

Summary
Choroid plexus carcinomas (CPCs) are poorly understood and frequently lethal brain tumors with few treatment options. Using a mouse model of the disease and a large cohort of human CPCs, we performed a cross-species, genome-wide search for oncogenes within syntenic regions of chromosome gain. TAF12, NFYC and RAD54L co-located on human chromosome 1p32-35.3 and mouse chromosome 4qD1-D3, were identified as oncogenes that are gained in tumors in both species and required for disease initiation and progression. TAF12 and NFYC are transcription factors that regulate the epigenome, while RAD54L plays a central role in DNA repair. Our data identify a group of concurrently gained oncogenes that cooperate in the formation of CPC and reveal potential avenues for therapy.
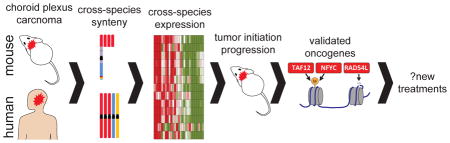
Graphical Abstract
INTRODUCTION
Various genetic alterations activate oncogenes or delete tumor suppressor genes (TSGs) in cancer. Recurrent mutations that disrupt the same gene can be highly informative, pinpointing oncogenic alterations that may serve as therapeutic targets (Baselga et al., 1996; Druker et al., 2001; Flaherty et al., 2010). But focal alterations are relatively infrequent in many cancers, particularly those arising in children (Alexandrov et al., 2013; Zhang et al., 2012). Rather, these tumors contain large DNA copy number alterations (CNAs) that presumably drive the overexpression of oncogenes or delete TSGs (Chen et al., 2014; Downing et al., 2012; Johnson et al., 2010; Wu et al., 2012). These CNAs are often chromosomal in scale, making it difficult to identify which genes are driving transformation. RNA silencing technologies have discovered TSGs within large deletions (Scuoppo et al., 2012; Xue et al., 2012; Zender et al., 2008); but approaches to screen the oncogenic capacity of genes located within large regions of gain are less well developed.
Choroid plexus carcinomas (CPCs) are highly malignant brain tumors that are characterized by gains of chromosomes 1, 2, 4, 7, 12, 14, 19, 20 and 21, and numerous autosomal losses (Paulus and Brandner, 2007; Rickert et al., 2002; Ruland et al., 2014). The great majority of CPCs are diagnosed in children aged less than three years, of whom two thirds die within five years (Wrede et al., 2009). Efforts to identify more effective treatments of CPC have been hindered by poor understanding of its pathogenesis. Germline alterations of TP53, and possibly hSNF5/INI1, predispose to CPC in humans (Garber et al., 1991; Malkin et al., 1990; Olivier et al., 2003; Sevenet et al., 1999; Tinat et al., 2009) and ablation of Tp53 and/or Rb function causes CPCs in mice (Brinster et al., 1984; Sáenz Robles et al., 1994). Deletion of PTEN has also been implicated in CPC, but the oncogenes that drive this cancer have not been identified (Morigaki et al., 2012; Rickert et al., 2002; Ruland et al., 2014). The goal of our study was to identify CPC oncogenes within large regions of chromosome gain.
RESULTS
Tp53, Rb and Pten suppress CPC
To better understand the cellular and molecular origin of CPC we first developed a mouse model of the disease (Figure 1). Using in utero electroporation, we introduced Cre Recombinase into the hindbrain choroid plexus epithelium (CPE) of embryonic day (E) 12.5 mice carrying various conditional alleles. At postnatal day (P) 0, efficient recombination was evident in the CPE of mice carrying the ROSA-yellow fluorescence protein (ROSAYFP) lineage tracing allele (Figure 1A,B). YFP+ CPE cells expressing Transthyretin (Ttr), a marker of mature CPE (Harms et al., 1991), were retained by adult RosaYFP mice that had been electroporated in utero with Cre Recombinase; but these mice never developed tumors (total mice n=7, Figure 1C–E). In stark contrast, electroporation of Cre Recombinase into the hindbrain CPE of Tp53flx/flx; Rbflx/flx; Ptenflx/flx; ROSAYFP E12.5 embryos resulted in CPCs within 220 days of postnatal life (penetrance, 38% [n=26/69]; Figure 1F–O). All tumors were YFP+, confirming their origin from in utero Cre-recombined cells (Figures 1F,G). Mouse CPCs recapitulated the morphology (admixed papillary and syncytial architecture and pleomorphic epithelioid cytology [Figure 1H]); differentiation state (relative decrease in Ttr expression and expression of cytokeratin 8 [Figure 1I,J]); proliferation (high Ki67 and BrDU labelling index [Figure 1K]), and ultrastructural features (microvilli, intracellular tight junctions, and extensive basal membrane folding [Figure 1L,M]) of the human disease. Allele specific polymerase chain reactions confirmed the deletion of Tp53, Rb and Pten from these tumors but not adjacent normal tissue (Figure 1N).
Figure 1. A mouse model of CPC.

Expression of the recombined ROSAYFP fluorescent lineage tracing allele in whole (A) and microscopic (B) preparations of postnatal day (P) 0 hindbrain choroid plexus following in utero electroporation with Cre-recombinase at embryonic day (E) 12.5 (scale bar=50μm). Recombined (C), but histologically normal (D) and Ttr+ (E) choroid plexus persisting in adult CPE (scale bar=15μm). In utero electroporation of the hindbrain choroid plexus of Tp53flx/flx; Rbflx/flx; Ptenflx/flx; ROSAYFP E12.5 embryos generated large YFP+ tumors (F,G) that recapitulate the histology (H); reduced Ttr (I) and increased Cytokeratin 8 (J) expression; high proliferation rate (K); and ultrastructural features (L,M) of human CPC (scale bar F to K=20μm; L to M=2μm). N, polymerase chain reactions of recombined (RC) alleles in mouse CPC (T) and intact (WT) alleles in normal tissue (N). O, Kaplan-Meier survival curves of E12.5 mice harbouring the indicate alleles that were Cre-electroporated in utero. P, unsupervised hierarchical clustering of gene expression profiles of mouse: CPC; E12.5 and adult CPE (eCP and aCP); WNT and Sonic Hedgehog (SHH) medulloblastomas; P7 dorsal brainstem (P7 DBS); P7 cerebellum (P7 CB); E12.5 cerebellum (eCB); and E12.5 lower rhombic lip (eLRL). Q, GSEA of ‘Lein choroid plexus markers’ in CPC reporting normalized enrichment score (NES) and the false discovery rate (FDR) Q value. See also Table S1.
Tp53 and Rb, but not Pten, have previously been shown to suppress CPC (Brinster et al., 1984; Sáenz Robles et al., 1994). Therefore, to better assess the role of Pten as a CPC TSG, we performed tumor surveillance studies of Tp53flx/flx; Rbflx/flx; ROSAYFP (n=68), Tp53flx;flx; Ptenflx/flx; ROSAYFP (n=36) and Rbflx;flx; Ptenflx/flx; ROSAYFP (n=24) mice electroporated with Cre Recombinase at E12.5. Only 10% (n=7/68) of Tp53flx/flx; Rbflx/flx; ROSAYFP and no Tp53flx;flx; Ptenflx/flx; ROSAYFP or Rbflx;flx;Ptenflx/flx; ROSAYFP mice developed CPCs (Figure 1O). Thus, in our model, loss of both Tp53 and Rb is required to generate CPC, and neither of these deletions can be substituted by loss of Pten. However, deletion of Pten together with loss of Tp53 and Rb significantly increases tumor penetrance.
