

Abstract
Background and Purpose
Endocannabinoids alter permeability at various epithelial barriers, and cannabinoid receptors and endocannabinoid levels are elevated by stroke, with potential neuroprotective effects. We therefore explored the role of endocannabinoids in modulating blood–brain barrier (BBB) permeability in normal conditions and in an ischaemia/reperfusion model.
Experimental Approach
Human brain microvascular endothelial cell and astrocyte co-cultures modelled the BBB. Ischaemia was modelled by oxygen-glucose deprivation (OGD) and permeability was measured by transepithelial electrical resistance. Endocannabinoids or endocannabinoid-like compounds were assessed for their ability to modulate baseline permeability or OGD-induced hyperpermeability. Target sites of action were investigated using receptor antagonists and subsequently identified with real-time PCR.
Key Results
Anandamide (10 μM) and oleoylethanolamide (OEA, 10 μM) decreased BBB permeability (i.e. increased resistance). This was mediated by cannabinoid CB2 receptors, transient receptor potential vanilloid 1 (TRPV1) channels, calcitonin gene-regulated peptide (CGRP) receptor (anandamide only) and PPARα (OEA only). Application of OEA, palmitoylethanolamide (both PPARα mediated) or virodhamine (all 10 μM) decreased the OGD-induced increase in permeability during reperfusion. 2-Arachidonoyl glycerol, noladin ether and oleamide did not affect BBB permeability in normal or OGD conditions. N-arachidonoyl-dopamine increased permeability through a cytotoxic mechanism. PPARα and γ, CB1 receptors, TRPV1 channels and CGRP receptors were expressed in both cell types, but mRNA for CB2 receptors was only present in astrocytes.
Conclusion and Implication
The endocannabinoids may play an important modulatory role in normal BBB physiology, and also afford protection to the BBB during ischaemic stroke, through a number of target sites.
Tables of Links
| TARGETS |
|---|
| GPCRsa |
| CB1 receptors |
| CB2 receptors |
| CGRP receptor |
| Ion channelsb |
| TRPV1 channels |
| Nuclear hormone receptorsc |
| PPARα (NR1C1) |
| PPARγ (NR1C3) |
| LIGANDS | |
|---|---|
| 2-AG, 2-arachidonoylglycerol | HU308 |
| AEA, anandamide | NADA, N-arachidonoyl-dopamine |
| AM251 | Noladin ether, 2-arachidonyl glyceryl ether |
| AM630 | OEA, N-oleoylethanolamide |
| Capsazepine | Oleamide |
| CGRP8–37 | PEA, N-palmitoylethanolamide |
| Dexamethasone | URB597 |
| GW6471 | Virodhamine, O-arachidonoyl ethanolamine |
| GW9662 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,cAlexander et al., 2013a,b,c).
Introduction
The blood–brain barrier (BBB) is formed by brain endothelial cells that line the cerebral microvasculature, capillary basement membranes and astrocyte end feet, which surround 99% of the BBB endothelia and play an important role in maintaining BBB integrity. Tight junctions restrict the paracellular pathway for diffusion of hydrophilic solutes, allowing the body to control which substances can gain access to the brain (Abbott, 2002). Cerebral reperfusion following ischaemia initiates a cascade of events such as inflammation, protease activation and oxidative and nitrosative stress, which increases the permeability of the BBB (Lo et al., 2003). The compromised state of the BBB aggravates haemorrhagic transformation and vasogenic oedema, which has profound neurological consequences (Latour et al., 2004). Indeed, uncontrolled cerebral oedema represents the leading cause of patient mortality within the first week following an ischaemic stroke (Hacke et al., 1996).
The endocannabinoid system (ECS) is comprised of cannabinoid receptors (CB1 and CB2), endogenous lipid ligands (the endocannabinoids) and enzymes that synthesize and degrade these compounds (Pertwee et al., 2010). Anandamide (AEA) and 2-arachidonoylglycerol (2-AG) are the best studied endocannabinoids, but other, chemically similar, compounds have been suggested as endocannabinoids or endogenous cannabinoid-like compounds, including N-arachidonoyl-dopamine (NADA), noladin ether, oleamide, N-oleoylethanolamide (OEA), N-palmitoylethanolamide (PEA) and virodhamine. CB1 and CB2 receptors are not the only pharmacological targets for cannabinoids, which also display activity at transient receptor potential vanilloid 1 (TRPV1) channels, GPR55, PPARα and PPARγ (Pertwee et al., 2010).
Some endocannabinoids have been shown to play a role in the regulation of BBB permeability in conditions other than ischaemia. Using in vivo and in vitro models, increased BBB permeability following chronic head injury and multiple sclerosis were decreased by the exogenous addition of 2-AG (Panikashvili et al., 2006) or AEA (Mestre et al., 2011) respectively. Oleamide has been found to inhibit gap junction coupling, thus increasing barrier permeability in vitro using pig brain microvascular endothelial cells (Nagasawa et al., 2006). Interestingly, the effect of any other endocannabinoid or endocannabinoid-like ligand on BBB permeability in normal conditions has not been investigated.
Components of the ECS are known to be altered by stroke. Human and animal in vivo data have shown increases in neurological levels of AEA (peripheral levels also elevated), 2-AG, OEA and PEA (Hillard, 2008; Naccarato et al., 2010). The expression of cannabinoid receptors is up-regulated in the rat brain following cerebral ischaemia, indicating that the ECS may play an important role in the endogenous response to stroke (see Hillard, 2008). Indeed, exogenous administration of 2-AG (Wang et al., 2009), AEA (Wang et al., 2009), OEA (Sun et al., 2007; Zhou et al., 2012) and PEA (Ahmad et al., 2012b) offer neuroprotection against ischaemic stroke using in vitro and in vivo models, but the impact on BBB permeability in stroke has only been assessed for OEA, where it was found to decrease in vivo BBB permeability via PPARα (Zhou et al., 2012).
Because endocannabinoids offer neuroprotection from stroke and alter BBB permeability in various neurological disorders, we hypothesized that endocannabinoids might regulate in vitro BBB permeability in normal and ischaemic conditions. Our results show for the first time that AEA and OEA decrease permeability in normal conditions. When given before oxygen-glucose deprivation (OGD), only OEA, PEA and virodhamine decreased BBB permeability. This study illustrates the important role the ECS plays in regulating BBB permeability via several target sites of action.
