length TOR1AIP1 (supplemental Fig. S3B). A mAb directed against the TOR1AIP1 N terminus fails to detect this cleavage product confirming the proteomics data (supplemental Fig. S1) and strengthening that TOR1AIP1 is proteolytically cleaved from its N-terminal membrane anchor by SPPL3 (supplemental Fig. S3B).
Fig. 2.

Immunoblot validation of novel SPPL3 candidate substrates identified in HEK293 cells overexpressing catalytically active SPPL3. Levels of secreted (s) and cellular XylT2 (A), HS6ST2 (B), HS6ST1 (C), β3GalT6 (D), Sgk196 (E), and EXTL3 (F) were analyzed by immunoblotting in TCA-precipitated conditioned supernatants (sup) and whole-cell lysates, respectively, obtained from HEK293 cells. Where indicated, cells were transiently transfected with nontargeting control siRNA pools (CTRL) or with siRNA pools specific for SPPL3 (20 nm). siRNA pools targeting the individual substrates were transfected to control for antibody specificity. SPPL3 was ectopically expressed in a HEK293 cell line stably transfected with SPPL3 under control of a doxycycline-sensitive repressor. To induce SPPL3 overexpression, media were supplemented with doxycycline (+ Dox) throughout the experiment, whereas control cells were left untreated (− Dox). In all panels calnexin was used as a loading control. **, Sgk196 specific band of unknown nature.
Because SPPL3 overexpression may lead to cleavage of substrates that are not subject to proteolysis by SPPL3 under physiological conditions, we investigated whether the basal secretion of the candidate substrates may be likewise dependent on endogenously expressed SPPL3. To this end, we transiently knocked down SPPL3 expression in HEK293 cells using specific siRNA pools (Fig. 2, supplemental Figs. S2 and S3). Indeed, in supernatants of cells with reduced endogenous SPPL3 expression, we observed a reduced secretion for most of the candidate substrates tested (HS6ST2 (Fig. 2B), HS6ST1 (Fig. 2C), β3GalT6 (Fig. 2D), Sgk196 (Fig. 2E), EXTL3 (Fig. 2F) and OGFOD3 (supplemental Fig. S2)). In most cases, loss of SPPL3 did not completely abrogate substrate secretion, indicating that, in addition to SPPL3, other proteases may likewise execute substrate proteolysis. In addition, some of the candidate substrates displayed an intracellular accumulation of the full-length protein (HS6ST2 (Fig. 2B), HS6ST1 (Fig. 2C) and β3GalT6 (Fig. 2D)) compared with cells transfected with control siRNA. Such an accumulation could be because of an impaired proteolytic turn-over as it was previously observed for other SPPL3 substrates (9).
Taken together, these experiments confirm our SPECS data and demonstrate that SPPL3 facilitates shedding of these newly identified substrates in an over-expression setting in cell culture. Although probably other proteases or compensatory effects also contribute to substrate shedding, endogenous SPPL3 activity contributes to a certain extent to substrate release under physiological conditions. Thus, our biochemical validation corroborates that the proteins identified constitute genuine SPPL3 substrates.
Identification of Novel SPPL3 Substrates in Sppl3-deficient Fibroblasts
Secretome analysis of cells ectopically expressing SPPL3 only identified one (ER α-mannosidase I, MAN1B1) of the previously described (9) SPPL3 substrates whereas GnT-V (gene name: MGAT5), β4GalT1 (gene name: B4GALT1), and β3GnT1 (gene name: B3GNT1, but recently renamed to B4GAT1 (29, 30)) were not identified. However, this is not surprising, as SPPL3 overexpression substantially reduced intracellular levels of these endogenous glycosyltransferases, yet did not boost glycosyltransferase secretion, which we consider an indication that cleaved glycosyltransferase ectodomains may be subject to intracellular degradation (9).
Hence, to uncover additional candidate SPPL3 substrates that undergo proteolytic processing in a manner similar to GnT-V, β4GalT1, and β3GnT1, we conducted a second SPECS analysis that compared the secretomes of immortalized murine embryonic fibroblasts (MEF) obtained from wild type (Sppl3+/+) and Sppl3−/− embryos (Fig. 3 and Table II). We detected 330 proteins in at least four (of five) replicates with a peptide and protein false discovery rate of ≤1%. The majority of proteins identified are secreted proteins (59%), whereas a smaller fraction of proteins are annotated to adopt a type I and type II topology in the membrane, respectively (26 and 11%, Fig. 3A and supplemental Table S4). As illustrated in a volcano plot (Fig. 3B), the quantitative comparison of the two secretomes revealed substantial differences between the secretome of Sppl3 knock out and the respective control cells. Secretion of numerous proteins was found to be reproducibly elevated (Fig. 3B, green dots) whereas secretion of many others was reduced (Fig. 3B, red dots). Importantly, the majority of secretome proteins with type II membrane protein annotation (Fig. 3B, yellow dots) displayed reduced secretion in Sppl3−/− MEF (Fig. 3B) as expected for genuine SPPL3 substrates (9). Among these candidate Sppl3 substrates the previously identified SPPL3 substrates GnT-V, β4GalT1 and β3GnT1 (reduced by 97%, 52%, and 74%, respectively) were found (Fig. 3 and Table II). Similar to the results observed in the overexpression approach, the list of type II membrane proteins with altered secretion in Sppl3−/− MEF includes predominantly Golgi-localized type II membrane proteins implicated in Golgi N- and also O-glycosylation but also other cellular processes (Table II). Interestingly, NDST1, HS6ST1, HS6ST2, MGAT1 and α-mannosidase 2x (gene name: Man2a2) can be found among the candidate substrates identified in both screening approaches (Tables I and II).
Fig. 3.