To further characterize our model, we compared the gene expression profiles of Tp53flx/flx; Rbflx/flx; Ptenflx/flx; ROSAYFP CPCs with those of other mouse hindbrain tumors and tissues (Figure 1P). The transcriptomes of CPC and normal adult and embryonic choroid plexuses co-clustered separately from those of medulloblastoma and normal embryonic and/or adult brainstem and cerebellum (Gibson et al., 2010; Uziel et al., 2005). Furthermore, gene set enrichment analysis (GSEA) identified ‘Markers of Choroid Plexus’ as the most enriched of 4,293 gene sets in CPCs relative to mouse medulloblastoma, brainstem and cerebellum (Figure 1Q; Table S1). Among the choroid plexus derived tissues, CPCs were more closely related to embryonic than adult choroid. Thus our mouse CPCs recapitulate the histological and ultrastructural features of the human disease and express an embryonic choroid plexus-like transcriptome.
Chromosome 1p31.3-ter encodes candidate CPC oncogenes
To identify genetic alterations that drive CPC, we used Affymetrix 6.0 DNA SNP microarrays to catalogue CNAs in human choroid plexus papillomas (CPPs, n=32) and human CPCs (n=23; Table S2). We also performed microarray comparative genomic hybridization (aCGH) of 47 Tp53flx/flx; Rbflx/flx; Ptenflx/flx; ROSAYFP mouse CPCs, including 12 primary tumors and serial secondary (n=20) and tertiary (n=15) orthotopic transplants of these tumors. Additionally, we sequenced the whole genomes (WGS) of four human CPCs and matched normal blood (>98% of the tumor genome and >91% of the normal genome had 20-fold coverage with high-quality sequence reads; see Supplementary Experimental Procedures). No recurrent, single nucleotide variations, insertion/deletions, or focal CNAs (<5 genes) were identified in the four human CPCs subject to WGS, although one of these cases had extensive chromothripsis of chromosomes 1 and 19 in the absence of a mutation in TP53 (Tables S3–S7; Supplementary Experimental Procedures). However, as expected, human CPCs contained numerous, recurrent, chromosomal gains and losses (Figure 2A). Similar non-random chromosomal alterations were observed in mouse tumors, suggesting large CNAs are a primary oncogenic driver of CPC (Figure 2B).
Figure 2. Cross-species analysis of syntenic chromosomal gains in human and mouse CPC.

Heat maps of genomewide DNA copy number alterations in human CPPs and CPCs (A) and mouse CPCs (B). Heatmaps of the copy number of chromosome 1 (C), 7 (D) and 12 (E) in human CPPs and CPCs (left in each figure) and the corresponding syntenic regions in mouse CPCs (right in each figure). See also Tables S2–7.
Since both human and mouse CPCs contained recurrent chromosomal CNAs, we looked for syntenic chromosome fragments that were gained in tumors in both species since these might be enriched for oncogenes. While 61% (n=14/23) of human CPCs gained at least one whole copy of chromosome 1 (encoding ~2,100 genes), only one syntenic fragment of chromosome 1 was gained in mouse CPCs (encoding 671 genes; mouse chromosome 4qC6-qE2 [94,608,732–155,608,945]; syntenic with human chromosome 1p31.3-ter [895,967- 67,594,220]; Figure 2C). Gain of 4qC6-qE2 was seen in 50% (n=6/12) of mouse CPCs and was retained through secondary and tertiary serial tumor transplants (Figure 2C). Only 3% (n=1/32) of the more benign human CPPs gained chromosome 1. Thus human chromosome 1p31.3-ter/mouse chromosome 4qC6-qE2 (hereon, chr1p31.3-ter/4qC6-qE2) may harbour oncogenes that drive aggressive choroid plexus tumors. Notably, gain of chr1p31.3-ter/4qC6-qE2 may be associated with dysfunctional TP53 since our mouse model is Tp53 null and chromosome 1 gain was significantly associated with mutant TP53 in human CPCs (Fisher’s Exact, p<0.05; Figure 3; Table S2). Similar analyses of other autosomal gains in human CPCs including chromosomes 7 and 12 that are among the most commonly gained chromosomes in the disease (Rickert et al., 2002; Rudland et al., 2014), failed to identify additional syntenic gains (Figure 2D,E).
Figure 3. A common set of 21 syntenic genes gained and overexpressed on 1p31.3-ter in human and 4qC6-qE2 in mouse, CPC.

Top: heat maps of human chromosome 1 copy number in 34 human CPCs and CPPs (left), and mouse chromosome 4qC6-qE2 in 27 mouse CPCs and normal choroid (right). Middle: TP53 status and tissue type of each sample. Bottom: heat maps reporting the expression of 21 copy-number driven orthologs located on human chromosome 1p31.3-ter in the 34 human tumors (left) and 27 mouse CPCs and normal choroid (right). Dotted lines demarcate the results of independent microarray probes for each human and mouse ortholog. Gene symbols shown right. See also Figure S1.
To further resolve which of the 671 genes on chr1p31.3-ter/4qC6-qE2 might be oncogenes, we integrated the copy number and expression of this region in human and mouse tumors to identify genes that were both gained and overexpressed (Figure 3). 26% of genes (n=176/671) on human chromosome 1p31.3-ter were significantly overexpressed in human tumors that gained this region (n=9), relative to those in which it was balanced or deleted (tumors, n=25; log ratio ≥2, p<0.05 with Bonferonni correction). Mouse CPCs that gained 4qC6-qE2 (n=15) overexpressed 11% of genes (n=64/579 Affymetrix 430 microarray) in this region relative to balanced mouse tumors (n=5) and normal mouse choroid plexus (n=7; log ratio ≥2, p<0.05 with Bonferonni correction). Comparison of these human and mouse data revealed a common set of 21 genes on chr1p31.3-ter/4qC6-qE2 that were both gained and overexpressed in human and mouse CPCs, pinpointing these as ‘lead candidate’ oncogenes (Figure 3).
Gain of chromosome 4qC6-qE2 is an early event in CPC tumorigenesis
Cancers accumulate genetic alterations sequentially, suggesting these defects play temporally distinct roles during transformation. Therefore, to enable appropriate investigation of our 21 lead candidate oncogenes, we first determined when 4qC6-qE2 is gained during mouse CPC development (Figure 4). Tp53flx/flx; Rbflx/flx; Ptenflx/flx; ROSAYFP mice were electroporated in utero with Cre Recombinase exactly as described above. Mice were then sacrificed at P0, P21 or P35 two hours following injection with BrdU and their hindbrains subject to histologic study (≥4 mice per time point).
Figure 4. Serial analysis of choroid plexus transformation in mice.

A, In utero electroporation of the hindbrain choroid plexus of Tp53flx/flx; Rbflx/flx; Ptenflx/flx; ROSAYFP E12.5 embryos resulted in progressive expansion of YFP+ choroid plexus (top row, scale bar=50μm) that displayed: dysplasia (second row, hematoxylin and eosin [H&E]; dotted line and arrows indicate disruption of normal single layer epithelium; scale bar=10μm); loss of Ttr expression (third row, in situ hybridization; scale bar=20μm) and increasing proliferation (fourth row, nuclear BrdU incorporation marked with arrows [note dotted line encompasses same region in P0 H&E stain] scale bar=10μm); accumulation of DNA DSB (γH2ax stain marked with arrows; scale bar=10μm); and gain of 4qC6-qE2 (note two separate FISH probes used targeting Stmn1 and Cdca8 relative to control Baat at 4qB1). Graphs to the right report the quantification of BrdU incorporation (B), γH2ax stain (C) and gain of 4qC6-qE2 (D) in recombined (YFP+) cells. Mann Whitney, *= P<0.05, ***= P<0.0005.
