

Abstract
Isolation and molecular characterization of rare cells (e.g. circulating tumor and stem cells) within biological fluids and tissues has significant potential in clinical diagnostics and personalized medicine. The present work describes an integrated platform of sample procurement, preparation, and analysis for deep proteomic profiling of rare cells in blood. Microfluidic magnetophoretic isolation of target cells spiked into 1 ml of blood at the level of 1000–2000 cells/ml, followed by focused acoustics-assisted sample preparation has been coupled with one-dimensional PLOT-LC-MS methodology. The resulting zeptomole detection sensitivity enabled identification of ∼4000 proteins with injection of the equivalent of only 100–200 cells per analysis. The characterization of rare cells in limited volumes of physiological fluids is shown by the isolation and quantitative proteomic profiling of first MCF-7 cells spiked into whole blood as a model system and then two CD133+ endothelial progenitor and hematopoietic cells in whole blood from volunteers.
Rare cells in blood and tissue have been shown to serve as specific indicators of disease status and progression, a source of adult stem cells, and a tool for patient stratification and monitoring. Previous reports (1–4), for example, have shown that the concentration of circulating tumor cells (CTCs) within a cancer patient's blood can act as a therapeutic monitoring tool (1–4). Additionally, the isolation of adult stem cells provides a needed cell source for tissue engineering and regenerative medicine treatments (5, 6). Finally, separation and genomic analysis of key cell populations from patients allows for targeted treatment regimens (7, 8).
Rare cells in blood or other body fluids represent a particularly challenging problem for discovery proteomic analysis as the volume of the fluid sample is limited and the concentration of cells within that sample is very low. For a blood sample containing rare cells of interest, this low level means capturing a subpopulation of target cells with high recovery and purity from a greatly heterogeneous mixture in only one or a few ml and then performing sample preparation with minimal sample loss. Furthermore, ultra-trace LC-MS needs to be conducted with specially prepared columns with highly sensitive MS, along with advanced data processing. Key to success is the full integration of all the steps in the workflow to achieve the detection level required. The present work combines a series of innovative steps leading to successful discovery proteomic analysis of rare cells.
Consider first rare cell isolation for which several approaches have recently been developed (9, 10). A particularly powerful approach is magnet-activated cell sorting (MACS) where antibody-functionalized magnetic beads are utilized to enrich a subset of cells in a complex sample such as whole blood (10, 11). Although magnet-activated cell sorting-based and other microfluidic approaches of cell separation have recently shown the ability to isolate rare cells (e.g. <10 cells per ml of whole blood) with high levels of purity (>90%) and efficiency (>95%)(12–14), the potential of these systems in enabling downstream molecular analyses has yet to be fully realized. Microfluidic channels, in comparison to traditional magnet-activated cell sorting, allow for improved control of the magnetic field for precise focusing in the microchannels, resulting in higher efficiency, recovery, and purity of isolation.
For proteomic analysis, rare cell isolation is followed by a series of sample preparation steps, for example cell lysis and protein extraction and digestion. Several approaches such as denaturant-assisted lysis, acetone precipitation, filter-aided sample preparation, and monolithic microreactor-based techniques have been developed for processing small amounts of sample, for example 500–1000 cultured cells (15–17). However, these methodologies only allow identification of a few hundred proteins at these levels. In this work, we describe a sample preparation approach that utilizes novel small volume focused acoustics-assisted cell lysis, followed by low volume serial reduction, proteolytic digestion and ultra-trace LC-MS analysis.
Although two-dimensional separations are often used for deep proteomic analysis, limited sample analysis is best conducted by high peak capacity separation in a single dimension, eliminating potential sample losses from the second dimension. Furthermore, it is known that ultra-low mobile phase flow rates (≤20 nL/min) dramatically improve electrospray signals, as a consequence of improved ionization efficiency (18–21). In prior work, we have shown that reduction of the LC column diameter in a high resolution porous layer open tube (PLOT)1 format utilizing ultra-low flow can generate a significant gain in limited sample proteomic profiling capabilities (22). As shown in the current paper, a combination of PLOT-LC with advanced MS instrumentation and data processing can lead to zeptomole detection sensitivity and quantitation. Furthermore, the integration of all the above steps yields thousands of proteins identified and quantitated from a small number of rare cells (less than one thousand) isolated from 1 ml whole blood. The developed technology opens up the possibility of deep proteomic analysis of rare cells in body fluids.
EXPERIMENTAL PROCEDURES
Reagents and Chemicals
All reagents and chemicals were purchased from Sigma-Aldrich (St. Louis, MO) at the highest purity unless otherwise stated.
Cell Handling
MCF-7 human breast adenocarcinoma cells (ATCC, Manassas, VA) were cultured in 75 cm2 tissue culture flasks at 37 °C, 5% CO2. MCF-7 cells were incubated in Eagle's Minimum Essential Medium (EMEM; ATCC) supplemented with 10% fetal bovine serum, 100 U ml−1 penicillin, 100 μg ml−1 streptomycin, and 0.01 mg ml−1 bovine insulin. Cells were grown to preconfluence and isolated for experiments by trypsinization using a 0.25% trypsin-EDTA solution. Prior to cell isolation or cell lysis, cells were centrifuged at 200 g for 10 min at 4 °C. The cell culture medium was removed, and MCF-7 cells were washed twice with ice cold 1× Dulbecco's phosphate-buffered saline (DPBS, Sigma-Aldrich) containing 0.5% EDTA (v/v). The cells were resuspended at a concentration of ∼1000 cells/μl, and the cell numbers were counted three times using a hemacytometer and/or flow cytometer (Cell Lab Quanta SC; Beckman Coulter, Brea, CA).
Microfluidic Device Design and Fabrication
Microfluidic channels were fabricated as previously described.(23, 24) Wire arrays were designed using PCB123® printed-circuit board design software and ordered from Sunstone Circuits (Mulino, OR). The wire dimensions were set to provide a gap encompassing the width of the microfluidic channel; the height and width of the wires were set to 35 μm and 178 μm, respectively. Teflon-insulated 18 gauge copper wires were soldered to the ends of each of the printed circuit board arrays, and the arrays were connected to a DC power supply (Elenco Electronics XP-4, Wheeling, IL) that provided three fixed-current settings of 0.25 A, 0.50 A, and 1.00 A via standard alligator clip connectors. The PDMS channels and wire arrays were visually aligned.
Functionalization of Magnetic Beads
DynaBeads® MyOneTM Carboxylic Acid magnetic particles (Life Technologies, Carlsbad, CA) were modified with antibodies, either against the epithelial cell adhesion molecule (mouse anti-human EpCAM; Santa Cruz Biotechnology, Santa Cruz, CA) or against CD133 (mouse anti-human CD133, Miltenyi Biotec Inc, Auburn, CA) using standard carbodiimide chemistry (25) in ratios suggested by the reagent manufacturer (1:1 molar ratio of beads to protein; Pierce Biotechnology, Rockford, IL).
Whole Blood Cancer Cell Isolation
Whole blood was drawn from healthy volunteers and collected in EDTA-coated Vacutainer® tubes (Becton Dickinson, Franklin Lakes, NJ). Approval from the Northeastern University Institutional Review Board was obtained for this purpose (NU IRB #: 11–07-19). The location of the interface between the injected blood and buffer was first evaluated (12, 26). A Coulter counter/flow cytometer (Cell Lab Quanta™ SC; Beckman Coulter, Brea, CA) was used to count the number of target (MCF-7) cells versus native polymorphonuclear cells that were separated. A protocol based on the distinct size difference of these two cells, was developed to identify each cell population. The cells were gated by their electronic volume and granularity, and the total number of cells within the recovered suspension was determined.