Secretome analysis of Sppl3-deficient MEF. A, Pie chart of all glycoproteins which have been identified in at least four out of five biological replicates of the MEF secretome subdivided according to their topology and the presence of a transmembrane domain. B, Volcano plot: Plotted is the values of the negative lg of the p value of log2 intensity ratios (y axis) versus the log2 ratio of intensity values of Sppl3 knockout MEF and wild type MEF (x axis) of a given identified protein. The interspaced light gray line defines the significance level of p = 0.05. Type II membrane proteins were almost exclusively reduced and thus clustered in the left half of the volcano. The previously identified substrates are given in bold, those also identified in the SPPL3 overexpression secretome analysis in italics.
Table II. Type II membrane proteins which are significantly altered in secretomes of Sppl3−/− MEF. Table contains all membrane proteins with type-II orientation which are significantly altered upon Sppl3 knockout. Those type II proteins that were less abundantly secreted from Sppl3−/− MEF compared to control cells are shown above the dotted line, those secreted more below. Indicated are the names of the proteins, the gene name, number of unique peptides, the mean of the ratio between Sppl3 knockout (KO) and control (wt) of five biological replicates and the p value calculated with a heteroscedastic, two-sided student's t-test based on the log2 transformed intensity ratios between the control and the SPPL3 overexpression condition. Previously published SPPL3 substrates are highlighted in bold, those identified also in the HEK293 SPPL3 overexpression screen (Fig. 1 and Table I) in italics.
| Protein names | Gene names | Unique peptides | Mean Sppl3 KO/wt | p value |
|---|---|---|---|---|
| Alpha-mannosidase 2 | Man2a1 | 25 | 0.03 | 1.19E-11 |
| Alpha-1,6-mannosylglycoprotein 6-beta-N-acetylglucosaminyltransferase A | Mgat5 | 13 | 0.06 | 6.60E-08 |
| Agrin | Agrn | 30 | 0.04 | 7.97E-07 |
| Soluble calcium-activated nucleotidase 1 | Cant1 | 6 | 0.11 | 3.26E-05 |
| Protein FAM198B | Fam198b | 14 | 0.21 | 8.12E-05 |
| Carbohydrate sulfotransferase 14 | Chst14 | 11 | 0.29 | 1.23E-04 |
| N-acetyllactosaminide beta-1,3-N-acetylglucosaminyltransferase | B3gnt1 | 5 | 0.26 | 1.36E-04 |
| Alpha-1,6-mannosyl-glycoprotein 2-beta-N-acetylglucosaminyltransferase | Mgat2 | 8 | 0.32 | 7.14E-04 |
| Heparan-sulfate 6-O-sulfotransferase 1 | Hs6st1 | 4 | 0.31 | 8.63E-04 |
| Beta-1,3-galactosyl-O-glycosyl-glycoprotein beta-1,6-N-acetylglucosaminyltransferase | Gcnt1 | 6 | 0.45 | 1.21E-03 |
| N-acetylated-alpha-linked acidic dipeptidase 2 | Naalad2 | 12 | 0.27 | 1.26E-03 |
| Alpha-1,3-mannosyl-glycoprotein 2-beta-N-acetylglucosaminyltransferase | Mgat1 | 5 | 0.28 | 2.74E-03 |
| Beta-galactoside alpha-2,6-sialyltransferase 1 | St6gal1 | 4 | 0.34 | 5.02E-03 |
| Carbohydrate sulfotransferase 12 | Chst12 | 3 | 0.21 | 5.04E-03 |
| UDP-GalNAc:beta-1,3-N-acetylgalactosaminyltransferase 1 | B3galnt1 | 2 | 0.35 | 6.25E-03 |
| Alpha-mannosidase 2x | Man2a2 | 6 | 0.51 | 9.39E-03 |
| Beta-1,4-galactosyltransferase 5 | B4galt5 | 4 | 0.33 | 1.02E-02 |
| 4F2 cell-surface antigen heavy chain | Slc3a2 | 11 | 0.40 | 1.14E-02 |
| Beta-1,4 N-acetylgalactosaminyltransferase 1 | B4galnt1 | 6 | 0.44 | 1.16E-02 |
| Mannosidase, alpha, class 1C | Man1c1 | 6 | 0.53 | 1.27E-02 |
| Beta-1,4-galactosyltransferase 1 | B4galt1 | 4 | 0.52 | 1.36E-02 |
| Beta-1,3-N-acetylglucosaminyltransferase lunatic fringe | Lfng | 4 | 0.46 | 1.62E-02 |
| Polypeptide N-acetylgalactosaminyltransferase 10 | Galnt10 | 8 | 0.27 | 1.69E-02 |
| UDP-GlcNAc:betaGal beta-1,3-N-acetylglucosaminyltransferase 9 | B3gnt9 | 4 | 0.34 | 2.13E-02 |
| Bifunctional heparan sulfate N-deacetylase/N-sulfotransferase 1 | Ndst1 | 5 | 0.63 | 2.37E-02 |
| Integral membrane protein 2B | Itm2b | 2 | 0.70 | 2.43E-02 |
| Exostosin-like 2 | Extl2 | 4 | 0.61 | 3.17E-02 |
| Golgi membrane protein 1 | Golm1 | 5 | 0.34 | 3.42E-02 |
| Heparan-sulfate 6-O-sulfotransferase 2 | Hs6st2 | 9 | 0.68 | 4.28E-02 |
| Aminopeptidase N | Anpep | 15 | 58.36 | 3.70E-10 |
| Collectin-12 | Colec12 | 10 | 4.44 | 2.98E-03 |
The reduced secretion of non-type II membrane proteins from Sppl3−/− MEF may not only be caused by the absence of SPPL3 but can also result from cell line-specific properties of the two distinct MEF cell lines (wild-type and Sppl3−/−) compared. In light of this, hits unveiled in this setup require careful validation to eventually be considered genuine SPPL3 substrates. Therefore, we analyzed selected candidate substrates in Sppl3+/+ and Sppl3−/− MEF as well as in the HEK293 cell culture model (Fig. 4). As observed previously (9), secretion of the Golgi glycosyltransferases GnT-V and β4GalT1 from Sppl3−/− MEF was strongly reduced compared with wild-type cells and was accompanied by an intracellular accumulation of full-length glycosyltransferase levels (Fig. 4A). The same behavior was observed for the calcium-activated nucleotidase 1 (CANT1) implicated in glycosaminoglycan biosynthesis (31), which was newly identified as candidate substrate in the mass spectrometry-based screen (Fig. 4A). To further substantiate these findings, we analyzed endogenous CANT1 in HEK293 cells treated with SPPL3-specific siRNA. Indeed, constitutive secretion of CANT1 was blocked upon SPPL3 knockdown and at the same time the protein accumulated intracellularly (Fig. 4B). Moreover, ectopic expression of catalytically active SPPL3 in HEK293 cells led to a strong reduction of intracellular CANT1 levels, whereas secreted CANT1 was slightly more abundant in conditioned supernatants of these cells. The polypeptide N-acetylgalactosaminyltransferase 10 (GalNAcT10), a Golgi glycosyltransferase implicated in the initiation of mucin-type O-glycosylation on serine/threonine residues, was analyzed in a similar manner (Fig. 4B). Although we were not able to detect MEF GalNAcT10 in Western blots of MEF cell lysates, in HEK293 cells endogenous human GalNAcT10 was readily detected in conditioned supernatants as well as in cell lysates and displayed all expected features of a SPPL3 substrate (Fig. 4B).