Methods
In vitro BBB co-culture model
Human brain microvascular endothelial cells isolated from human brain tissue (HBMECs; Catalog #1000, ScienCell, Carlsbad, CA, USA) and human astrocytes isolated from human cerebral cortex (HAs; Catalog #1800, ScienCell) were co-cultured in endothelial cell medium (Catalog #1001; concentrations, mL–1; 10 μg apo-transferrin, 10 μg BSA, 2 ng fibroblast growth factor (FGF)-2, 1 μg hydrocortisone, 2 ng insulin-like growth factor 1 (IGF-I), 7.5 μg insulin and 20 nM progesterone, 2% FBS, 5.55 mM glucose and 10 000 units·mL−1 of penicillin and streptomycin) and astrocyte medium (Catalog #1801; concentrations, mL–1; 10 μg apo-transferrin, 10 μg BSA, 10 ng EGF, 10 ng FGF-2, 1 μg hydrocortisone, 5 μg insulin, 2 ng IGF-I, 0.5 ng IGF LR3 10-8 M retinoic acid and 2 ng VEGF, 5% FBS, glucose 5.55 mM and 10 000 units·mL−1 of penicillin and streptomycin; both ScienCell). HAs were seeded on the outside of collagen-coated 0.4 μm pore PTFE membrane transwell inserts (12-well type; Corning Costar, Tewksbury, MA, USA) directed upside down and allowed to adhere to the membrane overnight. HBMECs were seeded on the inside of the insert and cells were grown to confluence to create a contact co-culture model (Allen and Bayraktutan, 2009; Mestre et al., 2011).
Measurement of BBB permeability
Transepithelial electrical resistance (TEER) was measured as a marker of co-culture integrity and as a measure of paracellular permeability. The resistance across the membrane was measured using STX2 electrodes linked to an EVOM2 resistance meter (World Precision Instruments, Hitchin, Hertfordshire, UK). Three readings were taken per insert and the average value was used. A baseline TEER reading was taken (i.e. 0 h) and the percentage change from this value was calculated for subsequent readings. The average TEER was 30.23 ± 0.24 Ω·cm−2, similar to that previously reported using the same methodology (Allen and Bayraktutan, 2009).
To assess their impact on permeability, endocannabinoids were added to the luminal (endothelial) chamber and TEER was measured at various time points over 48 h, at which point the media was changed, endocannabinoids were reapplied and TEER was measured for another 48 h. Endocannabinoids that significantly altered permeability had their mechanism of action probed using relevant receptor antagonists which were co-administered with the endocannabinoids.
Real-time PCR (RT-PCR)
Presence of predicted sites of action was investigated at the mRNA level using reverse transcription followed by PCR (RT-PCR). Total RNA was extracted from HA and HBMEC cells using Allprep DNA/RNA kit with on column DNaseI treatment (Qiagen, Hilden, Germany). Reverse transcription with and without reverse transcriptase was performed in 20 μL final volume using 2 μg of total RNA and random primers with the high capacity cDNA reverse transcription kit (Life Technologies, Paisley, UK) according to the manufacturer's instructions. PCR reactions were carried out in a final volume of 25 μL with Zymotaq (ZymoResearch, Irvine, CA, USA) using 2 μL of reverse transcription product as template. Primer pairs used to amplify PPARα and PPARγ fragments (99bp and 87bp, respectively) were as described in Reynders et al. (2006); those for 128bp hypoxanthine-guanine phosphotibosyl transferase (HPRT) were from Spinsanti et al. (2008); those for 303 bp CB1 receptor and 365 bp CB2 receptor were from Cencioni et al. (2010); those for 511 bp TRPV1 were from Luo et al. (2008); and finally the 380 bp CGRP receptor cDNA fragment was amplified using the primers reported in Dong et al. (1999). After 5 min at 95°C, PCRs were performed for 40 cycles except those for CGRP receptors and CB2 receptors which were carried out for 60 cycles. The cycles included 30 s at 95°C, 30 s at the annealing temperature optimal for each primer pair (56°C for CB1 and CB2 receptors; 60°C for PPARα, PPARγ and HPRT; 58°C for TRPV1 channel; 61°C for CGRP receptor) and a final extension step of 30 s at 72°C. Amplification products were separated by gel electrophoresis through ethidium bromide stained 2% agarose (CB1 receptor, CB2 receptor, TRPV1 channel, CGRP receptor and HPRT) or 3% metaphore (PPARα and PPARγ) gels and visualized using a Biorad Chemidoc (Life Science, Hemel Hampstead, UK).
OGD
Ischaemic conditions were simulated using an OGD protocol. Cell culture media were replaced with glucose-free RPMI medium (Invitrogen, Life Technologies) and the plates were placed into a GasPak EZ Anaerobe Pouch (Beckton Dickinson, Oxford, UK) with anaerobic conditions being achieved within 20 min, and the inserts were left in OGD conditions for a further 4 h in the incubator. No preconditioning was carried out on the cells. After OGD, TEER was read and the RPMI medium was replaced with the cells' normal medium and returned to the incubator. The permeability of the BBB was assessed throughout the reperfusion period. Medium samples were collected and stored at −80°C whenever the medium was changed. Endocannabinoids were added before the OGD protocol to mimic an endogenous response to ischaemia.
Lactate dehydrogenase (LDH) assay
LDH levels were measured using a commercially available kit according to the manufacturer's instructions (LDH-cytotoxicity assay kit II, Biovision, Milpitas, CA, USA). Medium samples were transferred into an optically clear 96-well plate and reaction mix (containing water soluble tetrazolium-1) was added to each well. After 30 min, absorbance was measured at 450 nm, subtracting the 650 nm reading to correct for optical imperfections in the plate.
Data analysis
Results are presented as means ± SEM. Data were analysed with GraphPad Prism software (La Jolla, CA, USA), using either Student's t-test or one-way anova with Dunnett's or Bonferroni's post hoc test. AUC values were calculated using the trapezoidal method. In experiments conducted in control conditions, the baseline was set to be at the lowest value in the data sets and the area above baseline was calculated. In the OGD experiments, the baseline was set to be highest value obtained in the data sets and the area below baseline was calculated. P < 0.05 was considered significant.
Materials
All endocannabinoids were purchased from Tocris (Bristol, UK) and dissolved in ethanol to a stock concentration of 10 mM, except 2-AG which was purchased from Abcam (Cambridge, UK) and dissolved in acetonitrile. AM251, AM630, GW6471, GW9662 (all 100 nM), capsazepine, O-1918 (both 1 μM) (all dissolved in dimethyl sulfoxide) and CGRP8–37 (2 μM, dissolved in distilled water) were all purchased from Tocris and URB597 (1 μM, dissolved in dimethyl sulfoxide) was purchased from Sigma (Dorset, UK). All were dissolved to a stock solution of 10 mM.