Various concentrations of MCF-7 cells (500–100,000 cells) were spiked into 1 ml of whole blood. Following this, 10 μl of modified antiEpCAM magnetic microbeads was added to 1 ml of unprocessed blood and allowed to incubate for 30 min on a rotary mixer. This experiment was conducted at optimized flow rates, as described in the theory section below. For all MCF-7 experiments, the flow rate of the samples was fixed at 240 μl/min, and a center stream of 1× RBC lysis buffer (Ebioscience Inc., San Diego, CA) flowed at 160 μl/ml. Target and nontarget cells were collected in separate methanol cleaned microcentrifuge tubes.
To establish an accurate gating of MCF-7 for subsequent cell counts, we first analyzed a homogeneous suspension of approx. 100,000 MCF7 cells. To identify the MCF-7 cells in the flow cytometer, we gated the electronic volume (EV) versus side scattering (SS). We also spiked 100,000 MCF-7 cells into whole blood and ran the sample through the flow cytometer to ensure an accurate gating of the target cells. MCF-7 are distinguishably larger than the surrounding blood cells and thus are the only cells located in the gate. The CV for these calibration samples was 1.2–2.6% (n = 15). On the other hand, gating for the EPCs and HSCs could not be based on cultured homogeneous suspensions. Therefore, buffy coat samples (via Ficoll-Paque density gradient centrifugation) were used to establish standard gates. Gates were first generated with unstained samples (EV and SS and CD34+ only staining to obtain the initial gating from the histograms (CV of 7.5%)). Also, kinase insert domain-containing receptor (KDR)-only (CV = 5.3%, n = 8) and CD45-only (WBC are CD45+; CV = 15.2%, n = 8) were analyzed for their subsequent histograms. These individual gates then allowed for accurate scatter plots based on three-color staining. From these studies, we can assume the CV was between 5.3 and 15.2% for the EPC and HCS cell counting.
Isolation of Hematopoietic Stem Cells and Endothelial Progenitor Cells from Whole Blood
To illustrate the utility of the magnetophoretic design for isolation of rare cells, we extracted hematopoietic stem cell (HSCs) and endothelial progenitor cells (EPCs) from whole blood using antiCD133 functionalized microparticles. Again, whole blood was drawn from healthy volunteers and collected in EDTA-coated Vacutainer® tubes (Becton Dickinson). Isolated cells were then labeled with antibodies to identify HSC and EPC populations. The HSCs were identified as labeling positive for mouse anti-human CD34 conjugated to fluorescein isothiocyanate (antiCD34-FITC; Santa Cruz) and mouse anti-human CD45 conjugated to phycoerythrin (antiCD45-PE; Santa Cruz), and negative for goat anti-human KDR (kinase insert domain receptor; Santa Cruz). The KDR was then conjugated to a secondary antibody donkey anti-goat peridinin chlorophyll protein (PerCP; R&D Systems, Minneapolis, MN). EPCs were identified as labeling positive for antiCD34-FITC and antiKDR-PerCP, and negative for antiCD45-PE. Both cell populations were distinguished via a flow cytometer (Fig. 6).
Fig. 6.

Target isolation and proteomic profiling of rare endothelial progenitor cells (EPC) and hematopoietic stem cells (HSC) from blood. A, Bright-field and fluorescent microscopy images of EPC and HSC cells labeled with magnetic microbeads functionalized with antiCD133 antibody. B, Characterization of EPC and HSC cell populations by flow cytometry. C, Protein identifications for EPC/HSC cells isolated from healthy donors (1 and 2 - smokers, 3–7 - nonsmokers). Approximately 1000 cells were analyzed in each injection per duplicate blood draws (n = 3). D, Box-plots of protein abundance (log10 iBAQ values) in HSC and EPC isolated from donors. Approximately 1000 cells were analyzed in each injection (n = 3). E, Pearson correlation between quantitative proteomic profiles of EPC/HSC isolates show high overall similarity of profiles with highest correlation between isolates from smoking donors. F, Assessment of cell isolation by immunoblottting. Samples: Lysed EPC/HSC isolates were probed in lanes 1–7, with smokers versus nonsmokers as above; red blood cells (8); red blood cell ghosts (9); BJAB lymphocytes (10) and blood plasma (11).
Sample Processing Prior to Lysis and LC-MS
After separation, the ∼650 μl samples (in RBC lysis buffer) were concentrated using a NdFeB permanent magnet. The samples were rinsed twice with 500 μl of PBS and transferred to a lysis mini-tube. The cell losses because of concentrating, rinsing, and transfer were monitored using at least one replicate isolation to assess starting and ending cell numbers. See supplemental Table S1 for a summary of these results. Other experimental procedures, including LC-MS conditions, are available in the Supplement in the online version of the paper.
Imaging of Cells
For the purposes of imaging and verification of cell marker presence, separated cells were concentrated using an NdFeB magnet and resuspended in 100 μl PBS. MCF-7 cells were labeled with mouse antiEpCAM-FITC (Santa Cruz). For the CD133+ cells, we labeled the cells with rabbit antiCD34-FITC, mouse antiCD45-PE, and goat antiKDR, along with a secondary stain with anti-goat Alexa Fluor 350.
Counting of Beads on Cells
Again, cells were concentrated using an NdFeB magnet. The cells were then fixed using 2% paraformaldehyde and placed onto a hemacytometer for enumeration of the cells. Approximately 100 cells were evaluated for the number of bead attached to the cells.
Microfluidic Capture of Spiked MCF-7 Cells
Sample levels of MCF-7 cells (1000, 2000, 3000, 5000, and 10,000 cells) were spiked into 1 ml of whole human blood and mixed with the Dynal MyOne EpCAM-functionalized magnetic microbeads. As negative controls, human blood, without spiked-in MCF-7 cells, was mixed with the EpCAM-functionalized microbeads. Cells functionalized with magnetic microbeads were isolated from human blood cells using a microfluidic magnitophoretic device, as described (12). A flow cytometer was used to count MCF-7 cells. The cells were washed twice with 50 μl of DPBS and processed immediately, as described in the supplement and below.
Other experimental procedures and any associated references are available in the Supplement in the online version of the paper.
RESULTS AND DISCUSSION
Experimental Platform
Fig. 1 presents an overview of the platform. For method development, samples consisting of cultured MCF-7 cells spiked into blood at levels of 1000–100,000 MCF-7 cells in 1 ml of whole blood were utilized (Fig. 1A). The magnetophoretic isolation of these cells included incubation of the sample with magnetic beads functionalized with antibodies against EpCAM followed by isolation (12). The collected target cells were rinsed and lysed using focused ultrasonication (Covaris), digested with trypsin and analyzed using PLOT nLC coupled to a Q-Exactive Orbitrap mass spectrometer (Thermo Fisher Scientific) (Fig. 1C). Stable isotope labeled (SIL) peptides were added to the samples after completion of tryptic digestion for targeted quantitative analysis of selected proteins. MS data were processed to enable quantitative proteomic profiling using both label-free and isotope reference-based techniques followed by gene ontology analysis. After development and validation of the platform using MCF-7 cells spiked into whole blood, the optimized workflow was then applied to high specificity isolation from the blood and proteomic characterization of the CD133+ cells from blood (Fig. 1B). A key attribute of this platform is the reduction of the minimum amount of blood necessary to study EPC and HSC and potentially other rare cell populations by at least 1–2 orders of magnitude relative to the current state of the art (27, 28). We first detail the individual steps that lead to the advanced workflow for deep proteomic analysis of rare cells in whole blood.
