

Abstract
Cowden syndrome (CS) is an infrequent autosomal dominant multisystem genodermatosis, generally involving the skin, oral mucosa, thyroid, breast and gastrointestinal tract. It is characterized by a late onset in the 2nd or 3rd decade of life, an extraordinary potential for malignant transformation, especially of breast and thyroid, and an identifiable germline mutation. In 80% cases, the human tumor suppressor gene, phosphatase and tensin homolog (PTEN) is mutated; mutations involving KILLIN, SDH B/D, PIK3CA and AKT1 genes account for the rest of the cases. Its clinical signs are not only the “essential pearls” for early and accurate diagnosis of CS but also help timely detection of neoplasia as they precede development of cancer by several years. We describe the first Indian and the third world report of polydactyly with CS, review this entity highlighting on recent clinical developments and emphasize on regular and thorough screening for prompt identification and management of the potentially malignant growths. We have also designed a baseline workup routine as well as a detailed screening program for these patients.
Keywords: Genodermatosis, multiple hamartoma syndrome, phosphatase and tensin homolog, KLLN (Killin), hereditary cancer predisposition syndrome
What was known?
Cowden syndrome (CS) is a rare autosomal dominant multisystem complex genodermatosis, mostly due to PTEN mutations
It has notable malignant associations which require aggressive screening.
Introduction
Cowden syndrome (CS), documented since 1963,[1] is a rare autosomal dominant multisystem hamartomatous, cancer predisposition syndrome with incomplete penetrance, variable expressivity and PTEN (phosphatase and tensin homolog) gene mutation[2] whose mucocutaneous manifestations, virtually universal, evolve slowly and may be followed by myriad afflictions of any/many organ/systems—frequent ones being thyroid, breast, skeleton and gastrointestinal tract—[3] eventuating in the development of cancer, at least one, in 40% patients.[4] Herein, reported is a case of CS with characteristic mucocutaneous papules, goiter, syndactyly and rare association of polydactyly, documented only twice earlier.[5] Salient features and recent advances of CS are briefly reviewed and a baseline workup and surveillance routine proposed.
Case Report
A 41-year-old female presented with numerous raised solid lesions over neck, axillae, groins and forehead since 5 years. She was born with an extra digit each on right hand and left foot, the former amputated surgically two decades back. She had underwent subtotal thyroidectomy for multinodular goiter 2 years ago. Physical examination revealed pallor, macrocephaly, syndactyly (right 2nd and 3rd toes; left middle and ring fingers) and polydactyly [Figure 1] of left foot and a post-amputation stump adjacent to the right little finger. Dermatological examination revealed multiple, forehead papules [Figure 2]; skin tags over neck [Figure 3], axillae and groin; one verrucous nodule each over dorsa of left middle and right ring fingers; a midline, cervical scar (of thyroidectomy) and “cobblestone” tongue with coalesced papules [Figure 4]. Diagnosed as a case of CS she was investigated: Hemotological workup was normal except hemoglobin (9 gm%); biopsy of an acral nodule: Marked hyperkeratosis, acanthosis and papillomatosis; imaging studies (ultrasound abdomen and pelvis, chest x-ray, ECG, MRI brain and echocardiography) and certain special tests (upper GI endoscopy, colonoscopy and mammography): Normal. Testing for PTEN gene mutation was not possible. Acrochordons and facial papules were electrocauterized, acral nodules were excised and yearly surveillance was advised.
Figure 1.
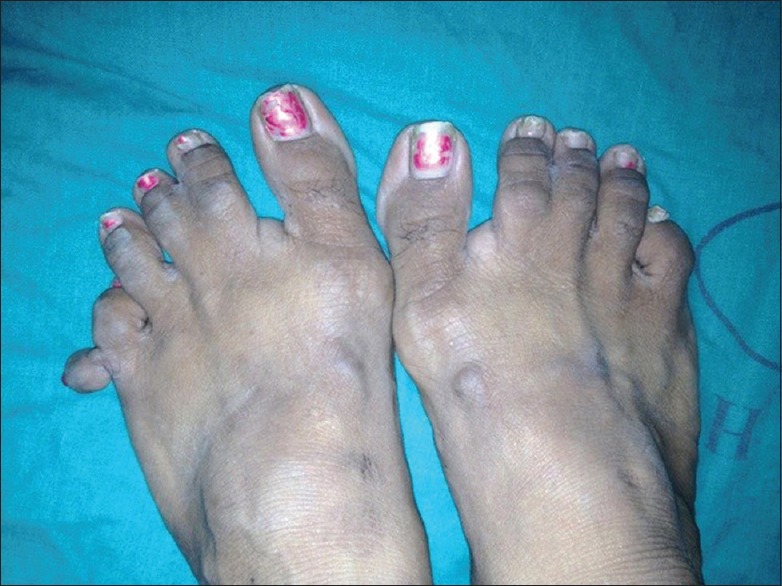
Bilateral syndactyly (2nd and 3rd toes) and unilateral polydactyly (left foot)
Figure 2.