Results
Effects of anandamide on BBB permeability
AEA is a well-characterized and frequently studied endocannabinoid displaying effects on epithelial barrier permeability in BBB and non-BBB sites, therefore, this was the first compound investigated. AEA at 10 μM, but not 100 nM or 1 μM, decreased permeability (i.e. increased TEER/monolayer resistance) (see Figure 1A,B). In all subsequent antagonist studies, AEA (10 μM) also significantly increased TEER compared with vehicle in the same experimental set up as the antagonists. In these studies, the effect of AEA on BBB permeability was not inhibited by AM251 (CB1), GW6471 (PPARα), GW9662 (PPARγ) or O1918 (novel endothelial receptor) (see Table 1 for AUC values). However, the effect of AEA (10 μM) was inhibited by the CB2 antagonist AM630, the TRPV1 antagonist capsazepine and the CGRP receptor antagonist CGRP8–37 (Figure 1C–E). The synthetic CB2 agonist HU308 and the steroid dexamethasone (as a positive control) were also able to significantly increase TEER in this BBB model (Figure 1F,G).
Figure 1.

The effects of increasing concentrations of AEA on BBB permeability measured by TEER (A) with corresponding AUC (B) (n = 9 inserts from three separate experiments). The effects of capsazepine (Cpz) (C) (n = 7 inserts from three separate experiments) or AM630 (D) (n = 7–8 inserts from three separate experiments) or CGRP (E) (n = 5–6 inserts from three separate experiments) on the effect of AEA (10 μM). The effects of dexamethasone (n = 6) and the CB2 agonist HU308 on BBB permeability over time (F) and expressed as AUC (G). Data are given as mean ± SEM. ***P < 0.001, **P < 0.01, *P < 0.05; AEA compared with vehicle-treated inserts; ††P < 0.01; AEA and antagonist compared with AEA alone; #P < 0.05, ##P < 0.01; HU308 compared with vehicle; one-way anova with Dunnett's (B) or Bonferroni's test (C,D,E).
Table 1.
AUC values for the effects of cannabinoids on TEER
| TEER (AUC) | ||
|---|---|---|
| Vehicle | Endocannabinoid (10 μM) | |
| AEA | 3027 ± 328 | 4315 ± 303* |
| +AM251 | 2355 ± 205 | 3380 ± 210† |
| +GW6471 | 2841 ± 129 | 3595 ± 99†† |
| +GW9662 | 2626 ± 148 | 3362 ± 145† |
| +O-1918 | 2369 ± 60 | 3359 ± 240†† |
| OEA | 1649 ± 124 | 2646 ± 133*** |
| +AM251 | 1356 ± 171 | 2205 ± 208†† |
| +AM630 | 1333 ± 164 | 2040 ± 50†† |
| +Capsazepine | 1274 ± 262 | 2060 ± 57† |
| +GW9662 | 1391 ± 182 | 2008 ± 129† |
| 2-AG | 2572 ± 104 | 2854 ± 351 |
| NADA | 2699 ± 104 | 2060 ± 204* |
| Noladin ether | 2342 ± 208 | 2718 ± 192 |
| Oleamide | 2320 ± 205 | 2399 ± 176 |
| PEA | 2004 ± 104 | 2349 ± 350 |
| Virodhamine | 1850 ± 104 | 2183 ± 179 |
All data are from experiments conducted in normal conditions (i.e. no OGD). Data are given as mean ± SEM.
P < 0.001
P < 0.05 compared with vehicle-treated inserts
P < 0.01
P < 0.05 endocannabinoid and antagonist compared with endocannabinoid alone; Student's t-test or one-way anova with Bonferroni's test (antagonist experiments). AM251, CB1 antagonist; capsazepine, TRPV1 antagonist; GW6471, PPARα antagonist; GW9662, PPARγ antagonist; O1918, novel endothelial receptor antagonist.
RT-PCR was carried out to profile the expression at the RNA level of these potential target sites of action in HAs and HBMECs. PPARs (α and γ), CB1 receptors, TRPV1 channels and CGRP receptors were found to be present in both cell types. By contrast, mRNA for CB2 receptors was only present in the astrocytes (Figure 2).
Figure 2.
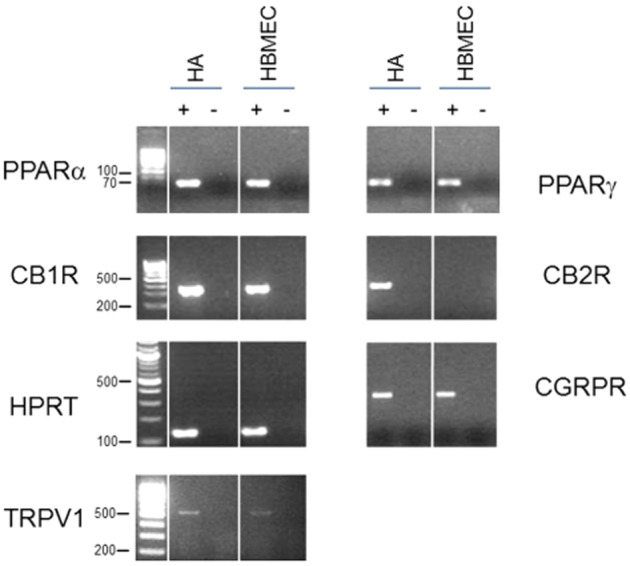
Expression profiling of potential target sites of action in HA and HBMEC cells. Shown are the ethidium bromide-stained gels of the products obtained by RT-PCR using primers specific for PPARα, PPARγ, CB1 receptors, CB2 receptors, TRPV1 channels, CGRP receptors and the control gene HPRT. cDNAs generated in the presence (+) or absence (−) of reverse transcriptase on total RNA from HA or HBMEC cells were used as template for the PCRs. The 100 bp DNA ladder was used in all gels except for PPARα and PPARγ where a 10 bp ladder was used. Sizes are in base pairs.
Inhibition of the degradation of AEA by the fatty acid amide hydrolase (FAAH) inhibitor URB597 blocked the effects of AEA such that the change in TEER was no longer significantly different to that observed in the vehicle control inserts (Figure 3).
Figure 3.
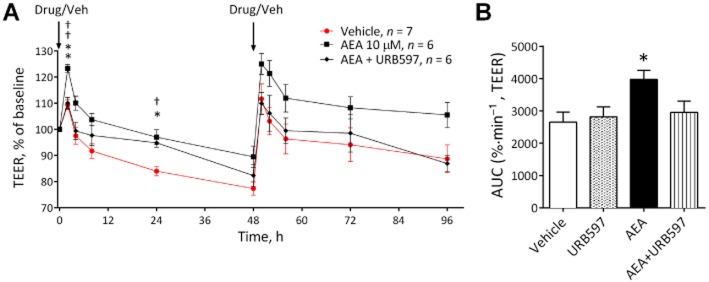
The effects of AEA (n = 6) in the presence and absence of the FAAH inhibitor URB597 (n = 6) on permeability in the BBB (A), with corresponding AUC (B). Data are given as mean ± SEM. **P < 0.01, *P < 0.05 AEA versus the vehicle; ††P < 0.01 AEA and antagonist compared with AEA alone; one-way anova with Dunnett's test.
