

Abstract
Background and Purpose
Angiotensin II (AngII) and IL-1β are involved in cardiovascular diseases through the induction of inflammatory pathways. HuR is an adenylate- and uridylate-rich element (ARE)-binding protein involved in the mRNA stabilization of many genes. This study investigated the contribution of HuR to the increased expression of COX-2 induced by AngII and IL-1β and its consequences on VSMC migration and remodelling.
Experimental Approach
Rat and human VSMCs were stimulated with AngII (0.1 μM) and/or IL-1β (10 ng·mL−1). Mice were infused with AngII or subjected to carotid artery ligation. mRNA and protein levels were assayed by quantitative PCR, Western blot, immunohistochemistry and immunofluorescence. Cell migration was measured by wound healing and transwell assays.
Key Results
In VSMCs, AngII potentiated COX-2 and tenascin-C expressions and cell migration induced by IL-1β. This effect of AngII on IL-1β-induced COX-2 expression was accompanied by increased COX-2 3′ untranslated region reporter activity and mRNA stability, mediated through cytoplasmic HuR translocation and COX-2 mRNA binding. These effects were blocked by ERK1/2 and HuR inhibitors. VSMC migration was reduced by blockade of ERK1/2, HuR, COX-2, TXAS, TP and EP receptors. HuR, COX-2, mPGES-1 and TXAS expressions were increased in AngII-infused mouse aortas and in carotid-ligated arteries. AngII-induced tenascin-C expression and vascular remodelling were abolished by celecoxib and by mPGES-1 deletion.
Conclusions and Implications
The synergistic induction of COX-2 by AngII and IL-1β in VSMCs involves HuR through an ERK1/2-dependent mechanism. The HuR/COX-2 axis participates in cell migration and vascular damage. HuR might be a novel target to modulate vascular remodelling.
Tables of Links
| LIGANDS | |
|---|---|
| 6,16-dimethyl PGE2 | PD98059 |
| AG1478 (tyrphostin) | PGE2 |
| Angiotensin II | PGH2 |
| Arachidonic acid | PGI2 |
| Calmodulin | SB203580 |
| Celecoxib | SP600125 |
| Chelerythrine | SQ29548 |
| IL-1β | U0126 |
| L798106 | U46619 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,cAlexander et al., 2013a,b,c,,).
Introduction
The constitutive and the inducible COXs, COX-1 and COX-2, respectively, are expressed in the vasculature where they convert arachidonic acid into an endoperoxide intermediate, PGH2, which is immediately metabolized by different prostaglandin synthases (PGS) into specific prostanoids. COX-2 is the dominant source of PGs in inflammatory vascular diseases, such as atherosclerosis (Cipollone and Fazia, 2006), balloon-injured arteries (Yang et al., 2004,2004) and hypertension (Martínez-Revelles et al., 2013). Angiotensin II (AngII) is a key player in vascular damage through its significant pro-inflammatory actions, including the production of cytokines (Marchesi et al., 2008) such as IL-1β. Both AngII and cytokines induce COX-2 expression at the vascular level (Ohnaka et al., 2000; Galán et al., 2011; Martín et al., 2012).
COX-2 expression in response to pro-inflammatory stimuli is initiated through transcriptional induction (Harper and Tyson-Capper, 2008). As a means of controlling COX-2 expression, post-transcriptional mechanisms are in place that target the mRNA for rapid degradation. A key feature present in the COX-2 3′ untranslated region (3′UTR) that is involved in the control of mRNA degradation is the adenylate- and uridylate-rich element (ARE; Dixon et al., 2000). HuR protein (Hu antigen R; ELAVL1) is an ARE RNA-binding protein that is primarily localized in the nucleus and contributes to the stabilization of the mRNA of many genes when exported to the cytoplasm (Abdelmohsen and Gorospe, 2010). Thus, HuR is involved in a wide variety of physiological and pathological processes, such as cell growth, differentiation and inflammation (Abdelmohsen and Gorospe, 2010). In previous work, we identified COX-2 mRNA as a HuR target in intestinal cancer cells and in myeloid leukocytes (Dixon et al., 2001; 2006,; Young et al., 2012). Similarly, in human mesangial cells, AngII increases the stability of COX-2 mRNA through HuR (Doller et al., 2008). However, the ability of HuR to influence COX-2 expression in vascular smooth muscle cells (VSMCs) has not yet been explored.
