

Abstract
Inhibitors of oncogenic B-RAFV600E and MKK1/2 have yielded remarkable responses in B-RAFV600E-positive melanoma patients. However, the efficacy of these inhibitors is limited by the inevitable onset of resistance. Despite the fact that these inhibitors target the same pathway, combination treatment with B-RAFV600E and MKK1/2 inhibitors has been shown to improve both response rates and progression-free survival in B-RAFV600E melanoma patients. To provide insight into the molecular nature of the combinatorial response, we used quantitative mass spectrometry to characterize the inhibitor-dependent phosphoproteome of human melanoma cells treated with the B-RAFV600E inhibitor PLX4032 (vemurafenib) or the MKK1/2 inhibitor AZD6244 (selumetinib). In three replicate experiments, we quantified changes at a total of 23,986 phosphosites on 4784 proteins. This included 1317 phosphosites that reproducibly decreased in response to at least one inhibitor. Phosphosites that responded to both inhibitors grouped into networks that included the nuclear pore complex, growth factor signaling, and transcriptional regulators. Although the majority of phosphosites were responsive to both inhibitors, we identified 16 sites that decreased only in response to PLX4032, suggesting rare instances where oncogenic B-RAF signaling occurs in an MKK1/2-independent manner. Only two phosphosites were identified that appeared to be uniquely responsive to AZD6244. When cells were treated with the combination of AZD6244 and PLX4032 at subsaturating concentrations (30 nm), responses at nearly all phosphosites were additive. We conclude that AZD6244 does not substantially widen the range of phosphosites inhibited by PLX4032 and that the benefit of the drug combination is best explained by their additive effects on suppressing ERK1/2 signaling. Comparison of our results to another recent ERK1/2 phosphoproteomics study revealed a surprising degree of variability in the sensitivity of phosphosites to MKK1/2 inhibitors in human cell lines, revealing unexpected cell specificity in the molecular responses to pathway activation.
Mitogen-activated protein kinase (MAPK)1 pathways orchestrate key intracellular responses to a variety of extracellular signals including mitogenic stimuli and cellular stress. In the case of the RAF/MKK/ERK pathway, receptor tyrosine kinases activate the small GTPase Ras, which then binds members of the RAF family of kinases (RAF1, B-RAF, ARAF) leading to their activation. Activated RAF kinases phosphorylate and activate MAP kinase kinases 1 and 2 (MKK1, MKK2), which in turn phosphorylate and activate extracellular signal-related kinases 1 and 2 (ERK1, ERK2). The specificity of this cascade is remarkable, as the only widely accepted targets of B-RAF are MKK1/2, and the only validated targets of MKK1/2 are ERK1/2 (1–4). Once activated, ERK1/2 mediates the effects of pathway activation by phosphorylating scores of cytoplasmic and nuclear targets. However, the full scope of cellular substrates of ERK1/2 remains unknown.
The importance of identifying targets of B-RAF/MKK/ERK signaling on a global scale is magnified by the fact that this pathway is constitutively activated in a number of human cancers, most notably melanoma, colorectal cancer, thyroid cancer, and glioblastoma (5). Reliance on ERK signaling is most pronounced in melanoma, where as many as 75% of tumors harbor activating mutations in either NRAS (20–25%) or B-RAF (40–50%) (6). Alternative driver mutations, such as those in CKIT (6), GNAQ/GNA11 (7, 8), and NF1 (9) also increase ERK1/2 activity and suggest that nearly all melanomas harbor constitutive ERK signaling. Inhibitors specific for oncogenic B-RAFV600E (vemurafenib (10), dabrafenib (11)) and MKK1/2 (trametinib (12), cobimetinib (13), selumetinib (14)) have been successful in clinical trials and several are now FDA-approved for treatment of metastatic melanoma. Interestingly, recent clinical trials treating patients with combinations of a B-RAFV600E and MKK1/2 inhibitor have reported improved response rates and progression-free survival when compared with single agent B-RAFV600E inhibitor therapy (13, 15–17).
It is not necessarily intuitive that two inhibitors that target the same pathway should lead to improved patient responses. It has been suggested that the combination of B-RAF and MKK1/2 inhibitors may be more effective because it provides a barrier to mechanisms of acquired resistance (MOR) that reactivate ERK1/2 signaling downstream of B-RAFV600E (16, 18). Another explanation for the improved patient response is that the combination is more effective at inhibiting ERK signaling below the threshold required to achieve a positive clinical response (19, 20). This could be because of either an additive or synergistic effect of the combination on ERK signaling. A third possibility is that, in addition to their shared targets, B-RAFV600E and MKK1/2 each have small number of unique targets and that inhibition of all MAPK pathway targets is therefore only possible with the combination. By profiling the changes in phosphorylation in response to B-RAFV600E and MKK1/2 inhibitors, phosphoproteomics can provide insight into which of these possibilities is most likely.
Several proteomics and phosphoproteomics strategies have been employed to identify ERK1/2 targets, including 2D-PAGE (21, 22), analog sensitive ERK1/2 kinases (23, 24), negative ionization mass spectrometry (MS) (25), and shotgun phosphoproteomics using stable isotope labeling by amino acids in cell culture (SILAC)-based MS (24, 26, 27) or label-free MS (28, 29). Surprisingly, the degree of overlap between the ERK1/2 targets identified in these studies has been reported to be very low (28). This may be reflective of low sampling and indicate that only a fraction of ERK targets are identified in each study. Alternatively, it may be reflective of the variability in responses between different cell types and/or different treatment conditions. Importantly, the degree of variability in phosphorylation responses to a single kinase inhibitor in different cell types remains largely unexplored. Delineating the degree to which cells respond similarly to ERK pathway inhibition will be critical for identifying universal biomarkers that can report activation or inhibition of the pathway.
Here we use large-scale phosphoproteomics to identify targets of the B-RAF/MKK/ERK pathway in a human metastatic melanoma cell line that is sensitive to the B-RAFV600E inhibitor PLX4032 (PLX; vemurafenib) and the MKK1/2 inhibitor, AZD6244 (AZD; selumetinib). In total, changes at 23,986 phosphosites on 4784 proteins could be quantified from replicate experiments and 1317 phosphosites showed reproducible decreases with inhibitor. Several new candidate substrates of ERK1/2 with potential roles in melanoma progression or melanoma cell survival were identified. Although the vast majority of phosphosites responded similarly to both AZD and PLX, 16 sites decreased only in response to PLX, suggesting that there are MKK1/2-independent targets of B-RAFV600E. Only two sites decreased in an AZD-specific manner, indicating that unique targets of MKK1/2 inhibitors are exceedingly rare and unlikely to explain the benefits of combination treatment. Combinatorial experiments using subsaturating concentrations of AZD and PLX showed additive responses at nearly all phosphosites, consistent with a Bliss independence model. Therefore, a more complete inhibition of ERK1/2 signaling is the most likely explanation for the clinical efficacy of combination treatment with B-RAFV600E and MKK1/2 inhibitors. Finally, we compared phosphosites that responded to AZD in our dataset to those in a prior study of an MKK1/2 inhibitor in two other cell lines. This comparison revealed a surprisingly large degree of variability in the phosphosite responses across cell lines, even for well-established targets of the ERK1/2 pathway.
EXPERIMENTAL PROCEDURES
Cell Culture
The WM239A cell line was a gift from Meenhard Herlyn, Wistar Institute. Cells were grown in SILAC-RPMI1640 media (Thermo Fisher Scientific) supplemented with 10% FBS (Gemini) and were maintained at 37 °C with 5% CO2.
Sample Preparation
Low passage (p10–15) WM239A cells were cultured for a minimum of five passages in SILAC-RPMI1640 (Thermo) supplemented with light, medium, or heavy lysine (Cambridge Isotopes; 40 μg/ml) and arginine (Cambridge Isotopes; 200 μg/ml). Following incorporation of labeled amino acids, 35 × 106 cells were seeded overnight in 15 cm dishes. Media was changed the following morning and cells were treated with AZD, PLX, or AZD+PLX for 2 h. Cells were rinsed twice with PBS and processed for MS analysis according to a modified FASP protocol (30). Cells were lysed in 750 μl 95° SDT buffer (0.1 m Tris, pH 7.5, 4% SDS, 0.1 m dithiothreitol) and returned to 95° for 10 min. Lysates were then sonicated with a microtip sonicator for 15 s. Light, medium, and heavy lysates were combined and brought to a final volume of 30 ml with buffer UA (8 m urea, 0.1 m Tris, pH 8.5), and then split evenly into two Amicon Ultra-15 10K filter units (Millipore) and centrifuged for 25 min at 4000 × g. Samples were washed with 10 ml UA, carbamidomethylated with 5 ml 50 mm iodoacetamide in UA, then centrifuged and washed three times with 5 ml UA. After transfer to new filter units (filter units exposed to the high concentrations of SDS in SDT buffer leaked overnight), samples were washed once with 7 ml 0.5 m ammonium bicarbonate to bring the urea concentration to 1 m. Samples were digested at 37 °C with 2% (w/w) sequence grade modified trypsin (Promega) in a total volume of 3 ml. After 16–18 h, digests were centrifuged and each filter unit was washed once with 3 ml H2O. Eluates from the two filter units were combined, acidified to pH ≤ 2 and desalted on Oasis HLB extraction cartridges (150 mg, Waters). Five micrograms of each sample was reserved for total protein measurements. The remainder was subject to TiO2 enrichment.
TiO2 Enrichment of Phosphopeptides
Samples from desalting columns were eluted with 65% (v/v) acetonitrile, 1% (v/v) TFA. Additional TFA was added to a final concentration of 2% and l-glutamate was added to a final concentration of 140 mm. Titanium beads were equilibrated with 1 min washes in 1 ml EB1 (20% ACN (acetonitrile), 1% NH4OH), WB1 (65% ACN, 0.5% TFA), and LB (65% ACN, 2% TFA, 140 mm glutamic acid). Batches of 2 mg lysate were rotated end-over-end with 20 mg TiO2 (GL Sciences) for 15 min at room temperature. The TiO2 resin was subjected to 1 ml washes with LB, WB1, and twice with WB2 (65% ACN, 0.1% TFA), then added to a 200 μl C8 stagetip (Proxeon). Phosphopeptides were eluted with 200 μl EB1 followed by 400 μl EB2 (65% ACN, 1% NH4OH) using a 1 ml syringe, and lyophilized overnight.