Exposing the BBB to 4 h OGD increased permeability as shown by a reduction in TEER of approximately 35% (Figure 4). AEA did not alter the BBB permeability response to OGD when applied before (Figure 4A and B) or after the OGD protocol (Figure 4C and D).
Figure 4.

The effects of various concentrations of AEA either before (A) (n = 7–12 inserts from four separate experiments) or after (C) (n = 9–10 inserts from four separate experiments) 4 h OGD on permeability in the BBB, with corresponding AUC (B and D). Data are given as mean ± SEM. #, * (10 μM) † (30 μM), P < 0.05; compared with vehicle; one-way anova with Dunnett's test.
Effects of other endocannabinoids and endocannabinoid-like compounds on BBB permeability given in normal conditions
2-AG, noladin ether, oleamide, PEA and virodhamine did not alter BBB permeability when given in normal conditions (see Table 1). However, OEA significantly increased TEER (decreased permeability) in normal conditions at 10 μM (P < 0.01, Figure 5A). A concentration-response curve showed that a significant response to OEA was only observed at 10 μM (Figure 5B). The effects of OEA (10 μM) were not inhibited by AM251, AM630, capsazepine or GW9662 (see Table 1), but were inhibited by GW6471, a PPARα agonist (P < 0.05, Figure 5C and D).
Figure 5.

The effect of OEA over time on BBB permeability in the initial endocannabinoid screening (A) (n = 6 inserts from three separate experiments) as a concentration-response curve (B) (n = 4–6 inserts from three separate experiments) and in the presence of the PPARα antagonist GW6471 (C and D) (n = 5 inserts from three separate experiments). (E) The effects of NADA (10 μM) on TEER values in the BBB model (n = 7–8 inserts from four separate experiments). (F) Absorbance values for LDH assay conducted on cell culture medium obtained from the luminal (endothelial) chamber of the inserts at 48 h (n = 6 inserts from three separate experiments). Data are given as mean ± SEM. ***P < 0.001, **P < 0.01, *P < 0.05; OEA compared to vehicles treated inserts; †††P < 0.001, ††P < 0.01; OEA and antagonist compared with OEA alone; one-way anova with Dunnett's or Bonferroni's test.
After 48 h exposure to a single application of NADA (10 μM), BBB permeability was significantly increased (Figure 5E). Following a second application of NADA, BBB permeability remained significantly below that of vehicle for another 24 h (see Figure 5E). Visual inspection using a light microscope showed apparent cellular damage, therefore levels of LDH in the luminal (endothelial) medium from NADA-treated inserts were measured and were found to be significantly greater than vehicle at 48 h (P < 0.001, Figure 5F).
Effects of endocannabinoids and endocannabinoid-like compounds on BBB permeability given before OGD
OEA (P < 0.01, Figure 6A and B), PEA (P < 0.05, Figure 6C and D) and virodhamine (P < 0.01, Figure 6E and F) given before OGD all significantly reduced the increase in permeability induced by the OGD protocol (see AUC values Figure 6B, D and F). The effects of these compounds were mainly observed in the reperfusion period rather than the initial increase in permeability (see Figure 6A, C and E). However, 2-AG, oleamide, NADA and noladin ether had no effect on the permeability response to OGD (Figure 6G–J). In a separate experiments, the protective effect of OEA and PEA given before OGD were inhibited by the PPARα antagonist GW6471 (Figure 7).
Figure 6.
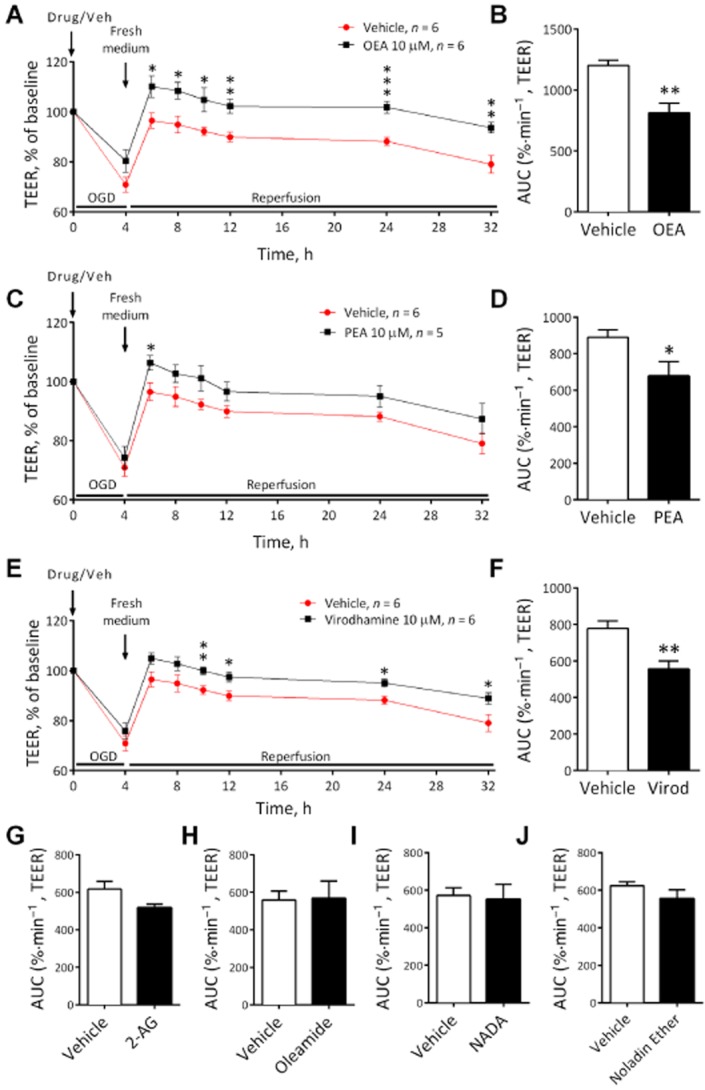
The effect of OEA (A and B) (n = 6 inserts from three separate experiments), PEA (C and D) (n = 5–6 inserts from three separate experiments), virodhamine (E and F) (n = 6 inserts from three separate experiments), 2-AG (G) (n = 6 inserts from three separate experiments), oleamide (H) (n = 6 inserts from three separate experiments), NADA (I) (n = 4–5 inserts from two separate experiments) and noladin ether (J) (n = 5 inserts from three separate experiments) administered before 4 h OGD on TEER. Data are given as mean ± SEM. ***P < 0.001, **P < 0.01, *P < 0.05; compared with vehicle-treated inserts; Student's t-test.
Figure 7.