Fig. 1.

Workflow for target isolation and proteomic profiling of limited amounts of rare cells from biological fluids. A, A model system where cultured MCF-7 cells were counted, spiked into blood, mixed with the antiEpCAM-functionalized magnetic microbeads and captured using a microfluidic magnetophoretic device. B, Blood collected from healthy donors was mixed with antiCD133-functionalized magnetic microbeads. Endothelial progenitor cells and hematopoietic stem cells with bound antiCD133 beads were isolated by the microfluidic device. C, Isolated target cells were lysed with assistance of focused ultrasonication, followed by reduction, alkylation, and enzymatic digestion performed in a single tube. Stable isotope labeled synthetic peptides were added into digested lysates prior to analysis. Aliquots of cell lysates were analyzed using SPE-nLC-PLOT MS. Acquired MS data were submitted for database searching, quantitation, and GO enrichment analysis.
Ultralow Flow (ULF) PLOT-nLC-MS/MS
A cornerstone of the platform for deep proteomic profiling of rare cells is the ultra-low flow (ULF) PLOT-nLC column technology. To capture the high sensitivity benefits of ultra-low flow, we used 4 m long 10 μm i.d. poly(styrene-divinylbenzene) (PS-DVB) PLOT columns with ∼1 μm thickness of the permeable layer (Fig. 2A). It has been previously shown that the PLOT columns offer excellent chromatographic efficiencies (22). The PLOT column was coupled to a monolithic trapping (microSPE) column using a zero dead volume connector. We optimized weight fractions of the PLOT polymerization mixture constituents that resulted in improved column-to-column reproducibility of the PLOT columns and increased hydrophobicity of the monolithic microSPE columns by copolymerizing PS-DVB with 1-decene. Furthermore, the coupling of the PLOT-nLC to an advanced fast duty cycle high resolution mass spectrometer (Q-Exactive) improved the sensitivity and accuracy of quantitation in comparison to our and other previous works (18–21) (see Supplemental Materials).
Fig. 2.

Performance assessment of PLOT nLC-MS. A, Scanning electron microscopy (S.E.M.) images of cross-sections of a 10 μm i.d. PLOT column. A thickness of the porous layer of ∼1.2 μm can be seen on the left image and a more detailed morphology of the porous layer could be observed by backscatter S.E. energy (right). B, Extracted ion chromatograms (XICs) of selected peptides from digested bovine protein standards (“Bovine 6 Protein Mix,” Michrom Bioresources, Auburn, CA) for injection levels from 10 zmol to 100 amol acquired using untargeted data-dependent data acquisition (DDA). Peptide ion intensities extracted using 2 ppm mass tolerance showed a linear response to the injected amount with R2 values typically of ≥0.95. The following selected peptides are shown in the XIC traces: 1) ALVYGEATSR, CA-II; 2) TPEVDDEALEK, Beta-LG; 3) YSTDVSVDEVK, GDH; 4) VLDALDSIK, CA-II; 5) VLVLDTDYK, Beta-LG; and 6) LVNELTEFAK, BSA. C, MS/MS spectra for ALVYGEATSR (m/z 533.7800) in panel B, demonstrating unambiguous identification even at low zmol levels. D, Targeted analysis of ALVYGEATSR at low zmol levels. E, XICs of selected target precursor ions by parallel product ion monitoring (PRM). XICs for different parent ion – fragment ion transitions are shown in different colors. See text for details.
As a baseline set of experiments to show the potential of the PLOT column-based LC-MS platform for high sensitivity proteomic analysis, the performance of the PLOT column was assessed using a split-injection approach (29). Analyzing an equimolar mixture of digested protein standards (Michrom Bioresources, Auburn, CA, “Bovine 6 Protein Mix,” P/N PTD/00001/63, containing beta lactoglobulin, lactoperoxidase, carbonic anhydrase, glutamate dehydrogenase, alpha casein, and serum albumin) with a nontargeted data dependent data acquisition (DDA) method resulted in detection limits down to 10–50 zmol level (S/N>5), based on single stage mass spectrometry (MS1) (Fig. 2B), and unambiguous MS/MS fragment matching (Fig. 2C). A linear MS response was recorded over a dynamic range of over four orders of magnitude, ranging from 10 zmol (S/N>25) to 100 amol (MS1) and higher, with a linear regression r2 > 0.99. The molar peptide amounts per analysis here and below were derived from the molar amounts of digested standards provided by the vendor (Michrom Bioresources). The quantities of the standard were checked by amino acid analysis and comparative LC-MS analysis of the Michrom standard and the same protein digests from other vendors.
As expected, improved levels of detection were found using a targeted approach, parallel reaction monitoring (PRM) with high-energy collisional dissociation (HCD) (30). Detection limits for tryptic peptides were determined to be less than 5 zmol in MS2 (S/N>25), an ∼fivefold increase compared with the DDA scanning mode for MS2 spectra (Fig. 2D). The HCD spectra at 5 zmol levels were still of high quality. Quantitation of targeted ions in the PRM mode was performed by integrating peak areas for the most prominent product ions (e.g. y7+, b2+, y6+, y8+, and b3+ in the case of peptide ALVYGEATSR, (Fig. 2D)). Excellent linear correlation between sample loads and experimental signal response with r2 ≥0.99 for a range of 5–500 zmol was obtained for both parent and fragment ions (Fig. 2D, E). Quantitation was also evaluated by spiking the same equimolar mixture of six digested bovine proteins (Michrom Bioresources, CA) into a complex background of the MCF-7 lysate. Based on monitoring of predominant parent ion–fragment ion transitions for targeted peptides using extracted ion chromatograms for parent ions and resulting fragment ions, and correspondence of their peak profiles, we were able to reliably quantify selected peptides at 50 zmol, and even 10 zmol, using one-dimensional separation of the unfractionated sample and a 40-min long gradient (Fig. 2E and Supplemental Fig. S1). As expected, noisier elution profiles and fewer reliably detectable transitions were observed at lower sample amounts.
To assess the performance of the microSPE-PLOT-nLC-MS platform in proteomic profiling of a limited number of cells, we prepared a lysate of 10 million human MCF-7 cells using conventional lysis and trypsin digestion techniques (see Supplemental Materials). Aliquots corresponding to 500 MCF-7 cells (∼50 ng of total protein based on our protein concentration measurements at higher levels and accounting for dilutions, and previous reports(31)) were loaded on the microSPE column and separated using a 4 h long gradient at a flow rate of 20 nL/min. Approximately 3700 unique protein groups and 22,645 unique peptides on average were identified (DDA) in a single analysis, and a combination of five replicate PLOT-nLC-MS runs resulted in identification of 5183 proteins and 41,020 peptides (Fig. 3A). The FDR≤1% was used for all peptide-level identifications. The replicate analyses resulted in an overlap in identification results of ∼80%, illustrating the high reproducibility of the analysis (Fig. 3C). The number of IDs decreased when lower amounts of cells were analyzed using the microSPE-PLOT column (Fig. 3B); nevertheless, we were still able to identify 1327 ±143 and 2026 ±98 protein groups from injections of 50 ±3 and 100 ±5 cells, respectively, (n = 3; Fig. 3B, D). The PLOT-nLC-based platform showed at least 4–5 fold improved profiling sensitivity in analysis of similar sample amounts if compared with other state-of-the-art LC/MS methods (15, 17, 32, 33).