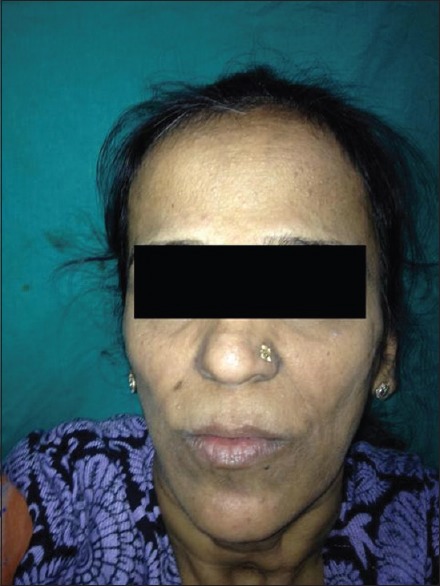
Numerous skin-colored papules over the forehead
Figure 3.
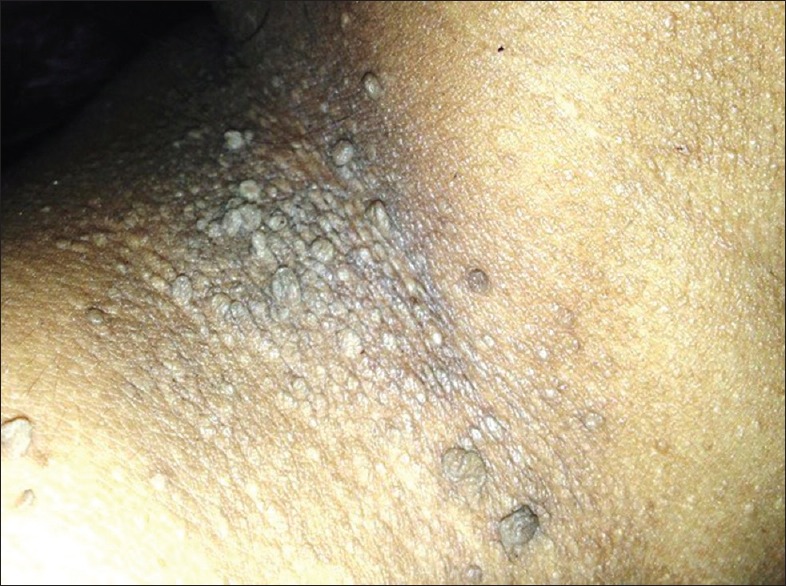
Multiple hyperpigmented skin tags over the neck
Figure 4.