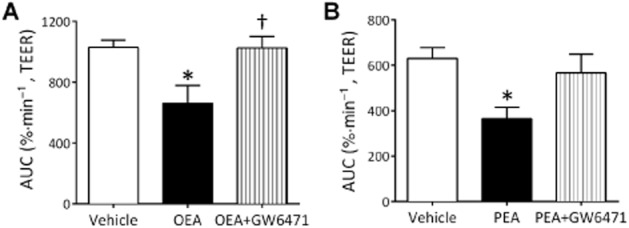
The effect of OEA (A) (n = 6 inserts from three separate experiments) or PEA (B) (n = 5–6 inserts from three separate experiments) alone or in combination with GW6471 before 4 h OGD on TEER. Data are given as mean ± SEM. *P < 0.05 compared with vehicle-treated inserts; †P < 0.05 OEA and antagonist compared with OEA alone; one-way anova with Bonferroni's test.
Discussion
The aim of this study was to investigate the potential roles that endocannabinoids play in the regulation of in vitro BBB permeability. Our results show that AEA (via TRPV1 channels, CB2 and CGRP receptors) and OEA (via PPARα) decreased permeability in normal conditions, while NADA increased permeability. When given before OGD, OEA, PEA (via PPARα) and virodhamine decreased BBB permeability. Overall, this study illustrates the important role the ECS plays in regulating BBB permeability, identifying a number of potential targets for future therapies.
Effects of AEA on BBB permeability
We found that AEA decreased BBB permeability (i.e. increases BBB resistance). This effect of AEA was observed within 2 h of application, unlike the steroid dexamethasone, where a time-dependent increase in BBB resistance was observed. AEA has been shown to reduce BBB permeability using mouse in vivo and in vitro models, through attenuation of vascular cell adhesion molecule (VCAM)-1 levels via CB1 receptor activation (Mestre et al., 2011). In contrast, we found that CB2 receptor activation by AEA was partly responsible for decreasing permeability and showed that a synthetic CB2 receptor agonist (HU308) was capable of inducing a similar acute increase in BBB resistance. PCR expression profiling revealed that the location of CB2 receptors is more likely to be on the astrocytes than endothelial cells. Activation of CB2 receptors has also been shown to decrease in vivo BBB permeability in mice following traumatic brain injury (Amenta et al., 2012) or inflammation (Ramirez et al., 2012), and following stroke, CB2 receptor agonists decrease infarct volume by reducing inflammatory infiltrate (Hillard, 2008). Therefore, activity at the BBB could account for some of the protective effects that CB2 receptor agonists have displayed in animal stroke models. It is worth noting that the lack of CB2 receptors in the human brain microvascular endothelial cells isolated from human brain tissue in the present study is in contrast to previous studies. This may be related to the source of human tissue. For example, in the work of Golech et al. (2004), the endothelial cells were isolated from brains of patients with idiopathic epilepsy. In the endothelium of human glioblastomas, CB2 receptors are expressed in about half of the cells (Schley et al., 2009). In multiple sclerosis, CB2 receptors have also been identified on the endothelium of cerebral arteries (Zhang et al., 2011). As CB2 receptors are known to be up-regulated in pathologies and in response to inflammation and stress, this may explain the expression of CB2 receptors in these studies and not in the cells in the present study, which are derived from normal tissue. In support of this, Ramirez et al. (2012) showed little CB2 receptor immunoreactivity in healthy brain endothelium or on human brain microvascular endothelial cells, but CB2 receptors were highly up-regulated in patients with encephalitis or after an inflammatory insult and were capable of reducing BBB permeability in these situations.
A recent study from our group showed that AEA activation of TRPV1 channels on the basolateral side of Caco-2 cells (human epithelial colorectal adenocarcinoma cells) reduced permeability, potentially via increases in tight junction proteins (Alhamoruni et al., 2010), and the present study further demonstrates the permeability reducing properties of TRPV1 channel activation at epithelial barriers. Activation of TRPV1 channels by AEA is known to increase CGRP release (Zygmunt et al., 1999); therefore, AEA may mediate its effects via increased CGRP levels, as suggested by the effect of the CGRP receptor antagonist in the present study. Both TRPV1 channel and CGRP receptor mRNA were demonstrated in brain endothelial cells and in astrocytes. This is consistent with a recent study showing that CGRP activation decreased cerebral oedema and BBB permeability following ischaemic stroke in the rat (Liu et al., 2011).
Inhibiting the degradation of AEA using the FAAH inhibitor URB597 also inhibited the effects of AEA, which suggests that metabolic products of AEA degradation are also involved in the effects of AEA effect on BBB permeability. This may seem contradictory to our data showing the effects of AEA through activation of CB2 receptors and TRPV1 channels, although this may not be the case. It has been shown for example that a cytochrome P450-derived epoxygenated metabolite of AEA, 5,6-epoxyeicosatrienoic acid ethanolamide, is a selective CB2 receptor agonist (Snider et al., 2009) and epoxyeicosatrienoic acids derived from AEA can activate TRPV4 channels (Watanabe et al., 2003), so the possibility exists that in our model, it is the metabolites of AEA that modulate BBB permeability through CB2 receptors and TRPV1 channesl.
Although AEA reduced BBB permeability in normal conditions, it had no effect when given either before or after OGD. The increase in permeability caused by OGD was greater than the increase in TEER induced by AEA, suggesting the effect of AEA is not strong enough to alter permeability overall. Alternatively, the expression or function of the targets involved, CB2 receptors and TRPV1 channels, might be altered by the OGD protocol. Perhaps, a negative effect of AEA on BBB permeability through activation of CB1 receptors is revealed in ischaemic conditions (Alhamoruni et al., 2012). Either way, protecting BBB permeability in ischaemia does not appear to underlie the neuroprotective effects of AEA observed in stroke.
Effects of other endocannabinoids and endocannabinoid-like compounds on BBB permeability
Like AEA, we found that OEA decreased permeability (increased TEER) when given in normal conditions or in the presence of OGD and both of these responses were via PPARα activation. PPARα mRNA was confirmed in both the endothelial cells and astrocytes. These data in human cells are consistent with in vivo mouse studies where OEA reduced infarct volume, oedema and BBB disruption following ischaemic stroke through activation of PPARα (Sun et al., 2007; Zhou et al., 2012). Fenofibrate, a synthetic PPARα agonist, also protected a rodent in vitro BBB model from hyperpermeability following OGD (Mysiorek et al., 2009). PPARα agonists inhibit expression of the adhesion molecules, VCAM-1 and ICAM-1, down-regulate MMPs and protease activity and up-regulate tight junction proteins (Marx et al., 1999; Deplanque et al., 2003; Huang et al., 2009), all of which are beneficial to BBB integrity. OEA is produced by neurons and glial cells following ischaemia (Hillard, 2008). Hence, OEA activity at PPARα could form an important component of the body's innate defence following stroke, with data from the present study suggesting actions at the BBB may be crucial to this beneficial effect of OEA.