ERLIC Chromatography
Samples were run on an Agilent 1100 Series HPLC using a 100 × 4.6-mm 5 μm polyWAX LP ERLIC column (PolyLC), prepared by passivation with 40 mm EDTA for 24 h at 0.5 ml/min, followed by H2O for 48 h at 1.0 ml/min. Lyophilized TiO2-enriched samples were resuspended in 65 μl Buffer A (16.7 mm ammonium formate, pH 2.2, 70% ACN) and placed in a bath sonicator for four pulses × 30 s. Particulate matter was removed by centrifugation for 1 min at 14,000 × g. The sample was then injected into a 50 μl sample loop and the ERLIC gradient was run at a flow rate of 1 ml/min and fractions were collected manually at 1-min intervals. The ERLIC gradient was 0–5 min: 100% Buffer A; 5–15 min: linear gradient to 100% Buffer B (16.7 mm ammonium formate, pH 2.2, 10% acetonitrile); 15–20 min: linear gradient to 100% Buffer C (1% TFA, 10% acetonitrile); 20–24 min: 100% Buffer C. Fractions were immediately frozen in liquid nitrogen, speedvac concentrated to <5 μl, and resuspended with 0.1% formic acid to a final volume of 12 μl. Samples were stored at −80 °C and 5 μl of each fraction was analyzed by LC/MS/MS.
LC/MS/MS Analysis
Peptide samples were separated using a Waters nanoACQUITY system. For phosphopeptide samples, 5 μl was loaded in direct injection mode onto a BEH130 C18 analytical column (1.7 μm, 75 μm × 250 mm, Waters) maintained at 40°C. Buffer A was 0.1% formic acid and buffer B was 80% acetonitrile, 0.1% formic acid. Samples were eluted with a 150-min gradient from 3–28% Buffer B at 0.3 μl/min. For total peptide samples, 4 μg of peptide was separated using 2D RP-RP UPLC. The 1st dimension separation was performed on an XBridge C18 NanoEase column (3 μm, 300 μm × 50 mm) equilibrated in 20 mm ammonium formate, pH 10 (buffer A1), and eluted with increasing concentrations of acetonitrile (buffer B1). The 2nd dimension separation was performed on a Symmetry C18 trap column (5 μm, 180 μm × 20 mm) and a BEH130 C18 analytical column (1.7 μm, 75 μm × 250 mm), equilibrated in 0.1% formic acid in water (buffer A2) and eluted with 0.1% formic acid, 80% acetonitrile (buffer B2). Total peptides were separated in the 1st dimension by an 18-step gradient (2 μl/min) (4% B1 in step 1, 6% B1 in step 2, with 1% B1 added with each step from step 3–17; 65% B1 for step 18). Each first dimension eluate was diluted 1:9 with 0.1% (v/v) formic acid in water (20 μl/min) before loading onto the 2nd dimension. Sample was eluted from the analytical column with a 2-min ramp to the starting percentage of buffer B2, followed by a 123-min gradient. Gradients were 8–32% B2 (steps 1–7), 12–33% B2 (steps 8–12), 15–33% B2 (step 13), and 16–34% B2 (steps 14–18).
MS/MS was performed using an LTQ Orbitrap Velos, scanning MS between 400–1600 m/z (1 × 106 ions, 60,000 resolution), and selecting the 10 most intense ions for MS/MS with 180 s dynamic exclusion, 10 ppm exclusion width (20 ppm for phosphopeptide samples), and a repeat count of one. Ions with unassigned charge states or a charge state of +1 were excluded. For phosphopeptide samples, neutral loss masses of 19.5948, 24.4937, 32.6584, and 48.9879 were used for data dependent MS3 scans. Maximal injection times were 500 ms for Orbitrap precursor scans (one microscan) and 150 ms for LTQ MS/MS (one microscan) with AGC 1 × 104. The normalized collision energy was 35%, with activation Q = 0.25 for 10 ms.
Data Analysis
Raw MS files for both phosphopeptide and total peptide data sets were uploaded together and searched against the Uniprot human proteome database (downloaded on 01/27/2014; 88,509 entries) in MaxQuant v.1.4.1.2 (31) using the Andromeda search engine (v1.4.0.0). Mass tolerances were 4.5 ppm for precursor ions and 0.5 Da for ITMS MS/MS ions. The minimum peptide length was seven amino acids. MaxQuant used default score cutoffs of zero for unmodified peptides and 40 for modified peptides. False discovery rates were 1% for both phosphopeptide and protein identifications. For protein quantification, the minimum number of total peptides was two (unique + razor). Phosphorylated peptides and their unmodified counterparts were excluded for protein quantification. Raw files for phosphopeptide data and total protein data were defined as separate groups. The multiplicity was set to three with medium labels of Arg6 and Lys4 and heavy labels of Arg10 and Lys8 for both groups. For phosphopeptide files, Phospho(STY) was set as a variable modification. All files were searched with carbamidomethylation (Cys) as a fixed modification and acetylation (N-term) and oxidation (Met) as variable modifications. Default settings were used except the “match between runs” feature was enabled with default settings and the site quantification was changed to “use highest change.” Localization probabilities were assessed with the MaxQuant PTM score. The enzyme specificity was trypsin/P and two missed cleavages were allowed. For phosphopeptide data, the phospho(STY) output file was uploaded into Perseus (v1.4.1.3). Reverse and contaminant rows were removed and the site table was expanded so that ratios for phosphosites with different multiplicities of phosphorylated residues [e.g. singly (one phosphate), doubly (two phosphates), and triply (three or more phosphates) phosphorylated peptides] were each treated as unique phosphosites. Rows not quantified in any of the experiments after expanding the site table were removed. Phosphosite positions mentioned in the text have been manually validated to match the reviewed human Uniprot entry for the gene of interest. Phosphosite positions in supplemental tables are directly from the MaxQuant output files. For total peptide samples, the protein groups output file was uploaded into the Perseus and rows designated as reverse, contaminant, and “only identified by site” were removed. Rows not quantified in any of the experiments were also removed. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (32) via the PRIDE partner repository with the data set identifier PXD001560 (10 μm SILAC experiment) and PXD001563 (30 nm SILAC experiment).
STRING and Gene Ontology Analyses
Genes containing Ser/Thr-Pro phosphosites observed to decrease with 10 μm AZD or 10 μm PLX (log2 < −0.84) were uploaded into STRING using the multiple names option, selecting human as the organism. The required confidence score was set to 0.900 and disconnected nodes were removed from the image.
The same genes were also uploaded into the PANTHER database selecting human as the organism. Functional classification of genes was viewed as a pie chart of Gene Ontology molecular function. The 49 genes identified as having nucleic acid binding transcription factor activity were manually validated to identify genes with known activity as transcription factors, transcriptional coactivators, or transcriptional repressors. This resulted in the final list of 39 genes.
Identifying Unique Targets of AZD and PLX
The 61 phosphosites with median PLX/AZD log2 ratios exceeding the significance threshold of log2 value ±0.840 were filtered to exclude sites where both the AZD/DMSO and PLX/DMSO ratios had log2 values < −0.840 (indicating differential regulation at a common target site). All phosphosites with PLX/AZD ratios that showed large variability in replicate experiments (i.e. both positive and negative PLX/AZD ratios in replicate experiments) were also removed.
Matching WM239A Data to Galan et al.
All quantified phosphosites in this study were first matched to Galan et al. based on common Uniprot identifier and amino acid position. For sites that could not be matched based on these criteria, an attempt was made to match them based on gene name and phosphopeptide sequence, or phosphopeptide sequence alone. Phosphopeptide sequences were the sequences in the phospho(STY) probabilities column with phosphorylations assigned at residues with probabilities ≥0.5. Sites were then filtered to accept only those in which the localization score in both studies was >0.75. For the significant sites identified in all three cell lines (879 total), 691 were matched by Uniprot identifier and amino acid position, 178 were matched by gene and phosphopeptide sequence, and 10 were matched by phosphopeptide sequence alone. Phosphosites were matched to regulatory sites in the PhosphositePlus database using the sequence window from the phospho(STY) text file. SILAC ratios reported for AZD/DMSO (this study) are reported as a median value for sites with replicate measurements.
Immunoblotting
Cells were lysed in SDT or RIPA buffer, and protein concentrations determined using the DC protein assay (Bio-Rad). Equal amounts of protein were separated on 10% SDS-PAGE gels or 4–20% protean-TGX precast gradient gels (Bio-Rad) and transferred to PVDF-SQ membranes (Millipore) for 1 h at 100 V. Membranes were incubated in primary antibody for 2 h at room temperature or overnight at 4 °C, and in secondary antibody for 1 h at room temperature.
Antibodies
All antibodies were from Cell Signaling: ERK1/2 (#4696), phospho-ERK1/2 (#4370), MKK1/2 (#9122), phospho-MKK1/2 (#9121), and tubulin (#5346).
RESULTS
Phosphoproteomics Responses to B-RAFV600E and MKK1/2 Inhibition
WM239A melanoma cells harbor a V600D BRAF mutation and are sensitive to both BRAFV600E and MKK1/2 inhibitors. RNA-seq data from these cells revealed no other mutations in BRAF, NRAS, MKK1, or MKK2, with the exception of a synonymous mutation in MKK2 at isoleucine 220 (data not shown). To compare the response of these cells to BRAFV600E and MKK1/2 inhibition, cells were labeled with light, medium, or heavy SILAC media (33) and treated with the MKK1/2 inhibitor, AZD6244 (AZD), or the B-RAFV600E inhibitor, PLX4032 (PLX), each at a concentration of 10 μm (Fig. 1). Phosphopeptides were enriched by TiO2 and separated into 24 fractions by electrostatic repulsion-hydrophilic interaction chromatography (ERLIC) using an ammonium formate buffer system (Experimental Procedures).
Fig. 1.
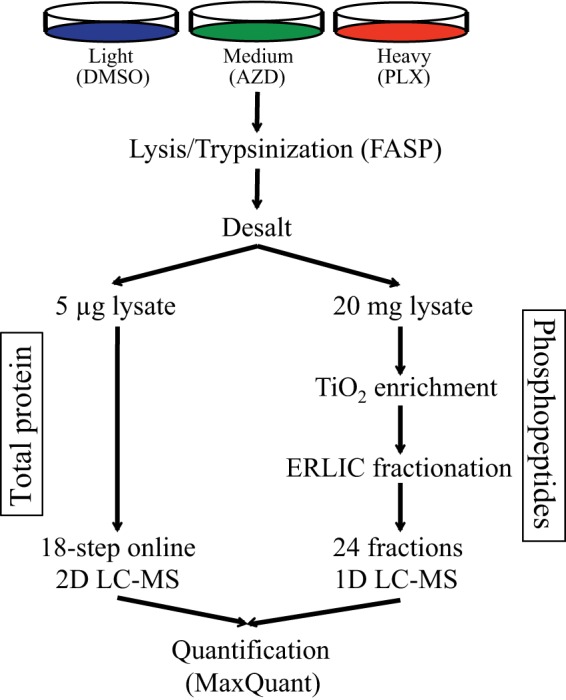
Experimental design for phosphoproteomics analysis of WM239A cells treated with AZD6244 and PLX4032. A schematic of the protocol used for enrichment and analysis of phosphopeptides in SILAC experiments.