Fig. 4.

Immunoblot validation of novel candidate Sppl3 substrates identified in Sppl3-deficient MEF. Selected type II membrane protein hits (A, B) and the type I membrane protein Integrin α5 (C, D) identified in the MEF secretome analysis (Fig. 3 and Table II) were validated by immunoblotting using specific antibodies in Sppl3-deficient MEF (A, C) and in HEK293 cells (B, D). Secreted (s) and cellular amounts of the respective protein hits were analyzed in TCA-precipitated conditioned supernatants (sup) and whole-cell lysates, respectively. Sppl3 knock-down and overexpression in HEK293 cells (B, D) were achieved as detailed in Fig. 2. The previously identified Sppl3 substrates GnT-V (gene name: MGAT5) and β4GalT1 (gene name: B4GALT1) were used as controls. In all panels calnexin was used as a loading control. *, nonspecific band.
To verify that altered secretion of type I transmembrane proteins is rather because of cell line-specific adaptations of Sppl3−/− MEF versus control cells than because of SPPL3 mediated proteolysis, the type I membrane protein Integrin α5, which displayed reduced secretion in the proteomic setup, was analyzed in more detail. Confirming the SPECS analysis, we observed a reduced secretion of Integrin α5 in conditioned supernatants of Sppl3−/− MEF compared with control cells (Fig. 4C). However, contrasting with most genuine SPPL3 substrates, no accumulation of the intracellular Integrin α5 was apparent (Fig. 4C). Instead, Integrin α5 expression levels were rather reduced in Sppl3−/− cells compared with Sppl3+/+ cells (Fig. 4C) accounting for the reduced Integrin α5 secretion in the Sppl3−/− cells. Moreover, SPPL3 knockdown neither impaired secretion of Integrin α5 nor resulted in intracellular accumulation of the protein in the human cell culture model (Fig. 4D). Similarly, overexpression of SPPL3 had no effect on Integrin α5 protein levels, hence, ruling out that SPPL3 facilitates proteolytic cleavage of Integrin α5. Of note, SPPL3 overexpression reduces the molecular weight of the glycoprotein Integrin α5, which is well in line with previous observations (9).
Taken together, secretome analysis of Sppl3-deficient MEF revealed a number of type II membrane proteins that constitute candidate SPPL3 substrates and we have thoroughly validated this for CANT1 and GalNAcT10.
Cleavage Site Analysis of SPPL3 Substrates
In order to determine general substrate requirements for SPPL3-mediated intramembrane proteolysis, for instance primary sequence determinants within the substrate, it is crucial to investigate cleavage sites in multiple substrates. To date, however, only the cleavage site of SPPL3 within GnT-V has been mapped in detail (9). Therefore, we searched peptide data acquired in the course of the secretome analyses (Fig. 1 and 3) for semitryptic peptides spanning the C-terminal part of the candidate substrates' transmembrane domain and their adjacent juxtamembrane domain. Among all identified semitryptic peptides of type II membrane proteins we found a number of peptides that originate within the C-terminal part of the TMD of glycan-modifying enzymes and therefore are well suited to infer the site of SPPL3-mediated endoproteolysis. The suitability of these peptides to determine the cleavage site of a protease is corroborated by the detection of the semi-tryptic peptide N-LLHFTIQQR-C (Fig. 5), which originates from GnT-V/MGAT5 and fully confirms the cleavage site of SPPL3 within GnT-V that was previously mapped by an independent approach (9). Moreover, this semitryptic peptide was identified both in samples from human and murine cells indicating that the SPPL3 cleavage pattern is conserved among species. Similar peptides originating from murine β4GalT1 and human ATP1B1 were detected (Fig. 5). Like for GnT-V, ATP1B1 cleavage occurs within a membrane-embedded Met_Leu-Leu motif and also the β4GalT1 cleavage site shares a Leu residue in the P1′ position (Fig. 5). In addition, semi-tryptic peptides of the type II transmembrane proteins B4GalT3 and A4GAT, which were not significantly altered in either of the two screening approaches, were detected (Fig. 5). Moreover, tryptic peptides of the candidate substrates GnT-I/MGAT1 and GnT-II/MGAT2 cover a region of the proteins ectodomain close to the luminal TMD boundary, suggesting that their original SPPL3 cleavage site locates N-terminal to the detected membrane proximal tryptic cleavage site, that is, to the predicted TMD.
