

Abstract
Background and Purpose
Hydrogen sulfide (H2S), an endogenous volatile mediator with pleiotropic functions, promotes vasorelaxation, exerts anti-inflammatory actions and regulates angiogenesis. Previously, the SH-containing angiotensin-converting enzyme inhibitor (ACEI), zofenopril, was identified as being effective in preserving endothelial function and inducing angiogenesis among ACEIs. Based on the H2S donor property of its active metabolite zofenoprilat, the objective of this study was to evaluate whether zofenoprilat-induced angiogenesis was due to increased H2S availability.
Experimental Approach
HUVECs were used for in vitro studies of angiogenesis, whereas the Matrigel plug assay was used for in vivo assessments.
Key Results
Zofenoprilat-treated HUVECs showed an increase in all functional features of the angiogenic process in vitro. As zofenoprilat induced the expression of CSE (cystathionine-γ-lyase) and the continuous production of H2S, CSE inhibition or silencing blocked the ability of zofenoprilat to induce angiogenesis, both in vitro and in vivo. The molecular mechanisms underlying H2S/zofenoprilat-induced angiogenesis were dependent on Akt, eNOS and ERK1/2 cascades. ATP-sensitive potassium (KATP) channels, the molecular target that mediates part of the vascular functions of H2S, were shown to be involved in the upstream activation of Akt and ERK1/2. Moreover, the up-regulation of fibroblast growth factor-2 was dependent on CSE-derived H2S response to H2S and KATP activation.
Conclusions and Implications
Zofenoprilat induced a constant production of H2S that stimulated the angiogenic process through a KATP channel/Akt/eNOS/ERK1/2 pathway. Thus, zofenopril can be considered as a pro-angiogenic drug acting through H2S release and production, useful in cardiovascular pathologies where vascular functions need to be re-established and functional angiogenesis induced.
Tables of Links
| TARGETS | |
|---|---|
| Ion channelsa | Enzymesb |
| KATP channel | Akt (PKB) |
| Cystathionine β-synthase (CBS) | |
| Cystathionine-γ-lyase (CSE) | |
| Endothelial (e) NOS | |
| ERK1 | |
| ERK2 | |
| MEK | |
| p38 MAPK | |
| PI3K |
| LIGANDS | |
|---|---|
| Captopril | Nitric oxide (NO) |
| Enalapril | Phalloidin |
| Enalaprilat | Propargylglycine (PAG) |
| FGF-2 | U0126 |
| Glibenclamide | VEGF |
| L-cysteine | Zofenopril |
| L-NAME | Zofenoprilat |
| NaHS |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a,b).
Introduction
Hydrogen sulfide (H2S) is endogenously synthesized in various tissues by two different enzymes: cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE), which use l-cysteine as a substrate (Kamoun, 2004). CSE is responsible for H2S production in the vasculature, although additional pathways are involved. H2S is increasingly recognized as a major player in several physiopathological conditions related to the cardiovascular and nervous systems, inflammation and cancer (Szabo, 2007; Szabo and Papapetropoulos, 2011; Vandiver and Snyder, 2012). Endogenous H2S is recognized as a mediator of the angiogenic response induced by VEGF (Papapetropoulos et al., 2009; Szabo and Papapetropoulos, 2011; Coletta et al., 2012). Recently, it has been demonstrated that H2S administration to cultured endothelial cells (ECs) promotes proliferation, migration and tube formation as well as in vivo angiogenesis (Papapetropoulos et al., 2009; Szabo and Papapetropoulos, 2011; Polhemus et al., 2013). This H2S-mediated pro-angiogenic effect is carried out both directly, by it acting on vascular endothelium through both Akt and MAPK (p38 and ERK1/2) activation (Cai et al., 2007; Papapetropoulos et al., 2009), and indirectly, due to the up-regulation of VEGF and its receptors associated with the down-regulation of angiogenesis inhibitors (endostatin, angiostatin and parstatin) in vascular smooth muscle cells under hypoxic conditions (Liu et al., 2010) and during myocardial infarction (Qipshidze et al., 2012).
We previously demonstrated a central role for fibroblast growth factor (FGF-2) and its receptor FGFR1 in mediating the acquisition of an angiogenic phenotype in coronary microvascular endothelium (Donnini et al., 2006) by a SH-containing ACEI (namely zofenoprilat), which has been reported to exhibit both potent antioxidant and scavenger effects, and an anti-inflammatory action (Cominacini et al., 2002). Zofenoprilat, but not other ACEI bearing (i.e. captopril) or not (i.e. enalaprilat) a SH group, was most potent and effective at promoting EC survival and ex vivo angiogenesis (Donnini et al., 2006; 2010,). In microvascular endothelium, zofenoprilat up-regulates eNOS, FGF-2 and telomerase mRNA, induces cell survival, restores damaged ECs and promotes angiogenesis (Donnini et al., 2006; 2010,). Moreover, zofenoprilat protected the coronary endothelium from doxorubicin-induced injury through the up-regulation of H2S/CSE (Monti et al., 2013).
Recently, H2S, spontaneously released by zofenoprilat (both the S-isomer and the R-isomer), has been demonstrated to account for the peripheral vascular effects of the drug zofenopril, independently of ACE inhibition (Bucci et al., 2014). The active drug metabolite behaved as a H2S donor in a cell-free environment, slowly releasing H2S into solution. Additionally, when the drug was administered in vivo and the H2S evaluation was performed ex vivo, an increased expression of CSE in vascular tissue (i.e. aorta) was registered. Moreover, increased levels of H2S were found in the plasma of SHRs, after zofenopril treatment (Bucci et al., 2014).
Here, we investigated the contribution of H2S to the pro-angiogenic effects induced by zofenoprilat in vascular endothelium and the molecular mechanisms involved. In our studies, zofenoprilat promoted angiogenesis in a H2S/CSE-dependent manner in both in vitro and in vivo models, through the activation of Akt, eNOS and ERK1/2 pathways downstream to ATP-sensitive potassium (KATP) channel opening. Moreover, zofenoprilat increased cell–cell junction organization and reduced the increase in endothelial permeability induced by VEGF, a feature dependent on CSE activity and H2S availability. Our data clearly indicate that the ACEI zofenoprilat is a pro-angiogenic drug producing functional vessels dependent on H2S release, a peculiarity that can explain its off-target protective effect, in addition to its ability to inhibit ACE and control vascular tone.
Methods
Cell cultures
HUVECs were purchased from PromoCell (Heidelberg, Germany) and were grown following the manufacturer's instructions. Cells were split 1:3 twice a week and used until passage 10.
siRNA transfection
The siRNA sequence of human CSE (ID: s3712) and the silencer negative control were from Ambion (Life Technologies, Paisley, UK) (Papapetropoulos et al., 2009; Suzuki et al., 2011). A total of 1.5 × 105 cells were seeded in 6-well plates and then transiently transfected using Lipofectamine 2000 (Life Technologies) according to the manufacturer's instructions. Cells were assayed 48 h after transfection.
H2S measurement in culture media
H2S release in the conditioned media was measured via the methylene blue assay (Stipanuk and Beck, 1982; Yang et al., 2005; Bucci et al., 2010). Confluent cells were stimulated with zofenoprilat (10 μM, 15 min, or 2, 4, 8 and 24 h) in the presence/absence of actinomycin D (5 μg·mL−1). Trichloroacetic acid (10%) was added to the supernatants collected. After protein precipitation, zinc acetate (1%) was added to the supernatants, and then N,N-dimethylphenylendiamine sulfate (20 mM) in 7.2 M HCl and FeCl3 (30 mM) in 1.2 M HCl were supplemented to the reaction mixture, and absorbance was measured after 20 min at a wavelength of 668 nm.