Fig. 3.

Proteomic profiling of limited amounts of MCF-7 cells. A, B, Cumulative protein and peptide identification for replicate injections of a digested MCF-7 lysate equivalent to 500 cells. Separation on a 4.2 m PLOT column and a 4 h long linear gradient at a flow rate of 20 nL/min. C, Numbers of proteins corresponding to 50, 100, and 500 MCF-7 cells injected. D, E, Overlaps in identification between replicate analyses of 500 injected cells D, and 50, 100, and 500 MCF-7 injected cells analyzed in triplicate E.
Cell Lysis and Protein Digestion for Limited Samples
The above experiments were performed with a starting sample of 10 million MCF-7 cells, injecting the equivalent of the specific low number of cells. Sample preparation of low cell numbers by direct down-scaling of the conventional lysis/digestion protocol led to five to tenfold decrease in the number of proteins identified because of multiple sample transfers and contact-surface adsorption losses. As a result, we developed a new single-tube sample preparation method based on the AFA (Adaptive Focused Acoustics™, Covaris, MA) assisted cell lysis. This technique allowed us to perform cell lysis followed by protein reduction, alkylation and digestion in the same glass microtube with minimal sample dilution, that is, 12–15 μl. The AFA technology is used for extraction of DNA in genomic applications but only previously reported for cell lysis for proteomic profiling (34, 35). We now add to this a key benefit of AFA of conducting cell lysis without chaotropes (e.g. urea, guanidinium chloride) or detergents along the standard steps of protein reduction, alkylation and digestion in the same tube. Eliminating chaotropes and detergents allowed us to avoid sample dilution and additional clean-up procedures, thus significantly improving sample recovery. Furthermore, we conducted cell lysis with the magnetic beads present in the tube, thus removing the step of release of the cells and improving the speed of lysis (2 to 3 min).
We then used the AFA approach to directly lyse 2000 MCF-7 cells (counted by flow cytometry) in a total volume of ∼20 μl, followed by reduction, alkylation and in-solution digestion. Proteomic profiling of an aliquot corresponding to 500 cells resulted in identification of 3370 ±119 (n = 3) protein IDs using a 4 h gradient, a number similar to the profiling depth for 10 million MCF-7 cells, again injecting a roughly equivalent of 500 cells. Using the same procedure 2061 ±39 proteins (n = 3) were identified for 100 cells injected, again similar to the number from 10 million cells, whereas the analysis of 50 cells resulted in 1802 ±18 (n = 3) protein groups, a 36% increase in comparison to the sample equivalent to 50 cells from the bulk sample preparation (10 million cells) (supplemental Fig. S2).
Target Microfluidic Magnetophoretic Cell Isolation Using a Model System
We combined the above miniaturized approaches with immmunoaffinity enrichment of target cells with magnetic beads followed by high specificity microfluidic magnetophoretic isolation of rare cells from biofluids (12). The microfluidic immunomagnetic cell separator device is designed to separate magnetically labeled target cells from two laminar streams adjacent to the walls of a straight channel into a central buffer stream (Fig. 4, supplemental Fig. S3). Postseparation, but before lysis, magnetically-tagged MCF-7 cells separated from blood were counted by flow cytometry via a gate of electronic volume versus side scatter (supplemental Fig. 3).
Fig. 4.

Microfluidic magnetophoretic cell isolation using MCF-7 cells spiked into blood. A, A schematic cross-sectional view of the magnetophoretic device with the strength of the magnetic field gradient shown as a heat-map. B, Principle of magnetophoretic cell separation: laminar flow is applied in the y-direction over a separation chamber, and a magnetic field in the x-direction. Nonmagnetic material follows the direction of the laminar flow, whereas magnetic particles/cells are deflected to a center collection stream. C, MCF-7 cells, labeled with an FITC-antiEpCAM antibody, illustrating the dense pack of microparticles to the cell surface. D, Assessment of cell isolation specificity from 1 ml of blood using immunoblotting. Lane: 1) Unlabeled MCF-7 cells (∼100,000 cells) spiked into 1 ml of blood prior (negative control 1); 2) ∼100,000 MCF-7 cells spiked and mixed with antiCD34 in 1 ml of blood (negative control 2); 3) Magnetic beads functionalized with antiEpCAM antibodies spiked into 1 ml of blood (no addition of MCF-7 cells) (negative control 3); 4) Suspension of ∼100,000 MCF-7 cells in DPBS mixed with antiEpCAM functionalized beads and subjected to cell isolation (positive control); 5–7) Lysate of ∼85,000 (5), 45,000 (6), or 25,000 (7) MCF-7 cells spiked into 1 ml of blood and isolated using antiEp-CAM functionalized beads; 8) Lysate of red blood cells (RBCs); 9) Lysate of RBC ghosts (∼20 ng); 10) Lysate of BJAB lymphocytes (∼20 ng); and 11) Human plasma, 0.2 μl. E, Protein identification for PLOT-nLC MS/MS of blank injections, negative controls (“no MCF-7 cells in blood,” equivalent to lane 3 in D), target isolation of MCF-7 cells from blood (“spiked-in MCF-7 cells in blood,” lane 5 in D; an equivalent of 500 cells was injected in each replicate) and positive control (“MCF-7 in DPBS,” lane 4 in D). Each result shows a mean value for six PLOT-nLC MS/MS experiments. Blue bar is average and red bar is total protein groups. were identified in each run on average and in all four performed injections, respectively. The negative control 2 resulted in identification of 103 ±25 proteins in each injection and 298 proteins in all four analyses (two independent isolations analyzed in duplicate), indicating a relatively low level of nonspecific binding. However, 3126 ±103 and 5117 protein groups were identified on average in each analysis of 500 MCF-7cells and in a total four analyses, respectively, where ∼3000 cells were spiked into 1 ml of blood and ∼1800 cells recovered by the microfluidic device. F, Venn diagrams illustrating overlap in protein identification for isolates of target cells from blood, blank runs and positive controls. G, Identified proteins in proteomic profiling of limited amount of isolated MCF-7 cells. Specifically, 1000, 2000, 3000, 5000, and 10000 MCF-7 cells were spiked in 1 ml aliquots of whole blood and captured using the magnetophoretic device. Aliquots of digested lysates corresponding to 122, 239, 409, 687, and 1440 cells (∼20% of total number of isolated and recovered cells) were analyzed in six replicates (two independent isolations analyzed in triplicates).
As a model system, various numbers of MCF-7 cultured cells ranging from 1000 to 10,000 cells were spiked into 1 ml of whole human blood that was then collected in heparin Vacutainers, incubated with magnetic microbeads functionalized with antiEpCAM antibodies (specific for an MCF-7 surface protein) and processed by the microfluidic cell capture device (Figs. 1, 4). Postenrichment, the recoveries were determined by flow cytometry to be consistently > 90%, with purities >85%. Two independent replicate isolations from the same 1 ml sample of blood were carried out at each cell concentration level. Isolated cells with bound magnetic microbeads were washed twice with DPBS buffer. Flow cytometry determined that ∼50–60% of cells were typically left after cell preparation, mainly because of concentration, rinse, and the transfer steps necessary for proteomic sample postprocessing (Supplemental Table S1).