Fig. 5.

Cleavage site analysis of SPPL3 substrates. Selected semitryptic (red) and selected tryptic peptides (green) identified in the secretome analyses are highlighted within the context of the respective full-length precursor and its TMD. An arrowhead indicates nontryptic cleavage sites. TMDs are shown by dashed lines with Uniprot annotations given in green and TMHMM v2.0 predictions (http://www.cbs.dtu.dk/services/TMHMM/) in blue. Nontryptic cleavage events are highlighted by a red arrowhead. MGAT5 and B4GALT1 were identified as SPPL3 substrates previously and were confirmed in this study (9) and ATP1B1, MGAT1, and MGAT2 were identified as novel candidate substrates. B4GALT3 and A4GAT peptides were detected in proteomic datasets but no statistically significant differences in secretion following SPPL3 overexpression or Sppl3 knockout were observed. hs, human; mm, murine.
Collectively, these data strongly corroborate the hypothesis that Golgi type II membrane proteins are liberated from their membrane anchors by intramembrane proteolysis and that SPPL3 is crucially involved in this process.
DISCUSSION
Although SPPL3 was discovered more than 10 years ago and is highly conserved in metazoans (1), initial studies on its proteolytic and physiological function have been published only very recently (8–10).
In the present study we provide the first systematic proteomic identification of substrates for the intramembrane protease SPPL3, for which only few substrates have been described before. We took advantage of SPPL3's capability to cleave full-length membrane proteins and mediate shedding (8, 9) that renders SPPL3 glycoprotein substrates accessible to proteomic analysis with the SPECS technology (16). The majority of candidate substrates identified in proteomic analyses of two distinct cell culture models localizes to the Golgi compartment and is implicated in protein glycosylation (Tables I and II), confirming the crucial role of SPPL3 in regulation of cellular glycosylation processes (9). Moreover, all previously identified physiological SPPL3 substrates (9) were confirmed in at least one of the two secretome analyses (Figs. 1 and 3, Tables I and II), underpinning the reliability of the experimental setups used.
Though we provide a much more extensive list of novel substrates (Tables I and II) than aforementioned SPP and SPPL2a/SPPL2b studies (14, 15), the outcome of proteome-wide substrate screens is always restricted by the cellular model system used, because the results only reflect the substrate spectrum of a certain protease in the particular setup and do not offer conclusions on substrate cleavage in other cellular contexts or tissues. In addition, cell line-specific adaptions, as it is for example, the case for MEF cell lines derived from litter-mate embryos, may result in a significant amount of false positive hits. Finally, the SPECS method only allows for enrichment of glycoproteins, that is, nonglycosylated candidate SPPL3 substrates could not be identified. Therefore, the list of SPPL3 candidate substrates is certainly not yet complete and future studies using other cellular or even in vivo models will be required to complement our analyses. Because of these limitations, it is moreover crucial to thoroughly validate the identified candidate substrates using independent detection technologies. In light of our previous observations (9), validation of selected candidate substrates was performed with a focus on proteins implicated in the cellular glycosylation machinery. Western blot analyses confirmed that secretion of XylT2, HS6ST1, HS6ST2, β3GalT6, Sgk196, EXTL3, OGFOD3, ASPH, TOR1AIP1, CANT1, and GalNAcT10 is mediated in a SPPL3-dependent fashion (Figs. 2 and 4, supplemental Figs. S2 and S3). However, comparison of substrates identified in the SPPL3 overexpression setting (Fig. 2) with those discovered in the Sppl3 knockout secretome analysis (Fig. 4) and previously identified substrates (9) revealed that individual SPPL3 substrates are affected in a variety of ways upon modulation of cellular SPPL3 activity in rather variable manners. SPPL3 overexpression for instance substantially boosted secretion of ER α-mannosidase 1 (9), XylT2, EXTL3, and others (Fig. 2 and supplemental Figs. S2 and S3). However, loss of cellular SPPL3 activity had differential effects on secretion of aforementioned substrates, for example, a mild reduction of XylT2 (Fig. 2A) and a strong reduction of EXTL3 (Fig. 2F). In contrast, secretion of GnT-V, β4GalT1, CANT1 and GalNAcT10 (Fig. 4) were substantially reduced following loss of SPPL3 activity. However, SPPL3 overexpression prominently reduced intracellular levels of these substrates, whereas their secretion was not substantially boosted pointing to an intracellular mechanism of ectodomain degradation acting on SPPL3 cleavage products prior to secretion. The latter is supported by the finding that secretion of the substrate is strongly increased by simultaneous overexpression of both protease and substrate, as previously shown for GnT-V (9).
These observations obviously raise questions regarding the underlying mechanisms explaining these distinct effects. Ectopically expressed SPPL3 could in principle localize to ER/Golgi subcompartments that normally do not harbor substantial amounts of endogenous SPPL3 activity and thus colocalize with substrates that are not efficiently cleaved under physiological conditions. Nonetheless, these substrates obviously harbor an intrinsic susceptibility to SPPL3-mediated shedding and, consequently, SPPL3 activity must be tightly regulated under physiological conditions. The proteolytic activity of rhomboid serine intramembrane proteases, which also are constitutively active toward full-length membrane proteins in the Golgi is for instance tightly regulated by subcellular compartmentalization of substrate and protease (32). Hence, future work with a particular focus on the intracompartment colocalization of endogenous SPPL3 and its substrates is required to understand how SPPL3 activity is regulated.