Adhesion assay
HUVECs were plated (4 × 104 cells per well) in 24-well plates with test substances and incubated at 37°C for 40 min. The culture medium was removed and adherent cells were fixed with methanol, stained and randomly counted at 20× magnification.
Wound assay
ECs were seeded into 24-well plates (1 × 105 cells per well) and, after confluence, were treated with test substances for 18 h. The assay was carried out as previously reported (Terzuoli et al., 2014). Results are expressed as percentage of wound area.
Proliferation assay
A total of 1 × 103 cells per well were seeded in 96 multiplates and, after adherence (3–4 h), cells, silenced or not for CSE, were treated with zofenoprilat (1–100 μM) or NaHS (6–60 μM) in the absence or presence of d-l-propargylglycine (PAG) (3 mM) or glibenclamide (10 μM). Cells were kept in culture for 5 consecutive days, and zofenoprilat or NaHS was freshly added every 2 days. In certain experiments, cells were exposed to VEGF (20 ng·mL−1) for 48 h in the absence or presence of zofenoprilat. Cells were then fixed, stained and randomly counted at 20× magnification (Donnini et al., 2006).
In vitro angiogenesis model on Matrigel
HUVECs were silenced for CSE or pretreated with PAG (3 mM, 30 min) and then treated with zofenoprilat and other ACEIs (10 μM, 18 h). Cells (7 × 104 cells per well of a 24 multi-well plate) were then plated onto a thin layer (300 μL) of basement membrane matrix (Matrigel; Becton Dickinson, Waltham, MA, USA). Quantification of the tubular structures after the staining with DY554-phalloidin (Thermo Fisher Scientific, Waltham, MA, USA) was performed after 18 h as previously published (Terzuoli et al., 2014).
Western blot
Cells (3 × 105 per 6 cm plate), at 90% of confluence, were treated with either zofenoprilat with or without the pretreatment with PAG (3 mM) or glibenclamide (10 μM) or L-NAME (200 μM) or U0126 (10 μM) or LY294002 (10 μM) for 30 min. Additionally, cells were stimulated with zofenoprilat (10 μM, 2, 4 and 24 h) in the presence/absence of actinomycin D (5 μg·mL−1). ERK1/2, Akt, eNOS-Ser1177 phosphorylation and CSE or FGF-2 expression were evaluated by Western blot (Donnini et al., 2006; Monti et al., 2010).
Measurement of permeability
Cells were seeded on collagen-coated insert membranes (Corning, Sigma-Aldrich, St. Louis, MO, USA) containing a high density of 0.4 μm-diameter pores, and the inserts were placed into a 12 multi-well plate. Cells were seeded at 8 × 104 cells per insert and cultured for 72 h. Confluent monolayers were pretreated for 18 h with zofenoprilat and then VEGF (20 ng·mL−1, 8 h) was added where indicated. FITC-dextran, 40 kDa, (10 μM) was used as fluorescent marker of permeability, which was evaluated after 20 min by measuring the fluorescence in a plate reader (Tecan, Mannedorf, Switzerland), at 485 and 535 nm excitation and emission respectively (Terzuoli et al., 2014).
Immunofluorescence analysis
Vascular endothelial cadherin (VE-cadherin) and zonula occludens-1 (ZO-1) protein, expressed at the cell surface, were visualized by confocal analysis. Cells, 5 × 104, were seeded onto 1 cm round glass coverslips. After 24 h, cells were washed and treated with the indicated stimuli. Immunofluorescence analysis was performed as previously reported (Monti et al., 2014; Terzuoli et al., 2014).
In vivo Matrigel angiogenesis assay
In vivo Matrigel angiogenesis assay was performed as previously described (Terzuoli et al., 2014). C57 male black mice (20–25 g, 25 animals) were kept in temperature- and humidity-controlled rooms (22°C, 50%) with lights on from 07:00 to 19:00 h, and water and food available ad libitum. Mice were s.c. injected in the dorsal midline region with 0.4 mL of Matrigel (Becton Dickinson, Franklin Lakes, NJ, USA) alone or with stimuli. After 15 days, mice were killed by CO2 inhalation and implants harvested. Plugs were re-suspended in 1 mL of Drabkin's reagent (Sigma-Aldrich) for 18 h on ice, and haemoglobin concentration was determined by absorbance at 540 nm and compared with a standard curve (Sigma-Aldrich).
Materials and reagents
Cell culture reagents, NaHS, PAG, L-NAME, glibenclamide and N,N-dimethylphenylendiamine sulfate, FeCl3, zinc acetate and actinomycin D were from Sigma-Aldrich, while U0126 and LY294002 were from Calbiochem (Milan, Italy). FBS was from Hyclone (Euroclone, Milan, Italy). Hoechst 33342, 40 kDa FITC-dextran, Lipofectamine 2000, scramble and CSE silencing sequences were from Life Technologies (Carlsbad, CA, USA). Trichloroacetic acid was purchased from Carlo Erba (Arese, Milan, Italy).
Anti-phospho-Akt (Ser473), anti-Akt, anti-phospho-ERK1/2, anti-ERK1/2 and anti-phospho-eNOS (Ser1177) antibodies were obtained from Cell Signaling (Celbio, Milan, Italy). Anti-CSE and anti-β-actin were from Sigma-Aldrich. Anti-FGF-2 antibody was from Upstate (Millipore, Vimodrone, Milan, Italy). Anti-ZO-1 was from BD Transduction Laboratories (Milan, Italy). Anti-VE-cadherin was from E-Bioscience (San Diego, CA, USA). VEGF was from R&D Systems (Milan, Italy). Zofenoprilat, captopril, enalapril and R-isomer were kindly provided by Menarini Group (Florence, Italy).
Animal welfare and ethical statements
All procedures were carried out in accordance with the Italian law (No. 116, 27 January 1992), which reflects the European Directive 2010/63/UE, were approved by the University of Siena Ethical Board and the Italian Ministry of Health. All efforts were made to minimize the number of animals used and their suffering. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Data analysis and statistical procedures
Results are either representative or average of at least three independent experiments performed in triplicate. Statistical analysis was performed using anova test, followed by Bonferroni's test and Student's t-test when appropriate (GraphPad, GraphPad Software, La Jolla, CA, USA). P < 0.05 was considered statistically significant.
Results
Zofenoprilat promotes angiogenesis in human ECs
As has been previously demonstrated, zofenoprilat induced angiogenesis in coronary post-capillary ECs (CVEC; Donnini et al., 2006). Exposure of HUVECs to increasing concentrations (1–100 μM) of zofenoprilat, consistent with that achieved in vivo (Evangelista and Manzini, 2005), promoted cell adhesion, migration and growth, with a maximal effect at 10 μM (Figure 1A–C). Analogue effects were obtained when an exogenous source of H2S was added, administered to the cells as NaHS (Figure 1D–F). However, VEGF, in the same experimental conditions, was more effective (Figure 1D–F). Similar data, in the proliferation assay, were obtained with the R-isomer, the inactive diastereoisomer of zofenoprilat on ACE activity (Supporting Information Fig. S1A). In the light of these results, we selected 10 μM as the concentration of zofenoprilat for the next experiments.
Figure 1.