One week following electroporation (P0), small areas of dysplastic YFP+ CPE were visible in which the epithelium lost its monolayer organization and decreased its expression of Ttr. However, significant proliferation (BrdU incorporation), DNA double strand breaks (DSB; γH2ax staining) and gain of 4qC6-qE2 (fluorescence in situ hybridization [FISH]) were not detected (Figure 4A–D). Three weeks later (P21), the amount of dysplastic YFP+/Ttr− CPE had clearly increased and significant numbers of aberrantly proliferating YFP+/BrdU+ CPE were now detected relative to control mice (p<0.05 Mann-Whitney; Figures 4A, B); however, neither DNA double stand breaks nor 4qC6-qE2 gain were apparent. By six weeks post-electroporation (P35) the choroid plexus had undergone a dramatic change: large regions of hyperplastic CPE were now visible, in which 20%±2.4SE of YFP+ CPE were proliferating (BrdU+, p<0.0005 Mann Whitney, relative to controls; Figures 4A,B). In addition, significant DNA DSBs and gain of 4qC6-qE2 were now detected in YFP+ CPE (both p<0.0005, Mann-Whitney; Figure 4A,C,D). Interestingly, while high levels of aberrant proliferation and gain of 4qC6-qE2 persisted in fully formed CPCs, DSBs were not detected in tumors (Figures 4B–D). Together, these data confirm that mouse CPCs develop from mutated CPE and that gain of 4qC6-qE2 is a relatively early event in the disease process, coinciding with choroid plexus hyperplasia and suggesting this region may contain oncogenes that play a role in tumor initiation.
Taf12, Nfyc and Rad54l promote aberrant proliferation of the developing choroid plexus
Since gain of 4qC6-qE2 is an early event in CPC development, we reasoned that developing choroid plexus would be an appropriate context in which to screen the transforming potential of our 21 lead candidate oncogenes. To do this, we tested the capacity of each candidate to drive dysplasia and proliferation of early postnatal CPE that is normally a single layer of post-mitotic cuboidal epithelium (Lehtinen et al., 2013). Plasmids expressing green fluorescence protein (GFP) and a single lead candidate oncogene each were co-electroporated into the hindbrain CPE of separate cohorts of E12.5 embryos (≥5 embryos/candidate, total n=107 embryos; Figure 5A). Proliferating CPE cells were labelled at E19.5 by maternal injection of BrdU. The proportion of targeted and proliferating (GFP+/BrdU+), relative to targeted but non-proliferating (GFP+/BrdU−), CPE cells was calculated at P0. Five control mice were electroporated with empty vector-GFP.
Figure 5. Functional in utero assessment of 1p31.3-ter/4qC6-qE2 candidate CPC oncogenes.

A. E12.5 choroid plexus was co-electroporated with plasmids encoding candidate and GFP to identify electroporated cells. Gels below show seven examples of 21 reverse-transcriptase (RT) PCR results of cells transfected with control plasmid or oncogene candidates with or without RT. B. Sections from each animal were analysed both for morphological change (top, H&E) and proliferation of electroporated cells (bottom, BrdU+/GFP+; scale bar=10μm). Only Taf12, Nfyc and Rad54l demonstrated dysplasia and aberrant proliferation relative to the other 18 candidates, Pgd shown as an example. C, Graph reporting the percentage of electroporated (GFP+) and proliferating choroid plexus epithelium cells. *P<0.05, **=P<0.005, Mann-Whitney.
GFP+ cells were detected in all electroporated mice. Fewer than 0.5% of electroporated (GFP+) CPE cells were proliferating (BrdU+) in embryos that were electroporated with control plasmid or 13 of 21 lead candidate oncogenes (Figure 5A–C). Expression of five other candidates (Cdc20, Stmn1, Atp6v0b, Clspn, Mier1) was associated with proliferation in 0.5–2% of GFP+ cells but did not alter choroid plexus morphology (Figure 5B,C). In marked contrast, Taf12, Nfyc and Rad54l induced CPE dysplasia and proliferation similar to that observed following 4qC6-qE2 gain in Cre-electroporated Tp53flx/flx; Rbflx/flx; Ptenflx/flx; ROSAYFP mice (compare Figures 4A and 5B). Both Taf12 (23.2%±8.4SE BrdU+YFP+, p<0.005 Mann-Whitney) and Nfyc (4.0%±2.0SE BrdU+YFP+, p<0.05 Mann-Whitney) induced significant levels of CPE proliferation relative to control transduced cells. Notably, we observed a worse prognosis among patients whose CPCs expressed relatively high levels of RAD54L (Figure S1). Therefore, in light of these data we selected Taf12, Nfyc and Rad54l for further study as potential CPC oncogenes.
Taf12, Nfyc and Rad54l maintain CPC
Next, we tested whether Taf12, Nfyc and/or Rad54l are required to maintain CPC cells both in vitro and in vivo. To ablate the expression of these candidate oncogenes, we generated three shRNA-green fluorescence protein (GFP) lentiviruses against each candidate. We also generated mismatch-control shRNAs (controlshRNA) for Taf12, Nfyc and Rad54l (one per candidate) in which five bases of each shRNA sequence were mutated to reduce target homology. Finally, shRNAs targeting two ‘control’ genes (Pqlc2 and Mier1) on chr1p31.3-ter/4qC6-qE2 that did not induce dysplasia or aberrant proliferation of developing CPE were also generated (Figure 5C).
Greater than 90% of mouse primary CPC cells were transduced following exposure to lentiviruses in vitro, resulting in ≥50% knockdown of the corresponding target gene relative to controlshRNA transduced cells (Figure 6A). Ablation of either Rad54lshRNA or Taf12shRNA induced significant Caspase 3/7 activity in mouse CPC cells and dramatically reduced the survival of these cells in culture relative to controlshRNA, Pqlc2shRNA or Mier1shRNA transduced cells (Figure 6B, C). In contrast, knockdown of Nfyc did not affect apoptosis or survival of CPC cells in vitro.
Figure 6. Taf12, Nfyc and Rad54l are required to initiate and maintain CPC in mice.

A. Reverse-transcriptase PCR of CPC cells transduced with control or gene targeted shRNA lentivirus. B. In vitro survival of CPC cells transduced with the indicated shRNA lentivirus. ***=P<0.0005, relative to controlshRNA cells. C. Caspase 3/7 assays of control and shRNA-transduced CPC cells. *=P<0.05, relative to controlshRNA cells. D. Kaplan-Meyer survival curves of mice implanted with CPC cells transduced with the indicated shRNA lentiviruses. *=P<0.05, **=P<0.005, ***=P<0.0005, Log-Rank relative to controlshRNA mice. E. Kaplan-Meyer survival curves of Tp53flx/flx; Rbflx/flx; Ptenflx/flx; ROSAYFP mice electroporated at E12.5 with Cre-recombinase and simultaneously injected in the hindbrain with the indicated shRNA (P<0.05, Log-Rank comparison of the three survival curves). F. Morphology (H&E), lentiviral transduction (GFP), and Cre-recombination (RosaYFP) of hyperplastic choroid plexus mass in adult Tp53flx/flx; Rbflx/flx; Ptenflx/flx; ROSAYFP mouse electroporated at E12.5 with Cre-recombinase and simultaneously injected in the hindbrain with Taf1shRNA (scale bar=20μm).
To test if Taf12, Nfyc and Rad54l maintain CPC cells in vivo, we transduced primary CPC cells with Taf12shRNA, NfycshRNA, Rad54lshRNA or controlshRNA and transplanted them orthotopically into the hindbrains of immunocompromised mice (Figure 6D). In agreement with our in vitro data, the median survival of mice harbouring Taf12shRNA (n=6, median survival 34 days) or Rad54lshRNA (n=8, median survival 55.5 days) transduced CPC cells was significantly longer than mice injected with controlshRNA transduced CPC cells (n=19, median survival 19 days; P<0.005 Log Rank; Figure 6D). Mice harbouring NfycshRNA transduced CPC cells also survived longer than control animals (n=5, median survival 29 days, p=0.05 Log Rank). Thus expression of Taf12, Nfyc and Rad54l appears to support optimal growth of CPC cells, supporting the notion that they are oncogenes.