Besides identifying numerous novel SPPL3 substrates, our proteomic analyses provides a first glimpse on SPPL3 physiological function that will be of crucial importance for future research. Previous work delineated the impact of SPPL3-mediated cleavage of glycosyltransferases on the nature and extent of cellular N-glycosylation (9). In line with these findings, we here report additional SPPL3 candidate substrates implicated in N-glycosylation. However, our list of novel substrates now suggests that SPPL3 may similarly modulate other Golgi glycosylation pathways. We for instance identified GalNAcT10 as SPPL3 substrate (Fig. 4B), which is a polypeptide-N-acetylgalactosaminyltransferase that catalyzes the transfer of N-acetylgalactosamine residues to serine and threonine residues, and in this way initiates mucin-type O-glycosylation (33). SPPL3 may also impact O-mannosylation as we identified the recently characterized Sgk196 (27) as novel SPPL3 substrate. Likewise the SPPL3 substrate β3GnT1 (9) was recently implicated in this pathway (29, 30). However, most prominent is the link of the newly identified SPPL3 substrates to glycosaminoglycan biosynthesis. Several substrates identified in the HEK293 (Fig. 1 and Table I) and/or the MEF (Fig. 3 and Table II) secretome screen performed are directly involved in cellular heparan sulfate biosynthesis (Fig. 6 and supplemental Table S7). CANT1, which we also extensively validated as SPPL3 substrate by immunoblot analyses (Fig. 4A and 4B), has recently also been linked to proteoglycan biosynthesis and mutations in CANT1 were linked to chondrodysplasia (31). Hence, in the light of these results, future functional studies are required to study whether alterations in SPPL3 levels in vitro and most importantly in vivo affect these various types of Golgi O-glycosylation in a similar fashion like for N-glycosylation (9).
Fig. 6.
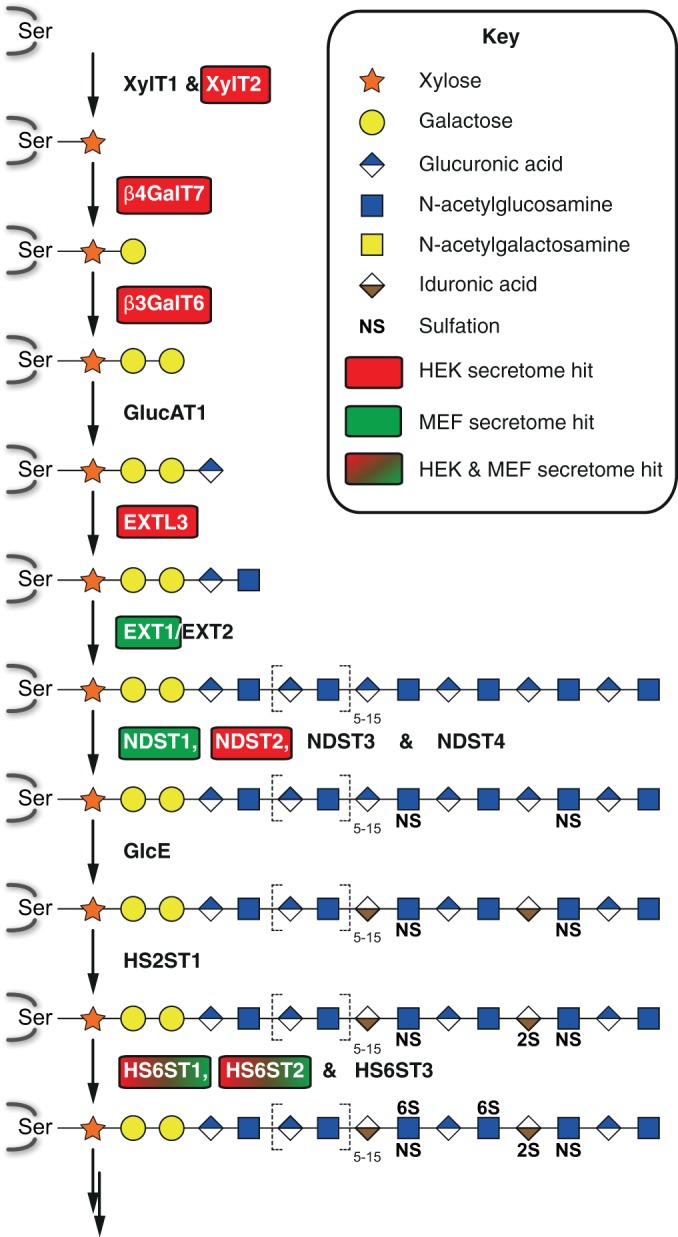
Schematic of the heparan sulfate biosynthesis. Heparan sulfate biosynthesis on serine residues of proteoglycans is illustrated according to (41). Enzymes involved in the individual steps are indicated. The tri-saccharide precursor generated by GlucAT1 activity can also give rise to chondroitin and dermatan sulfate glycosaminoglycans (not depicted). Heparan sulfate polymerization is catalyzed by successive addition of β1,4GlcA and α1,4GlcNAc by the EXT1/EXT2 heterodimer. SPPL3 substrates identified in the secretome analysis in HEK293 cells overexpressing SPPL3 (red boxes), in Sppl3-deficient MEF (green boxes), and in both (box filled with red-to-green gradient) are highlighted.
Shedding of Golgi glycosyltransferases and other glycan-modifying enzymes is a well-documented phenomenon, yet the physiological purpose, the destiny of the secreted ectodomains and the proteases catalyzing the ectodomain release have remained largely enigmatic (34). Cleavage and subsequent secretion of these enzymes has been discussed to constitute a mechanism that allows for regulation of Golgi glycosylation. Nonetheless, it cannot be excluded that secreted glycosyltransferase ectodomains fulfill extracellular functions or contribute to pathological processes. In that regard, it is noteworthy that dysregulated secretion of xylosyltransferases has been linked with distinct pathological conditions, such as for instance infertility in males (35) or skin disorders (36). Results from the present study suggest that SPPL3 is a major player in ectodomain release of these enzymes and thus regulates a variety of important physiological processes.