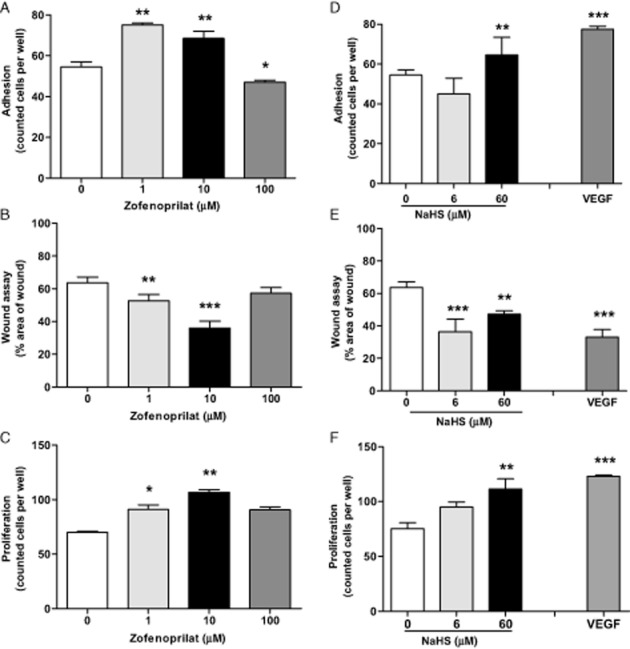
Effect of zofenoprilat and NaHS on EC adhesion, migration and proliferation. Evaluation of adhesion (40 min) (A and D), migration (18 h) (B and E) and proliferation (5 days) (C and F) in HUVECs in the presence of zofenoprilat (1–100 μM) (A–C) or NaHS (6–60 μM) (D–F). Data in panels (A), (C), (D) and (F) represent counted cells per well. Data in panels (B) and (E) are % area of wound, representing the complement of healing/migration at 18 h with respect to time 0. VEGF (20 ng·mL−1) was used as a control. n = 3. *P < 0.05, **P < 0.01, ***P < 0.001 versus untreated cells.
CSE-dependent H2S production mediates zofenoprilat-induced in vitro angiogenic activity
We evaluated the expression of CSE in HUVECs, and the ability of zofenoprilat or its R-isomer to modulate its expression was also assessed. As shown in Figure 2A and Supporting Information Fig. S1B, the endothelium constitutively expressed CSE, and incubation with zofenoprilat further increased it in a time-dependent manner, with a 3- to 3.5-fold increase versus control at 18 and 24 h respectively (Figure 2A). Enhanced CSE expression was confirmed when the R-isomer was used (Supporting Information Fig. S1B). As reported by Monti et al. (2013), captopril did not reproduce this effect (0.125-fold increase vs. control). Therefore, as zofenoprilat has been shown to have spontaneous H2S donor activity (Bucci et al., 2014), exposure of HUVECs to zofenoprilat resulted in the accumulation of H2S in the culture medium after 15 min, which was maintained for up to 8 h and further increased at 24 h (Figure 2B).
Figure 2.

Zofenoprilat increases CSE protein expression and H2S production. Expression of CSE (A) in HUVECs in control condition and after incubation with zofenoprilat (10 μM) for the indicated times. The blot is representative of three with similar results. The ratio between CSE and actin is presented below the blot. **P < 0.01, ***P < 0.001 compared with untreated cells. Production of H2S (B) was determined in control ECs and in response to zofenoprilat (10 μM) stimulation for 15 min, 2, 4, 8 and 24 h. n = 3. **P < 0.01 versus untreated cells. (C) Expression of CSE in HUVECs in control (Ctr) condition and in response to zofenoprilat (10 μM) in the presence and absence of actinomycin D (5 μg·mL−1) for 2 (a), 4 (b) and 18 (c) h. The blot is representative of three with similar results. The ratio between CSE and actin is presented below the blot. ***P < 0.001 compared with untreated cells. ###P < 0.01 versus zofenoprilat-treated cells. (D) Production of H2S was determined in response to zofenoprilat stimulation (10 μM) in the presence and absence of actinomycin D (5 μg·mL−1) for 2 (a), 8 (b) and 24 (c) h. n = 3. **P < 0.01, ***P < 0.001 versus untreated cells; #P < 0.05, ##P < 0.01 versus zofenoprilat-treated cells.
Moreover, to determine the importance of the modulation of CSE expression in acute and chronic treatment, measurement of H2S in the presence of zofenoprilat was performed in the presence and absence of the transcriptional inhibitor actinomycin D at different time points from 2 to 24 h. As shown in Figure 2C and D, expression of CSE and accumulation of H2S by zofenoprilat was reduced in a time-dependent manner in the presence of actinomycin D at 4 and 18 h and at 8 and 24 h respectively. These data document the continuous availability of H2S resulting from zofenoprilat initially behaving as a H2S donor and later through an up-regulation of CSE.
In the next experiments, the biosynthesis of H2S was prevented through the pharmacological inhibition of CSE by PAG or CSE silencing (Papapetropoulos et al., 2009). The pre-incubation of HUVECs with PAG (3 mM) completely impaired zofenoprilat-induced angiogenic effects (adhesion, migration and proliferation) (Figure 3A–C). Also, the activity of the R-isomer of zofenoprilat on cell proliferation was dependent on H2S formation (Supporting Information Fig. S1A). In addition, the anti-angiogenic effect of PAG on zofenoprilat-mediated angiogenesis was evident in a three-dimensional differentiation model. When plated in a thin layer of Matrigel and stimulated with zofenoprilat, HUVECs assembled into a network of capillary-like tubes (Figure 3D, panel b vs. panel a, and panel e for quantification). Pretreatment of ECs with PAG completely inhibited all these effects (Figure 3D). Similar effects were observed with the R-isomer of zofenoprilat (Supporting Information Fig. S1C). The effects of other ACEIs, such as captopril and enalaprilat (10 μM), on the main angiogenic function (proliferation and tube formation) were also evaluated. Both compounds were less effective at inducing angiogenic features than zofenoprilat (Supporting Information Fig. S2A and B). Of note, the SH-containing ACEI captopril partially acted through CSE-derived H2S, as shown from the pretreatment with PAG (Supporting Information Fig. S2A and B).
Figure 3.

Role of H2S production on in vitro angiogenesis in response to zofenoprilat. ECs were incubated with zofenoprilat (10 μM, 40 min or 5 days, respectively), pretreated or not with PAG (3 mM, 30 min), and adhesion (A) or proliferation (B) were assessed. Data represent counted cells per well. n = 3. *P < 0.05, **P < 0.01 versus untreated cells. #P < 0.05, ##P < 0.01 versus zofenoprilat-treated cells. Cell migration (C) was evaluated using scratch wound healing assay on ECs treated with 0.1% FBS (a), zofenoprilat (10 μM, 18 h, b), PAG (3 mM, 30 min pretreatment, c) or zofenoprilat with PAG (d). Representative pictures of wounded EC monolayers are shown. n = 3. Quantification (e) of cell migration presented as % area of wound. ***P < 0.001 versus untreated cells; ##P < 0.01 versus zofenoprilat. (D) Pseudocapillary formation in Matrigel by ECs exposed to 0.1% FBS (a), zofenoprilat (10 μM, b), PAG (3 mM, 30 min pretreatment, c) and PAG + zofenoprilat (d) after 18 h. Cells were labelled with DY554-phalloidin, and quantification (e) of pseudocapillaries was performed by counting the numbers of complete circles per well. n = 3. **P < 0.01 versus untreated cells; ##P < 0.01 versus zofenoprilat.
To corroborate the role of endogenous H2S on angiogenesis, we next used a siRNA approach to selectively impair H2S production in ECs. The CSE mRNA silencing blunted CSE protein expression (Figure 4A), leading to an attenuation of the proliferation (Figure 4B) and capillary-like formation in Matrigel (Figure 4C, panel d vs. panel b) in response to zofenoprilat treatment.
Figure 4.