Taf12, Nfyc and Rad54l are required to initiate CPC
Since gain of 4qC6-qE2 coincided with the onset of severe hyperplasia in our mouse model (Figure 4) we next tested whether the expression of our candidates is required to initiate CPC. Cre Recombinase was electroporated into the hindbrain choroid plexus of E12.5 Tp53flx/flx; Rbflx/flx; Ptenflx/flx; ROSAYFP mouse embryos exactly as described above, but this time we simultaneously injected the IV ventricle of these embryos with either Taf12shRNA (n=6 mice) NfycshRNA (n=13 mice), Rad54lshRNA (n=10 mice) or controlshRNA (n=12 mice) lentivirus. Remarkably, while 42% (n=5/12) of Cre-electroporated mice injected with controlshRNA developed CPCs, injection of either Taf12shRNA or NfycshRNA lentiviruses completely abolished CPC development and only 10% (n=1/10) of mice receiving Rad54lshRNA lentiviruses developed tumors (P<0.05 Log Rank; Figure 6E). To confirm that the choroid plexus of mice injected with active shRNAs underwent Cre-recombination and viral transduction, we reviewed the hindbrains of these animals after ≥100 days. Several of these mice contained hyperplastic but histologically benign choroid plexus masses containing numerous YFP+ (Cre-recombined) and GFP+ (shRNA transduced) cells (Figure 6F), suggesting that expression of shRNAs had arrested CPC development. These data provide compelling evidence that expression of Taf12, Nfyc or Rad54l is critical for the initiation of CPC.
Upregulation of Taf12, Nfyc and Rad54l drives CPC
Having shown that loss of Taf12, Nfyc and Rad54l expression markedly impairs CPC initiation and maintenance, we conducted two sets of experiments to test whether overexpression of these genes might promote disease initiation and/or progression. First, we tested if IV ventricular co-injections of Taf12-cop(c)GFP, Nfyc-cGFP and Rad54l-cGFP expressing lentiviruses (1.67×105 lentiviral particles each per mouse, total=5×105 particles) alters CPC disease course in Cre-electroporated Tp53flx/flx; Rbflx/flx; Ptenflx/flx; ROSAYFP E12.5 embryos. The tumor free survival of Cre-electroporated and lentivirus injected mice was significantly reduced relative to that of mice that were Cre-electroporated alone (Figure 7A; Log Rank p<0.05). Mice that were injected with Taf12-cGFP, Nfyc-cGFP and Rad54l-cGFP lentiviruses but were not Cre-electroporated contained numerous cGFP+ CPE cells but did not develop CPCs (total mice n=7, median follow up 374 days; Figure 7A). Thus, increased expression of Taf12, Nfyc and Rad54l significantly accelerates CPC development in Tp53, Rb, Pten deleted CPE, but is not sufficient to initiate the disease. These data are compatible with the observation that 4qC6-qE2 is gained following deletion of Tp53, Rb, Pten, but precedes CPE transformation (Figure 4A,D).
Figure 7. Taf12, Nfyc and Rad54l expression promote CPC in mice.

A, Kaplan-Meyer survival curves of Tp53flx/flx; Rbflx/flx; Ptenflx/flx; ROSAYFP mice injected in the hindbrain at E12.5 with Taf12, Nfyc and Rad54l expressing lentiviruses following sham (no Cre) or Cre-electroporation (Cre). Survival of Cre-electroporated mice not receiving lentivirus is also shown (Cre only). B, Left: macroscopic, direct YFP fluorescence of the hindbrains of adult Tp53flx/flx; Rbflx/flx; Ptenflx/flx; ROSAYFP mice receiving hindbrain injections of the indicated lentivirus at E12.5. Right panels show images of H&E stained or immunofluorescence stained sections of the corresponding hindbrain, left (scale bars=50μm). Kaplan-Meyer survival curves of Tp53flx/flx; Rbflx/flx; Ptenflx/flx; ROSAYFP mice injected in the hindbrain at E12.5 (C) or P1 (D) with the indicated lentiviruses. Statistics report the Log-Rank comparison of the survival curves. D, in situ hybridization (top and middle) and immunofluorescence (bottom) of Taf12, Nfyc and Rad54l expression in CPCs induced by lentiviral injection. All scale bars=50μm.
As a further test of whether upregulation of Taf12, Nfyc and Rad54l can cooperate with TSG deletion to drive CPC, we generated a variant of our mouse model in which we titrated the amount of Tp53, Rb and Pten deletion using a Cre Recombinase-cGFP (Cre-cGFP) lentivirus. In utero injection of 5.0×105 Cre-eGFP lentiviral particles into the IV ventricle of E12.5 Tp53flx/flx; Rbflx/flx; Ptenflx/flx; ROSAYFP mice resulted in highly penetrant YFP+ (recombined) cGFP+ (lentiviral transduced) CPCs (86%, n=6/7; Figure 7B,C). In contrast, injection of just 1.25×105 Cre-cGFP lentiviral particles yielded no tumors; although these mice did retain YFP+/cGFP+ co-expressing CPE cells and some developed hindbrain choroid plexus hyperplasia (n=8, median follow up 381 days; Figure 7B,C). Thus, lentiviral delivery of Cre-recombinase serves as a titratable alternate to electroporation for inducing CPCs in our model system.
Since 1.25×105 Cre-cGFP viral particles failed to generate CPCs in E12.5 Tp53flx/flx; Rbflx/flx; Ptenflx/flx; ROSAYFP mouse embryos, we tested if concurrent delivery of Taf12-cGFP, Nfyc-cGFP and Rad54l-cGFP lentiviruses might cooperate with this level of recombination to promote tumorigenesis. 75% (n=9/12) of E12.5 Tp53flx/flx; Rbflx/flx; Ptenflx/flx; ROSAYFP mouse embryos injected with 1.25×105 Cre-eGFP lentiviral particles and an equal mix of Taf12-cGFP, Nfyc-cGFP and Rad54l-cGFP (3.75×105 particles) developed YFP+/cGFP+ CPCs (total viral particles 5.0×105 per mouse; Figure 7B,C). In situ hybridization and immunofluorescence analysis of these tumors confirmed high-level co-expression of Taf12, Nfyc, and Rad54l (Figure 7E). To control for the possibility that high viral titres (5×105 particles) are non-specifically transforming, we repeated our experiments but this time co-injected 1.25×105 Cre-cGFP lentiviral particles with 3.75×105 particles of a single candidate (Taf12-cGFP, total viral particles 5×105 per mouse). These mice also developed CPC, but at a significantly lower penetrance than animals receiving all three oncogene candidates (22%, n=2/9; Figure 7C). Similar experiments showed that Taf12, Nfyc and Rad54l can also drive CPC in postnatal choroid plexus (Figure 7D). Together with our cross-species genomic and in vivo gene knockdown studies, these data comprehensively validate Taf12, Nfyc and Rad54l as CPC oncogenes and support the notion that concurrent gain of these three genes on chr1p31.3-ter/4qC6-qE2 cooperates with deletion of Tp53, Rb and Pten in the initiation and progression of CPC.
Aberrant DNA metabolism is a significant feature of CPC
TAF12, NFYC and RAD54L are key regulators of DNA metabolism, suggesting that dysregulation of DNA maintenance and/or repair are necessary for CPC formation (Benatti et al., 2011; Nardini et al., 2013; Schmitz et al., 2009; Wright and Heyer, 2014). As a first step to test this, we used GSEA to compare the transcriptomes of mouse E12.5 CPE with those of daughter CPCs, and measured the sensitivity of CPC cells to two separate inhibitors of the ataxia telangiectasia and Rad3-related protein (ATR) kinase that regulates DNA repair, including DSBs (Somyajit et al., 2013). In keeping with our hypothesis, numerous gene sets that maintain the integrity of the genome and epigenome were significantly upregulated in CPC (Figure 8A,B; Table S1). As expected, Rad54l was the most upregulated homologous repair gene (Figure 8B). Notably, DNA replication and repair gene sets were also enriched in human CPCs that are diploid for chromosome 1, suggesting dysregulation of these cell functions are a general feature of CPC (Table S8).