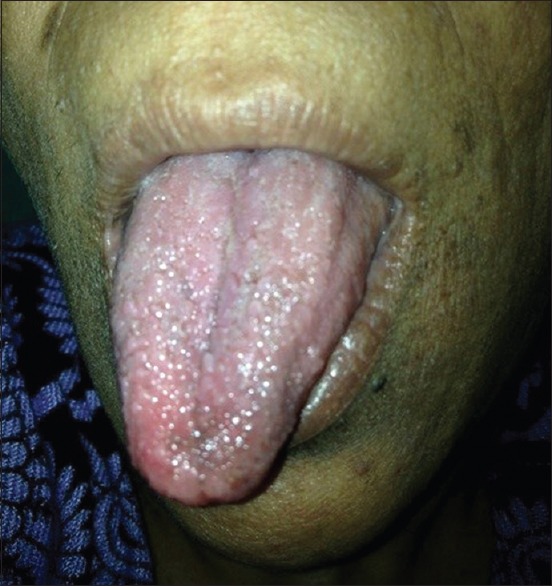
“Cobblestone appearance” of the tongue
Discussion
Though originally described by Costello, Lloyd and Dennis defined this entity, christening it after Rachel Cowden, their patient who died of breast cancer.[1]
Detectable germline mutation of PTEN tumor suppressor present on chromosome 10q23.3, and loss of function of the resultant protein causing uncontrolled cell growth via the phosphoinositol-3-kinase/AKT pathway occur in 80% of the patients who meet the diagnostic criteria for CS. Succinate dehydrogenase B/D (SDHB/D) gene is involved in ˜ 10% of patients.[6] Underexpression of PTEN or KILLIN—latter a TP53-regulated novel tumor suppressor gene, co-located with and functioning similar to the PTEN gene but transcribed in the opposite direction—genes due to loss of function, by other mechanisms such as hypermethylation, may account for the majority of remaining CS cases.[6] With the recent discovery of phosphatidylinositol 4,5-bisphosphate-3 kinase catalytic subunit alpha (PIK3CA) and AKT1 genes, the CS-susceptibility genes are presently six in number; however, PTEN, KILLIN and SDHB/D account for 92% of the cases.[6,7]
Diagnostic criteria for CS were initially proposed by Salem and Steck in 1983.[3] International Cowden Consortium (1995) recommended a revised classification to ascertain CS families, to which were added endometrial and renal cell carcinoma in 2000.[8]
The plethora of lesions, particularly mucocutaneous which evolve slowly, described ahead enjoins upon the dermatologist to suspect this syndrome and initiate timely workup and aggressive screening.[1]
Mucocutaneous manifestations, apparent in 99-100% of patients,[8] range from facial papules, customarily trichilemmomas,[2] acral keratoses (70%), pinpoint palmoplantar pits (40%)[4,9] and acrochordons (15%)[9] to subcutaneous lipomas; less characteristic dyspigmentory-, atrophic-, cystic- and nail dystrophic lesions, as also benign tumors occur.[2,3,10] Linear Cowden nevus, a non-organoid epidermal nevus characteristically associated with Type 2 segmental CS was recently (2007) established as a distinct entity.[4] Basal cell, Merkel cell and squamous cell carcinoma occur.[10] Oral lesions (in 83%)[7] generally occur as coalesced white lingual papules giving a “cobblestone appearance.”[10]
Extracutaneous manifestations occur in approximately 90%, most frequently (67%) involving thyroid gland[11] in colloid goiter, thyroglossal duct cyst, thyroiditis (3%), hypo-/hyperthyroidism, adenoma, and carcinoma (12%).[1,3,9,11]
Skeletal abnormalities manifest in one-third patients; macrocephaly, most widespread (80%) and polydactyly, unusual reported just twice previously;[5,11] others being high arched palate (15%), adenoid facies, kyphoscoliosis, bone cyst and syndactyly.[1,2]
Breast afflictions have been reported in 50-76%;[1,2,11] mostly fibroadenoma (80%) and fibrocystic disease (60%), latter usually pre-cancerous eventuated in 36% of affected females as tumors.[9] High (85%) lifetime risk of acquisition at young (mean, forty years) age and frequently bilateral (25%) occurrence of breast cancer mandates strict surveillance and prophylactic mastectomy, if required.[1,11]
Gastrointestinal (GI) endoscopy visualizes polyps (in 85%), more frequently colorectal;[10] pathologically: Predominantly hamartomatous[9,10] but with a 16% lifetime risk of acquiring colorectal carcinoma.[12]
Management can only be symptomatic or aesthetic after a high index of suspicion and thorough work up (proposed in Table 1) enables an early diagnosis of CS. Mucocutaneous lesions respond promptly to systemic acitretin but recur on discontinuation.[2] Facial papules have been tried with all modalities of treatment for warts. Future treatment options aim to restore PTEN-associated molecular pathways. An early targeting of mammalian target of rapamycin (mTOR) protein[13] and suppression of the exaggerated PI3K/Akt pathway by a timely administration of rapamycin may be salutary as per a study, completed in October 2012, by the U.S. National Cancer Institute.[2] The role of vitamin E as an anti-cancer adjunct and preventive agent in CS patients with SDH mutations has been established.[14]
Table 1.