Substrate requirements that determine SPPL3's specificity toward certain substrates have so far not been investigated in more detail. Besides the type II topology of SPPL3 substrates, SPPL3-substrate interaction may be critically influenced by the primary sequence flanking the protease cleavage site. However, so far, a precise SPPL3 cleavage site has only been mapped within GnT-V (9). Hence, to learn more about the cleavage mechanism of SPPL3, we analyzed semitryptic peptides identified in both proteomic analyses. All semitryptic peptides identified matched to the C-terminal region of the predicted TMD or the luminal juxtamembrane domain of the respective SPPL3 candidate substrate. Moreover, methionine and tyrosine in the P1 position of the cleavage site were the most abundant amino acids found (Fig. 5). Therefore, our observations strongly corroborate that these substrates are indeed liberated from their membrane anchors by intramembrane protease activity. In addition, secretome analysis, in particular of human cells overexpressing SPPL3 revealed a selective enrichment of candidate substrates that adopt a type II topology within the membrane and localize to the ER and/or Golgi network like SPPL3 (Fig. 1 and Table I). Together, these findings strongly suggest that the substrates identified in the secretome analysis are indeed genuine SPPL3 substrates.
Although until now GxGD aspartyl intramembrane proteases were believed to depend on helix-destabilizing residues in the substrates' TMD and so far unidentified determinants within the juxtamembrane domain for substrate recognition and cleavage, rhomboid intramembrane proteases seem to require a distinct consensus cleavage site in or close to the luminal juxtamembrane domain of the substrate (13, 37–39). Our findings point toward a putative SPPL3 cleavage consensus motif localized in a transmembrane domain part embedded adjacent to the luminal plasma membrane boundary, which at least partially might influence SPPL3 substrate selectivity. The fact that SPPL3 acts as a sheddase for type II full-length substrates together with the finding that SPPL3 cleavage may depend on primary sequence constraints supported by the repetitive detection of similar cleavage site consensus motifs indicate that SPPL3 from a mechanistic perspective could rather be compared with the rhomboid family than to the GxGD protease family. However, the cleavage site analysis based on semitryptic peptides has to be treated with caution, as detected cleavage products could have undergone trimming by unrelated proteolytic activities masking the original cleavage sites which is a well-documented phenomenon for ST6Gal1 (40). In the future, studies mutagenizing these SPPL3 cleavage sites could provide important clues on substrate selectivity of SPPL3.
The present study provides the first systematic and comprehensive analysis of the SPPL3 degradome in cellular model systems. Taken together, this not only complements recent progress (9) on the physiological function of SPPL3 as a regulator of Golgi-localized glycan-modifying enzymes, but also raises additional novel fundamental physiological implications. In light of the newly identified SPPL3 substrates, a much more general role in Golgi homeostasis can be attributed to SPPL3, because SPPL3's function is not restricted to the regulation of cellular N-glycosylation, but likewise extends to O-glycosylation pathways in the Golgi apparatus and beyond. Finally, the study once more demonstrates the power of the SPECS method in combination with mass spectrometry to identify novel substrates of sheddases and paves the way for further in-depth functional studies of SPPL3.
Supplementary Material
Acknowledgments
The Sppl3 knockout mouse line was kindly provided by Genentech, Inc., and Lexicon Pharmaceuticals, Inc., and was obtained through the MMRRC at the University of California at Davis.
Footnotes
Author contributions: P.K., M.V., S.F.L., and R.F. designed research; P.K., M.V., and M.H. performed research; B.S., U.S., and S.B. contributed new reagents or analytic tools; P.K., M.V., S.F.L., and R.F. analyzed data; P.K., M.V., S.F.L., and R.F. wrote the paper. C.H., S.F.L. and R.F. supervised research.
* This work was supported by the Deutsche Forschungsgemeinschaft (HA 1737/11-2 and FL 635/2-1) and by start-up funding for female researchers provided by the Center for Integrated Protein Science Munich (CIPSM) (to RF) as well as by JPND-RiMoD, the Helmholtz Israel program and the Breuer foundation award (to SFL). MV was generously supported by a PhD fellowship of the Hans und Ilse Breuer Stiftung and by the Elitenetwork of Bavaria within the Graduate Program “Protein Dynamics in Health and Disease”. PHK was generously supported by the Carl von Linde Junior Fellowship, Institute for Advanced Study, Technische Universität München.
 This article contains supplemental Figs. S1 to S3 and Tables S1 to S7.
This article contains supplemental Figs. S1 to S3 and Tables S1 to S7.
1 The abbreviations used are:
- SPP
- signal peptide peptidase
- ASPH
- aspartyl/asparaginyl β-hydroxylase
- β3GalT6
- β1,3 galactosyltransferase 6
- β3GnT1
- β1,3 N-acetylglucosaminyltransferase 1
- β4GalT1
- β1,4 galactosyltransferase 1
- BACE1
- β-site amyloid precursor protein converting enzyme 1
- CANT1
- calcium-activated nucleotidase 1
- EXTL3
- exostosin-like 3
- GalNAcT-10
- polypeptide N-acetylgalactosaminyltransferase 10
- GnT-V
- β1,6 N-acetylglucosaminyltransferase V
- HS6ST1
- heparan sulfate 6-O-sulfotransferase 1
- HS6ST2
- heparan sulfate 6-O-sulfotransferase 2
- ManAz
- tetraacetyl-N-azidoacetyl mannosamine
- MEF
- murine embryonic fibroblasts
- NDST1
- bifunctional heparin sulfate N-deacetylase/N-sulfotransferase 1
- NFAT
- nuclear factor of activated T cells
- OGFOD3
- 2-oxoglutarate and iron-dependent oxygenase domain-containing protein 3
- QARIP
- quantitative analysis of regulated intramembrane proteolysis
- Sgk196
- protein kinase-like protein Sgk196
- SPECS
- secretome protein enrichment with click sugars
- SPPL
- signal peptide peptidase-like
- ST6Gal1
- β-galactoside α2,6 sialyltransferase 1
- TMD
- transmembrane domain
- TOR1AIP1
- torsin 1A-interacting protein 1
- XylT2
- xylosyltransferase 2.