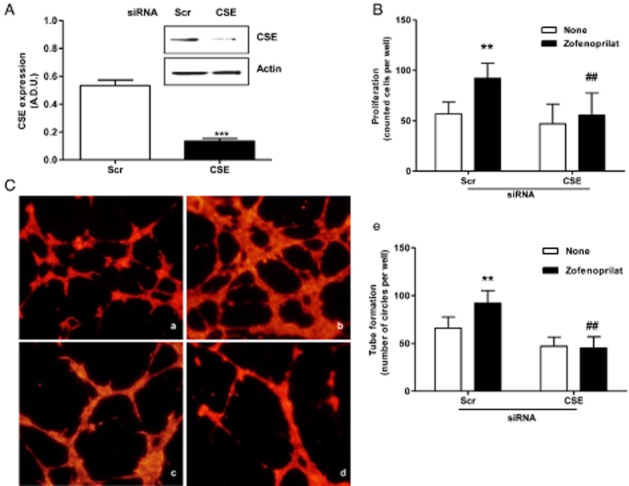
Effect of CSE silencing on in vitro angiogenesis induced by zofenoprilat. (A) Western blot analysis of CSE expression in ECs silenced for CSE and treated for 24 h with zofenoprilat (10 μM). The ratio between CSE and actin is presented. ***P < 0.001 compared with scramble (Scr) cells. (B) Proliferation of ECs transfected with Scr or CSE siRNA and stimulated with zofenoprilat. Data are expressed as cell number counted per well. **P < 0.01 versus Scr siRNA-untreated cells; ##P < 0.01 versus Scr siRNA cells treated with zofenoprilat. (C) Network formation of ECs seeded on Matrigel after transfection with Src siRNA in 0.1% FBS (a), or plus zofenoprilat (b), or with CSE siRNA in 0.1% FBS (c), or plus zofenoprilat (d) for 8 h. Cells were labelled with DY554-phalloidin, and quantification (e) of pseudocapillaries was performed by counting the number of complete circles per well. n = 3. **P < 0.01 versus Scr siRNA-untreated cells; ##P < 0.01 versus Scr siRNA zofenoprilat-treated cells.
These results demonstrate that endothelial CSE contributes to H2S release from zofenoprilat-treated endothelium and that zofenoprilat-derived H2S is necessary to stimulate angiogenesis.
Effect of zofenoprilat-derived H2S on the activation of angiogenic signalling pathways
In coronary post-capillary venules, it has been demonstrated that zofenoprilat acts through Akt/eNOS/ERK1/2/FGF-2 activation (Donnini et al., 2006; 2010,). Here, we confirmed the activation of this pathway by zofenoprilat in HUVECs. In particular, zofenoprilat was able to induce, in a time-dependent manner, the phosphorylation of Akt, eNOS (Ser1177) and ERK1/2, with a maximal effect at 15 min, while there was no activation of p38 (data not shown).
Pre-incubation with PAG (3 mM, 30 min) completely inhibited the activation of both phospho-Akt and ERK1/2 (Figure 5A), demonstrating the crucial role of zofenoprilat-induced released of H2S in the promotion of these early signals. Accordingly, the genetic ablation of endogenous CSE protein significantly inhibited Akt and ERK1/2 phosphorylation (Figure 5B).
Figure 5.
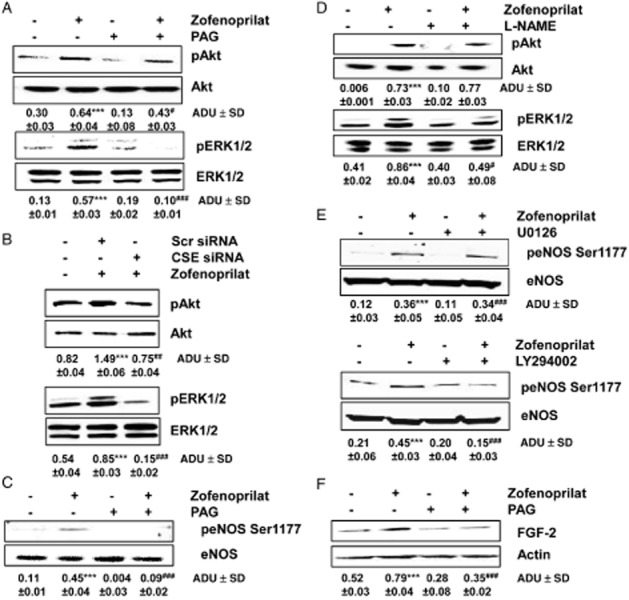
Zofenoprilat-derived H2S activates signalling pathways associated with angiogenesis. (A and B) Western blot analysis of phosphorylated Akt and ERK1/2 in ECs treated for 15 min with zofenoprilat (10 μM), pretreated or not with PAG (3 mM, 30 min) (A) or transfected with Src or CSE siRNA (B). n = 3. The ratio between pAKT or pERK1/2 and AKT or ERK1/2, respectively, is presented below the blots. ***P < 0.001 compared with untreated cells; #P < 0.05, ##P < 0.01, ###P < 0.001 compared with zofenoprilat-treated cells. (C) Western blot analysis of phosphorylated eNOS in Ser1177 in ECs treated for 15 min, with zofenoprilat (10 μM), pretreated or not with PAG (3 mM, 30 min). The ratio between peNOS Ser1177 and eNOS is reported. ***P < 0.001 compared with untreated cells; ###P < 0.001 compared with zofenoprilat-treated cells. (D) Akt and ERK1/2 phosphorylation evaluated in cells pretreated with L-NAME (200 μM, 30 min) and then stimulated with zofenoprilat (10 μM, 15 min). Blots are representative of three with overlapping results. The ratio between pAKT or pERK1/2 and AKT or ERK1/2, respectively, is presented below the blots. ***P < 0.001 compared with untreated cells; #P < 0.05 compared with zofenoprilat-treated cells. (E) Western blot analysis of phosphorylated eNOS in Ser1177 in ECs treated for 15 min with zofenoprilat (10 μM), pretreated or not with U0126 (10 μM, 30 min, top blot) or LY294002 (10 μM, 30 min, bottom blot). The ratio between peNOS Ser1177 and eNOS is presented below the blot. ***P < 0.001 compared with untreated cells; ###P < 0.001 compared with zofenoprilat-treated cells. (F) Western blot analysis of FGF-2 expression in ECs treated for 18 h with zofenoprilat (10 μM), pretreated or not with PAG (3 mM, 30 min). The ratio between FGF-2 and actin is presented below the blot. ***P < 0.001 compared with untreated cells; ###P < 0.001 compared with zofenoprilat-treated cells.
As previously demonstrated in CVEC (Donnini et al., 2006; 2010,), zofenoprilat promoted activation of the Akt-dependent eNOS pathway in HUVECs. eNOS phosphorylation induced by zofenoprilat was under the control of CSE activity (Figure 5C). Then, eNOS activation governed ERK1/2, but not Akt phosphorylation, as demonstrated by the treatment with L-NAME (200 μM, Figure 5D). This signalling pathway was confirmed by the finding that pretreatment of cells with U0126 (10 μM), a MEK inhibitor, did not modify eNOS Ser1177 phosphorylation (Figure 5E). Conversely, pretreatment with LY294002 (10 μM), a PI3K inhibitor, significantly reduced eNOS Ser1177 phosphorylation induced by zofenoprilat (Figure 5E).
Finally, zofenoprilat, as previously demonstrated in microvascular endothelium (Donnini et al., 2010), induced an up-regulation FGF-2, which we demonstrated here to be under the control of CSE/H2S availability (Figure 5F).
Since the vascular functions of H2S have been associated with the ATP-dependent K+ channel (Zhao et al., 2001; Papapetropoulos et al., 2009), its role in the occurrence of angiogenesis was investigated. HUVECs, expressing KATP channels (Papapetropoulos et al., 2009), were pretreated for 30 min with glibenclamide (10 μM), and Akt and ERK1/2 phosphorylation was evaluated by Western blot. Figure 6A shows that the pharmacological block of KATP channels partially impaired Akt and ERK1/2 activation, suggesting the involvement of this transport system in H2S-induced actions. Furthermore, the involvement of KATP in FGF-2 expression was evaluated and the results showed that this endogenous growth factor is also under the partial control of this channel (Figure 6B). From a functional point of view, blockade of KATP channels in ECs by glibenclamide reduced the proliferation of ECs induced by zofenoprilat as well as NaHS (Figure 6C).