ERLIC has been reported to separate phosphopeptides relatively evenly across fractions, permitting higher numbers of phosphopeptide identifications (34, 35). Consistent with these studies, we observed that phosphopeptides separated quite evenly across the 24 ERLIC fractions (Fig. 2A). The resolution was also excellent, with the vast majority of phosphopeptides (85%) eluting in only one or two fractions (Fig. 2B). The majority of phosphopeptides identified were singly phosphorylated, with multiply phosphorylated peptides abundant only in fractions 22–24 (Fig. 2C). In three biological replicates, we were able to quantify 16,890, 18,833, and 18,103 phosphosites (Fig. 2D). In all, we quantified 23,986 phosphosites, 20,553 (86%) of which were class I sites (localization probability >0.75 and delta score >5) (36), showing that that most phosphosites could be localized with high confidence (supplemental Tables S1 and S2).
Fig. 2.

Performance of the ERLIC fractionation method for separating complex phosphopeptide mixtures. A, Bar chart indicating the unique phosphorylated and nonphosphorylated peptides identified in each ERLIC fraction. Numbers report the sum of peptides identified in three replicate experiments. B, Bar chart showing the number of ERLIC fractions in which individual phosphopeptides and nonphosphopeptides were detected. The data are from one replicate experiment with similar results obtained in each experiment. C, Bar chart showing the number of phosphorylated residues on phosphopeptides in each ERLIC fraction. Numbers report the sum of phosphopeptides in three replicate experiments. D, Area-proportional Euler diagram (73) showing the overlap of phosphosites identified in the three replicate experiments.
We performed three biological replicates, alternating SILAC labeling between experiments (Fig. 3A). At concentrations of 10 μm, both AZD and PLX reduced phosphorylation of ERK1/2 at its regulatory phosphosites to levels undetectable by Western blotting (Fig. 3B). In addition, the SILAC peptide containing these phosphosites was undetectable under drug-treated conditions confirming that both inhibitors achieve nearly complete pathway inhibition at these concentrations (Fig. 3C). Changes in phosphorylation after inhibitor treatment were considered significant at a given phosphosite when the SILAC ratio reflected more than a 1.8-fold change (log2 value ±0.840). This threshold is the log2 value exceeded by 1% of phosphosite ratio measurements in a control experiment in which light-, medium-, and heavy-labeled cells with were each treated with DMSO (supplemental Fig. S1). These values correspond to z-scores of −3.09 and +3.13.
Fig. 3.
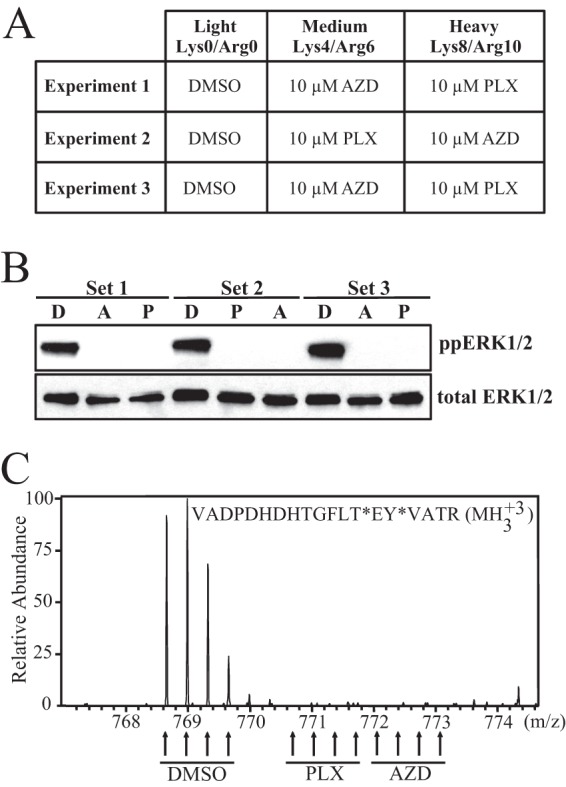
Experimental strategy for AZD and PLX treatment of WM239A cells. A, Summary of the treatment conditions for light, medium, and heavy SILAC media for each biological replicate experiment. B, Western blot of phospho-ERK1/2 in cell lysates from each experiment. C, Representative MS spectrum of the diphosphopeptide harboring the activating Thr and Tyr phosphorylation sites in ERK2.
In each biological replicate, 7–8% of phosphosites decreased significantly upon treatment with either AZD or PLX (Table I). As would be expected for treatment with kinase inhibitors, a smaller percentage of phosphosites were observed to increase in response to inhibitor treatment (1.6–3.6%). There was a high degree of overlap between experimental replicates, and we focused our analysis on the 17,790 phosphosites that were quantified in at least two experiments (Fig. 2D). Responses at these sites were considered significant if the median value of the SILAC ratios from individual experiments exceeded the significance threshold of a 1.8-fold change (for sites quantified in only two replicates, the median value is the average of the two ratios) (Table I).
Table I. Summary of phosphosite identifications in replicate experiments.
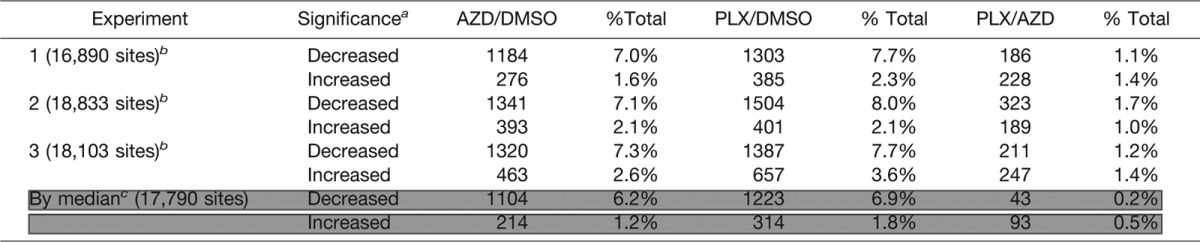
a Ratios were considered significant at a log2 value exceeding ±0.84.
b Total number of phosphosites quantified in each experiment after removing reverse hits and contaminants and expanding site table to separate phosphosites identified in multiply phosphorylated forms.
c Significance for all phosphosites identified in replicate experiments was determined using the log2 value of the median SILAC ratio (calculated as an average in the case of duplicate measurements).
Histograms of the median SILAC ratios (AZD/DMSO, PLX/DMSO, and PLX/AZD) for these phosphosites centered at log2 = 0, and AZD/DMSO and PLX/DMSO histograms showed a clear enrichment for phosphosites that decreased following drug treatment (Fig. 4A). In total, 1317 phosphosites on 763 proteins decreased in response to either AZD or PLX. These included 664 phosphosites that contained the minimal ERK1/2 consensus phosphorylation motif (Ser/Thr-Pro), of which 277 sites contained the full consensus motif (Pro-Xxx-Ser/Thr-Pro). Nearly all drug-responsive phosphosites reflected true changes in phosphorylation. Only three phosphosites that decreased in response to inhibitor changed in a manner that could easily be explained by altered protein abundance (supplemental Fig. S2, supplemental Table S3). These were pSer74, pThr189, and pSer191 on FOSL1, which is destabilized by the inhibitor-dependent dephosphorylation of pSer265 (37). Overall, we observed very high overlap between the phosphosites that changed in response to AZD and PLX (Fig. 4B and 4C, blue symbols). Many phosphosites that scored as significant with only one inhibitor actually responded to both, but fell just below the 1% FDR cutoff with the other inhibitor (Fig. 4C, green symbols). Thus, the responses to inhibition of B-RAF and MKK1/2, representing different tiers of the B-RAF/MKK/ERK pathway, were remarkably uniform.
Fig. 4.

Overlap between phosphosites that respond to AZD or PLX. A, Log2 values of the median SILAC ratios for all phosphosites identified in at least two replicate experiments. Histograms are plotted for AZD/DMSO, PLX/DMSO, and PLX/AZD ratios. B, Area-proportional Venn diagrams showing the overlap in phosphosites with significant AZD/DMSO or PLX/DMSO ratios based on median values. Diagrams distinguish between phosphosites that increase or decrease in response to inhibitor and between phosphosites that do or do not contain the minimal ERK1/2 phosphorylation motif (Ser/Thr-Pro). C, Scatter plot of the log2 values of the median AZD/DMSO and PLX/DMSO ratios for individual phosphosites. Colors reflect phosphosites that change significantly with both AZD and PLX (cyan), either AZD or PLX (green), or neither inhibitor (gray), using a significance threshold of log2 value ±0.840. Many phosphosites with apparent responses to only one inhibitor actually change with both, but narrowly miss the significance cutoff with the other inhibitor. Dashed box indicates the significance threshold.
Cellular Networks Responding to AZD and PLX
Proteins with phosphosites that decrease in response to AZD and PLX were examined for high confidence interactions using the STRING database (38, 39). We focused on proteins containing phosphosites with the minimal ERK1/2 consensus motif (Ser/Thr-Pro), a subset that should be enriched in direct ERK1/2 substrates. Analysis of these proteins identified subnetworks representing the nuclear pore complex, regulators of transcription, growth factor signaling, small GTPase guanine nucleotide exchange factors (GEFs) and hydrolysis-activating proteins (GAPs), and proteins associated with the centrosome and mitotic regulation (Fig. 5).
Fig. 5.

STRING network analysis of proteins harboring Ser-Thr/Pro phosphosites that decrease in response to inhibitor. STRING network of high-confidence interactions (minimum confidence score of 0.900) among proteins containing Ser/Thr-Pro phosphosites that decrease in response to AZD or PLX. Major subnetworks are highlighted in blue. Disconnected nodes are not shown. Color of the nodes is arbitrary. Large nodes indicate that structural information is available for that protein.
The most interconnected network was the nuclear pore complex, of which NUP50, NUP98, NUP153, NUP214, and TPR have previously been identified as ERK1/2 substrates (22, 28). In addition to these components, we identified sites that decreased on NUP35 and NUP188. Interestingly, the phosphosites that decreased in response to ERK pathway inhibition clustered to the cytoplasmic and nuclear FG nucleoporins (NUPs) as well as NUPs of the inner ring (Fig. 6A). Sites within the transmembrane ring, the linker, and outer ring NUPs were unaffected by inhibitor. Thus, there appears to be spatial clustering of the phosphosites regulated by AZD and PLX within the nuclear pore complex.
Fig. 6.