The high specificity of MCF-7 isolation was further assessed by immunoblotting and 1D PAGE using a panel of spiked cells and control blood samples (Fig. 4D and supplemental Fig. 5F). A number of negative control microfluidic cell isolation experiments indicated the absence of immunoblot or gel detectable contamination of isolates from high abundance blood constituents, e.g. plasma proteins, erythrocytes, and leukocytes (Fig. 4D and supplemental Fig. 5F). On the other hand, the isolates of cells spiked at different immunoblot detectable levels (25,000–85,000 cells in 1 ml of blood) showed a proportional response in blot signal and detection of identical to the positive control (neat MCF-7 in DPBS) bands (Fig. 4D).
To assess the level of baseline contamination of the microSPE-PLOT-nLC-MS system and buffers, we injected aliquots of 0.1% aqueous formic acid into the SPE column and ran blanks, prior to analysis of cell lysates, after rinsing the system (Fig. 4E). Two separate quality control experiments were performed to assess the isolation efficiency and specificity using the PLOT-based nLC-MS platform: identical aliquots of either (1) negative control: whole human blood without added MCF-7 cells or (2) positive control: 2000 counted MCF-7 cells in DPBS processed without passing through the magnetophoretic device. Each aliquot of the AFA-processed and in-solution digested cell lysates, corresponding to 500 cells, was loaded on the SPE and separated by the PLOT column. In the target cell isolation experiments, where ∼3000 cells were spiked into 1 ml of blood and ∼1800 cells were recovered from the microfluidic device, on average 3126±103 and 5117 protein groups were identified in each analysis of 500 MCF-7 cells and in a total four analyses (two independent isolations analyzed in duplicate), respectively. These numbers and the identified constituents of the MCF-7 proteome were similar to the numbers found in the positive control (neat MCF-7 in DPBS, 3449±204 protein IDs on average and 5223 IDs in total, n = 4) (Fig. 4E, 4F and supplemental Fig. S4A). Based on our comparative analysis of the above samples, we estimated that only ∼3% of proteins identified in MCF-7 cells isolated from the blood corresponded to nonspecifically bound blood proteins, which is in agreement with previous findings where ≥95% purity of isolation was observed (26). The coefficient of variation (CV) of 3% in the number of protein groups identified in the four analyses showed good reproducibility of both cell capture and analysis. We also examined the level of carryover between injections for different amounts of cells (e.g. 50, 100, and 500 cells) and determined that it was negligible (≤1–2% of total protein IDs, see Supplemental Fig. 6A). Additionally, we determined that the presence of the magnetic beads during sample processing aided efficiency as nonfunctionalized beads resulted in a ∼20% gain in protein IDs compared with the sample processing without addition of beads to MCF-7 cells.
Further investigation of the differences in the proteome results from the above experiments was performed using comparative Gene Ontology (GO) enrichment analysis. The proteomic profiles of the isolated MCF-7 cells from blood and the negative control isolates from blood showed very significant differences in distributions of the corresponding cellular localization GO terms (supplemental Fig. S4B, S4C). It is noteworthy that many high or moderate abundance plasma proteins, such as immunoglobulins and kallikrein (supplemental Table S1), were unique for the negative control experiment. For the positive control, where the cultured MCF-7 cells in DPBS were analyzed, the GO profiles were very similar to those in the analysis of the MCF-7 cells isolated from blood by the microfluidic device for both unique and total proteins that verified the high specificity of cell capture (supplemental Fig. S4C).
Proteomic Profiling of Isolated Target Cells in Model Systems
To evaluate the microfluidic platform in efficient isolation of rare cells, we first spiked 1000, 2000, 3000, 5000, and 10,000 MCF-7 cells (counted by flow cytometry, CV≤2.6% for all MCF-7 cell counts) in 1 ml aliquots of whole blood and enriched using the magnetophoretic device. Approximately 90–95% of the spiked-in cells were isolated by the device and ∼50–60% of the cells remained (again by flow cytometry) after concentration and transfer into tubes for AFA-assisted lysis (supplemental Table S1). The cells were processed, as described above, and aliquots corresponding to 1/5th of the resulting digests were subjected to microSPE-PLOT- nLC MS analysis in each replicate. Profiling of a sample aliquot equivalent to 122 cells resulted in identification of 2512 ±246 protein groups on average and 3752 proteins total in six replicates (two independent isolations analyzed in triplicate) when 1000 cells were spiked in whole blood and ∼600 cells recovered (Fig. 4G). The depth of proteomic profiling increased proportionally with the number of injected cells. Ultimately, when ∼1440 cells were analyzed, 3402 ±169 and 5074 protein groups were identified per injection and in total, respectively (Fig. 4G, supplemental Table S2). The number of the identified peptides followed a similar trend (supplemental Fig. S6B).
To assess the quantitative performance, we spiked in 1 ml of blood MCF-7 cells at five different concentration levels (1000 to 10,000 cells) in triplicate (Fig. 5). Isolates from one replicate were used to count cells, and the two remaining replicates were subjected to proteomic profiling in triplicate, where 1/5th of each isolate was injected in each analysis (i.e. 122, 239, 409, 687, and 1440 cells according to the flow cytometry counts). We estimated protein abundance using the iBAQ approach (MaxQuant) (36). Changes in dynamic ranges by proteomic profiling for the five levels of MCF-7 cells showed a log-linear correlation between sample amount and log10 protein intensity with r2 of 0.84 (Fig. 5A). Both isolation and proteomic profiling replicates showed good reproducibility in profiling depths, dynamic ranges of MS signal intensity measurements and distribution of protein abundance values (supplemental Fig. S5).
Fig. 5.
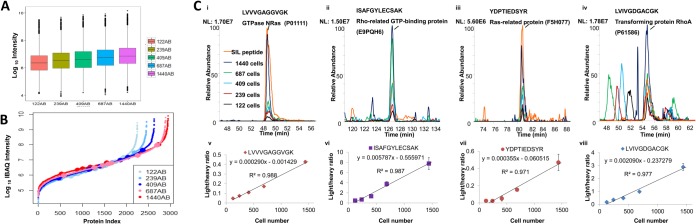
Quantitative proteomic analysis of isolated MCF-7 cells. A, Box plot distributions of protein abundances (log10 iBAQ values) for determined in analyses of different amounts of MCF-7 cells. Cell amounts per injection are shown in the labels on the right. B, Proteins ranked according to their abundance levels (log10 iBAQ values) for in proteomes revealed in analyses of 122, 239, 409, 687, and 1440 cells per injection. Synthetic stable isotope labeled peptides spiked in as internal standards. C, (i–iv) Extracted ion chromatograms corresponding to endogenous MCF-7 peptides detected at different cell numbers and spiked SIL internal standards (orange trace); (v–viii) correlation of normalized peptide abundances calculated as ratios of peptide peak areas of endogenous (“light”) to SIL (“heavy”) ratios for different cell numbers. In all experiments (A–C), sample aliquots were analyzed in six replicates (two independent isolations in triplicate).
Isotopically labeled peptides with sequences corresponding to endogenous proteins of different abundances were next spiked in the digested lysates of isolated cells for absolute quantitative analysis (see Fig. 5B shown in dots on protein distribution S-curves). Areas of labeled peptide peaks were used to normalize abundance measurements for peptides of endogenous proteins. As expected, abundance levels of sample peptides increased relative to the labeled standards when the number of cells increased (Fig. 5C, i–iv). Excellent linear correlation (r2 = 0.97–0.99) was achieved within the concentration range of 1000–10,000 cells/ml (Fig. 5C, v–viii).