2-AG, noladin ether and oleamide did not affect BBB permeability. These endocannabinoids are known to activate receptors that we have shown to modify the permeability of the BBB in the present study (i.e. CB2 receptors and PPARα), so it is unclear why their effects are dissimilar to AEA and OEA. However, endocannabinoids are known to have complicated pharmacology which may be explained by a number of phenomena including mechanisms of cell transport and trafficking, metabolism and pharmacologically active metabolites, agonist bias, allosteric modulation and activation of other target sites that might oppose any effects at CB2 receptors or PPARα (Alexander and Kendall, 2007; Kenakin, 2009; Console-Bram et al., 2012; Fowler, 2013).
We found that PEA and virodhamine did not alter the permeability of the BBB in normal conditions, but they did decrease permeability following OGD. Similar to OEA, the permeability-lowering effects of PEA were inhibited by the presence of a PPARα antagonist, further confirming the role for this receptor in modulating BBB permeability in ischaemia. PEA has been shown to reduce oedema and brain infarct size in mice using models of two separate diseases, both of which cause BBB damage, traumatic brain injury (Ahmad et al., 2012a) and ischaemic stroke (Ahmad et al., 2012b). Neurological PEA levels in human stroke patients are increased following ischaemia (Schabitz et al., 2002), which suggests a protective role for PEA in stroke, with permeability-reducing effects on the BBB potentially forming part of this.
To date, no studies have investigated the effects of virodhamine on epithelial barrier permeability or stroke. However, virodhamine does inhibit neutrophil migration through CB1 receptor activation (McHugh et al., 2008). Neutrophil accumulation and infiltration into the cerebral microvasculature plays a critical role in neuronal injury following cerebral ischaemia, especially during reperfusion (Sughrue et al., 2004). Attenuation of neutrophil migration may be of benefit in treating hyperacute stroke, but previous trials assessing compounds that inhibit neutrophilic function have been ineffective (Krams et al., 2003).
NADA was found to increase BBB permeability with evidence of cell damage. In support of these findings, NADA has been shown to cause concentration-dependent cytotoxicity in human, murine or rat hepatic stellate cells (Wojtalla et al., 2012) and human peripheral blood mononuclear cells (Saunders et al., 2009). Interestingly, although NADA increased the permeability of the BBB in normal conditions, it did not further increase the permeability of inserts that were exposed to 4 h OGD. This is most likely because of the fact that in this protocol, the cells were only exposed to NADA for 4 h compared with 96 h in the non-OGD experiments, and that the cytotoxic effects of NADA are revealed with longer exposure times. NADA is known to be produced in bovine and rat brains (Walker et al., 2002), but no studies have investigated whether stroke alters the levels of NADA and what consequence this might have.
Of note, the effects of endocannabinoids were only observed in the high micromolar concentrations. This may be partly because of technical issues such as binding of endocannabinoids to cell culture plastic (see Fowler et al., 2004), interactions with FBS or BSA in culture medium or the transport of endocannabinoids into and across the cell, which is particularly relevant for the PPARα-activating endocannabinoids (Kaczocha et al., 2012). In addition, although studies have investigated the levels of endocannabinoid in the brain, it is not known what the exact intracellular concentration of endocannabinoids is when their synthesis is stimulated. Despite this, the concentration of endocannabinoids required in the present study are in line with the known receptor affinity for AEA at CB2 receptors (Ki up to 2 μM) and TRPV1 channels (EC50 ranging from 0.63 to 4.9 μM depending on assay) (Pertwee et al., 2010) and for OEA and PEA at PPARα (EC50 3–4 μM, O'Sullivan, 2007).
In conclusion, this study demonstrates that AEA, OEA, PEA and virodhamine (all 10 μM) decrease BBB permeability in vitro in human cells. Roles for CB2 receptors, TRPV1 channels, CGRP receptors and PPARα activation are presented, which, in conjunction with published data, identify them as potential targets to modulate BBB permeability.
Author contributions
W. H. H. was involved in the acquisition and analysis of data and the preparation of the manuscript. C. T., S. I. A. and M. N. were involved in the acquisition and analysis of data. S. E. O. S. contributed to the conception and design of the study and preparation of the manuscript. T. J. E. contributed to the conception and design of the study and critical revision of the manuscript. All authors approved the final version to the published.
Conflict of interest
There are no conflicts of interest.
Glossary
- 2-AG
2-arachidonoylglycerol
- AEA
anandamide
- BBB
blood–brain barrier
- CB1
cannabinoid receptor 1
- CB2
cannabinoid receptor 2
- CGRP
calcitonin gene-related peptide
- ECS
endocannabinoid system
- FAAH
fatty acid amide hydrolase
- LDH
lactate dehydrogenase
- NADA
N-arachidonoyl-dopamine
- OEA
N-oleoylethanolamide
- OGD
oxygen-glucose deprivation
- PEA
N-palmitoylethanolamide
- RT-PCR
real-time PCR
- TEER
transepithelial electrical resistance
- TRPV1
transient receptor potential vanilloid 1
References
- Abbott NJ. Astrocyte-endothelial interactions and blood-brain barrier permeability. J Anat. 2002;200:629–638. doi: 10.1046/j.1469-7580.2002.00064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad A, Crupi R, Impellizzeri D, Campolo M, Marino A, Esposito E, et al. Administration of palmitoylethanolamide (PEA) protects the neurovascular unit and reduces secondary injury after traumatic brain injury in mice. Brain Behav Immun. 2012a;26:1310–1321. doi: 10.1016/j.bbi.2012.07.021. [DOI] [PubMed] [Google Scholar]