PGs downstream of COX-2 participate in pathological vascular remodelling (Wang et al., 2011; Sparks et al., 2013; Zhang et al., 2013). The main COX-derived prostanoids responsible for this effect are PGE2, derived from inducible microsomal PGE2 synthase-1 (mPGES-1), and TXA2, as they promote cell proliferation and migration whereas PGI2 has an opposing effect (Nie et al., 2000; Bos et al., 2004; Wang et al., 2011; Zhang et al., 2013). The influence of PGE2 on cell migration appears to be mediated, in part, by the inducible extracellular matrix (ECM) glycoprotein, tenascin-C (TN-C) (Wang et al., 2011). However, whether HuR is involved in these effects of COX-2-derived prostanoids upon cell migration and vascular damage is currently unknown.
In the present study we investigated: (i) the participation of HuR in AngII and IL-1β-induced COX-2 expression in VSMCs and (ii) the contribution of HuR to the effects of COX-2-derived prostanoids on cell migration and vascular remodelling. We demonstrated that the combination of AngII + IL-1β induces HuR-dependent stabilization of COX-2 mRNA leading to increased prostanoid-mediated cell migration. This effect could contribute to the potentiation of the vascular remodelling triggered by AngII in inflamed vascular beds.
Methods
All experimental procedures were approved by the Ethical Committees of Research of the Universidad Autónoma de Madrid and University of Kansas Medical Center and by the Reviewer Institutional Committee on Human Research of the Hospital de la Santa Creu i Sant Pau and conform to the Declaration of Helsinki. These studies were conducted in accordance with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996) and with guidelines for ethical care of experimental animals of the European Community, the current Spanish and European laws (RD 223/88 MAPA and 609/86). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Cell culture
Primary cultures of VSMCs were obtained from aortas isolated from Sprague Dawley rats and grown in DMEM-F12 medium supplemented with 10% FBS containing 100 U·mL−1 of penicillin, 100 μg·mL−1 of streptomycin as previously reported (Aguado et al., 2013). Human VSMCs were obtained from coronary arteries of hearts removed in transplant operations using a modification of the explant technique (Orriols et al., 2014). Human VSMCs were grown in M199 supplemented with 20% FBS, 1% human serum, 2 mM of L-glutamine, 100 U·mL−1 of penicillin and 100 μg·mL−1 of streptomycin. Cells were serum-starved in serum-free media for 24 h before being stimulated. All products were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Cells were stimulated with vehicle (control), AngII (0.1 μM; Sigma-Aldrich), IL-1β (10 ng·mL−1; Sigma-Aldrich), AngII + IL-1β, 16,16-dimethyl PGE2 (Sigma-Aldrich) or U46619 (Cayman Chemical, Ann Arbor, MI, USA) at the times and concentrations indicated in the Results section. The effects of the following inhibitors were analysed by adding them 30 min (or 4 h in the case of MS-444) before and throughout stimulation: L798106, SC19220, actinomycin D (Sigma-Aldrich); U0126, SP600125, LY294002, SB203580, N-(6-aminohexyl)-5-chloro-1-naphthalenesulfonamide (W7), N-(4-aminobutyl)-1-naphthalenesulfonamide (W12), KN93 and PD98059 (Calbiochem, Darmstadt, Germany); tyrphostin AG 1478 (AlomoneLabs, Jerusalem, Israel); chelerythrine (Tocris, Biogen Científica, Spain); SQ29548 (ICN Iberica, Barcelona, Spain); furegrelate (Cayman Chemical); celecoxib (generously provided by Pfizer Inc., Groton, CT, USA), ozagrel (generously provided by Kissei Pharmaceutical CO, Matsumoto, Japan); MS-444 (generously provided by Novartis, Basel, Switzerland). Compounds used for stimuli and ozagrel were dissolved in distilled water; the remaining compounds were dissolved in DMSO. None of these inhibitors affected the basal expression of COX-2, mPGES-1, TXA2 synthase (TXAS), prostacyclin synthase (PGIS) or TN-C (data not shown). MS-444 was originally characterized as a myosin light chain (MLC) kinase inhibitor in the μM range (Aotani and Saitoh, 1995); however, the concentrations used in these experiments did not affect myosin light chain phosphorylation induced by AngII + IL-1β or cell viability (data not shown).