Selected targets of B-RAFV600E signaling. A, The nuclear pore network, showing nuclear pore proteins harboring Ser/Thr-Pro phosphosites identified in this study (circles) and their positions within the nuclear pore complex (center). Phosphosites indicated in yellow were significantly inhibited by AZD or PLX, and phosphosites in black were unchanged. B, Transcriptional regulators harboring Ser/Thr-Pro phosphosites significantly inhibited by AZD or PLX. Top: Phosphosites on known substrates for ERK1/2. Bottom: Phosphosites on candidate substrates for ERK1/2. Color indicates the log2 value of the median SILAC ratio for the phosphosite. For proteins in which multiple phosphosites decrease, the color reports the site with the largest change. Asterisks indicate validated regulatory sites where phosphorylation affects transcriptional activity, protein localization, or protein stability. Double asterisks indicate phosphosites located within or immediately adjacent to functional domains.
The largest interaction network identified by STRING analysis was comprised of transcriptional activators, coactivators, and repressors (Fig. 5). This network included many well characterized ERK1/2 substrates, such as ELK1 (40), JUN (41), FOSL1 (42), MYC (43), and STAT3 (4), and prompted us to interrogate the data set for additional transcriptional regulators that are responsive to MAPK pathway inhibition in melanoma. To do this, proteins harboring Ser/Thr-Pro phosphosites that decreased upon inhibitor treatment were sorted by Gene Ontology molecular function using the PANTHER classification system (44). Proteins classified as having nucleic acid transcription factor activity were examined further. This analysis identified 39 proteins with bona fide roles in transcription, 19 of which have previously been characterized as downstream targets of MAPK signaling (Fig 6B, supplemental Table S4). Importantly, of the 30 phosphosites that decreased in response to inhibitor on these 19 known targets, 21 have previously been shown to affect transcriptional activity, protein localization, or protein stability, showing that many of the sites identified have regulatory functions.
The remaining 20 transcriptional regulators are new candidates for ERK1/2 substrates. Fourteen of these proteins have at least one consensus docking motif for ERK1/2, which strongly predicts direct substrates (supplemental Table S4). Additionally, several of the phosphosites that decrease on these candidate targets occur in regions of the protein likely to affect protein function (Fig. 6B). The pSer73 site in PTOV1 is immediately adjacent to the PTOV-A domain, a domain that can affect transcriptional activity, although its role in PTOV1 function is currently unclear (45). The drug-responsive phosphosites in ATF6 (pSer13/pSer16) and TCF12 (pSer67) lie within transactivation domains (46, 47) suggesting B-RAFV600E may control their transcriptional activity. Similarly, pSer353 on NFIL3 occurs in a previously characterized repression domain in which phosphorylation might affect protein activity (48). Finally, the inhibitor-responsive phosphosite in LIMD1 (pSer424) lies within a region necessary for its interaction with Rb, suggesting that phosphorylation may affect LIMD1:Rb binding and repression of E2F target genes (49).
Mapping Inhibitor Responses onto the Human Kinome
We next examined the 17,790 phosphosites for which we had replicate measurements with the goal of addressing which protein kinases are controlled by B-RAFV600E signaling. In total, there were 904 phosphosites that matched 217 enzymes in the human kinome (50) (supplemental Table S5). Of these, 120 sites on 72 kinases showed significant responses to AZD or PLX. Nineteen of these sites have validated functions in regulating enzyme activity, localization, molecular interactions, or protein stability (supplemental Table S5). The majority of these sites occurred on kinases within the RAF/MKK/ERK pathway, including A-RAF, B-RAF, RAF1, MKK1/2, ERK1/2, and p90RSK1/2/3. These included activating phosphorylation sites on MKK1 (pSer218, pSer222), MKK2 (pSer222, pSer226), ERK1 (pThr202, pTyr204), ERK2 (pThr185, pTyr187), and RSK2 (pSer369, pSer577). Each of these was inhibited in response to drug, as expected. A negative feedback site on RAF1 (pThr642) that is targeted by ERK1/2 (51) also decreased with inhibitor, reflecting loss of feedback inhibition following drug treatment.
Some responsive phosphosites occurred within protein kinases associated with other signaling pathways. Those with known regulatory function were linked to the activation of CHK1 (pSer280) and MKK4 (pSer257). These decreased with inhibitor treatment, indicating that B-RAFV600E enhances signaling through stress-response pathways. Likewise, activating sites in EPHA2 (pSer897) and p70S6K (pSer427) were inhibited by drug treatment, revealing the role for B-RAFV600E in promoting growth-regulatory mechanisms downstream of cell adhesion receptors and mTOR pathways. Drug-responsive phosphosite inhibition of GRK2/ADRBK1 (pSer670) suggests suppression of beta-adrenergic receptor signaling by B-RAFV600E. These results show crosstalk between oncogenic B-RAFV600E and other signaling pathways in melanoma.
Phosphorylation Events Uniquely Responsive to PLX or AZD
Although a handful of proteins have been implicated as MKK-independent targets of RAF1, the only known targets of B-RAF are MKK1/2 (1, 2). Similarly, the only known targets of MKK1/2 are ERK1 and ERK2 (3). Nevertheless, the existence of unique targets of B- RAFV600E and MKK1/2 could potentially explain the improved clinical responses seen with the combination of B-RAFV600E and MKK1/2 inhibitors. Therefore, we examined phosphosites that were uniquely responsive to PLX or AZD. Of the 1317 phosphosites that decreased in response to either drug, 61 had PLX/AZD ratios that surpassed the significance threshold of a ±1.8-fold change. After removing sites that decreased with both AZD and PLX but with different magnitudes, and sites with large variability between replicate experiments (“Experimental Procedures”), we identified 16 phosphosites that reproducibly decreased only in response to PLX, and two phosphosites that decreased only in response to AZD (Table II). Thus, although the overwhelming majority of phosphosites responded to both PLX and AZD, a small minority showed clear evidence for differential regulation.
Table II. Stuart et al.
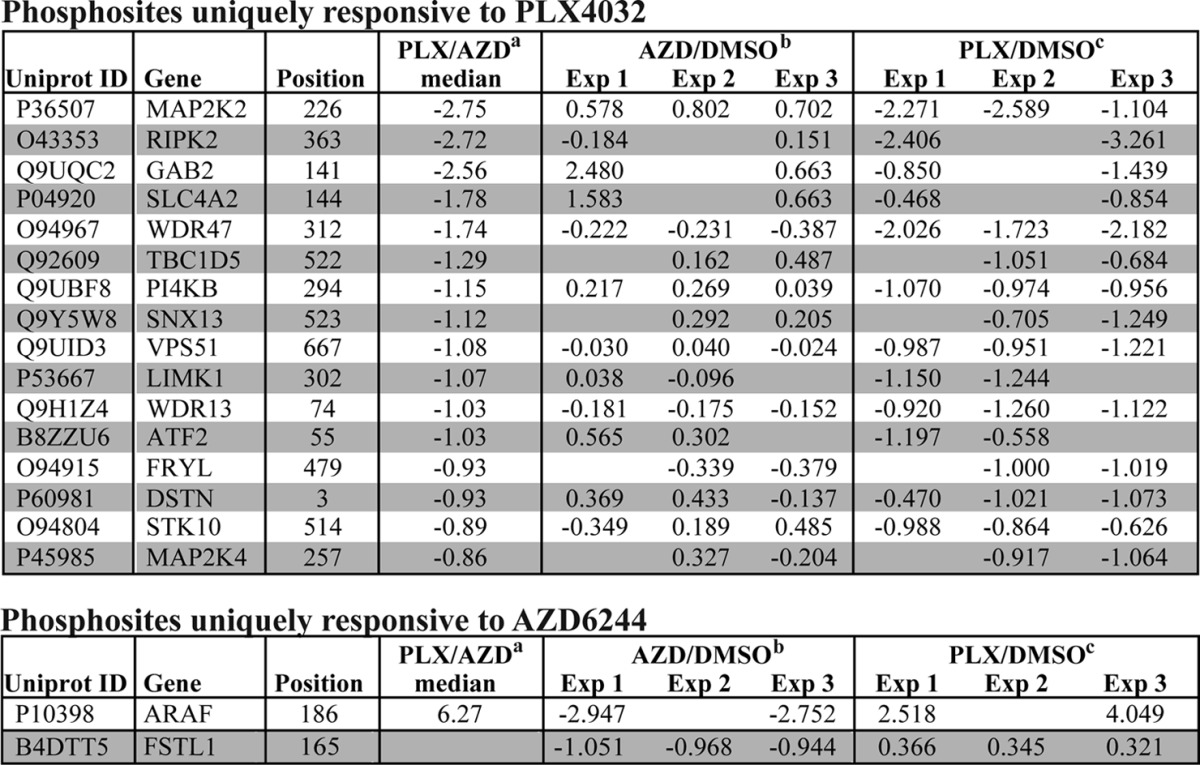
a log2 values of the median PLX/AZD SILAC ratio.
b log2 values of the median AZD/DMSO ratios in individual SILAC experiments.
c log2 values of the median PLX/DMSO ratios in individual SILAC experiments.
The one phosphopeptide that would have been expected to respond differently to AZD and PLX is the MKK1/2 phosphopeptide harboring its activating sites, which is directly phosphorylated by B-RAFV600E, and would be expected to decrease only in response to PLX. Indeed, Western blotting showed that phosphorylation at this site decreased in response to PLX but increased slightly in response to AZD (Table II, Fig 7A). The other 15 sites identified as unique targets of PLX include four protein kinases (RIPK2, LIMK1, MAP2K4, STK10), a lipid kinase (PI4KB), a transcription factor (ATF2), and two membrane trafficking proteins (SNX13, VPS51) (Table II). A particularly intriguing target was RIPK2, a dual-specificity kinase best characterized for its role in innate immune signaling downstream of NOD1 and NOD2 (52). Previous work has shown that RAF1 activates RIPK2, which in turn directly phosphorylates and activates ERK1/2 (53). Identification of RIPK2 as a unique target of PLX suggests that B-RAFV600E may be capable of activating ERK1/2 in an MKK1/2-independent manner by regulating RIPK2. Sequence alignment of the 16 phosphosites identified as unique targets of PLX failed to identify a consensus motif around the phosphorylated residue (Fig. 7B). Thus, to the extent that these sites represent direct targets of B-RAFV600E, no easily identifiable consensus sequence was indicated.
Fig. 7.