Proteomic Profiling of Hematopoietic Stem Cells (HSCs) and Endothelial Progenitor Cells (EPCS) Isolated from Whole Blood
Based on the above results, we turned to actual rare cell analysis in whole blood using the developed platform. Specifically, two rare and important CD133+ cell populations in whole blood (found at <1% of the total cell population), namely endothelial progenitor cells (EPCs) and hematopoietic stem cells (HSCs) (37) were isolated, followed by deep proteomic analysis. EPCs possess the ability to differentiate into endothelial cells that make up the lining of blood vessels, and their concentration in blood is known to correlate with cardiovascular risk and clinical outcome (38, 39). EPCs also play a role in tumor growth, metastasis and angiogenesis, and therefore, these poorly understood cells are of interest as targets for development of novel therapeutics in cancer research (40, 41). In addition, HSCs are a circulating stem cell population that gives rise to most cell types in the blood. HSCs can be derived from whole blood, bone marrow, and umbilical cord blood. This particular stem cell population has shown great promise in autologous transplantation for autoimmune disorders (42) and treatment of blood-origin cancer (43) but is not well characterized with regard to deep proteomics.
EPCs and HSCs were separately isolated from 1 ml of whole blood drawn from seven healthy donors using magnetic beads against the CD133 antigen. The cells were stained for CD34, KDR, and CD45 and then counted first via a CD34+ gate, followed by a dual gating for CD45−/KDR+ and CD45+/KDR− (Fig. 6). Cells isolated using the antiCD133 antibody, that subsequently stain CD34+/CD45-/KDR+, indicate an EPC phenotype, and CD34+/CD45+/KDR− cells are an HSC phenotype (44) (Fig. 6A, 6B). The efficiency of CD133+ cell isolation was determined to be >90%, with >60% cell purity. The specificity of cell isolation was confirmed by immunoblotting (Fig. 6F). Additionally, proteomic analysis revealed the presence of proteins indicative of EPC cells, including caveolin-1, angiopoietin-1 receptor, endothelial nitric oxide synthase and ephrin type-A receptors (45, 46). Similarly, proteins of a specific extracellular matrix repertoire reported to be indicative of HSCs and other multipotent stem cells (47, 48), including SPARC, integrins alpha-5 and 6, CD44, and ADAM17, among others, were identified in our analysis of the EPC/HSC isolates but not in the MCF-7 isolates.
The number of proteins identified in CD133+ EPC and HSC cells (∼ 2000 proteins identified in ∼1000 cells) was somewhat lower than for MCF-7 cells (Fig. 6C), as expected, based on observed cell sizes (Figs. 4C, 6A) (49, 50). Furthermore, the protein content expressed in stem cells is expected to be lower than in cultured cancer cells (51). Interestingly, in two subjects who were known to be smokers, the isolates contained ∼20% fewer EPCs and HSCs than those from nonsmokers (supplemental Fig. 6B), which is consistent with previously published data (52, 53). CD133+ EPC/HSC cells showed high levels of correlation in their quantitative proteomic profiles (r2 ≥ 0.8), with the highest similarity observed for cell isolates from the two smokers (Fig. 6D, 6E).
In summary, a combination of the immunoaffinity microfluidic magnetophoretic cell isolation, focused ultrasonication-assisted cell lysis, reduction, alkylation, and digestion, followed by 1D PLOT-nLC-MS profiling and advanced data processing resulted in the identification of ∼4000 proteins from the injection of only 100–200 cells per analysis, a level of at least 5–10 times better than reported to date. High specificity of the cell isolation technique in combination with advanced sample procurement and preparation and ultrasensitive PLOT-nLC-MS enabled differential profiling of limited levels of model samples and isolates collected from donors. Furthermore, we reduced the amount of blood necessary to conduct deep proteomic analysis of EPCs and HSCs by at least 1–2 orders of magnitude in comparison to other reported methods (27, 28). These results show the clear potential for in-depth discovery proteomic characterization of circulating rare cells for a variety of clinical and biological applications. Both targeted and discovery proteomic profiling of blood (liquid biopsy) or tissue samples using our approach may further enable therapeutic monitoring of individual patients from only small quantities of clinical samples. In addition, proteomic profiling of rare cell populations such as stem and progenitor cells has the potential to provide mechanistic insights into both normal (e.g. hematopoiesis (54)) as well as disease processes (55).
Supplementary Material
Acknowledgments
We thank JPT Peptides Technologies for providing stable isotope labeled peptide standards and Covaris for access to the S220X instrument. We also thank Drs. Brett Phinney and John M Schulze, and the Molecular Structure Facility at UC Davis for performing amino acid analysis of the standards.
This is contribution number 1052 from the Barnett Institute.
Footnotes
Author contributions: A.R.I., B.L.K. and S.K.M. conceived the experimental design of the presented approach. B.D.P. and S.K.M. designed and performed target cell isolation. A.R.I., B.L.K. and S.L. performed PLOT LC column design and analysis. A.R.I., S.L. and X.W.carried out sample processing, PLOT LC column preparation and proteomic profiling experiments. A.R.I, S.L. and S.R. performed data analysis and interpretation. A.M.B. performed gel electrophoresis and western blotting experiments. A.R.I, B.D.P., B.L.K., S.K.M. and S.L. wrote the manuscript.
* This work was supported by the US National Institutes of Health (NIH) grant GM15847 to B.L.K.
 This article contains supplemental Figs. S1 to S6, Tables S1 and S2, and Materials.
This article contains supplemental Figs. S1 to S6, Tables S1 and S2, and Materials.
1 The abbreviations used are:
- PLOT
- porous layer open tubular
- CTC
- circulating tumor cell
- DPBS
- Dulbecco's phosphate-buffered saline
- EMEM
- Eagle's Minimum Essential Medium
- EPC
- endothelial progenitor cell
- HSC
- hematopoietic stem cell
- KDR
- kinase insert domain receptor.