Figure 6.
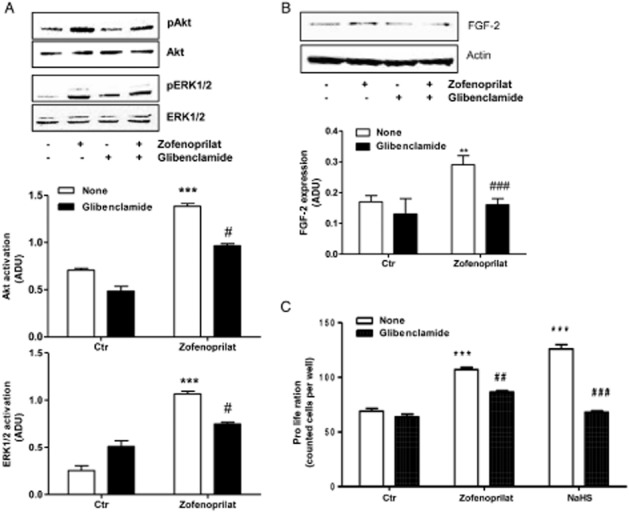
KATP channels are involved in signalling pathways and proliferation induced by zofenoprilat. (A) Western blot analysis of phosphorylated Akt and ERK1/2 in HUVECs treated for 15 min with zofenoprilat (10 μM) pretreated or not with glibenclamide (10 μM, 30 min). n = 3. The ratio between pAKT or pERK1/2 and AKT or ERK1/2, respectively, is presented. ***P < 0.001 compared with untreated cells; #P < 0.05 compared with zofenoprilat-treated cells. (B) Western blot analysis of FGF-2 in HUVECs treated for 18 h with zofenoprilat (10 μM) pretreated or not with glibenclamide (10 μM, 30 min). n = 3. The ratio between FGF-2 and actin is presented. **P < 0.01 compared with untreated cells; ###P < 0.001 compared with zofenoprilat-treated cells. (C) Proliferation of ECs treated with zofenoprilat (10 μM) or NaHS pretreated or not with glibenclamide (10 μM, 30 min). n = 3. ***P < 0.001 versus control (Ctr); ##P < 0.01, ###P < 0.001 versus zofenoprilat or H2S-treated cells.
All these data suggest that zofenoprilat induces the release of H2S, which, in turn, activates KATP channels, which, in part, control Akt and ERK1/2 phosphorylation, FGF-2 expression and endothelial angiogenic behaviour.
Zofenoprilat, via CSE activation, promotes in vivo angiogenesis and normalizes endothelial monolayers
To further study the H2S-mediated angiogenic activity of zofenoprilat in vivo, we used the Matrigel implant assay in mice. Matrigel plugs, either with zofenoprilat alone or in the presence of PAG, were s.c. injected in C57 black mice and harvested after 15 days. Implants containing zofenoprilat showed several branched structures throughout the implant (Figure 7A, panel b vs panel a). Conversely, in implants in which zofenoprilat and PAG were concomitantly administered, total vascular volume was markedly reduced (Figure 7A, panel d vs. panel b). Quantitative analysis of functioning vessels by haemoglobin determination revealed that zofenoprilat produced a 2.3-fold increase in blood content compared with control, whereas administration of PAG reduced it by 42% (Figure 7A, panel e).
Figure 7.

Zofenoprilat/H2S promotes in vivo angiogenesis and controls endothelial hyperpermeability. (A) Representative plugs isolated after 15 days from implant of Matrigel containing the following stimuli: none (a), 10 μM zofenoprilat (b), 3 mM PAG (c), zofenoprilat + PAG (d). (e) Quantitative analysis of haemoglobin/angiogenesis in implants. For each condition (n = 6), the means ± SD are shown. **P < 0.01 versus untreated plugs; ##P < 0.01 versus zofenoprilat-treated plugs. (B) Cell permeability was evaluated in HUVECs pretreated overnight with zofenoprilat (10 μM) or PAG (3 mM), where indicated, and then treated with VEGF (20 ng·mL−1, 8 h). Results are expressed as fluorescent units ± SD. ***P < 0.001 versus untreated cells; #P < 0.05, ###P < 0.001 versus VEGF; §P < 0.01 versus VEGF + zofenoprilat. (C and D) Immunofluorescence analysis of VE-cadherin (C) and ZO-1 protein (D) in confluent HUVECs treated with the following stimuli: none (a), 20 ng·mL−1 VEGF (b), 10 μM zofenoprilat (18 h) (c), zofenoprilat (18 h) + VEGF (8 h) (d), pretreatment for 30 min with 3 mM PAG + zofenoprilat (18 h) + VEGF for 8 h (e). Confocal images were taken at 60× magnification. Scale bar = 10 μm.
However, when designing a pro-angiogenic strategy, the quality and the morphology of newly formed vessels are crucial parameters as well as the interaction of the stimulus with known angiogenic factors. To pursue this aim, we performed a proliferation assay to evaluate whether zofenoprilat could interfere with VEGF. Exposing ECs treated with zofenoprilat (10 μM, 48 h) to a maximal concentration of VEGF, expected to be found in an angiogenic environment (20 ng·mL−1), ) did not enhance the pro-angiogenic response (Table 1).
Table 1.
Effect of zofenoprilat on the angiogenic activation of endothelium by VEGF
| VEGF alone | +Zofenoprilat | |
|---|---|---|
| Proliferation | 50 ± 8 | 52 ± 4 |
HUVECs were exposed to 20 ng·mL−1 VEGF in the absence or presence of 10 μM zofenoprilat for 48 h. Proliferation data are reported as % increase over control response, with 100% representing the cell number counted in the basal condition (n = 3).
Since VEGF stimulated vasculature is characterized by elevated permeability and leakage (Ferrara et al., 2003), there is a strong demand for pharmacological strategies that are able to produce functional angiogenesis (Cai et al., 2011; Anisimov et al., 2013). In our experiments, zofenoprilat inhibited VEGF-induced permeability in a H2S-dependent manner (Figure 7B), indicating a potential beneficial role in the normalization of vasculature. To confirm the capability of zofenoprilat to reduce VEGF-mediated permeability, VE-cadherin and ZO-1 protein expression and localization at the cell–cell junction were evaluated in vitro. As shown in Figure 7C and D (panels a), confluent HUVEC monolayers expressed VE-cadherin and ZO-1 protein mainly at the cell–cell contact. In the presence of VEGF, the contacts disappeared (panels b), while VE-cadherin and ZO-1 labelling was restored by zofenoprilat (panels d of Figure 7C and D). The protective effect of zofenoprilat was related to the availability of CSE-dependent H2S, since PAG pre-incubation reversed the protective effect of zofenoprilat, inducing cell retraction and a reduction in VE-cadherin and ZO-1 protein labelling (panels e of Figure 7C and D).
These data demonstrate that zofenoprilat has pro-angiogenic activity mediated through the production of H2S, which controls all the signalling pathways and functional responses of the angiogenic vascular endothelium to produce functional and normalized vessels.
Discussion and conclusions
In the last decade, our attention has been focused on a clinically available highly lipophilic ACEI, zofenopril, whose active metabolite zofenoprilat bears a SH group and displays the unique properties, among other ACEIs, of promoting EC survival and angiogenesis, beyond its vasoactive properties (Donnini et al., 2006; 2010,; Monti et al., 2013). Recently, it has been demonstrated that zofenoprilat, the active metabolite of zofenopril, spontaneously releases H2S in solution, and that in an in vivo pathological model, such as the SHR, zofenopril increases H2S levels in the vascular tissue and plasma (Bucci et al., 2014).