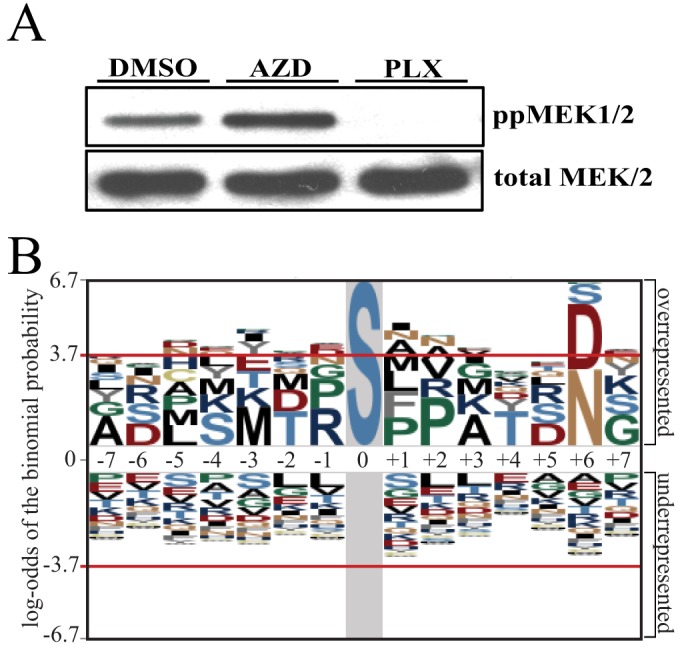
Phosphosites uniquely responsive to PLX do not show a consensus sequence. A, Western blot of phospho-MKK1/2 showing a decrease in phosphorylation in response to PLX but not AZD. B, pLOGO sequence motif for the 16 phosphosites found to decrease in response to PLX but not AZD.
The two phosphosites identified as unique targets of AZD occurred in the follistatin-related protein, FSTL1, and the RAF family member, A-RAF. Interestingly, the ARAF phosphopeptide exhibits a differential response to AZD and PLX only in its diphosphorylated form. Although both monophosphorylated peptides decrease in response to AZD and PLX, the diphosphorylated form decreases eightfold with AZD but increases as much as 16-fold with PLX. This behavior suggests that PLX either facilitates A-RAF phosphorylation or protects it from dephosphorylation, perhaps by promoting A-RAF:B-RAF dimerization.
Responses to Combination Treatment with AZD and PLX
We next examined the effects of combination treatment with B-RAFV600E and MKK1/2 inhibitors by designing a triple-labeled SILAC experiment to compare the responses of melanoma cells to AZD, PLX, or the combination of AZD + PLX (Fig. 8A). At 10 μm concentrations, we observed no difference in phosphosite responses to the combination of AZD + PLX versus AZD or PLX alone (supplemental Fig. S3). Thus, at 10 μm concentrations, both AZD and PLX completely inhibit ERK signaling. To identify concentrations at which combinatorial effects might be observed, we treated cells with different concentrations of either inhibitor and monitored the decrease in phospho-ERK1/2 levels. At concentrations of 30 nm, each inhibitor reduced ERK1/2 phosphorylation well below the maximum level and within an intermediate range in which additive or synergistic interactions could be observed (Fig. 8B). SILAC experiments were then conducted at this concentration, measuring phosphosite responses to AZD, PLX, and the combination in duplicate (Fig. 8A). Because most phosphosite responses were small at 30 nm inhibitor (supplemental Tables S6 and S7), we evaluated responses at those sites most likely to represent real decreases, based on their overlap with the 1317 sites that clearly decreased with 10 μm AZD or PLX (Fig. 8C). This yielded 319 sites where responses could be quantified in duplicate, and where inhibition was measurable with each drug individually. We used a Bliss independence model (54) to predict the additive response to the combination for each of these sites, based on the response of that site to each single drug. We calculated the predicted response using the equation:
where RE is the predicted response and RA and RP are the individual responses measured for AZD and PLX, respectively. A histogram of the differences between the predicted responses to the combination and the measured responses showed a normal distribution centered at zero (Fig. 8C). Only four phosphosites exhibited differences that exceeded a 3σ threshold. Manual inspection of the measured ratios for each of these four sites showed large variability between duplicate measurements suggesting these instances of significance may be spurious. Thus, at 30 nm concentrations, the molecular responses to the combination of AZD and PLX are additive, showing independence between the two inhibitors.
Fig. 8.

Combination treatment with AZD + PLX shows additive responses. A, Summary of treatment conditions for light, medium, and heavy SILAC media for experiments comparing AZD and PLX treatment to combination treatment. B, Western blots showing the decrease in phospho-ERK1/2 with varying concentrations of AZD (top) and PLX (bottom). C, Scatter plot of the log2 values of the AZD/DMSO and PLX/DMSO SILAC ratios, averaged from biological duplicate experiments. Only phosphosites identified in both duplicates were included. Red shows phosphosites that were used to test additivity in D. These sites were identified as ERK targets in response to 10 μm AZD or PLX and decrease with both single inhibitors and the combination at 30 nm concentrations. D, Histogram of the differences between the observed and predicted SILAC ratios for the 319 phosphosites for which a predicted ratio could be calculated using the Bliss independence model. Dashed lines show the 3σ significance threshold.
Variability in the Phosphorylation of RAF/MKK/ERK Signaling Targets
Finally, we examined the extent to which phosphorylation events targeted by inhibitors vary between different cell systems. To address this, we compared the phosphosite responses to 10 μm AZD in our study against responses to 10 μm PD184532, a MKK1/2 inhibitor used in large-scale phosphoproteomics experiments performed in human A375 cells expressing B-RAFV600E and HEK293 cells stimulated with PMA (27). In total, there were 4119 phosphosites that were quantified in all three cell lines from these two studies (supplemental Table S8). From these, we identified 879 class I phosphosites on 616 proteins that were inhibited by drug in at least one cell line (>1.8-fold decrease). These included 213 sites inhibited by AZD in WM239A cells, 360 sites inhibited by PD184352 in A375 cells, and 760 sites inhibited by PD184352 in HEK293 cells (Fig. 9A, supplemental Table S8). Surprisingly, only 142 of these 879 phosphosites passed the significance threshold in all three cell lines (Fig. 9A). This reveals that the majority of inhibitor-responsive phosphorylation events vary between cells. Because some of these differences might reflect phosphosites that narrowly miss the significance threshold in one or two cell types, we identified sites that were significantly responsive in one cell line (greater than 1.8-fold change; log2 < −0.84) but unresponsive in others (less than 1.2-fold change; log2 > −0.28). Using these criteria, we identified 217, 27, and 15 phosphosites that were uniquely responsive in HEK293 cells, A375 cells, and WM239A cells, respectively (Fig. 9B). The large number of phosphosite responses unique to HEK293 cells suggest that the spectrum of phosphorylation events regulated by the ERK1/2 pathway may be largely dependent on the cell type and/or mechanism of pathway activation. These unique responses do not appear to be because of different off-target effects of the two inhibitors given that both HEK293 and A375 cells were treated with PD184352.
Fig. 9.

Variability in phosphorylation responses to MKK1/2 inhibitors. A, Area-proportional Euler diagram (73) showing the overlap between phosphosites with significant responses to MKK1/2 inhibitor in WM239A cells (this study), A375 cells (27), or HEK293 cells (27). B, Area-proportional Euler diagram showing the overlap between phosphosites with a significant response in at least one cell line (log2 < −0.84) and unchanged (log2 > −0.28) in all lines where the response is not significant. C, Examples of proteins that display variability in phosphosite responses to MKK1/2 inhibitor. Charts above each example indicate the response in each cell line (displayed as log2 values of SILAC ratios). The horizontal red line within each chart indicates the significance threshold of log2 = −0.84.
Specific examples of phosphosite variation were instructive. NUP153 and NUP214 are both direct substrates for phosphorylation by ERK1/2 (22). Of the 10 phosphosites with inhibitor/control ratios that could be quantified in all three cell lines, seven showed significant responses in at least one cell line, but only two sites showed a significant response in all three cell lines (Fig. 9C). This reveals unexpected variability in nucleoporin phosphorylation, and implies that there is variation in the mechanism by which ERK1/2 regulates nuclear import in different cell systems. Other examples of proteins displaying variability in phosphorylation included the well-characterized ERK1/2 substrates, stathmin and cortactin. Here, three of five sites in stathmin, and three of four sites in cortactin showed varying responses to drug across the three cell lines (Fig. 9C). Two of these phosphorylation sites (pSer38 in STMN1 and pSer418 in CTTN) are direct substrates for ERK1/2 and also are key regulators of protein function. Taken together, our findings indicate that targets of a single pathway can show significantly different responses in different cell systems.
DISCUSSION
This study is the first of its scale to compare phosphoproteome responses to BRAFV600E and MKK1/2 inhibitors in melanoma cells. Our first objective was to evaluate the overlap between the molecular responses to B-RAFV600E and MKK1/2 inhibitors. Our results show that nearly all phosphosites that respond to AZD also respond to PLX. This high degree of overlap reinforces the idea that signaling from B-RAFV600E to MKK1/2 is remarkably linear. Our second objective was to use phosphoproteomics to understand the interactions between B-RAFV600E and MKK1/2 inhibitors and to provide molecular insight into why combination treatments lead to improved outcomes in clinical trials. Our results show that at subsaturating concentrations of inhibitors, phosphosite responses to combination treatment are additive, such that the magnitude of the responses to the combination can be predicted from the responses to each inhibitor individually in nearly all cases. This shows independence between BRAFV600E and MKK1/2 inhibitors and additive interactions in suppressing ERK signaling.
Collectively, these results provide insight into the clinical benefit of adding a MKK1/2 inhibitor to the B-RAFV600E inhibitor regimen. The fact that nearly all phosphosites that respond to AZD also respond to PLX make it unlikely that unique targets of MKK1/2 inhibitors account for the enhanced benefit of the combination. Instead, the interactions between the two drugs suggest that the benefit of the combination is best explained by the additive inhibition of ERK signaling. It has been reported that >80% inhibition of phospho-ERK1/2 is needed for an effective clinical response (20), and that this level of inhibition is difficult to achieve in many patients receiving single agent therapy with B-RAFV600E or MKK1/2 inhibitors (19). Our data suggest that the combination is likely more effective at reaching this level of inhibition than either drug individually.
Another common hypothesis used to explain the enhanced benefit of combination therapy is that the combination of inhibitors prevents the emergence of resistance mechanisms that still require MKK1/2 signaling (16, 18). This would include mechanisms of resistance such as N-RAS mutations, B-RAFV600E amplification, B-RAF splice variants, or receptor tyrosine kinase up-regulation. However, it was recently reported that two of five patients developing acquired resistance to combination treatment harbored resistance mechanisms that reactivated ERK signaling upstream of MKK1/2 (55). One resistant tumor showed B-RAFV600E amplification, whereas the other expressed a B-RAFV600E splice variant (56) known to confer resistance to B-RAFV600E inhibition. This suggests that the MKK1/2 inhibition achieved at the doses used in combination treatments is not sufficient for complete inhibition of MKK1/2 signaling and reinforces the notion that the enhanced benefit of the combination is likely because of the additive inhibition of ERK signaling.