REFERENCES
- 1. Nagrath S., Sequist L. V., Maheswaran S., Bell D. W., Irimia D., Ulkus L., Smith M. R., Kwak E. L., Digumarthy S., Muzikansky A., Ryan P., Balis U. J., Tompkins R. G., Haber D. A., Toner M. (2007) Isolation of rare circulating tumor cells in cancer patients by microchip technology. Nature 450, 1235–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cristofanilli M., Budd G. T., Ellis M. J., Stopeck A., Matera J., Miller M. C., Reuben J. M., Doyle G. V., Allard W. J., Terstappen L. W., Hayes D. F. (2004) Circulating tumor cells, disease progression, and survival in metastatic breast cancer. New Engl. J. Med. 351, 781–791 [DOI] [PubMed] [Google Scholar]
- 3. Gleghorn J. P., Pratt E. D., Denning D., Liu H., Bander N. H., Tagawa S. T., Nanus D. M., Giannakakou P. A., Kirby B. J. (2010) Capture of circulating tumor cells from whole blood of prostate cancer patients using geometrically enhanced differential immunocapture (GEDI) and a prostate-specific antibody. Lab Chip 10, 27–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Riethdorf S., Fritsche H., Müller V., Rau T., Schindlbeck C., Rack B., Janni W., Coith C., Beck K., Jänicke F., Jackson S., Gornet T., Cristofanilli M., Pantel K. (2007) Detection of circulating tumor cells in peripheral blood of patients with metastatic breast cancer: a validation study of the cellsearch system. Clin. Cancer Res. 13, 920–928 [DOI] [PubMed] [Google Scholar]
- 5. Green J. V., Radisic M., Murthy S. K. (2009) Deterministic lateral displacement as a means to enrich large cells for tissue engineering. Anal. Chem. 81, 9178–9182 [DOI] [PubMed] [Google Scholar]
- 6. Plouffe B. D., Kniazeva T., Mayer J. E., Jr., Murthy S. K., Sales V. L. (2009) Development of microfluidics as endothelial progenitor cell capture technology for cardiovascular tissue engineering and diagnostic medicine. FASEB J. 23, 3309–3314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hamburg M. A., Collins F. S. (2010) The path to personalized medicine. New Engl. J. Med. 363, 301–304 [DOI] [PubMed] [Google Scholar]
- 8. Yu M., Bardia A., Wittner B. S., Stott S. L., Smas M. E., Ting D. T., Isakoff S. J., Ciciliano J. C., Wells M. N., Shah A. M., Concannon K. F., Donaldson M. C., Sequist L. V., Brachtel E., Sgroi D., Baselga J., Ramaswamy S., Toner M., Haber D. A., Maheswaran S. (2013) Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science 339, 580–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pratt E. D., Huang C., Hawkins B. G., Gleghorn J. P., Kirby B. J. (2011) Rare cell capture in microfluidic devices. Chem. Eng. Sci. 66, 1508–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zborowski M., Chalmers J. J. (2011) Rare cell separation and analysis by magnetic sorting. Anal. Chem. 83, 8050–8056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Miltenyi S., Müller W., Weichel W., Radbruch A. (1990) High gradient magnetic cell separation with MACS. Cytometry 11, 231–238 [DOI] [PubMed] [Google Scholar]
- 12. Plouffe B. D., Mahalanabis M., Lewis L. H., Klapperich C. M., Murthy S. K. (2012) Clinically relevant microfluidic magnetophoretic isolation of rare-cell populations for diagnostic and therapeutic monitoring applications. Anal. Chem. 84, 1336–1344 [DOI] [PubMed] [Google Scholar]
- 13. Ozkumur E., Shah A. M., Ciciliano J. C., Emmink B. L., Miyamoto D. T., Brachtel E., Yu M., Chen P. I., Morgan B., Trautwein J., Kimura A., Sengupta S., Stott S. L., Karabacak N. M., Barber T. A., Walsh J. R., Smith K., Spuhler P. S., Sullivan J. P., Lee R. J., Ting D. T., Luo X., Shaw A. T., Bardia A., Sequist L. V., Louis D. N., Maheswaran S., Kapur R., Haber D. A., Toner M. (2013) Inertial focusing for tumor antigen-dependent and -independent sorting of rare circulating tumor cells. Science Trans. Med. 5, 179ra147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Karabacak N. M., Spuhler P. S., Fachin F., Lim E. J., Pai V., Ozkumur E., Martel J. M., Kojic N., Smith K., Chen P.-i., Yang J., Hwang H., Morgan B., Trautwein J., Barber T. A., Stott S. L., Maheswaran S., Kapur R., Haber D. A., Toner M. (2014) Microfluidic, marker-free isolation of circulating tumor cells from blood samples. Nat. Protocols 9, 694–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wang N., Xu M., Wang P., Li L. (2010) Development of mass spectrometry-based shotgun method for proteome analysis of 500 to 5000 cancer cells. Anal. Chem. 82, 2262–2271 [DOI] [PubMed] [Google Scholar]
- 16. Tian R., Wang S., Elisma F., Li L., Zhou H., Wang L., Figeys D. (2011) Rare cell proteomic reactor applied to stable isotope labeling by amino acids in cell culture (SILAC)-based quantitative proteomics study of human embryonic stem cell differentiation. Mol. Cell. Proteomics 10, M110.000679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maurer M., Müller A. C., Wagner C., Huber M. L., Rudashevskaya E. L., Wagner S. N., Bennett K. L. (2013) Combining filter-aided sample preparation and pseudoshotgun technology to profile the proteome of a low number of early passage human melanoma cells. J. Proteome Res. 12, 1040–1048 [DOI] [PubMed] [Google Scholar]
- 18. Valaskovic G. A., Kelleher N. L., McLafferty F. W. (1996) Attomole protein characterization by capillary electrophoresis-mass spectrometry. Science 273, 1199–1202 [DOI] [PubMed] [Google Scholar]
- 19. Zhou F., Lu Y., Ficarro S. B., Adelmant G., Jiang W., Luckey C. J., Marto J. A. (2013) Genome-scale proteome quantification by DEEP SEQ mass spectrometry. Nat. Commun. 4, 2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Luo Q., Tang K., Yang F., Elias A., Shen Y., Moore R. J., Zhao R., Hixson K. K., Rossie S. S., Smith R. D. (2006) More sensitive and quantitative proteomic measurements using very low flow rate porous silica monolithic LC columns with electrospray ionization-mass spectrometry. J. Proteome Res. 5, 1091–1097 [DOI] [PubMed] [Google Scholar]
- 21. Ivanov A. R., Zang L., Karger B. L. (2003) Low-attomole electrospray ionization MS and MS/MS analysis of protein tryptic digests using 20-microm-i.d. polystyrene-divinylbenzene monolithic capillary columns. Anal. Chem. 75, 5306–5316 [DOI] [PubMed] [Google Scholar]
- 22. Yue G., Luo Q., Zhang J., Wu S.-L., Karger B. L. (2007) Ultratrace LC/MS proteomic analysis using 10-microm-i.d. Porous layer open tubular poly(styrene-divinylbenzene) capillary columns. Anal. Chem. 79, 938–946 [DOI] [PubMed] [Google Scholar]
- 23. Plouffe B. D., Njoka D. N., Harris J., Liao J., Horick N. K., Radisic M., Murthy S. K. (2007) Peptide-mediated selective adhesion of smooth muscle and endothelial cells in microfluidic shear flow. Langmuir 23, 5050–5055 [DOI] [PubMed] [Google Scholar]
- 24. Xia Y., Whitesides G. M. (1998) Soft lithography. Angew. Chem. Int. Ed. 37, 550–575 [DOI] [PubMed] [Google Scholar]
- 25. Hermanson G. T. (1996) Bioconjugate Techniques, Academic Press, Boston [Google Scholar]
- 26. Plouffe B. D., Lewis L. H., Murthy S. K. (2011) Computational design optimization for microfluidic magnetophoresis. Biomicrofluidics 5, 013413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim J., Jeon Y. J., Kim H. E., Shin J. M., Chung H. M., Chae J. I. (2013) Comparative proteomic analysis of endothelial cells progenitor cells derived from cord blood- and peripheral blood for cell therapy. Biomaterials 34, 1669–1685 [DOI] [PubMed] [Google Scholar]
- 28. Medina R. J., O'Neill C. L., Sweeney M., Guduric-Fuchs J., Gardiner T. A., Simpson D. A., Stitt A. W. (2010) Molecular analysis of endothelial progenitor cell (EPC) subtypes reveals two distinct cell populations with different identities. BMC Med. Genomics 3, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yue G., Luo Q., Zhang J., Wu S. L., Karger B. L. (2007) Ultratrace LC/MS proteomic analysis using 10-microm-i.d. Porous layer open tubular poly(styrene-divinylbenzene) capillary columns. Anal. Chem. 79, 938–946 [DOI] [PubMed] [Google Scholar]