The increased availability of H2S, induced by zofenoprilat, prompted us to analyse the involvement of this gaseous transmitter in the pro-angiogenic and protective effect of this ACEI on endothelium using in vitro and in vivo experimental models associated with angiogenesis. In HUVECs, all the functional activities related to angiogenesis in response to zofenoprilat were analysed. HUVECs exposed to zofenoprilat showed an increase in cell adhesion, migration and proliferation, with maximal effects at 10 μM, as previously demonstrated in coronary post-capillary endothelium (Donnini et al., 2006; 2010,). H2S, given exogenously as NaHS, reproduced all the effects seen with zofenoprilat. The zofenoprilat R-isomer, a molecule that maintains the SH group but is ineffective as an ACEI, replicated these pro-angiogenic effects. This last finding supports the hypothesis that the angiogenic effect induced by zofenoprilat is dependent upon the presence of the SH group in its structure and its ability to release H2S.
Experiments were carried out to further dissect the relationship between zofenoprilat-released H2S and the biosynthetic pathway of this gaseous transmitter. The release of H2S from vascular endothelium mainly involves CSE (Kamoun, 2004; Kimura, 2010). HUVECs constitutively express CSE, which is further up-regulated by zofenoprilat and its R-isomer, but not by captopril, as previously demonstrated in CVEC (Monti et al., 2013). Moreover, experiments with the transcriptional inhibitor actinomycin D showed how the accumulation of H2S by zofenoprilat is related to gene transcription as it is reduced in the presence of actinomycin D.
Another H2S donor drug has also been reported to have this peculiar property (Ma et al., 2011). Furthermore, it has recently been demonstrated that exogenous H2S significantly increases the transcription and expression of CSE (Wang et al., 2013). Zofenoprilat reacts with l-cysteine, the endogenous substrate of CSE, to form adducts that are substrates of CSE (Subissi et al., 1999). Thus, zofenoprilat acts not only as a H2S donor but also as a substrate for CSE to produce H2S, and is responsible for CSE up-regulation in endothelium, as also recently demonstrated by Bucci et al. (2012,2014). So, the release of H2S and the peculiar structure of zofenoprilat create a tonic release of the gaseous transmitter, an effect sustained by the up-regulation of CSE. This mechanism maintains high levels of H2S over time (15 min–24 h), justifying its involvement in the complex angiogenic process. Nevertheless, the exact mechanism for the up-regulation of CSE remains to be elucidated.
By inhibiting or ablating CSE, all the functional responses induced by zofenoprilat in HUVECs are impaired. EC proliferation, migration and organization of networks are indeed blocked, as well as all the signalling pathways associated with them. These findings strongly support the notion that the off-target actions of this drug are exerted by the SH moiety directly on vascular endothelium.
It is now clear that exogenous and endogenous H2S, which can be induced by VEGF, is a stimulator of the angiogenic responses in cultured endothelium and in different angiogenic models in vivo. From a mechanistic point of view, previous reports demonstrated that H2S stimulates Akt, ERK1/2 and p38 in ECs and induces eNOS activation through phosphorylation of Ser1177 and dephosphorylation of Thr495 (Cai et al., 2007; Osipov et al., 2009; Papapetropoulos et al., 2009; Coletta et al., 2012). In HUVECs and in rat aortic vasorelaxation, the two gaseous transmitters H2S and NO cooperate in the fine control of vascular functions (Bucci et al., 2012; 2014,; Coletta et al., 2012). In our experiments, the molecular mechanisms underlying the H2S/zofenoprilat-induced angiogenic phenotype in HUVECs are dependent on Akt, eNOS-Ser1177 and ERK1/2. These results confirm data previously obtained in CVEC (Donnini et al., 2006; 2010,), indicating that H2S released by zofenoprilat is responsible for the sequential activation of the Akt/eNOS/ERK1/2 signalling pathway, which, in turn, controls the endogenous up-regulation of FGF-2.
All the signalling cascades involved in the effect of zofenoprilat seem to be dependent on activation of CSE, since silencing or pharmacological blockade of CSE blunts the activation of Akt, eNOS-Ser1177 and ERK1/2 and expression of FGF-2. Some of the vascular functions of H2S have been attributed to the opening of the KATP channel (Zhao et al., 2001; Zanardo et al., 2006; Li et al., 2011). ECs express KATP channels both in the plasma membrane and in the intracellular organelles (Katnik and Adams, 1997; Malester et al., 2007). The influence of KATP channels on the angiogenic effect of H2S is evident from the data showing that glibenclamide abolishes p38 activation and EC mobility (Papapetropoulos et al., 2009; Szabo and Papapetropoulos, 2011). In our model, KATP channels were shown to be partially involved in the upstream activation of Akt and ERK1/2 and the expression of FGF-2, including the sulfhydration modification of cysteine residues in the target downstream proteins, demonstrated to enhance their activity (Mustafa et al., 2009; Yang et al., 2013).
In addition to these findings, zofenoprilat was found to promote neovascularization of Matrigel plugs s.c. implanted in mice. This effect was dependent on a functioning CSE since the presence of PAG impaired the occurrence of angiogenesis. The stimulation of an angiogenic response might, however, acquire a negative feature when it is induced within the tumour environment or in ischaemic diseases, resulting in disorganized and highly permeable vessels (Jain, 2003; Cai et al., 2011; Anisimov et al., 2013). We thus aimed to ascertain whether zofenoprilat could cooperate or synergize with angiogenic factors such as VEGF and how it could correct endothelial hyperpermeability. Zofenoprilat did not potentiate the EC responses in the presence of VEGF; the concentration of VEGF used was as high as that usually found in an angiogenic environment. On the contrary, in the presence of VEGF, zofenoprilat restored endothelial permeability to basal levels. Inhibition of endothelial leakage (measured as permeability induced by VEGF and expression/localization of VE-cadherin and ZO-1 protein) is thus an additional positive feature of zofenoprilat's effect in the intervention against cardiovascular diseases. All the related vessel normalization parameters were dependent on H2S production, but the exact molecular mechanisms that control the restoration of transendothelial junctions remain to be elucidated. Recently, H2S has been demonstrated to exert a protective effect on lung vascular hyperpermeability by scavenging reactive oxygen species and activation of Akt (Wang et al., 2012); these mechanisms cannot be excluded in our model.
In conclusion, H2S released from zofenoprilat stimulates all the angiogenic properties of ECs through a KATP channel/Akt/eNOS/ERK1/2/FGF-2 pathway. Moreover, zofenopril/zofenoprilat induced CSE to generate H2S, a property exclusive to this drug with respect to other SH-containing ACEI. This extra beneficial effect, beyond ACE inhibition, could be helpful in cardiovascular pathologies where EC functions and integrity need to be re-established and functional angiogenesis induced.
Acknowledgments
This work was financed by Menarini Group and by the COST Action BM1005 (ENOG: European Network on Gasotransmitters). Part of this work was funded by Agenzia Spaziale Italiana (ASI) and Istituto Toscano Tumori (ITT). M. M. was a Noxamet srl fellow. E. T. was a FIRC (Fondazione Italiana per la Ricerca sul Cancro) fellow.
We are grateful to Dr Cinzia Della Giovampaola for the support at confocal microscopy and Dr Valerio Ciccone for technical assistance.
Glossary
- ACEI
angiotensin-converting enzyme inhibitor
- CSE
cystathionine-γ-lyase
- ECs
endothelial cells
- FGF-2
fibroblast growth factor-2
- KATP
ATP-sensitive potassium
- PAG
d-l-propargylglycine
Author contributions
E. T. and M. M. performed the research. E. T., M. M., M. Z. and L. M. designed the research study. V. V., M. B. and G. C. contributed essential reagents or tools. E. T., M. M. and L. M. analysed the data. E. T., M. M., M. Z., M. B. and L. M. wrote the paper.