Proposed baseline workup for Cowden syndrome

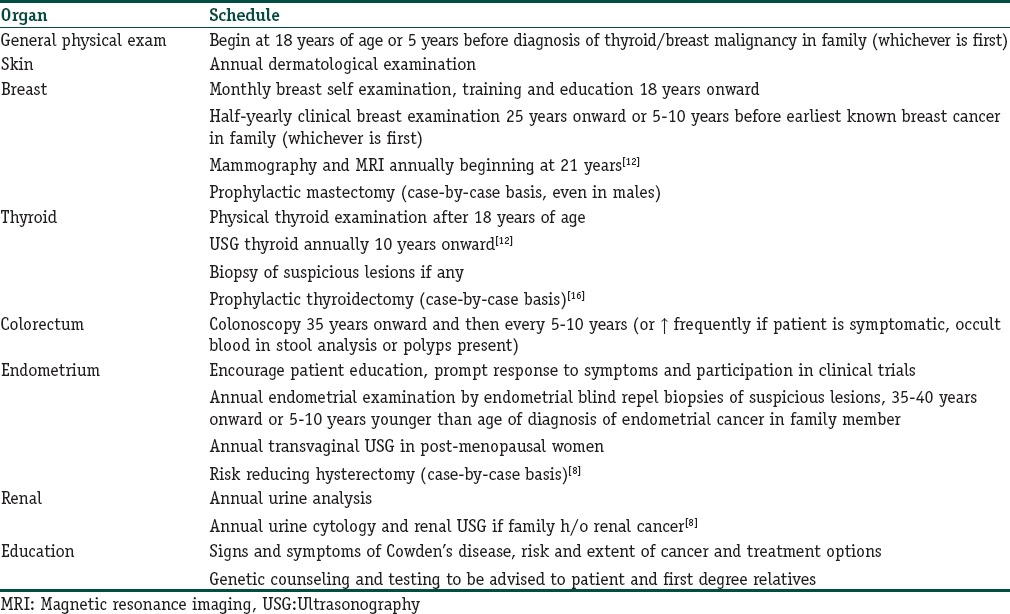
As the mucocutaneous manifestations of CS precede cancer development by several years, dermatological counseling can provide these patients with an opportunity for expert screening for cancer prevention.[3] We propose [Table 2] a screening routine adapted from the National Comprehensive Cancer Network (NCCN) guidelines (updated 2010),[15] modified by an earlier—at 21 years—commencement of annual mammography/MRI breast, USG thyroid from 10 years onward in agreement with the recommendations by Reigert-Johnson et al.,[12] and prophylactic hysterectomy[4] and thyroidectomy.[16]
Table 2.
Proposed screening routine for Cowden syndrome
Genetic testing for gene aberrations will help cancer management, e.g. patients with KILLIN hypermethylation, in whom exists higher rate of cancer breast (3-fold) and renal (>2-fold) than those with PTEN mutations, can make informed decisions to the patients about when they should start cancer surveillance and whether they should consider prophylactic surgeries.[6] Those with SDH mutations can potentially benefit by adjunctive vitamin E.[14] Despite a high incidence of sporadic mutations in CS—as also in our case—first degree relatives should also be advised genetic testing.
Conclusion
Like our patient, most of them present merely for aesthetic management of their mucocutaneous lesions providing the dermatologist with a window of opportunity. Hence, raising our suspicion, appropriate workup and aggressive screening as proposed will help arrive at a timely diagnosis, thereby facilitating successful management.
What is new?
Following the discovery of new CS-susceptibility genes, their number has been raised to six, now including PTEN, KILLIN, SDHB/D, PIK3CA and AKT1
The unfolding of novel genes in the pathogenesis of CS has lead the path to new treatment modalities like vitamin E and will help point out patients suitable for genetic counseling
Sirolimus for treatment of hamartomas in CS
Baseline workup routine for newly diagnosed CS patients
Aggressive screening routine based on recent incidences of various cancers in CS
The first Indian and the third world report of polydactyly with CS.
Footnotes
Source of support: Nil
Conflict of Interest: Nil.