REFERENCES
- 1. Voss M., Schröder B., Fluhrer R. (2013) Mechanism, specificity, and physiology of signal peptide peptidase (SPP) and SPP-like proteases. Biochim. Biophys. Acta 1828, 2828–2839 [DOI] [PubMed] [Google Scholar]
- 2. Weihofen A., Binns K., Lemberg M. K., Ashman K., Martoglio B. (2002) Identification of signal peptide peptidase, a presenilin-type aspartic protease. Science 296, 2215–2218 [DOI] [PubMed] [Google Scholar]
- 3. Ponting C. P., Hutton M., Nyborg A., Baker M., Jansen K., Golde T. E. (2002) Identification of a novel family of presenilin homologues. Hum. Mol. Genet. 11, 1037–1044 [DOI] [PubMed] [Google Scholar]
- 4. Grigorenko A. P., Moliaka Y. K., Korovaitseva G. I., Rogaev E. I. (2002) Novel class of polytopic proteins with domains associated with putative protease activity. Biochemistry Mosc. 67, 826–835 [DOI] [PubMed] [Google Scholar]
- 5. Struhl G., Adachi A. (2000) Requirements for presenilin-dependent cleavage of notch and other transmembrane proteins. Mol. Cell 6, 625–636 [DOI] [PubMed] [Google Scholar]
- 6. Lemberg M. K., Martoglio B. (2002) Requirements for signal peptide peptidase-catalyzed intramembrane proteolysis. Mol. Cell 10, 735–744 [DOI] [PubMed] [Google Scholar]
- 7. Martin L., Fluhrer R., Haass C. (2009) Substrate requirements for SPPL2b-dependent regulated intramembrane proteolysis. J. Biol. Chem. 284, 5662–5670 [DOI] [PubMed] [Google Scholar]
- 8. Voss M., Fukumori A., Kuhn P.-H., Künzel U., Klier B., Grammer G., Haug-Kröper M., Kremmer E., Lichtenthaler S. F., Steiner H., Schröder B., Haass C., Fluhrer R. (2012) Foamy Virus Envelope Protein Is a Substrate for Signal Peptide Peptidase-like 3 (SPPL3). J. Biol. Chem. 287, 43401–43409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Voss M., Künzel U., Higel F., Kuhn P.-H., Colombo A., Fukumori A., Haug-Kröper M., Klier B., Grammer G., Seidl A., Schröder B., Obst R., Steiner H., Lichtenthaler S. F., Haass C., Fluhrer R. (2014) Shedding of glycan-modifying enzymes by signal peptide peptidase-like 3 (SPPL3) regulates cellular N-glycosylation. EMBO J. 33, 2890–2905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Makowski S. L., Wang Z., Pomerantz J. L. (2015) A Protease-Independent Function for SPPL3 in NFAT Activation. Mol. Cell. Biol. 35, 451–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Steiner H., Fluhrer R., Haass C. (2008) Intramembrane proteolysis by gamma-secretase. J. Biol. Chem. 283, 29627–29631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. López-Otín C., Overall C. M. (2002) Protease degradomics: a new challenge for proteomics. Nat. Rev. Mol. Cell Biol. 3, 509–519 [DOI] [PubMed] [Google Scholar]
- 13. Hemming M. L., Elias J. E., Gygi S. P., Selkoe D. J. (2008) Proteomic profiling of gamma-secretase substrates and mapping of substrate requirements. PLoS Biol. 6, e257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boname J. M., Bloor S., Wandel M. P., Nathan J. A., Antrobus R., Dingwell K. S., Thurston T. L., Smith D. L., Smith J. C., Randow F., Lehner P. J. (2014) Cleavage by signal peptide peptidase is required for the degradation of selected tail-anchored proteins. J. Cell Biol. 205, 847–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Videm P., Gunasekaran D., Schröder B., Mayer B., Biniossek M. L., Schilling O. (2014) Automated peptide mapping and protein-topographical annotation of proteomics data. BMC Bioinformatics 15, 207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kuhn P.-H., Koroniak K., Hogl S., Colombo A., Zeitschel U., Willem M., Volbracht C., Schepers U., Imhof A., Hoffmeister A., Haass C., Roβner S., Bräse S., Lichtenthaler S. F. (2012) Secretome protein enrichment identifies physiological BACE1 protease substrates in neurons. EMBO J. 31, 3157–3168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Martin L., Fluhrer R., Reiss K., Kremmer E., Saftig P., Haass C. (2008) Regulated intramembrane proteolysis of Bri2 (Itm2b) by ADAM10 and SPPL2a/SPPL2b. J. Biol. Chem. 283, 1644–1652 [DOI] [PubMed] [Google Scholar]
- 18. Fürniss D., Mack T., Hahn F., Vollrath S. B. L., Koroniak K., Schepers U., Bräse S. (2013) Peptoids and polyamines going sweet: Modular synthesis of glycosylated peptoids and polyamines using click chemistry. J. Org. Chem. 9, 56–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shevchenko A., Tomas H., Havlis J., Olsen J. V., Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