Conflict of interest
The authors declare no conflict of interest.
Supporting Information
Figure S1 Effect of R-isomer of zofenoprilat on endothelial cell functionality. (A) Cell proliferation evaluated in HUVECs treated with zofenoprilat R-isomer (10 μM, 5 days) in presence/absence of PAG (3 mM, 30 min). Data are expressed as counted cells/well. n = 3; **P < 0.01 versus untreated cells and #P < 0.05 versus R-isomer. (B) CSE expression was evaluated in HUVECs treated with R-isomer (10 μM) for indicated times. The blot is representative of 3 with similar results. The ratio between CSE over actin is reported. ***P < 0.001, compared to untreated cells. (C) Pseudocapillary formation in Matrigel by ECs exposed to 0.1% FBS (a), R-isomer (10 μM, b), PAG (3 mM, pretreatment for 30 min, c), and PAG + R-isomer (d) after 18 h. Quantification (e) of pseudocapillaries was performed by counting the numbers of complete circles/well. n = 3; ***P < 0.01 versus untreated cells, ###P < 0.01 versus R-isomer.
Figure S2 Effect of captopril and enalaprilat on angiogenic parameters. (A) HUVECs were treated with captopril or enalaprilat (10 μM) with or without PAG (3 mM, 30 min pretreatment). Data are expressed as counted cells/well after 5 days of incubation. n = 3; *P < 0.01 versus untreated cells and #P < 0.05 versus R-isomer. (B) Evaluation of network formation of ECs seeded on Matrigel: 0.1% FBS (a), captopril (b), PAG (c), enalaprilat (d) and captopril + PAG (e). Quantification (f) of pseudocapillaries was performed by counting the numbers of complete circles/well. n = 3; **P < 0.01 versus untreated cells.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ion channels. Br J Pharmacol. 2013a;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013b;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anisimov A, Tvorogov D, Alitalo A, Leppänen VM, An Y, Han EC, et al. Vascular endothelial growth factor-angiopoietin chimera with improved properties for therapeutic angiogenesis. Circulation. 2013;127:424–434. doi: 10.1161/CIRCULATIONAHA.112.127472. [DOI] [PubMed] [Google Scholar]
- Bucci M, Papapetropoulos A, Vellecco V, Zhou Z, Pyriochou A, Roussos C, et al. Hydrogen sulfide is an endogenous inhibitor of phosphodiesterase activity. Arterioscler Thromb Vasc Biol. 2010;30:1998–2004. doi: 10.1161/ATVBAHA.110.209783. [DOI] [PubMed] [Google Scholar]
- Bucci M, Papapetropoulos A, Vellecco V, Zhou Z, Zaid A, Giannogonas P, et al. cGMP-dependent protein kinase contributes to hydrogen sulfide-stimulated vasorelaxation. PLoS ONE. 2012;7:e53319. doi: 10.1371/journal.pone.0053319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucci M, Vellecco V, Cantalupo A, Brancaleone V, Zhou Z, Evangelista S, et al. Hydrogen sulfide accounts for the peripheral vascular effects of zofenopril independently of ACE inhibition. Cardiovasc Res. 2014;102:138–147. doi: 10.1093/cvr/cvu026. [DOI] [PubMed] [Google Scholar]
- Cai J, Wu L, Qi X, Shaw L, Li Calzi S, Caballero S, et al. Placenta growth factor-1 exerts time-dependent stabilization of adherens junctions following VEGF-induced vascular permeability. PLoS ONE. 2011;6:e18076. doi: 10.1371/journal.pone.0018076. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Cai WJ, Wang MJ, Moore PK, Jin HM, Yao T, Zhu YC. The novel proangiogenic effect of hydrogen sulfide is dependent on Akt phosphorylation. Cardiovasc Res. 2007;76:29–40. doi: 10.1016/j.cardiores.2007.05.026. [DOI] [PubMed] [Google Scholar]
- Coletta C, Papapetropoulos A, Erdelyi K, Olah G, Módis K, Panopoulos P, et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium-dependent vasorelaxation. Proc Natl Acad Sci U S A. 2012;109:9161–9166. doi: 10.1073/pnas.1202916109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cominacini L, Pasini A, Garbin U, Evangelista S, Crea AE, Tagliacozzi D, et al. Zofenopril inhibits the expression of adhesion molecules on endothelial cells by reducing reactive oxygen species. Am J Hypertens. 2002;15:891–895. doi: 10.1016/s0895-7061(02)02995-3. [DOI] [PubMed] [Google Scholar]
- Donnini S, Solito R, Giachetti A, Granger HJ, Ziche M, Morbidelli L. Fibroblast growth factor-2 mediates angiotensin-converting enzyme inhibitor-induced angiogenesis in coronary endothelium. J Pharmacol Exp Ther. 2006;319:515–522. doi: 10.1124/jpet.106.108803. [DOI] [PubMed] [Google Scholar]
- Donnini S, Terzuoli E, Ziche M, Morbidelli L. Sulfhydryl angiotensin converting enzyme inhibitor promotes endothelial cell survival through nitric oxide synthase, fibroblast growth factor-2 and telomerase cross-talk. J Pharmacol Exp Ther. 2010;332:776–784. doi: 10.1124/jpet.109.159178. [DOI] [PubMed] [Google Scholar]
- Evangelista S, Manzini S. Antioxidant and cardioprotective properties of the sulphydryl angiotensin-converting enzyme inhibitor zofenopril. J Int Med Res. 2005;33:42–54. doi: 10.1177/147323000503300103. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Gerber HP, Le Couter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–676. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- Jain RK. Molecular regulation of vessel maturation. Nat Med. 2003;9:685–693. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- Kamoun P. Endogenous production of hydrogen sulfide in mammals. Amino Acids. 2004;26:243–254. doi: 10.1007/s00726-004-0072-x. [DOI] [PubMed] [Google Scholar]
- Katnik C, Adams DJ. Characterization of ATP-sensitive potassium channels in freshly dissociated rabbit aortic endothelial cells. Am J Physiol. 1997;272:H2507–H2511. doi: 10.1152/ajpheart.1997.272.5.H2507. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura H. Hydrogen sulfide: its production, release and functions. Amino Acids. 2010;41:113–121. doi: 10.1007/s00726-010-0510-x. [DOI] [PubMed] [Google Scholar]
- Li L, Rose P, Moore P. Hydrogen sulfide and cell signaling. Annu Rev Pharmacol Toxicol. 2011;51:169–187. doi: 10.1146/annurev-pharmtox-010510-100505. [DOI] [PubMed] [Google Scholar]
- Liu X, Pan L, Zhou Y, Gong Q, Rose P, Zhu Y. Hypoxia-inducible factor-1α is involved in the pro-angiogenic effect of hydrogen sulfide under hypoxic stress. Biol Pharm Bull. 2010;33:1550–1554. doi: 10.1248/bpb.33.1550. [DOI] [PubMed] [Google Scholar]
- Ma K, Liu Y, Zhu Q, Liu CH, Duan JL, Tan BK, et al. H2S donor,S-propargyl-cysteine, increases CSE in SGC-7901 and cancer-induced mice: evidence for a novel anti-cancer effect of endogenous H2S? PLoS ONE. 2011;6:e20525. doi: 10.1371/journal.pone.0020525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malester B, Tong X, Ghiu I, Kontogeorgis A, Gutstein DE, Xu J, et al. Transgenic expression of a dominant negative K(ATP) channel subunit in the mouse endothelium: effects on coronary flow and endothelin-1 secretion. FASEB J. 2007;21:2162–2172. doi: 10.1096/fj.06-7821com. [DOI] [PubMed] [Google Scholar]