- Ahmad A, Genovese T, Impellizzeri D, Crupi R, Velardi E, Marino A, et al. Reduction of ischemic brain injury by administration of palmitoylethanolamide after transient middle cerebral artery occlusion in rats. Brain Res. 2012b;1477:45–58. doi: 10.1016/j.brainres.2012.08.006. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G Protein-Coupled Receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ion Channels. Br J Pharmacol. 2013b;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL. Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Nuclear Hormone Receptors. Br J Pharmacol. 2013c;170:1652–1675. doi: 10.1111/bph.12448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Kendall DA. The complications of promiscuity: endocannabinoid action and metabolism. Br J Pharmacol. 2007;152:602–623. doi: 10.1038/sj.bjp.0707456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alhamoruni A, Lee AC, Wright KL, Larvin M, O'Sullivan SE. Pharmacological effects of cannabinoids on the Caco-2 cell culture model of intestinal permeability. J Pharmacol Exp Ther. 2010;335:92–102. doi: 10.1124/jpet.110.168237. [DOI] [PubMed] [Google Scholar]
- Alhamoruni A1, Wright KL, Larvin M, O'Sullivan SE. Cannabinoids mediate opposing effects on inflammation-induced intestinal permeability. Br J Pharmacol. 2012;165:2598–2610. doi: 10.1111/j.1476-5381.2011.01589.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen CL, Bayraktutan U. Antioxidants attenuate hyperglycaemia-mediated brain endothelial cell dysfunction and blood-brain barrier hyperpermeability. Diabetes Obes Metab. 2009;11:480–490. doi: 10.1111/j.1463-1326.2008.00987.x. [DOI] [PubMed] [Google Scholar]
- Amenta PS, Jallo JI, Tuma RF, Elliott MB. A cannabinoid type 2 receptor agonist attenuates blood-brain barrier damage and neurodegeneration in a murine model of traumatic brain injury. J Neurosci Res. 2012;90:2293–2305. doi: 10.1002/jnr.23114. [DOI] [PubMed] [Google Scholar]
- Cencioni MT1, Chiurchiù V, Catanzaro G, Borsellino G, Bernardi G, Battistini L, et al. Anandamide suppresses proliferation and cytokine release from primary human T-lymphocytes mainly via CB2 receptors. PLoS ONE. 2010;5:e8688. doi: 10.1371/journal.pone.0008688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Console-Bram L, Marcu J, Abood ME. Cannabinoid receptors: nomenclature and pharmacological principles. Prog Neuropsychopharmacol Biol Psychiatry. 2012;38:4–15. doi: 10.1016/j.pnpbp.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deplanque D, Gele P, Petrault O, Six I, Furman C, Bouly M, et al. Peroxisome proliferator-activated receptor-alpha activation as a mechanism of preventive neuroprotection induced by chronic fenofibrate treatment. J Neurosci. 2003;23:6264–6271. doi: 10.1523/JNEUROSCI.23-15-06264.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong YL, Fang L, Kondapaka S, Gangula PR, Wimalawansa SJ, Yallampalli C. Involvement of calcitonin gene-related peptide in the modulation of human myometrial contractility during pregnancy. J Clin Invest. 1999;104:559–565. doi: 10.1172/JCI6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler CJ. Transport of endocannabinoids across the plasma membrane and within the cell. FEBS J. 2013;280:1895–1904. doi: 10.1111/febs.12212. [DOI] [PubMed] [Google Scholar]
- Fowler CJ, Tiger G, Ligresti A, Lopez-Rodriguez ML, Di Marzo V. Selective inhibition of anandamide cellular uptake versus enzymatic hydrolysis–a difficult issue to handle. Eur J Pharmacol. 2004;492:1–11. doi: 10.1016/j.ejphar.2004.03.048. [DOI] [PubMed] [Google Scholar]
- Golech SA, McCarron RM, Chen Y, Bembry J, Lenz F, Mechoulam R, et al. Human brain endothelium: coexpression and function of vanilloid and endocannabinoid receptors. Brain Res Mol Brain Res. 2004;132:87–92. doi: 10.1016/j.molbrainres.2004.08.025. [DOI] [PubMed] [Google Scholar]
- Hacke W, Schwab S, Horn M, Spranger M, De Georgia M, von Kummer R. ‘Malignant’ middle cerebral artery territory infarction: clinical course and prognostic signs. Arch Neurol. 1996;53:309–315. doi: 10.1001/archneur.1996.00550040037012. [DOI] [PubMed] [Google Scholar]
- Hillard CJ. Role of cannabinoids and endocannabinoids in cerebral ischemia. Curr Pharm Des. 2008;14:2347–2361. doi: 10.2174/138161208785740054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Eum SY, Andras IE, Hennig B, Toborek M. PPARalpha and PPARgamma attenuate HIV-induced dysregulation of tight junction proteins by modulations of matrix metalloproteinase and proteasome activities. FASEB J. 2009;23:1596–1606. doi: 10.1096/fj.08-121624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczocha M, Vivieca S, Sun J, Glaser SJ, Deutsch DG. Fatty acid-binding proteins transport n-acylethanolamines to nuclear receptors and are targets of endocannabinoid transport inhibitors. J Biol Chem. 2012;287:3415–3424. doi: 10.1074/jbc.M111.304907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T. Biased agonism. F1000 Biol Rep. 2009;1:87. doi: 10.3410/B1-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krams M, Lees KR, Hacke W, Grieve AP, Orgogozo JM, Ford GA. Acute Stroke Therapy by Inhibition of Neutrophils (ASTIN): an adaptive dose-response study of UK-279,276 in acute ischemic stroke. Stroke. 2003;34:2543–2548. doi: 10.1161/01.STR.0000092527.33910.89. [DOI] [PubMed] [Google Scholar]
- Latour LL, Kang DW, Ezzeddine MA, Chalela JA, Warach S. Early blood-brain barrier disruption in human focal brain ischemia. Ann Neurol. 2004;56:468–477. doi: 10.1002/ana.20199. [DOI] [PubMed] [Google Scholar]
- Liu Z, Liu Q, Cai H, Xu C, Liu G, Li Z. Calcitonin gene-related peptide prevents blood-brain barrier injury and brain edema induced by focal cerebral ischemia reperfusion. Regul Pept. 2011;171:19–25. doi: 10.1016/j.regpep.2011.05.014. [DOI] [PubMed] [Google Scholar]
- Lo EH, Dalkara T, Moskowitz MA. Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci. 2003;4:399–415. doi: 10.1038/nrn1106. [DOI] [PubMed] [Google Scholar]
- Luo D, Zhang YW, Peng WJ, Peng J, Chen QQ, Li D, et al. Transient receptor potential vanilloid 1-mediated expression and secretion of endothelial cell-derived calcitonin gene-related peptide. Regul Pept. 2008;150:66–72. doi: 10.1016/j.regpep.2008.05.007. [DOI] [PubMed] [Google Scholar]
- Marx N, Sukhova GK, Collins T, Libby P, Plutzky J. PPARalpha activators inhibit cytokine-induced vascular cell adhesion molecule-1 expression in human endothelial cells. Circulation. 1999;99:3125–3131. doi: 10.1161/01.cir.99.24.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh D, Tanner C, Mechoulam R, Pertwee RG, Ross RA. Inhibition of human neutrophil chemotaxis by endogenous cannabinoids and phytocannabinoids: evidence for a site distinct from CB1 and CB2. Mol Pharmacol. 2008;73:441–450. doi: 10.1124/mol.107.041863. [DOI] [PubMed] [Google Scholar]