Figure 8. DNA repair is upregulated in CPC.

A. GSEA of ‘Kauffmann DNA Repair Gene’ set in CPC versus E12.5 CPE. B. Heat map reporting the GSEA of the ‘Kegg Homologous Repair Gene’ set in CPC versus E12.5 CPE. C. Growth inhibition assays of mouse CPC and ependymoma (Johnson et al., 2010) cells following 72 hour exposure to the ATR inhibitors AZ-20 and VE-821. D. Quantification of DNA DSBs in the CPC and ependymoma cells shown in C, both before (time 0) and at the indicated times following exposure to 0.4μM AZ-20. See also Table S8.
Finally, CPC cells proved remarkably sensitive to ATR inhibitors in vitro relative to cells from an unrelated mouse brain tumor model of ependymoma (Figure 8C). Exposure of CPC cells to 0.4μM AZ-20 that inhibited their growth by 90%, increased the induction of DNA DSB in these cells >3-fold after only six hours; however, this concentration of AZ-20 had no impact on ependymoma cell proliferation or DSB formation (Figure 8C,D). Importantly, while sensitivity to ATR inhibitors may vary with basal levels of replicative stress; CPC cells proliferated slightly slower that ependymoma cells (doubling time 25.7 hours ±1.2SD CPC vs. 19.6 hours ±1.7SD ependymoma) and no significant difference in replication stress was detected between untreated CPC and ependymoma cells (Figure 8D). These data support the hypothesis that gain of TAF12, NFYC and RAD54L on chr1p31.3-ter/4qC6-qE2 promotes aberrant DNA maintenance and repair during CPC formation and suggest that inhibition of this activity may have therapeutic potential.
DISCUSSION
Using a combination of cross-species genomics and in vivo mouse modelling, we describe a mouse model of CPC; demonstrate that postnatal CPE from which TSGs have been deleted in utero can generate these tumors; identify three oncogenes – TAF12, NFYC and RAD54L – that are required to initiate and maintain the disease; and implicate upregulation of DNA maintenance and repair as a critical requirement for CPC formation.
How might concurrent gain of TAF12, NFYC and RAD54L contribute to CPC formation? Both TAF12 and NFYC are histone-fold domain containing transcription factors (Bieniossek et al., 2013; Nardini et al., 2013). TAF12 recruits GADD45, and thereby the nucleotide excision repair machinery, to the promoter of active genes, removing methylated cytosines and maintaining a hypomethylated active state (Schmitz et al., 2009). Transcriptional regulation by TAF12 is thought to contribute to transformation, possibly by promoting an invasive phenotype (Voulgari et al., 2008). NFYC is also engaged in chromatin remodeling, establishing permissive chromatin modifications at CCAAT promoters, including those of cell cycle regulators (Benatti et al., 2011; Nardini et al., 2013). Deletion of NFYC or the related gene NFYB, halts cell cycle progression, predominantly by causing a G2/M arrest (Benatti et al., 2011). Thus, upregulation of TAF12 and NFYC might promote an aberrant epigenome during CPE transformation, maintaining or re-activating the expression of proto-oncogenes that are normally silenced in post-mitotic, postnatal choroid plexus. This notion is supported by our observation that the CPC transcriptome most closely matches embryonic CPE. RAD54L plays a central role in homologous recombination in which damaged DNA – particularly DSBs – are repaired by copying an intact homologous sequence (Wright and Heyer, 2014). Loss of Rad54l from Tp53 and LigIV null cells results in the accumulation of DSBs, severe chromosomal defects, and failed cell proliferation (Mills et al., 2004). Therefore, upregulation of RAD54L may be required for CPE to tolerate the severe genotoxic stress that is associated with rampant proliferation in the context of accumulating chromosomal insults. Our observations that Tp53/Rb/Pten null CPE accumulate DSBs that apparently resolve following the gain of 4qC6-qE2, and that CPC transcriptomes are markedly enriched for DNA repair genes, supports this concept.
In light of our data, we propose a hypothesis for the development of CPC; particularly in patients with germline mutations in TP53 (Figure 8E). We propose that the loss of TP53 (and potentially RB and PTEN) from CPE results in the accumulation of genetic and epigenetic changes that disrupt terminal differentiation and senescence. Early in postnatal life, these aberrantly proliferating CPE cells develop increasingly abnormal genomes leading to genotoxic crisis. While most of these cells undergo cell death, presumably through TP53-independent mechanisms, a fraction acquire aberrant DNA repair and epigenome remodelling capacity, including gain of TAF12, NFYC and RAD54L. This enables them to tolerate an aberrant but stable genome and drive further transformation. The advantage afforded by these genes provides a selective pressure for retention of chr1p31.3-ter/4qC6-qE2 gain. These mutant CPE cells go on to form CPCs that can tolerate the recurrent and extensive chromosome changes associated with the disease. Thus, the concurrent gain of TAF12, NFYC and RAD54L may serve to model an aberrant epigenome that promotes a proliferative, and relatively undifferentiated state (TAF12 and NFYC), while tolerating genotoxic stress (RAD54L). Interestingly, recent data indicate that CPCs that do not gain chromosome 1 retain both copies of this chromosome in an otherwise hypodiploid genome (Merino et al., 2014). Thus absolute or relative gain of this chromosome might be required for CPC development.
Our demonstration that increased expression of Taf12, Nfyc and Rad54l induce dysplasia and aberrant proliferation in otherwise wild-type CPE, support the notion that these are CPC oncogenes. However, the dependency of transforming cells on aberrant DNA repair may also occur in the context of non-oncogene addiction (Luo et al., 2009). Thus, CPCs might be ‘addicted’ to RAD54L, and/or the targets of TAF12 and NFYC chromatin remodelling, as non-oncogenes. Regardless of the underlying mechanism, validation of aberrant DNA repair as a requirement for CPC formation and maintence could lead to new therapies, since inhibition of DNA repair enzymes should result in intolerable DNA damage. Precedent for this approach exists in the addiction of BRCA2 mutant cells to the DNA repair enzyme PARP1 and our demonstration that CPC cells are sensitive to ATR inhibitors (Sakai et al., 2008). As well as suggesting potential drug targets within DNA repair pathways, our data show that inhibitors of phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) signaling (suppressed by PTEN) might also serve as new CPC treatments. But translating these and other potential new therapies to the clinic for CPC has proved immensely challenging since fewer than 50 children are diagnosed with this disease in the United States each year (CBTRUS, 2006; Paulus and Brandner, 2007). Therefore, rigorous preclinical studies using our faithful model of CPC could help discover, triage and prioritize potential new therapies for clinical trial.
Our model also provides a useful system to study the origin of CPC. There are at least two candidate cells of origin of CPC: CPE progenitors located near the anterior lower rhombic lip (Awatramani et al., 2003; Currle et al., 2005; Huang et al., 2009; Hunter and Dymecki, 2007; Lehtinen et al., 2013) and fully differentiated, post-mitotic, Ttr+ progeny that populate the CPE. Our serial histologic review of Cre-recombined CPE revealed multiple stochastic regions of proliferative and dysplastic CPE sandwiched between otherwise normal regions of mature CPE, favouring mature, post-mitotic CPE as the origin of tumors.