- 20. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 21. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 22. Eden E., Navon R., Steinfeld I., Lipson D., Yakhini Z. (2009) GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics 10, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ivankov D. N., Bogatyreva N. S., Hönigschmid P., Dislich B., Hogl S., Kuhn P.-H., Frishman D., Lichtenthaler S. F. (2013) QARIP: a web server for quantitative proteomic analysis of regulated intramembrane proteolysis. Nucleic Acids Res. 41, W459–W464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Humphries D. E., Sullivan B. M., Aleixo M. D., Stow J. L. (1997) Localization of human heparan glucosaminyl N-deacetylase/N-sulphotransferase to the trans-Golgi network. Biochem. J. 325, 351–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bai X., Zhou D., Brown J. R., Crawford B. E., Hennet T., Esko J. D. (2001) Biosynthesis of the linkage region of glycosaminoglycans: cloning and activity of galactosyltransferase II, the sixth member of the beta 1,3-galactosyltransferase family (beta 3GalT6). J. Biol. Chem. 276, 48189–48195 [DOI] [PubMed] [Google Scholar]
- 26. Pan S., Wang S., Utama B., Huang L., Blok N., Estes M. K., Moremen K. W., Sifers R. N. (2011) Golgi localization of ERManI defines spatial separation of the mammalian glycoprotein quality control system. Mol. Biol. Cell 22, 2810–2822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yoshida-Moriguchi T., Willer T., Anderson M. E., Venzke D., Whyte T., Muntoni F., Lee H., Nelson S. F., Yu L., Campbell K. P. (2013) SGK196 is a glycosylation-specific O-mannose kinase required for dystroglycan function. Science 341, 896–899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Goodchild R. E., Dauer W. T. (2005) The AAA+ protein torsinA interacts with a conserved domain present in LAP1 and a novel ER protein. J. Cell Biol. 168, 855–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Praissman J. L., Live D. H., Wang S., Ramiah A., Chinoy Z. S., Boons G.-J., Moremen K. W., Wells L. (2014) B4GAT1 is the priming enzyme for the LARGE-dependent functional glycosylation of α-dystroglycan. Elife 3, doi: 10.7554/eLife.03943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Willer T., Inamori K.-I., Venzke D., Harvey C., Morgensen G., Hara Y., Beltrán Valero de Bernabé D., Yu L., Wright K. M., Campbell K. P. (2014) The glucuronyltransferase B4GAT1 is required for initiation of LARGE-mediated α-dystroglycan functional glycosylation. Elife 3, doi: 10.7554/eLife.03941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nizon M., Huber C., De Leonardis F., Merrina R., Forlino A., Fradin M., Tuysuz B., Abu-Libdeh B. Y., Alanay Y., Albrecht B., Al-Gazali L., Basaran S. Y., Clayton-Smith J., Désir J., Gill H., Greally M. T., Koparir E., van Maarle M. C., MacKay S., Mortier G., Morton J., Sillence D., Vilain C., Young I., Zerres K., Le Merrer M., Munnich A., Le Goff C., Rossi A., Cormier-Daire V. (2012) Further delineation of CANT1 phenotypic spectrum and demonstration of its role in proteoglycan synthesis. Hum. Mutat. 33, 1261–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Freeman M. (2008) Rhomboid proteases and their biological functions. Annu. Rev. Genet. 42, 191–210 [DOI] [PubMed] [Google Scholar]
- 33. Brockhausen I., Schachter H., Stanley P. (2009) in Essentials of Glycobiology, eds Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME. (Cold Spring Harbor Laboratory Press, Cold Spring Harbor: ), pp 115–127. 2nd Ed [PubMed] [Google Scholar]
- 34. Varki A., Esko J. D., Colley K. J. (2009) in Essentials of Glycobiology, eds Varki A, Cummings RG, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME. (Cold Spring Harbor Laboratory Press, Cold Spring Harbor: ), pp 37–46. 2nd Ed [PubMed] [Google Scholar]
- 35. Götting C., Kuhn J., Brinkmann T., Kleesiek K. (2002) Xylosyltransferase activity in seminal plasma of infertile men. Clin. Chim. Acta 317, 199–202 [DOI] [PubMed] [Google Scholar]
- 36. Götting C., Sollberg S., Kuhn J., Weilke C., Huerkamp C., Brinkmann T., Krieg T., Kleesiek K. (1999) Serum xylosyltransferase: a new biochemical marker of the sclerotic process in systemic sclerosis. J. Invest. Dermatol. 112, 919–924 [DOI] [PubMed] [Google Scholar]
- 37. Beel A. J., Sanders C. R. (2008) Substrate specificity of gamma-secretase and other intramembrane proteases. Cell. Mol. Life Sci. 65, 1311–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Strisovsky K., Sharpe H. J., Freeman M. (2009) Sequence-specific intramembrane proteolysis: identification of a recognition motif in rhomboid substrates. Mol. Cell 36, 1048–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fluhrer R., Martin L., Klier B., Haug-Kröper M., Grammer G., Nuscher B., Haass C. (2012) The α-helical content of the transmembrane domain of the British dementia protein-2 (Bri2) determines its processing by signal peptide peptidase-like 2b (SPPL2b). J. Biol. Chem. 287, 5156–5163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kitazume S., Tachida Y., Oka R., Kotani N., Ogawa K., Suzuki M., Dohmae N., Takio K., Saido T. C., Hashimoto Y. (2003) Characterization of alpha 2,6-sialyltransferase cleavage by Alzheimer's beta-secretase (BACE1). J. Biol. Chem. 278, 14865–14871 [DOI] [PubMed] [Google Scholar]
- 41. Esko J. D., Kimata K., Lindahl U. (2009) in Essentials of Glycobiology, eds Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME. (Cold Spring Harbor Laboratory Press, Cold Spring Harbor: ), pp 229–248 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.