- Mestre L, Inigo PM, Mecha M, Correa FG, Hernangomez-Herrero M, Loria F, et al. Anandamide inhibits Theiler's virus induced VCAM-1 in brain endothelial cells and reduces leukocyte transmigration in a model of blood brain barrier by activation of CB(1) receptors. J Neuroinflammation. 2011;8:102. doi: 10.1186/1742-2094-8-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysiorek C, Culot M, Dehouck L, Derudas B, Staels B, Bordet R, et al. Peroxisome-proliferator-activated receptor-alpha activation protects brain capillary endothelial cells from oxygen-glucose deprivation-induced hyperpermeability in the blood-brain barrier. Curr Neurovasc Res. 2009;6:181–193. doi: 10.2174/156720209788970081. [DOI] [PubMed] [Google Scholar]
- Naccarato M, Pizzuti D, Petrosino S, Simonetto M, Ferigo L, Grandi FC, et al. Possible anandamide and palmitoylethanolamide involvement in human stroke. Lipids Health Dis. 2010;9:47. doi: 10.1186/1476-511X-9-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagasawa K, Chiba H, Fujita H, Kojima T, Saito T, Endo T, et al. Possible involvement of gap junctions in the barrier function of tight junctions of brain and lung endothelial cells. J Cell Physiol. 2006;208:123–132. doi: 10.1002/jcp.20647. [DOI] [PubMed] [Google Scholar]
- O'Sullivan SE. Cannabinoids go nuclear: evidence for activation of peroxisome proliferator-activated receptors. Br J Pharmacol. 2007;152:576–582. doi: 10.1038/sj.bjp.0707423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panikashvili D, Shein NA, Mechoulam R, Trembovler V, Kohen R, Alexandrovich A, et al. The endocannabinoid 2-AG protects the blood-brain barrier after closed head injury and inhibits mRNA expression of proinflammatory cytokines. Neurobiol Dis. 2006;22:257–264. doi: 10.1016/j.nbd.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG, Howlett AC, Abood ME, Alexander SP, Di Marzo V, Elphick MR, et al. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB(1) and CB(2) Pharmacol Rev. 2010;62:588–631. doi: 10.1124/pr.110.003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez SH, Hasko J, Skuba A, Fan S, Dykstra H, McCormick R, et al. Activation of cannabinoid receptor 2 attenuates leukocyte-endothelial cell interactions and blood-brain barrier dysfunction under inflammatory conditions. J Neurosci. 2012;32:4004–4016. doi: 10.1523/JNEUROSCI.4628-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynders V, Loitsch S, Steinhauer C, Wagner T, Steinhilber D, Bargon J. Peroxisome proliferator-activated receptor alpha (PPAR alpha) down-regulation in cystic fibrosis lymphocytes. Respir Res. 2006;30:104. doi: 10.1186/1465-9921-7-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders CI, Fassett RG, Geraghty DP. Up-regulation of TRPV1 in mononuclear cells of end-stage kidney disease patients increases susceptibility to N-arachidonoyl-dopamine (NADA)-induced cell death. Biochim Biophys Acta. 2009;1792:1019–1026. doi: 10.1016/j.bbadis.2009.07.008. [DOI] [PubMed] [Google Scholar]
- Schabitz WR, Giuffrida A, Berger C, Aschoff A, Schwaninger M, Schwab S, et al. Release of fatty acid amides in a patient with hemispheric stroke: a microdialysis study. Stroke. 2002;33:2112–2114. doi: 10.1161/01.str.0000023491.63693.18. [DOI] [PubMed] [Google Scholar]
- Schley M, Ständer S, Kerner J, Vajkoczy P, Schüpfer G, Dusch M, et al. Predominant CB2 receptor expression in endothelial cells of glioblastoma in humans. Brain Res Bull. 2009;79:333–337. doi: 10.1016/j.brainresbull.2009.01.011. [DOI] [PubMed] [Google Scholar]
- Snider NT1, Nast JA, Tesmer LA, Hollenberg PF. A cytochrome P450-derived epoxygenated metabolite of anandamide is a potent cannabinoid receptor 2-selective agonist. Mol Pharmacol. 2009;75:965–972. doi: 10.1124/mol.108.053439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinsanti G, Zannolli R, Panti C, Ceccarelli I, Marsili L, Bachiocco V, et al. Quantitative real-time PCR detection of TRPV1-4 gene expression in human leukocytes from healthy and hyposensitive subjects. Mol Pain. 2008;4:51. doi: 10.1186/1744-8069-4-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sughrue ME, Mehra A, Connolly ES, Jr, D'Ambrosio AL. Anti-adhesion molecule strategies as potential neuroprotective agents in cerebral ischemia: a critical review of the literature. Inflamm Res. 2004;53:497–508. doi: 10.1007/s00011-004-1282-0. [DOI] [PubMed] [Google Scholar]
- Sun Y, Alexander SP, Garle MJ, Gibson CL, Hewitt K, Murphy SP, et al. Cannabinoid activation of PPAR alpha; a novel neuroprotective mechanism. Br J Pharmacol. 2007;152:734–743. doi: 10.1038/sj.bjp.0707478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JM, Krey JF, Chu CJ, Huang SM. Endocannabinoids and related fatty acid derivatives in pain modulation. Chem Phys Lipids. 2002;121:159–172. doi: 10.1016/s0009-3084(02)00152-4. [DOI] [PubMed] [Google Scholar]
- Wang Q, Peng Y, Chen S, Gou X, Hu B, Du J, et al. Pretreatment with electroacupuncture induces rapid tolerance to focal cerebral ischemia through regulation of endocannabinoid system. Stroke. 2009;40:2157–2164. doi: 10.1161/STROKEAHA.108.541490. [DOI] [PubMed] [Google Scholar]
- Watanabe H1, Vriens J, Prenen J, Droogmans G, Voets T, Nilius B. Anandamide and arachidonic acid use epoxyeicosatrienoic acids to activate TRPV4 channels. Nature. 2003;424:434–438. doi: 10.1038/nature01807. [DOI] [PubMed] [Google Scholar]
- Wojtalla A, Herweck F, Granzow M, Klein S, Trebicka J, Huss S, et al. The endocannabinoid N-arachidonoyl dopamine (NADA) selectively induces oxidative stress-mediated cell death in hepatic stellate cells but not in hepatocytes. Am J Physiol Gastrointest Liver Physiol. 2012;302:G873–G887. doi: 10.1152/ajpgi.00241.2011. [DOI] [PubMed] [Google Scholar]
- Zhang H1, Hilton DA, Hanemann CO, Zajicek J. Cannabinoid receptor and N-acyl phosphatidylethanolamine phospholipase D–evidence for altered expression in multiple sclerosis. Brain Pathol. 2011;21:544–557. doi: 10.1111/j.1750-3639.2011.00477.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Yang L, Ma A, Zhang X, Li W, Yang W, et al. Orally administered oleoylethanolamide protects mice from focal cerebral ischemic injury by activating peroxisome proliferator-activated receptor alpha. Neuropharmacology. 2012;63:242–249. doi: 10.1016/j.neuropharm.2012.03.008. [DOI] [PubMed] [Google Scholar]
- Zygmunt PM1, Petersson J, Andersson DA, Chuang H, Sørgård M, Di Marzo V, et al. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400:452–457. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]