Finally, our syntenic mapping approach holds promise to identify oncogenes located within large regions of chromosomal gain in other cancers. Indeed, emerging evidence suggests that large CNAs characteristic of other pediatric solid tumors are replicated when these tumors develop in other species (Chen et al., 2013; Frappart et al., 2009). Similar studies of syntenic regions of loss coupled with RNA silencing technologies could be applied to discover TSGs (Scuoppo et al., 2012; Xue et al., 2012; Zender et al., 2008).
EXPERIMENTAL PROCEDURES
Mouse tumorigenesis studies
All in vivo experiments were performed in accordance with protocols that were reviewed and approved by the St. Jude Children’s Research Hospital Animal Care and Usage Committee. Mice harbouring conditional floxed alleles of Tp53, Rb, Pten and ROSAYFP (Chow et al., 2011) were housed and studied under St. Jude IACUC approved protocols. The choroid plexuses of E12.5 mouse embryos were electroporated in utero using pcDNA3.1 plasmids encoding Cre-Recombinase, GFP and/or candidate oncogenes exactly as we described previously (Gibson et al., 2010). Electroporation was performed using a CUY-21 electroporator (Nepa Gene Do., LTD, Tokyo, Japan) with five 50-ms pulses of 50 V with 950-ms intervals. Actively proliferating CPE and CPC cells were labelled in vivo by injection of 0.1mg/kg Bromodeoxyuridine (Brdu, Sigma-Aldrich, St. Louis, MO) two hours prior to euthanizing animals. Lentiviruses encoding the cDNA of Cre-Recombinase, Taf12, Rad54l or Nfyc, or shRNAs targeting these genes (three shRNAs per gene designed using www.dharmacon.gelifesciences.com/design-center), were cloned into lentiviruses, packaged, titrated and injected in utero into the embryonic IV ventricle exactly as we described previously (Robinson et al., 2012). Mismatch control shRNAs were generated by mutating five bases within the 21mer sequence of each shRNA. Orthotopic implants of CPCs were generated by injecting the hindbrains of five to six week-old female CD1 nude mice (www.criver.com) with 5×106 CPC cells transduced with control or shRNA-lentivirus suspended in 10μl matrigel. CPC cells were injected into IV ventricle through cisterna magna using Hamilton syringe.
Immunohistochemistry and in situ hybridization
Formalin fixed and paraffin embedded mouse CPCs (or for ATR inhibitor studies, cytospins of cultured CPC cells) were stained with standard immunohistochemical approaches using rabbit polyclonal antibodies to Ki67 (Vector Labs), Phospho-Histone H2A.XSer139 (Cell signaling, Danvers, MA), GFP (Invitrogen, Carlsbad, CA), copGFP (Evrogen, Moscow); or mouse monoclonal antibodies to BrdU (Sigma-Aldrich, St. Louis, MO), Taf12, Nfyc or Rad54l (Abcam, Cambridge, MA). For immunofluorescence staining, second antibodies were conjugated with either Alexa488 or Alexa 594 and sections mounted with Vectashield mounting medium containing DAPI (Vector laboratories, Inc, Burlingame, CA, USA).
In situ hybridization (ISH) was performed using standard approaches and probes generated using the openreading frames of Taf12 (cDNA clone MGC:30438 IMAGE:3493047), Nfyc (cDNA clone MGC:28940 IMAGE:4009737), Rad54l (cDNA clone MGC:13963 IMAGE:3987790) and Ttr (generous gift of Dr. Edwin Monuki, IMAGE clone 1078224, Accession Number AA822938).
Dual-color fluorescence in situ hybridization (FISH) to estimate DNA copy number was performed on 5 μm paraffin embedded tissue sections with BAC clones encoding Baat, Cdca8 and Stmn1 (BACPAC Resources, Oakland, CA). Probes were labelled with either AlexaFluor-488 or AlexaFluor-555 fluorochromes and nuclei counterstained with DAPI (Vector Labs).
RNA and DNA microarray analysis
Human CPCs and CPPs and associated clinical data were collected from institutions in Canada, USA, Brazil, Israel, and Germany in accordance with each institution’s Research Ethics Board. Informed consent was obtained from the parents/legal guardians of all patients. In addition, the research conducted using these samples was reviewed by St. Jude Children’s Research Hospital Institutional Review Board and/or were appropriate the Hospital for Sick Children’s Research Ethics Board. DNA was isolated from both human and mouse tumors and tissues using standard phenol-chloroform. RNA was isolated using TRIzol (Invitrogen, Carlsbad, USA). Gene expression profiles of human and mouse RNAs were generated using GeneChip® Human Exon 1.0ST and 430v2 microarrays, respectively (Affymetrix, Santa Clara, USA). Gene expression data were normalized and analyzed to detected significantly differentially expressed genes exactly as described previously (Parker et al., 2014).
Human and mouse DNA samples were hybridized to Genome-Wide Human SNP Array 6.0 (Affymetrix, Santa Clara, USA) and the Mouse Genome CGH 244K (Agilent), respectively. Human gene copy number was inferred using Partek® Genomics Suite™ 6.5 and genomic segmentation algorithm (Partek, St Louis, MO). Segmented regions were those significantly different from neighbouring regions (p<0.001) and contained 10 or more probes, with a signal to noise ratio of 0.3. Mouse aCGH tumor data were compared to a common control as reference DNA and the circular binary segmentation algorithm [1] implemented in the DNAcopy package from Bioconductor used to identify copy number alterations for each tumor sample. Aberrant segments contained at least 5 probes with a mean log2 ratio greater than 0.3 or less than −0.3.
Whole Genome Sequencing Data Analysis
Detection of somatic single nucleotide variations (SNVs), indels, structural variations and focal copy number changes from whole genome sequencing (WGS) was performed as previously described and are described in detail in Supplementary Experimental Procedures (Parker et al., 2014).
Cell culture
Fresh, primary, CPC tumor tissue was dissected from the IV ventricle of mice, minced and disaggregated using hyaluronidase (Atlanta Biologicals, Lawrenceville, GA) and collagenase type IV (Invitrogen, Carlsbad, CA). Disaggregated cells were filtered through a 40 μm cell strainer and transduced with control or shRNA-lentiviruses. The proliferation of single cell suspensions cultured in neurobasal medium, in the presence or absence of the indicated concentration of ATR inhibitors was recorded using CellTiter-Glo as described (Gibson et al., 2010). Apoptosis was assessed in single cell suspensions using the CaspaseGlo assay (see Supplementary Experimental Procedures).
Quantitative Reverse Transcriptase PCR (qRT-PCR)
Total cDNA was generated from cells and analysed using qRT-PCR and standard approaches. Primers pairs employed in assays included TAF12 (AGCCATCTCATCCTTGGATTTTT : ACGACCAAAATGCTCTTCCTACA), NFYC (AGGACAGATCCAGACACTTGCTAC : GATTGGCTGACTGAATAAACATGG), Rad54l (CCTGGTGAAGAACTGGTACAATG : ATGACCAGTCCAACATTTCCTTT) and Gapdh (AGGTCGGTGTGAACGGATTTG : TGTAGACCATGTAGTTGAGGTCA).
Supplementary Material
HIGHLIGHTS.
TAF12, NFYC and RAD54L identified as CPC oncogenes.
CPC arises from post-mitotic, differentiated choroid plexus epithelium.
Dysregulation of DNA metabolism is a critical requirement for CPC development.
CPC mouse model to study the biology and treatment of this aggressive disease.
Significance.
Few recurrent point mutations or focal amplifications have been identified in whole genome sequencing studies of pediatric cancers. Rather, these tumors contain large chromosomal alterations encompassing tens to thousands of genes, rendering the identification of oncogenes difficult. Choroid plexus carcinomas (CPC) frequently gain chromosome 1 that encodes ~2,100 genes. Using a combination of cross-species genomics and in vivo functional studies, we identified a syntenic fragment of chromosome 1p that is gained early in mouse CPC development, and that contains three oncogenes - Taf12, Nfyc and Rad54l - required to initiate and progress the disease. This approach holds promise to pinpoint oncogenes within large regions of chromosomal gain and the associated dysregulated pathways that might serve as therapeutic targets.