- Monti M, Donnini S, Giachetti A, Mochly-Rosen D, Ziche M. deltaPKC inhibition or epsilonPKC activation repairs endothelial vascular dysfunction by regulating eNOS post-translational modification. J Mol Cell Cardiol. 2010;48:746–756. doi: 10.1016/j.yjmcc.2009.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti M, Solito R, Puccetti L, Pasotti L, Roggeri R, Monzani E, et al. Protective effects of novel metal-nonoates on the cellular components of the vascular system. J Pharmacol Exp Ther. 2014;351:500–509. doi: 10.1124/jpet.114.218404. [DOI] [PubMed] [Google Scholar]
- Monti M, Terzuoli E, Ziche M, Morbidelli L. The sulphydryl containing ACE inhibitor zofenoprilat protects coronary endothelium from doxorubicin-induced apoptosis. Pharmacol Res. 2013;76:171–181. doi: 10.1016/j.phrs.2013.08.003. [DOI] [PubMed] [Google Scholar]
- Mustafa AK, Gadalla MM, Sen N, Kim S, Mu W, Gazi SK, et al. H2S signals through protein S-sulfhydration. Sci Signal. 2009;2:ra72. doi: 10.1126/scisignal.2000464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osipov RM, Robich MP, Feng J, Liu Y, Clements RT, Glazer HP, et al. Effect of hydrogen sulfide in the porcine model of myocardial ischemia-reperfusion: comparison of different administration regimens and characterization of the cellular mechanisms of protection. J Cardiovasc Pharmacol. 2009;54:287–297. doi: 10.1097/FJC.0b013e3181b2b72b. [DOI] [PubMed] [Google Scholar]
- Papapetropoulos A, Pyriochou A, Altaany Z, Yang G, Marazioti A, Zhou Z, et al. Hydrogen sulfide is an endogenous stimulator of angiogenesis. Proc Natl Acad Sci U S A. 2009;106:21972–21977. doi: 10.1073/pnas.0908047106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl. Acids Res. 2014;42:D1098–1106. doi: 10.1093/nar/gkt1143. (Database Issue): [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polhemus DJ, Kondo K, Bhushan S, Bir SC, Kevil CG, Murohara T, et al. Hydrogen sulfide attenuates cardiac dysfunction after heart failure via induction of angiogenesis. Circ Heart Fail. 2013;6:1077–1086. doi: 10.1161/CIRCHEARTFAILURE.113.000299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qipshidze N, Metreveli N, Mishra PK, Lominadze D, Tyagi SC. Hydrogen sulfide mitigates cardiac remodeling during myocardial infarction via improvement of angiogenesis. Int J Biol Sci. 2012;8:430–441. doi: 10.7150/ijbs.3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stipanuk MH, Beck PW. Characterization of the enzymic capacity for cysteine desulphhydration in liver and kidney of the rat. Biochem J. 1982;206:267–277. doi: 10.1042/bj2060267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subissi A, Evangelista S, Giachetti A. Preclinical profile of zofenopril: an angiotensin converting enzyme inhibitor with peculiar cardioprotective properties. Cardiovasc Drug Rev. 1999;2:115–133. [Google Scholar]
- Suzuki K, Olah G, Modis K, Coletta C, Kulp G, Gerö D, et al. Hydrogen sulfide replacement therapy protects the vascular endothelium in hyperglycemia by preserving mitochondrial function. Proc Natl Acad Sci U S A. 2011;108:13829–13834. doi: 10.1073/pnas.1105121108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov. 2007;6:917–935. doi: 10.1038/nrd2425. [DOI] [PubMed] [Google Scholar]
- Szabo C, Papapetropoulos A. Hydrogen sulphide and angiogenesis: mechanisms and applications. Br J Pharmacol. 2011;164:853–865. doi: 10.1111/j.1476-5381.2010.01191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzuoli E, Meini S, Cucchi P, Catalani C, Cialdai C, Maggi CA, et al. Antagonism of bradykinin B2 receptor prevents inflammatory responses in human endothelial cells by quenching the NF-κB pathway activation. PLoS ONE. 2014;9:e84358. doi: 10.1371/journal.pone.0084358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandiver M, Snyder SH. Hydrogen sulfide: a gasotransmitter of clinical relevance. J Mol Med (Berl) 2012;90:255–263. doi: 10.1007/s00109-012-0873-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Guo Z, Wang S. The effect of certain conditions in the regulation of cystathionine γ-lyase by exogenous hydrogen sulfide in mammalian cells. Biochem Genet. 2013;51:503–513. doi: 10.1007/s10528-013-9581-1. [DOI] [PubMed] [Google Scholar]
- Wang T, Wang L, Zaidi SR, Sammani S, Siegler J, Moreno-Vinasco L, et al. Hydrogen sulfide attenuates particulate matter-induced human lung endothelial barrier disruption via combined reactive oxygen species scavenging and Akt activation. Am J Respir Cell Mol Biol. 2012;47:491–496. doi: 10.1165/rcmb.2011-0248OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Wu L, Wang R. Proapoptotic effect of endogenous H2S on human aorta smooth muscle cells. FASEB J. 2005;20:553–555. doi: 10.1096/fj.05-4712fje. [DOI] [PubMed] [Google Scholar]
- Yang G, Zhao K, Ju Y, Mani S, Cao Q, Puukila S, et al. Hydrogen sulfide protects against cellular senescence via s-sulfhydration of Keap1 and activation of Nrf2. Antioxid Redox Signal. 2013;18:1906–1919. doi: 10.1089/ars.2012.4645. [DOI] [PubMed] [Google Scholar]
- Zanardo RC, Brancaleone V, Distrutti E, Fiorucci S, Cirino G, Wallace JL. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006;20:2118–2120. doi: 10.1096/fj.06-6270fje. [DOI] [PubMed] [Google Scholar]
- Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H2S as a novel endogenous gaseous KATP channel opener. EMBO J. 2001;20:6008–6016. doi: 10.1093/emboj/20.21.6008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Effect of R-isomer of zofenoprilat on endothelial cell functionality. (A) Cell proliferation evaluated in HUVECs treated with zofenoprilat R-isomer (10 μM, 5 days) in presence/absence of PAG (3 mM, 30 min). Data are expressed as counted cells/well. n = 3; **P < 0.01 versus untreated cells and #P < 0.05 versus R-isomer. (B) CSE expression was evaluated in HUVECs treated with R-isomer (10 μM) for indicated times. The blot is representative of 3 with similar results. The ratio between CSE over actin is reported. ***P < 0.001, compared to untreated cells. (C) Pseudocapillary formation in Matrigel by ECs exposed to 0.1% FBS (a), R-isomer (10 μM, b), PAG (3 mM, pretreatment for 30 min, c), and PAG + R-isomer (d) after 18 h. Quantification (e) of pseudocapillaries was performed by counting the numbers of complete circles/well. n = 3; ***P < 0.01 versus untreated cells, ###P < 0.01 versus R-isomer.
Figure S2 Effect of captopril and enalaprilat on angiogenic parameters. (A) HUVECs were treated with captopril or enalaprilat (10 μM) with or without PAG (3 mM, 30 min pretreatment). Data are expressed as counted cells/well after 5 days of incubation. n = 3; *P < 0.01 versus untreated cells and #P < 0.05 versus R-isomer. (B) Evaluation of network formation of ECs seeded on Matrigel: 0.1% FBS (a), captopril (b), PAG (c), enalaprilat (d) and captopril + PAG (e). Quantification (f) of pseudocapillaries was performed by counting the numbers of complete circles/well. n = 3; **P < 0.01 versus untreated cells.