Our high coverage of phosphosites was made possible by the use of the TiO2-ERLIC enrichment protocol, where batch TiO2 enrichment prior to ERLIC fractionation significantly decreased sample processing time with no loss of phosphopeptide identifications. In addition, the formate buffer system used for ERLIC fractionation yielded phosphopeptides that were predominantly singly phosphorylated (>96%). This contrasts with triethylammonium phosphate (TEAP)-based ERLIC separations, in which as many as 75% of phosphopeptides are multiply phosphorylated (57–60). Identification of singly phosphorylated peptides is advantageous for phosphoproteomics studies using MaxQuant because phosphosite ratios for these peptides reflect phosphorylation only at the site of interest and are easily interpreted. In contrast, phosphosite ratios that arise from multiply phosphorylated peptides can be derived from the SILAC ratios of more than one multiply phosphorylated peptide in which the site of interest is phosphorylated. Thus, quantifying changes in phosphosites present in multiply phosphorylated peptides requires manual analysis, which can be difficult with large data sets.
Our analysis of phosphosites that decreased in response to AZD and PLX led to the identification of potentially novel ERK sites on 20 transcription factors. Several of the 20 candidate target genes have previously been linked to melanoma survival or associated with melanoma progression (supplemental Table S4). PTOV1 (61) and the short isoform of PHF19 (62) are both overexpressed in advanced melanoma indicating that their regulation by B-RAFV600E might be important for disease progression. ATF6 has been shown to contribute to the intrinsic resistance of many melanoma cell lines to ER-stress induced apoptosis by upregulating antiapoptotic genes. Thus, phosphorylation of ATF6 within its transactivation domain by B-RAFV600E might increase transcription of antiapoptotic genes and promote cell survival (63, 64). CRTC2 is a coactivator of ATF6 (65) and its regulation by B-RAFV600E may also modulate ATF6 activity to affect transcription and promote survival. Finally, the transcriptional repressor, TSC22D4, promotes the bypass of B-RAFV600E-mediated oncogene-induced senescence when overexpressed in fibroblasts (66). It is tempting to speculate that regulation of TSC22D4 by B-RAFV600E might affect a similar process in melanocytes to allow melanomas to bypass senescence.
Our identification of phosphosites that respond to PLX, but not to AZD, suggests that oncogenic B-RAFV600E has cellular targets that are independent of MKK1/2 and that signaling branch points between B-RAFV600E and MKK1/2 may be more prevalent than currently appreciated. Some of these targets may be therapeutically relevant. RIPK2, for example, has been shown to bypass MKK1/2 and directly phosphorylate ERK1/2 downstream of RAF1 (53). Thus, RIPK2 may play a role similar to COT1, a kinase that when overexpressed also directly phosphorylates and activates ERK1/2 (67). Further studies are needed to address the extent to which the unique responses to PLX or AZD represent direct substrates of B-RAFV600E and MKK1/2, and whether these phosphorylation events affect melanoma survival or progression.
In our analysis of phosphosite responses, all available ratios for phosphosites on multiply phosphorylated peptides were treated as unique. This increased the number of phosphosite ratios by 16.0% (from 20,760 to 23,986) and made it possible to compare phosphosite ratios for singly and multiply phosphorylated peptides to assess potential hierarchal dependences between neighboring phosphosites. This strategy revealed that the unique sensitivity of ARAF pSer186 to AZD was specific to the diphosphorylated form of the peptide, suggesting regulation at this site is influenced by phosphorylation of ARAF at pThr181.
The degree to which phosphorylation events targeted by specific pathways vary between different cell systems, or in response to different treatment conditions, is undetermined. Courcelles et al. recently reported a striking lack of overlap in responses to MKK1/2 inhibitors among five published studies (28). However, these comparisons were made at the level of proteins rather than phosphosites, and the five studies compared used cell lines from different species and differed widely in their methods for quantifying changes. Therefore, it was difficult to conclusively determine the extent of variation in phosphosite regulation. To better address this question, we compared our study against that of Galan et al. (27), who examined human HEK293 cells and A375 melanoma cells treated with 10 μm PD184352. Both AZD and PD184352 bind to the same allosteric pocket in MKK1/2, and show similar effects in preclinical cancer models (68–70). Despite this, we observed a surprising degree of heterogeneity in the inhibitor responses between these three cell lines. Of the phosphosites that could be quantified across all three cell lines, only 16% (142) passed our threshold for significance in all cells. In contrast, nearly 37% of sites (321) showed clear variation between cell lines. In addition, 25% (217) were responsive to MKK1/2 inhibition only in PMA-stimulated HEK293 cells suggesting that activated B-RAFV600E in A375 and WM239A cells regulates only a small subset of potential ERK targets. These comparisons reveal that even within a single species, the molecular responses to activation of a pathway depend heavily on both the cell line and the means of pathway activation.
Recent analyses of phosphoproteomics data sets have shown that orthologous proteins show large variations in phosphosite localization between species, even after accounting for sequence conservation (71, 72). These analyses also showed that conserved phosphosites have regulatory functions more often than nonconserved sites. Therefore, we asked whether phosphosites that consistently respond to MKK1/2 inhibition across cell lines were also more likely to have regulatory function. To do this, we examined the 4119 phosphosites that could be quantified in all three cell lines, and found 237 that matched known human regulatory sites in the PhosphositePlus database (50). Sites with regulatory function accounted for 13 of the 142 sites (9.1%) that responded in all three cell lines, and 20 of the 333 sites (6.2%) that varied in at least one cell line. Thus, phosphosites that show consistent responses in all cell lines are not enriched for sites with regulatory function. Even with well-characterized ERK1/2 substrates such as NUP153/NUP214, STMN1, and CTTN, phosphorylation events controlling nuclear translocation, microtubule binding, and cell motility, respectively, show variability in their responses across cell systems. Determining the mechanisms by which phosphorylation responses vary is key to predicting responses to activation of a specific pathway. This is needed for the long-term goal of defining phosphorylation signatures that can serve as biomarkers for pathway activation.
Supplementary Material
Acknowledgments
We thank Johannes Rudolph and Mary Tarrant Connacher for thoughtful feedback on the manuscript.
Footnotes
Author contributions: S.A.S., S.H., and N.G.A. designed research; S.A.S. performed research; S.A.S., T.L., W.M.O., and N.G.A. analyzed data; S.A.S. and N.G.A. wrote the paper; N.W. ran ms samples.
* This study was funded by ACS PF-11-071-01-TBG and NIH 1F32CA154052-01 to SAS, R01 CA118972 to NGA, and R21 CA175448 to WMO.
 This article contains supplemental Figs. S1 to S3 and Tables S1 to S8.
This article contains supplemental Figs. S1 to S3 and Tables S1 to S8.
DATA AVAILABILITY: The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium (32) via the PRIDE partner repository with the data set identifiers PXD001560 (10 μm SILAC experiment) and PXD001563 (30 nm SILAC experiment).
1 The abbreviations used are:
- MAPK
- Mitogen-activated protein kinase
- 2D-PAGE
- 2-dimensional polyacrylamide gel electrophoresis
- AZD
- AZD6244
- ER
- endoplasmic reticulum
- ERLIC
- electrostatic repulsion-hydrophilic interaction chromatography
- FASP
- filter assisted sample preparation
- FDR
- false discovery rate
- FG
- nucleoporins - Phe-Gly nucleoporins
- GAP
- GTPase activating protein
- GEF
- guanine nucleotide exchange factor
- LTQ
- linear trap quadrupole
- NUP
- nucleoporin
- PANTHER
- protein analysis through evolutionary relationships
- PLX
- PLX4032
- PMA
- phorbol 12-myristate 13-acetate
- SILAC
- stable isotope labeling by amino acids in cell culture
- STRING
- search tool for the retrieval of interacting genes/proteins
- TFA
- trifluoroacetic acid
- TEAP
- triethylamine phosphate
- TiO2
- titanium dioxide.