Acknowledgments
This work was supported by grants from the National Institutes of Health (R.J.G., R01CA129541, P01CA96832 and P30CA021765), the Department of Defence (W81XWH-10-1-0674) and by the American Lebanese Syrian Associated Charities. We are grateful to the staff of the Hartwell Center for Bioinformatics and Biotechnology, the Cell and Tissue Imaging Shared Resource and the ARC at St Jude Children’s Research Hospital for technical assistance.
Footnotes
Accession numbers
Human WGS data associated with these studies are available from the European Bioinformatics Institute (EGAS00001000961). Human and mouse RNA and DNA microarray data are available from GEO (GSE60899).
AUTHOR CONTRIBUTIONS
Yiai Tong conducted the great majority of the in vivo mouse work and mouse tumor studies. Diane Marino completed the great majority of the human genomic studies that were supervised by David Malkin. Birgit Nimmervoll was responsible for the CPC tumor serial transfer experiment. Kirti Gupta and David Ellison performed all neuropathological reviews of CPCs. Yong-Dong Wang and David Finkelstein contributed to the mouse genomic studies. James Dalton conducted all FISH analyses. Xiaotu Ma completed the next generation sequencing assays and analyses. Richard Gilbertson conceived and supervised the entire research project including the cross-species genomics analyses. All authors participated in the preparation of the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awatramani R, Soriano P, Rodriguez C, Mai JJ, Dymecki SM. Cryptic boundaries in roof plate and choroid plexus identified by intersectional gene activation. Nat Genet. 2003;35:70–75. doi: 10.1038/ng1228. [DOI] [PubMed] [Google Scholar]
- Baselga J, Tripathy D, Mendelsohn J, Baughman S, Benz CC, Dantis L, Sklarin NT, Seidman AD, Hudis CA, Moore J, et al. Phase II study of weekly intravenous recombinant humanized anti-p185HER2 monoclonal antibody in patients with HER2/neu-overexpressing metastatic breast cancer. J Clin Oncol. 1996;14:737–744. doi: 10.1200/JCO.1996.14.3.737. [DOI] [PubMed] [Google Scholar]
- Benatti P, Dolfini D, Viganò A, Ravo M, Weisz A, Imbriano C. Specific inhibition of NF-Y subunits triggers different cell proliferation defects. Nucleic Acids Research. 2011;39:5356–5368. doi: 10.1093/nar/gkr128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieniossek C, Papai G, Schaffitzel C, Garzoni F, Chaillet M, Scheer E, Papadopoulos P, Tora L, Schultz P, Berger I. The architecture of human general transcription factor TFIID core complex. Nature. 2013;493:699–702. doi: 10.1038/nature11791. [DOI] [PubMed] [Google Scholar]
- Brinster RL, Chen HY, Messing A, van Dyke T, Levine AJ, Palmiter RD. Transgenic mice harboring SV40 t-antigen genes develop characteristic brain tumors. Cell. 1984;37:367–379. doi: 10.1016/0092-8674(84)90367-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CBTRUS. Statistical Report: Primary Brain Tumors in the United States, 1995–1999. Central Brain Tumor Registry of the United States; Hinsdale, IL: 2006. 2002. [Google Scholar]
- Chen EY, Dobrinski KP, Brown KH, Clagg R, Edelman E, Ignatius MS, Chen JY, Brockmann J, Nielsen GP, Ramaswamy S, et al. Cross-species array comparative genomic hybridization identifies novel oncogenic events in zebrafish and human embryonal rhabdomyosarcoma. PLoS Genet. 2013;9:e1003727. doi: 10.1371/journal.pgen.1003727. doi:1003710.1001371/journal.pgen.1003727. Epub 1002013 Aug 1003729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Bahrami A, Pappo A, Easton J, Dalton J, Hedlund E, Ellison D, Shurtleff S, Wu G, Wei L, et al. Recurrent Somatic Structural Variations Contribute to Tumorigenesis in Pediatric Osteosarcoma. Cell Reports. 2014;7:104–112. doi: 10.1016/j.celrep.2014.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow Lionel ML, Endersby R, Zhu X, Rankin S, Qu C, Zhang J, Broniscer A, Ellison David W, Baker Suzanne J. Cooperativity within and among Pten, p53, and Rb Pathways Induces High-Grade Astrocytoma in Adult Brain. Cancer Cell. 2011;19:305–316. doi: 10.1016/j.ccr.2011.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currle DS, Cheng X, Hsu C-m, Monuki ES. Direct and indirect roles of CNS dorsal midline cells in choroid plexus epithelia formation. Development. 2005;132:3549–3559. doi: 10.1242/dev.01915. [DOI] [PubMed] [Google Scholar]
- Downing JR, Wilson RK, Zhang J, Mardis ER, Pui CH, Ding L, Ley TJ, Evans WE. The Pediatric Cancer Genome Project. Nat Genet. 2012;44:619–622. doi: 10.1038/ng.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, Sawyers CL. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O’Dwyer PJ, Lee RJ, Grippo JF, Nolop K, Chapman PB. Inhibition of Mutated, Activated BRAF in Metastatic Melanoma. New England Journal of Medicine. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frappart PO, Lee Y, Russell HR, Chalhoub N, Wang YD, Orii KE, Zhao J, Kondo N, Baker SJ, McKinnon PJ. Recurrent genomic alterations characterize medulloblastoma arising from DNA double-strand break repair deficiency. Proceedings of the National Academy of Sciences. 2009;106:1880–1885. doi: 10.1073/pnas.0806882106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garber JE, Goldstein AM, Kantor AF, Dreyfus MG, Fraumeni JF, Jr, Li FP. Follow-up study of twenty-four families with Li-Fraumeni syndrome. Cancer Res. 1991;51:6094–6097. [PubMed] [Google Scholar]
- Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C, Kranenburg TA, Hogg T, Poppleton H, Martin J, et al. Subtypes of medulloblastoma have distinct developmental origins. Nature. 2010;468:1095–1099. doi: 10.1038/nature09587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms PJ, Tu GF, Richardson SJ, Aldred AR, Jaworowski A, Schreiber G. Transthyretin (prealbumin) gene expression in choroid plexus is strongly conserved during evolution of vertebrates. Comp Biochem Physiol B. 1991;99:239–249. doi: 10.1016/0305-0491(91)90035-c. [DOI] [PubMed] [Google Scholar]
- Huang X, Ketova T, Fleming JT, Wang H, Dey SK, Litingtung Y, Chiang C. Sonic hedgehog signaling regulates a novel epithelial progenitor domain of the hindbrain choroid plexus. Development. 2009;136:2535–2543. doi: 10.1242/dev.033795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter NL, Dymecki SM. Molecularly and temporally separable lineages form the hindbrain roof plate and contribute differentially to the choroid plexus. Development. 2007;134:3449–3460. doi: 10.1242/dev.003095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RA, Wright KD, Poppleton H, Mohankumar KM, Finkelstein D, Pounds SB, Rand V, Leary SE, White E, Eden C, et al. Cross-species genomics matches driver mutations and cell compartments to model ependymoma. Nature. 2010;466:632–636. doi: 10.1038/nature09173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen MK, Bjornsson CS, Dymecki SM, Gilbertson RJ, Holtzman DM, Monuki ES. The Choroid Plexus and Cerebrospinal Fluid: Emerging Roles in Development, Disease, and Therapy. The Journal of Neuroscience. 2013;33:17553–17559. doi: 10.1523/JNEUROSCI.3258-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Solimini NL, Elledge SJ. Principles of Cancer Therapy: Oncogene and Non-oncogene Addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malkin D, Li FP, Strong LC, Fraumeni JF, Jr, Nelson CE, Kim DH, Kassel J, Gryka MA, Bischoff FZ, Tainsky MA, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science. 1990;250:1233–1238. doi: 10.1126/science.1978757. [DOI] [PubMed] [Google Scholar]