RNA analysis
Cells were harvested in TRI reagent (Sigma-Aldrich) or TRIzol (Life Technologies, Inc., Gaithersburg, MD, USA) according to the manufacturer's recommendations to obtain total RNA and reverse transcribed using a high capacity cDNA archive kit (Life Technologies, Inc.) with random hexamers. Quantitative PCR (qPCR) was performed in a 7500 Fast ABI System (Life Technologies, Inc.). qPCR for human COX-2, mPGES-1, PGIS, TXAS and 18S rRNA, along with rat and mouse TXAS, were performed using Taqman gene expression assays (COX-2: Hs00153133_m1; mPGES-1: Hs00610420_m1; PGIS: Hs00168766_m1; TXAS: Hs01022706_m1; 18S rRNA: Hs99999901_s1 in humans; TXAS: Rn00562160_m1 in rats, TXAS: Mm00495553_m1 in mice; Life Technologies, Inc.). qPCR for COX-2, mPGES-1, HuR, TN-C and β2-microglobulin in rats and mice and PGIS in rats were performed using SYBR green PCR master mix (iTaq FAST SYBRGreen Supermix with ROX, Bio-Rad, Hercules, CA, USA). Primer sequence information is listed on Supporting Information Table S1. PCR cycles proceeded as described (Martínez-Revelles et al., 2013). Data analyses used the 2−ΔΔCt method, where β2-microglobulin or 18S rRNA served as the internal control.
Determination of COX-2 mRNA stability in stimulated cells was initiated by adding 5 μg·mL−1 actinomycin D (Sigma-Aldrich) to the medium during the specified times. Total RNA was isolated and COX-2 and β2-microglobulin mRNA expression levels were measured by qPCR as indicated earlier.
Western blot analysis
Whole-cell lysates were harvested in RIPA buffer containing: 50 mM Tris pH 7.5, 150 mM NaCl, 1 mM MgCl2, 1 mM EDTA, 1% Nonidet-P40 (NP-40), 0.5% deoxycholate Na, 1% SDS, a protease inhibitor cocktail (Roche Applied Science, Barcelona, Spain) and a mix of phosphatase inhibitors (1 mM orthovanadate, 20 mM β-glycerophosphate, 10 mM NaF from Sigma-Aldrich). For cellular fractionation, cells were lysed in hypotonic lysis buffer (10 mM HEPES pH 8, 3 mM MgCl2, 40 mM KCl, 0.2% NP-40, 10% glycerol, 0.1 mM DTT with protease and phosphatase inhibitors) for 15 min at 4°C. Samples were centrifuged 2 min at 9300× g to separate cytoplasmic supernatant from nuclei. Nuclei were washed four times in hypotonic buffer and lysed in RIPA buffer. Samples were then centrifuged 10 min at 9300× g to obtain the nuclear fraction. Protein content was determined with Lowry (Bio-Rad) or bicinchoninic acid protein assay reagent (Pierce, Rockford, IL, USA), using BSA (Sigma-Aldrich or Pierce) as standard. Lysates (20–40 μg) were separated by 10% SDS-PAGE, transferred to PVDF or nitrocellulose membranes (Amersham, GE Healthcare, Buckinghamshire, UK) and probed with antibodies against HuR [1:1000 (sc5261); Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA], Nucleoporin p62 (1:5000; BD Bioscience, San Jose, CA, USA), COX-2 and mPGES-1 (1:200; Cayman Chemical), PGIS (1:1000; Cayman Chemical), MLC kinase, p-MLC kinase, p-ERK1/2, ERK1/2, p-p38, p38 MAPK, p-JNK, JNK, p-Akt and Akt (1:1000; Cell Signaling, Boston, MA, USA). Blots were stripped and then probed with antibodies against β-actin (1:50 000; Sigma-Aldrich) or tubulin (1:10 000; Sigma-Aldrich). Detection was accomplished using HRP-coupled anti-rabbit (1:2000; Bio-Rad) or anti-mouse (1:5000; Stressgen, Victoria, Canada) IgG antibodies for 1 h at room temperature. The signal was detected using the Luminata Forte (Millipore Corporation, Billerica, MA, USA) detection system. Immunoblot signals were quantified using NIH ImageJ using β-actin or tubulin expressions as loading controls.