REFERENCES
- 1. Matallanas D., Birtwistle M., Romano D., Zebisch A., Rauch J., von Kriegsheim A., Kolch W. (2011) Raf family kinases: Old dogs have learned new tricks. Genes Cancer 2, 232–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hindley A., Kolch W. (2002) Extracellular signal regulated kinase (ERK)/mitogen activated protein kinase (MAPK)-independent functions of Raf kinases. J. Cell Sci. 115, 1575–1581 [DOI] [PubMed] [Google Scholar]
- 3. Roskoski R. (2012) MEK1/2 dual-specificity protein kinases: structure and regulation. Biochem. Biophys. Res. Commun. 417, 5–10 [DOI] [PubMed] [Google Scholar]
- 4. Yoon S., Seger R. (2006) The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors 24, 21–44 [DOI] [PubMed] [Google Scholar]
- 5. Davies H., Bignell G. R., Cox C., Stephens P., Edkins S., Clegg S., Teague J., Woffendin H., Garnett M. J., Bottomley W., Davis N., Dicks E., Ewing R., Floyd Y., Gray K., Hall S., Hawes R., Hughes J., Kosmidou V., Menzies A., Mould C., Parker A., Stevens C., Watt S., Hooper S., Wilson R., Jayatilake H., Gusterson B. A., Cooper C., Shipley J., Hargrave D., Pritchard-Jones K., Maitland N., Chenevix-Trench G., Riggins G. J., Bigner D. D., Palmieri G., Cossu A., Flanagan A., Nicholson A., Ho J. W. C., Leung S. Y., Yuen S. T., Weber B. L., Seigler H. F., Darrow T. L., Paterson H., Marais R., Marshall C. J., Wooster R., Stratton M. R., Futreal P. A. (2002) Mutations of the BRAF gene in human cancer. Nature 417, 949–954 [DOI] [PubMed] [Google Scholar]
- 6. Sullivan R. J., Lorusso P. M., Flaherty K. T. (2013) The intersection of immune-directed and molecularly targeted therapy in advanced melanoma: where we have been, are, and will be. Clin. Cancer Res. 19, 5283–5291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Van Raamsdonk C. D., Bezrookove V., Green G., Bauer J., Gaugler L., O'Brien J. M., Simpson E. M., Barsh G. S., Bastian B. C. (2009) Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 457, 599–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Van Raamsdonk C. D., Griewank K. G., Crosby M. B., Garrido M. C., Vemula S., Wiesner T., Obenauf A. C., Wackernagel W., Green G., Bouvier N., Sozen M. M., Baimukanova G., Roy R., Heguy A., Dolgalev I., Khanin R., Busam K., Speicher M. R., O'Brien J., Bastian B. C. (2010) Mutations in GNA11 in uveal melanoma. N. Engl. J. Med. 363, 2191–2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Andersen L. B., Fountain J. W., Gutmann D. H., Tarlé S. A., Glover T. W., Dracopoli N. C., Housman D. E., Collins F. S. (1993) Mutations in the neurofibromatosis 1 gene in sporadic malignant melanoma cell lines. Nat. Genet. 3, 118–121 [DOI] [PubMed] [Google Scholar]
- 10. Chapman P. B., Hauschild A., Robert C., Haanen J. B., Ascierto P., Larkin J., Dummer R., Garbe C., Testori A., Maio M., Hogg D., Lorigan P., Lebbe C., Jouary T., Schadendorf D., Ribas A., O'Day S. J., Sosman J. a, Kirkwood J. M., Eggermont A. M. M., Dreno B., Nolop K., Li J., Nelson B., Hou J., Lee R. J., Flaherty K. T., McArthur G. A. (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 364, 2507–2516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hauschild A., Grob J.-J., Demidov L. V, Jouary T., Gutzmer R., Millward M., Rutkowski P., Blank C. U., Miller W. H., Kaempgen E., Martín-Algarra S., Karaszewska B., Mauch C., Chiarion-Sileni V., Martin A.-M., Swann S., Haney P., Mirakhur B., Guckert M. E., Goodman V., Chapman P. B. (2012) Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet 380, 358–365 [DOI] [PubMed] [Google Scholar]
- 12. Flaherty K. T., Robert C., Hersey P., Nathan P., Garbe C., Milhem M., Demidov L. V, Hassel J. C., Rutkowski P., Mohr P., Dummer R., Trefzer U., Larkin J. M. G., Utikal J., Dreno B., Nyakas M., Middleton M. R., Becker J. C., Casey M., Sherman L. J., Wu F. S., Ouellet D., Martin A.-M., Patel K., Schadendorf D. (2012) Improved survival with MEK inhibition in BRAF-mutated melanoma. N. Engl. J. Med. 367, 107–114 [DOI] [PubMed] [Google Scholar]
- 13. Larkin J., Ascierto P. A., Dréno B., Atkinson V., Liszkay G., Maio M., Mandalà M., Demidov L., Stroyakovskiy D., Thomas L., Merino L. D. L. C., Dutriaux C., Garbe C., Sovak M. A., Chang I., Choong N., Hack S. P., McArthur G. A., Ribas A. (2014) Combined Vemurafenib and Cobimetinib in BRAF -Mutated Melanoma. N. Engl. J. Med. 371, 1867–1876 [DOI] [PubMed] [Google Scholar]
- 14. Carvajal R. D., Sosman J. A., Quevedo J. F., Milhem M. M., Joshua A. M., Kudchadkar R. R., Linette G. P., Gajewski T. F., Lutzky J., Lawson D. H., Lao C. D., Flynn P. J., Albertini M. R., Sato T., Lewis K., Doyle A., Ancell K., Panageas K. S., Bluth M., Hedvat C., Erinjeri J., Ambrosini G., Marr B., Abramson D. H., Dickson M. A., Wolchok J. D., Chapman P. B., Schwartz G. K. (2014) Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: a randomized clinical trial. Jama 311, 2397–2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Robert C., Karaszewska B., Schachter J., Rutkowski P., Mackiewicz A., Stroiakovski D., Lichinitser M., Dummer R., Grange F., Mortier L., Chiarion-Sileni V., Drucis K., Krajsova I., Hauschild A., Lorigan P., Wolter P., Long G. V., Flaherty K., Nathan P., Ribas A., Martin A.-M., Sun P., Crist W., Legos J., Rubin S. D., Little S. M., Schadendorf D. (2014) Improved Overall Survival in Melanoma with Combined Dabrafenib and Trametinib. N. Engl. J. Med., [DOI] [PubMed] [Google Scholar]
- 16. Flaherty K. T., Infante J. R., Daud A., Gonzalez R., Kefford R. F., Sosman J., Hamid O., Schuchter L., Cebon J., Ibrahim N., Kudchadkar R., Burris H. A., Falchook G., Algazi A., Lewis K., Long G. V, Puzanov I., Lebowitz P., Singh A., Little S., Sun P., Allred A., Ouellet D., Kim K. B., Patel K., Weber J. (2012) Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N. Engl. J. Med. 367, 1694–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Long G. V., Stroyakovskiy D., Gogas H., Levchenko E., de Braud F., Larkin J., Garbe C., Jouary T., Hauschild A., Grob J. J., Sileni V. C., Lebbe C., Mandalà M., Millward M., Arance A., Bondarenko I., Haanen J. B. a. G., Hansson J., Utikal J., Ferraresi V., Kovalenko N., Mohr P., Probachai V., Schadendorf D., Nathan P., Robert C., Ribas A., DeMarini D. J., Irani J. G., Casey M., Ouellet D., Martin A.-M., Le N., Patel K., Flaherty K. (2014) Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. N. Engl. J. Med. 371, 1877–8188 [DOI] [PubMed] [Google Scholar]
- 18. Paraiso K. H. T., Fedorenko I. V, Cantini L. P., Munko A. C., Hall M., Sondak V. K., Messina J. L., Flaherty K. T., Smalley K. S. M. (2010) Recovery of phospho-ERK activity allows melanoma cells to escape from BRAF inhibitor therapy. Br. J. Cancer 102, 1724–1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bollag G., Hirth P., Tsai J., Zhang J., Ibrahim P. N., Cho H., Spevak W., Zhang C., Zhang Y., Habets G., Burton E. A., Wong B., Tsang G., West B. L., Powell B., Shellooe R., Marimuthu A., Nguyen H., Zhang K. Y. J., Artis D. R., Schlessinger J., Su F., Higgins B., Iyer R., D'Andrea K., Koehler A., Stumm M., Lin P. S., Lee R. J., Grippo J., Puzanov I., Kim K. B., Ribas A., McArthur G. A., Sosman J. A., Chapman P. B., Flaherty K. T., Xu X., Nathanson K. L., Nolop K. (2010) Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 467, 596–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hartsough E., Shao Y., Aplin A. E. (2014) Resistance to RAF inhibitors revisited. J. Invest. Dermatol. 134, 319–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Lewis T. S., Hunt J. B., Aveline L. D., Jonscher K. R., Louie D. F., Yeh J. M., Nahreini T. S., Resing K. A., Ahn N. G. (2000) Identification of Novel MAP Kinase Pathway Signaling Targets by Functional Proteomics and Mass Spectrometry. Mol. Cell 6, 1343–1354 [DOI] [PubMed] [Google Scholar]
- 22. Kosako H., Yamaguchi N., Aranami C., Ushiyama M., Kose S., Imamoto N., Taniguchi H., Nishida E., Hattori S. (2009) Phosphoproteomics reveals new ERK MAP kinase targets and links ERK to nucleoporin-mediated nuclear transport. Nat. Struct. Mol. Biol. 16, 1026–1035 [DOI] [PubMed] [Google Scholar]
- 23. Eblen S. T., Kumar N. V., Shah K., Henderson M. J., Watts C. K. W., Shokat K. M., Weber M. J. (2003) Identification of novel ERK2 substrates through use of an engineered kinase and ATP analogs. J. Biol. Chem. 278, 14926–14935 [DOI] [PubMed] [Google Scholar]
- 24. Carlson S. M., Chouinard C. R., Labadorf A., Lam C. J., Schmelzle K., Fraenkel E., White F. M. (2011) Large-scale discovery of ERK2 substrates identifies ERK-mediated transcriptional regulation by ETV3. Sci. Signal. 4, rs11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Old W. M., Shabb J. B., Houel S., Wang H., Couts K. L., Yen C.-Y., Litman E. S., Croy C. H., Meyer-Arendt K., Miranda J. G., Brown R. A., Witze E. S., Schweppe R. E., Resing K. A., Ahn N. G. (2009) Functional proteomics identifies targets of phosphorylation by B-Raf signaling in melanoma. Mol. Cell 34, 115–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pan C., Olsen J. V, Daub H., Mann M. (2009) Global effects of kinase inhibitors on signaling networks revealed by quantitative phosphoproteomics. Mol. Cell. Proteomics 8, 2796–2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Galan J. A., Geraghty K. M., Lavoie G., Kanshin E., Tcherkezian J., Calabrese V., Jeschke G. R., Turk B. E., Ballif B. A., Blenis J., Thibault P., Roux P. P. (2014) Phosphoproteomic analysis identifies the tumor suppressor PDCD4 as a RSK substrate negatively regulated by 14–3-3. Proc. Natl. Acad. Sci. U. S. A. 111, E2918–2927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Courcelles M., Frémin C., Voisin L., Lemieux S., Meloche S., Thibault P. (2013) Phosphoproteome dynamics reveal novel ERK1/2 MAP kinase substrates with broad spectrum of functions. Mol. Syst. Biol. 9, 669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Parker R., Clifton-Bligh R., Molloy M. P. (2014) Phosphoproteomics of MAPK inhibition in BRAF-mutated cells and a role for the lethal synergism of dual BRAF and CK2 inhibition. Mol. Cancer Ther. 13, 1894–1906 [DOI] [PubMed] [Google Scholar]
- 30. Wiśniewski J. R., Zougman A., Nagaraj N., Mann M. (2009) Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 [DOI] [PubMed] [Google Scholar]
- 31. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 32. Vizcaíno J. A., Deutsch E. W., Wang R., Csordas A., Reisinger F., Ríos D., Dianes J. A., Sun Z., Farrah T., Bandeira N., Binz P.-A., Xenarios I., Eisenacher M., Mayer G., Gatto L., Campos A., Chalkley R. J., Kraus H.-J., Albar J. P., Martinez-Bartolomé S., Apweiler R., Omenn G. S., Martens L., Jones A. R., Hermjakob H. (2014) ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ong S.-E., Mann M. (2006) A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC). Nat. Protoc. 1, 2650–2660 [DOI] [PubMed] [Google Scholar]