- Merino DM, Shlien A, Villani A, Pienkowska M, Mack SC, Ramaswamy V, Shih DJ, Tatevossian R, Novokmet A, Choufani S, et al. Molecular characterization of choroid plexus tumors reveals novel clinically relevant subgroups. Clinical Cancer Research. 2014 doi: 10.1158/1078-0432.CCR-14-1324. [DOI] [PubMed] [Google Scholar]
- Mills KD, Ferguson DO, Essers J, Eckersdorff M, Kanaar R, Alt FW. Rad54 and DNA Ligase IV cooperate to maintain mammalian chromatid stability. Genes & Development. 2004;18:1283–1292. doi: 10.1101/gad.1204304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morigaki R, Pooh KH, Shouno K, Taniguchi H, Endo S, Nakagawa Y. Choroid plexus papilloma in a girl with hypomelanosis of Ito. Journal of Neurosurgery: Pediatrics. 2012;10:182–185. doi: 10.3171/2012.5.PEDS11556. [DOI] [PubMed] [Google Scholar]
- Nardini M, Gnesutta N, Donati G, Gatta R, Forni C, Fossati A, Vonrhein C, Moras D, Romier C, Bolognesi M, Mantovani R. Sequence-Specific Transcription Factor NF-Y Displays Histone-like DNA Binding and H2B-like Ubiquitination. Cell. 2013;152:132–143. doi: 10.1016/j.cell.2012.11.047. [DOI] [PubMed] [Google Scholar]
- Olivier M, Goldgar DE, Sodha N, Ohgaki H, Kleihues P, Hainaut P, Eeles RA. Li-Fraumeni and related syndromes: correlation between tumor type, family structure, and TP53 genotype. Cancer Res. 2003;63:6643–6650. [PubMed] [Google Scholar]
- Parker M, Mohankumar KM, Punchihewa C, Weinlich R, Dalton JD, Li Y, Lee R, Tatevossian RG, Phoenix TN, Thiruvenkatam R, et al. C11orf95-RELA fusions drive oncogenic NF-kB signaling in ependymoma. Nature. 2014;506:451–455. doi: 10.1038/nature13109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulus W, Brandner S. Choroid plexus tumours. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, editors. WHO Classification of Tumours of the Central Nervous System. Lyon, France: IARC Press; 2007. pp. 81–85. [Google Scholar]
- Rickert CH, Wiestler OD, Paulus W. Chromosomal Imbalances in Choroid Plexus Tumors. The American Journal of Pathology. 2002;160:1105–1113. doi: 10.1016/S0002-9440(10)64931-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L, Phoenix TN, Hedlund E, Wei L, Zhu X, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012;488:43–48. doi: 10.1038/nature11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruland V, Hartung S, Kordes U, Wolff JE, Paulus W, Hasselblatt M. Choroid plexus carcinomas are characterized by complex chromosomal alterations related to patient age and prognosis. Genes, Chromosomes and Cancer. 2014;53:373–380. doi: 10.1002/gcc.22148. [DOI] [PubMed] [Google Scholar]
- Sáenz Robles MT, Symonds H, Chen J, Van Dyke T. Induction versus progression of brain tumor development: differential functions for the pRB- and p53-targeting domains of simian virus 40 T antigen. Molecular and Cellular Biology. 1994;14:2686–2698. doi: 10.1128/mcb.14.4.2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, Villegas E, Jacquemont C, Farrugia DJ, Couch FJ, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451:1116–1120. doi: 10.1038/nature06633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz KM, Schmitt N, Hoffmann-Rohrer U, Schäfer A, Grummt I, Mayer C. TAF12 Recruits Gadd45a and the Nucleotide Excision Repair Complex to the Promoter of rRNA Genes Leading to Active DNA Demethylation. Molecular Cell. 2009;33:344–353. doi: 10.1016/j.molcel.2009.01.015. [DOI] [PubMed] [Google Scholar]
- Scuoppo C, Miething C, Lindqvist L, Reyes J, Ruse C, Appelmann I, Yoon S, Krasnitz A, Teruya-Feldstein J, Pappin D, et al. A tumour suppressor network relying on the polyamine-hypusine axis. Nature. 2012;487:244–248. doi: 10.1038/nature11126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevenet N, Sheridan E, Amram D, Schneider P, Handgretinger R, Delattre O. Constitutional mutations of the hSNF5/INI1 gene predispose to a variety of cancers. Am J Hum Genet. 1999;65:1342–1348. doi: 10.1086/302639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somyajit K, Basavaraju S, Scully R, Nagaraju G. ATM- and ATR-Mediated Phosphorylation of XRCC3 Regulates DNA Double-Strand Break-Induced Checkpoint Activation and Repair. Molecular and Cellular Biology. 2013;33:1830–1844. doi: 10.1128/MCB.01521-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinat J, Bougeard G, Baert-Desurmont S, Vasseur S, Martin C, Bouvignies E, Caron O, Bressac-de Paillerets B, Berthet P, Dugast C, et al. 2009 version of the Chompret criteria for Li Fraumeni syndrome. J Clin Oncol. 2009;27:e108–109. doi: 10.1200/JCO.2009.22.7967. author reply e110. [DOI] [PubMed] [Google Scholar]
- Uziel T, Zindy F, Xie S, Lee Y, Forget A, Magdaleno S, Rehg JE, Calabrese C, Solecki D, Eberhart CG, et al. The tumor suppressors Ink4c and p53 collaborate independently with Patched to suppress medulloblastoma formation. Genes Dev. 2005;19:2656–2667. doi: 10.1101/gad.1368605. Epub 2005 Oct 2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voulgari A, Voskou S, Tora L, Davidson I, Sasazuki T, Shirasawa S, Pintzas A. TATA Box-Binding Protein–Associated Factor 12 Is Important for RAS-Induced Transformation Properties of Colorectal Cancer Cells. Molecular Cancer Research. 2008;6:1071–1083. doi: 10.1158/1541-7786.MCR-07-0375. [DOI] [PubMed] [Google Scholar]
- Wrede B, Hasselblatt M, Peters O, Thall P, Kutluk T, Moghrabi A, Mahajan A, Rutkowski S, Diez B, Wang X, et al. Atypical choroid plexus papilloma: clinical experience in the CPT-SIOP-2000 study. Journal of Neuro-Oncology. 2009;95:383–392. doi: 10.1007/s11060-009-9936-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright William D, Heyer WD. Rad54 Functions as a Heteroduplex DNA Pump Modulated by Its DNA Substrates and Rad51 during D Loop Formation. Molecular Cell. 2014;53:420–432. doi: 10.1016/j.molcel.2013.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet. 2012;44:251–253. doi: 10.1038/ng.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue W, Kitzing T, Roessler S, Zuber J, Krasnitz A, Schultz N, Revill K, Weissmueller S, Rappaport AR, Simon J, et al. A cluster of cooperating tumor-suppressor gene candidates in chromosomal deletions. Proceedings of the National Academy of Sciences. 2012;109:8212–8217. doi: 10.1073/pnas.1206062109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zender L, Xue W, Zuber J, Semighini CP, Krasnitz A, Ma B, Zender P, Kubicka S, Luk JM, Schirmacher P, et al. An Oncogenomics-Based In Vivo RNAi Screen Identifies Tumor Suppressors in Liver Cancer. Cell. 2008;135:852–864. doi: 10.1016/j.cell.2008.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481:157–163. doi: 10.1038/nature10725. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.