References
- 1.Lee HR, Moon YS, Yeom CH, Kim KW, Chun JY, Kim HK, et al. Cowden's disease-a report on the first case in Korea and literature review. J Korean Med Sci. 1997;12:570–5. doi: 10.3346/jkms.1997.12.6.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Masmoudi A, Chermi ZM, Marrekchi S, Raida BS, Boudaya S, Mseddi M, et al. Cowden syndrome. J Dermatol Case Rep. 2011;5:8–13. doi: 10.3315/jdcr.2011.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guimaraes PB, Branco AA, Carvalho E, Lima FE, Almeida JR, Santos JB, et al. Cowden's syndrome: A new case report. An Bras Dermatol. 2002;77:711–20. [Google Scholar]
- 4.Ngan V. Cowden disease. 2004. [Last updated on 2013 Jan 14; cited 2013 Feb 28]. Available from: http://www.dermnetnz.org/systemic/cowden .
- 5.Buxbaum JD, Cai G, Chanste P, Nygren G, Goldsmith J, Reichert J, et al. Mutation screening of the PTEN gene in patients with autism spectrum disorders and macrocephaly. Am J Med Genet B Neuropsychiatr Genet. 2007;144B:484–91. doi: 10.1002/ajmg.b.30493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bennett KL, Mester J, Eng C. Germline epigenetic regulation of KILLIN in Cowden and Cowden-like syndrome. JAMA. 2010;304:2724–31. doi: 10.1001/jama.2010.1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orloff MS, He X, Peterson C, Chen F, Chen JL, Mester JL, et al. Germline PIK3CA and AKT1 Mutations in Cowden and Cowden-like Syndromes. Am J Hum Genetics. 2013;92:76–80. doi: 10.1016/j.ajhg.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eng C. Will the real Cowden syndrome please stand up: Revised diagnostic criteria. J Med Genet. 2000;37:828–30. doi: 10.1136/jmg.37.11.828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Williard W, Borgen P, Bol R, Tiwari R, Osborne M. Cowden's disease. A case report with analyses at the molecular level. Cancer. 1992;69:2969–74. doi: 10.1002/1097-0142(19920615)69:12<2969::aid-cncr2820691217>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 10.Trufant JW, Greene L, Cook DL, McKinnon W, Greenblatt M, Bosenberg MW. Colonic ganglioneuromatous polyposis and metastatic adenocarcinoma in the setting of Cowden syndrome: A case report and literature review. Hum Pathol. 2012;43:601–4. doi: 10.1016/j.humpath.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 11.Capitan Canadas LM, Salinas Sanchez JL, Martinez Castillo SL, Labrot Moleón IL, Durán Moreno D, Sánchez López D, et al. Multiple oral fibropapillomatosis as an initial manifestation of Cowden Syndrome. Case report. Med Oral Patol Oral Cir Bucal. 2006;11:E319–24. [PubMed] [Google Scholar]
- 12.Riegert-Johnson DL, Gleeson FC, Roberts M, Tholen K. Cancer and Lhermitte-Duclos disease are common in Cowden syndrome patients. Hered Cancer Clin Pract. 2010;8:6. doi: 10.1186/1897-4287-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leao JC, Batista V, Guimaraes PB, Belo J, Porter SR. Cowden's syndrome affecting the mouth, gastrointestinal, and central nervous system: A case report and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;99:569–72. doi: 10.1016/j.tripleo.2004.08.032. [DOI] [PubMed] [Google Scholar]
- 14.Ni Y, Eng C. Vitamin E protects against lipid peroxidation and rescues tumorigenic phenotypes in cowden/cowden-like patient-derived lymphoblast cells with germline SDHx variants. Clin Cancer Res. 2012;18:4954–61. doi: 10.1158/1078-0432.CCR-12-1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.National Comprehensive Cancer Network. Genetic/Familial High Risk Assessment. Breast and Ovarian v. 1. 2010. [Last accessed on 2010]. Available from: http://www.jnccn.org/content/8/5/562.full.pdf+html .
- 16.Milas M, Mester J, Metzger R, Shin J, Mitchell J, Berber E, et al. Should patients with Cowden syndrome undergo prophylactic thyroidectomy? Surgery. 2012;152:1201–10. doi: 10.1016/j.surg.2012.08.055. [DOI] [PubMed] [Google Scholar]