- 34. Alpert A., Mitulovi G., Mechtler K. in HPLC 2008 Conference (Baltimore, MD: ), p Poster P–2412–W. [Google Scholar]
- 35. Boichenko A. P., Govorukhina N., van der Zee A. G. J., Bischoff R. (2013) Multidimensional separation of tryptic peptides from human serum proteins using reversed-phase, strong cation exchange, weak anion exchange, and fused-core fluorinated stationary phases. J. Sep. Sci. 36, 3463–3470 [DOI] [PubMed] [Google Scholar]
- 36. Olsen J. V, Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., Mann M. (2006) Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
- 37. Basbous J., Chalbos D., Hipskind R., Jariel-Encontre I., Piechaczyk M. (2007) Ubiquitin-independent proteasomal degradation of Fra-1 is antagonized by Erk1/2 pathway-mediated phosphorylation of a unique C-terminal destabilizer. Mol. Cell. Biol. 27, 3936–3950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Snel B., Lehmann G., Bork P., Huynen M. A. (2000) STRING: a web-server to retrieve and display the repeatedly occurring neighbourhood of a gene. Nucleic Acids Res. 28, 3442–3444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Von Mering C., Jensen L. J., Kuhn M., Chaffron S., Doerks T., Krüger B., Snel B., Bork P. (2007) STRING 7–recent developments in the integration and prediction of protein interactions. Nucleic Acids Res. 35, D358–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gille H., Kortenjann M., Thomae O., Moomaw C., Slaughter C., Cobb M. H., Shaw P. E. (1995) ERK phosphorylation potentiates Elk-1-mediated ternary complex formation and transactivation. EMBO J. 14, 951–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pulverer B. J., Kyriakis J. M., Avruch J., Nikolakaki E., Woodgett J. R. (1991) Phosphorylation of c-jun mediated by MAP kinases. Nature 353, 670–674 [DOI] [PubMed] [Google Scholar]
- 42. Young M. R., Nair R., Bucheimer N., Tulsian P., Brown N., Chapp C., Hsu T., Colburn N. H. (2002) Transactivation of Fra-1 and consequent activation of AP-1 occur extracellular signal-regulated kinase dependently. Mol. Cell. Biol. 22, 587–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Alvarez E., Northwood I. C., Gonzalez F. A., Latour D. A., Seth A., Abate C., Curran T., Davis R. J. (1991) Pro-Leu-Ser/Thr-Pro is a consensus primary sequence for substrate protein phosphorylation. Characterization of the phosphorylation of c-myc and c-jun proteins by an epidermal growth factor receptor threonine 669 protein kinase. J. Biol. Chem. 266, 15277–15285 [PubMed] [Google Scholar]
- 44. Mi H., Muruganujan A., Casagrande J. T., Thomas P. D. (2013) Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 8, 1551–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Benedit P., Paciucci R., Thomson T. M., Valeri M., Nadal M., Càceres C., de Torres I., Estivill X., Lozano J. J., Morote J., Reventós J. (2001) PTOV1, a novel protein overexpressed in prostate cancer containing a new class of protein homology blocks. Oncogene 20, 1455–1464 [DOI] [PubMed] [Google Scholar]
- 46. Yoshida H., Okada T., Haze K., Yanagi H., Yura T., Negishi M., Mori K. (2000) ATF6 activated by proteolysis binds in the presence of NF-Y (CBF) directly to the cis-acting element responsible for the mammalian unfolded protein response. Mol. Cell. Biol. 20, 6755–6767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Massari M. E., Jennings P. A., Murre C. (1996) The AD1 transactivation domain of E2A contains a highly conserved helix which is required for its activity in both Saccharomyces cerevisiae and mammalian cells. Mol. Cell. Biol. 16, 121–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cowell I. G., Hurst H. C. (1994) Transcriptional repression by the human bZIP factor E4BP4: definition of a minimal repression domain. Nucleic Acids Res. 22, 59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sharp T. V, Munoz F., Bourboulia D., Presneau N., Darai E., Wang H.-W., Cannon M., Butcher D. N., Nicholson A. G., Klein G., Imreh S., Boshoff C. (2004) LIM domains-containing protein 1 (LIMD1), a tumor suppressor encoded at chromosome 3p21.3, binds pRB and represses E2F-driven transcription. Proc. Natl. Acad. Sci. U. S. A. 101, 16531–16536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hornbeck P. V, Kornhauser J. M., Tkachev S., Zhang B., Skrzypek E., Murray B., Latham V., Sullivan M. (2012) PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 40, D261–270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dougherty M. K., Müller J., Ritt D. A., Zhou M., Zhou X. Z., Copeland T. D., Conrads T. P., Veenstra T. D., Lu K. P., Morrison D. K. (2005) Regulation of Raf-1 by direct feedback phosphorylation. Mol. Cell 17, 215–224 [DOI] [PubMed] [Google Scholar]
- 52. Tigno-Aranjuez J. T., Asara J. M., Abbott D. W. (2010) Inhibition of RIP2's tyrosine kinase activity limits NOD2-driven cytokine responses. Genes Dev. 24, 2666–2677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Navas T. A., Baldwin D. T., Stewart T. A. (1999) RIP2 is a Raf1-activated mitogen-activated protein kinase kinase. J. Biol. Chem. 274, 33684–33690 [DOI] [PubMed] [Google Scholar]
- 54. Zhao W., Sachsenmeier K., Zhang L., Sult E., Hollingsworth R. E., Yang H. (2014) A New Bliss Independence Model to Analyze Drug Combination Data. J. Biomol. Screen. 19, 817–821 [DOI] [PubMed] [Google Scholar]
- 55. Wagle N., Van Allen E. M., Treacy D. J., Frederick D. T., Cooper Z. A., Taylor-Weiner A., Rosenberg M., Goetz E. M., Sullivan R. J., Farlow D. N., Friedrich D. C., Anderka K., Perrin D., Johannessen C. M., McKenna A., Cibulskis K., Kryukov G., Hodis E., Lawrence D. P., Fisher S., Getz G., Gabriel S. B., Carter S. L., Flaherty K. T., Wargo J. A., Garraway L. A. (2014) MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 4, 61–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Poulikakos P. I., Persaud Y., Janakiraman M., Kong X., Ng C., Moriceau G., Shi H., Atefi M., Titz B., Gabay M. T., Salton M., Dahlman K. B., Tadi M., Wargo J. A., Flaherty K. T., Kelley M. C., Misteli T., Chapman P. B., Sosman J. A., Graeber T. G., Ribas A., Lo R. S., Rosen N., Solit D. B. (2011) RAF inhibitor resistance is mediated by dimerization of aberrantly spliced BRAF(V600E). Nature 480, 387–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gan C. S., Guo T., Zhang H., Lim S. K., Sze S. K. (2008) A comparative study of electrostatic repulsion-hydrophilic interaction chromatography (ERLIC) versus SCX-IMAC-based methods for phosphopeptide isolation/enrichment. J. Proteome Res. 7, 4869–4877 [DOI] [PubMed] [Google Scholar]
- 58. Zarei M., Sprenger A., Metzger F., Gretzmeier C., Dengjel J. (2011) Comparison of ERLIC-TiO2, HILIC-TiO2, and SCX-TiO2 for global phosphoproteomics approaches. J. Proteome Res. 10, 3474–3483 [DOI] [PubMed] [Google Scholar]
- 59. Zarei M., Sprenger A., Gretzmeier C., Dengjel J. (2012) Combinatorial use of electrostatic repulsion-hydrophilic interaction chromatography (ERLIC) and strong cation exchange (SCX) chromatography for in-depth phosphoproteome analysis. J. Proteome Res. 11, 4269–4276 [DOI] [PubMed] [Google Scholar]
- 60. Zarei M., Sprenger A., Gretzmeier C., Dengjel J. (2013) Rapid combinatorial ERLIC-SCX solid-phase extraction for in-depth phosphoproteome analysis. J. Proteome Res. 12, 5989–5995 [DOI] [PubMed] [Google Scholar]
- 61. Fernández S., Mosquera J. L., Alaña L., Sanchez-Pla A., Morote J., Ramón Y Cajal S., Reventós J., de Torres I., Paciucci R. (2011) PTOV1 is overexpressed in human high-grade malignant tumors. Virchows Arch. 458, 323–330 [DOI] [PubMed] [Google Scholar]
- 62. Wang S., Robertson G. P., Zhu J. (2004) A novel human homologue of Drosophila polycomblike gene is up-regulated in multiple cancers. Gene 343, 69–78 [DOI] [PubMed] [Google Scholar]
- 63. Jiang C. C., Chen L. H., Gillespie S., Wang Y. F., Kiejda K. A., Zhang X. D., Hersey P. (2007) Inhibition of MEK sensitizes human melanoma cells to endoplasmic reticulum stress-induced apoptosis. Cancer Res. 67, 9750–9761 [DOI] [PubMed] [Google Scholar]
- 64. Jiang C. C., Lucas K., Avery-Kiejda K. A., Wade M., DeBock C. E., Thorne R. F., Allen J., Hersey P., Zhang X. D. (2008) Up-regulation of Mcl-1 is critical for survival of human melanoma cells upon endoplasmic reticulum stress. Cancer Res. 68, 6708–6717 [DOI] [PubMed] [Google Scholar]
- 65. Wang Y., Vera L., Fischer W. H., Montminy M. (2009) The CREB coactivator CRTC2 links hepatic ER stress and fasting gluconeogenesis. Nature 460, 534–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Hömig-Hölzel C., van Doorn R., Vogel C., Germann M., Cecchini M. G., Verdegaal E., Peeper D. S. (2011) Antagonistic TSC22D1 variants control BRAF(E600)-induced senescence. EMBO J. 30, 1753–1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Johannessen C. M., Boehm J. S., Kim S. Y., Thomas S. R., Wardwell L., Johnson L. A., Emery C. M., Stransky N., Cogdill A. P., Barretina J., Caponigro G., Hieronymus H., Murray R. R., Salehi-Ashtiani K., Hill D. E., Vidal M., Zhao J. J., Yang X., Alkan O., Kim S., Harris J. L., Wilson C. J., Myer V. E., Finan P. M., Root D. E., Roberts T. M., Golub T., Flaherty K. T., Dummer R., Weber B. L., Sellers W. R., Schlegel R., Wargo J. A., Hahn W. C., Garraway L. A. (2010) COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 468, 968–972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Haass N. K., Sproesser K., Nguyen T. K., Contractor R., Medina C. A., Nathanson K. L., Herlyn M., Smalley K. S. M. (2008) The mitogen-activated protein/extracellular signal-regulated kinase kinase inhibitor AZD6244 (ARRY-142886) induces growth arrest in melanoma cells and tumor regression when combined with docetaxel. Clin. Cancer Res. 14, 230–239 [DOI] [PubMed] [Google Scholar]
- 69. Collisson E. A., De A., Suzuki H., Gambhir S. S., Kolodney M. S. (2003) Treatment of metastatic melanoma with an orally available inhibitor of the Ras-Raf-MAPK cascade. Cancer Res. 63, 5669–5673 [PubMed] [Google Scholar]
- 70. Wang D., Boerner S. A., Winkler J. D., LoRusso P. M. (2007) Clinical experience of MEK inhibitors in cancer therapy. Biochim. Biophys. Acta 1773, 1248–1255 [DOI] [PubMed] [Google Scholar]
- 71. Beltrao P., Bork P., Krogan N. J., van Noort V. (2013) Evolution and functional cross-talk of protein post-translational modifications. Mol. Syst. Biol. 9, 714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Beltrao P., Albanèse V., Kenner L. R., Swaney D. L., Burlingame A., Villén J., Lim W. A., Fraser J. S., Frydman J., Krogan N. J. (2012) Systematic functional prioritization of protein posttranslational modifications. Cell 150, 413–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Micallef L., Rodgers P. (2014) eulerAPE: drawing area-proportional 3-Venn diagrams using ellipses. PLoS One 9, e101717. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.