- 30. Gallien S., Duriez E., Crone C., Kellmann M., Moehring T., Domon B. (2012) Targeted proteomic quantification on quadrupole-orbitrap mass spectrometer. Mol. Cell. Proteomics 11, 1709–1723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Drabovich A. P., Pavlou M. P., Dimitromanolakis A., Diamandis E. P. (2012) Quantitative analysis of energy metabolic pathways in MCF-7 breast cancer cells by selected reaction monitoring assay. Mol. Cell. Proteomics 11, 422–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Maurer M., Muller A. C., Wagner C., Huber M. L., Rudashevskaya E. L., Wagner S. N., Bennett K. L. (2013) Combining filter-aided sample preparation and pseudoshotgun technology to profile the proteome of a low number of early passage human melanoma cells. J. Proteome Res. 12, 1040–1048 [DOI] [PubMed] [Google Scholar]
- 33. Wisniewski J. R., Dus K., Mann M. (2013) Proteomic workflow for analysis of archival formalin-fixed and paraffin-embedded clinical samples to a depth of 10,000 proteins. Proteomics. Clin. Appl. 7, 225–233 [DOI] [PubMed] [Google Scholar]
- 34. Li S., Wang X., Karger B. L., Ivanov A. R. (2013) High Sensitivity Microproteomic Analysis of Rare Samples by Porous Layer Open Tubular (PLOT) Columns Coupled with Mass Spectrometry. ASMS 2013, 61st Conference on Mass Spectrometry and Allied Topics, Springer, Minneapolis, Minnesota [Google Scholar]
- 35. Martin J. G., Rejtar T., Martin S. A. (2013) Integrated microscale analysis system for targeted liquid chromatography mass spectrometry proteomics on limited amounts of enriched cell populations. Anal. Chem. 85, 10680–10685 [DOI] [PubMed] [Google Scholar]
- 36. Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M. (2011) Global quantification of mammalian gene expression control. Nature 473, 337–342 [DOI] [PubMed] [Google Scholar]
- 37. Yoder M. C., Mead L. E., Prater D., Krier T. R., Mroueh K. N., Li F., Krasich R., Temm C. J., Prchal J. T., Ingram D. A. (2007) Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood 109, 1801–1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Van Craenenbroeck E. M., Conraads V. M., Van Bockstaele D. R., Haine S. E., Vermeulen K., Van Tendeloo V. F., Vrints C. J., Hoymans V. Y. (2008) Quantification of circulating endothelial progenitor cells: a methodological comparison of six flow cytometric approaches. J. Immunol. Methods 332, 31–40 [DOI] [PubMed] [Google Scholar]
- 39. Werner N., Kosiol S., Schiegl T., Ahlers P., Walenta K., Link A., Bohm M., Nickenig G. (2005) Circulating endothelial progenitor cells and cardiovascular outcomes. N. Engl. J. Med. 353, 999–1007 [DOI] [PubMed] [Google Scholar]
- 40. Nolan D. J., Ciarrocchi A., Mellick A. S., Jaggi J. S., Bambino K., Gupta S., Heikamp E., McDevitt M. R., Scheinberg D. A., Benezra R., Mittal V. (2007) Bone marrow-derived endothelial progenitor cells are a major determinant of nascent tumor neovascularization. Genes Dev. 21, 1546–1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Roodhart J. M., Langenberg M. H., Vermaat J. S., Lolkema M. P., Baars A., Giles R. H., Witteveen E. O., Voest E. E. (2010) Late release of circulating endothelial cells and endothelial progenitor cells after chemotherapy predicts response and survival in cancer patients. Neoplasia 12, 87–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gratwohl A., Passweg J., Bocelli-Tyndall C., Fassas A., van Laar J. M., Farge D., Andolina M., Arnold R., Carreras E., Finke J., Kotter I., Kozak T., Lisukov I., Lowenberg B., Marmont A., Moore J., Saccardi R., Snowden J. A., van den Hoogen F., Wulffraat N. M., Zhao X. W., Tyndall A. (2005) Autologous hematopoietic stem cell transplantation for autoimmune diseases. Bone Marrow Transplant. 35, 869–879 [DOI] [PubMed] [Google Scholar]
- 43. Michallet M., Dreger P., Sutton L., Brand R., Richards S., van Os M., Sobh M., Choquet S., Corront B., Dearden C., Gratwohl A., Herr W., Catovsky D., Hallek M., de Witte T., Niederwieser D., Leporrier M., Milligan D. (2011) Autologous hematopoietic stem cell transplantation in chronic lymphocytic leukemia: results of European intergroup randomized trial comparing autografting versus observation. Blood 117, 1516–1521 [DOI] [PubMed] [Google Scholar]
- 44. Yoder M. C., Mead L. E., Prater D., Krier T. R., Mroueh K. N., Li F., Krasich R., Temm C. J., Prchal J. T., Ingram D. A. (2007) Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood 109, 1801–1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hofer E., Schweighofer B. (2007) Signal transduction induced in endothelial cells by growth factor receptors involved in angiogenesis. Thromb. Haemost. 97, 355–363 [PMC free article] [PubMed] [Google Scholar]
- 46. Briasoulis A., Tousoulis D., Antoniades C., Papageorgiou N., Stefanadis C. (2011) The role of endothelial progenitor cells in vascular repair after arterial injury and atherosclerotic plaque development. Cardiovasc. Ther. 29, 125–139 [DOI] [PubMed] [Google Scholar]
- 47. Bonardi F., Fusetti F., Deelen P., van Gosliga D., Vellenga E., Schuringa J. J. (2013) A proteomics and transcriptomics approach to identify leukemic stem cell (LSC) markers. Mol. Cell. Proteomics 12, 626–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Klimmeck D., Hansson J., Raffel S., Vakhrushev S. Y., Trumpp A., Krijgsveld J. (2012) Proteomic cornerstones of hematopoietic stem cell differentiation: distinct signatures of multipotent progenitors and myeloid committed cells. Mol. Cell. Proteomics 11, 286–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Asahara T., Kawamoto A., Masuda H. (2011) Concise review: circulating endothelial progenitor cells for vascular medicine. Stem Cells 29, 1650–1655 [DOI] [PubMed] [Google Scholar]
- 50. Arya S. K., Lee K. C., Bin Dah'alan D., Daniel, Rahman A. R. A. (2012) Breast tumor cell detection at single cell resolution using an electrochemical impedance technique. Lab Chip 12, 2362–2368 [DOI] [PubMed] [Google Scholar]
- 51. Di Palma S., Stange D., van de Wetering M., Clevers H., Heck A. J. R., Mohammed S. (2011) Highly sensitive proteome analysis of facs-sorted adult colon stem cells. Journal Proteome Res. 10, 3814–3819 [DOI] [PubMed] [Google Scholar]
- 52. Ludwig A., Jochmann N., Kertesz A., Kuhn C., Mueller S., Gericke C., Baumann G., Stangl K., Stangl V. (2010) Smoking decreases the level of circulating CD34+ progenitor cells in young healthy women – a pilot study. BMC Womens Health 10, 1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chiang C. H., Huang P. H., Chung F. P., Chen Z. Y., Leu H. B., Huang C. C., Wu T. C., Chen J. W., Lin S. J. (2012) Decreased circulating endothelial progenitor cell levels and function in patients with nonalcoholic fatty liver disease. PLoS One 7, e31799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Adamo L., Naveiras O., Wenzel P. L., McKinney-Freeman S., Mack P. J., Gracia-Sancho J., Suchy-Dicey A., Yoshimoto M., Lensch M. W., Yoder M. C., Garcia-Cardena G., Daley G. Q. (2009) Biomechanical forces promote embryonic haematopoiesis. Nature 459, U1131-consis consisfluo-U1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wood S. L., Westbrook J. A., Brown J. E. (2013) Omic-profiling in breast cancer metastasis to bone: implications for mechanisms, biomarkers, and treatment. Cancer Treat. Rev. 40, 139–152 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.