Ribonucleoprotein complex immunoprecipitation
Ribonucleoprotein complex immunoprecipitation was performed as described previously (Lal et al., 2004). Briefly, cells were lysed in polysome lysis buffer (20 mM Tris-HCl pH 7.5, 150 mM NaCl, 5 mM MgCl2, 1 mM DTT, 0.5% NP-40, 1 mM PMSF, protease inhibitors, 100 U RNAse inhibitor). Two hundred micrograms of cytoplasmic lysate were incubated with anti-HuR antibody or control IgG precoated to protein A/G PLUS agarose (Santa Cruz Biotechnology) overnight at 4°C in NT2 buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM MgCl2, 0.05% NP-40) adding 0.1 mM DTT, 40 U RNAse inhibitor (Promega, Mannheim, Germany), 5 mM EDTA and 100 μg·mL−1 tRNA (Life Technologies, Inc.). Immunoprecipitates were collected by centrifugation and washed four times with NT2 buffer. Total RNA was isolated from immunoprecipitates using 1 mL TRIzol reagent and then used for cDNA synthesis and qPCR as described earlier.
DNA and siRNA transfections
Luciferase reporter constructs containing 3.9 kb of human COX-2 promoter (pGL3-COX-2P) or the COX-2 3′UTR (from 3074 to 3236) (pGL3P-UTR2) were kindly provided by Dr W. Eberhardt (Klinikum der Johann Wolfgang Goethe-Universitat Frankfurt am Main, Germany). Cells were seeded in 12-well culture plates at a density of 6 × 104 cells per well and transiently transfected with 0.5 μg luciferase reporter gene construct per well using the Lipofectamine LTX Plus™ Reagent (Life Technologies, Inc.). After 6 h, media were changed and cells were transferred to depletion medium overnight. After stimulation, cells were harvested in reporter lysis buffer and luciferase activities were measured using a luminometer (Orion I, Berthold Detection Systems GmbH, Titertek-Berthold, Pforzheim, Germany) according to the manufacturer's instructions. Relative luciferase activities were normalized to total protein.
In another set of experiments, cells seeded in 6-well plates or transwells at a density of 1.2 × 105 or 3 × 104 cells per well, respectively, were transfected with a predesigned siRNA against HuR (Life Technologies Inc.) or a negative control siRNA (Qiagen-Izasa, Barcelona, Spain) using the Lipofectamine LTX Plus reagent (Life Technologies Inc.) according to the manufacturer's protocol. Cells were transfected for 48 h with 20 nM siRNA and silencing efficiency was determined by Western blot.
Migration assays
VSMC migration was examined using a wound healing assay. VSMC monolayers were wounded using a sterile 10 μL pipette tip. Phase contrast images were taken immediately after wounding and at 24 h post-stimulation using a Nikon microscope (Tokyo, Japan) connected to a video camera (Sony Corporation, Tokyo, Japan). To measure migration, wound area was quantified using Adobe Photoshop (Adobe Systems Inc., San Jose, CA, USA) and expressed as percentage of wound closure.
Cell migration was also determined using a 6.5 mm transwell chamber with an 8 μm pore size (Corning Costar Inc., Corning, New York, NY, USA). Cells, 3 × 104, were seeded in the upper compartment of each chamber and after 16 h of serum deprivation, inhibitors were added in the upper chamber and the stimuli were added in the bottom chamber. After 5 h, cells were removed from the upper membrane surface by a cotton swab and washed with PBS. Migration values were determined by counting three fields per chamber after fixing the membrane in 4% paraformaldehyde and staining with Hoechst 33342.
Animal models
The following animal models were used: (i) C57BL/6J male mice with permanent ligation of the left common carotid artery for 3 weeks, as described previously (Rodríguez-Calvo et al., 2013); (ii) C57BL/6J male mice infused with saline, with AngII (1.44 mg·kg−1·day−1, 2 weeks, s.c. by osmotic minipumps, Alza Corp., Cupertino, CA, USA) or with AngII plus celecoxib (25 mg·kg−1·day−1 i.p. started 24 h before AngII infusion), as described previously (Martínez-Revelles et al., 2013); and (iii) mPGES-1 wild-type (WT) and mPGES-1-deficient (mPGES-1−/−) mice infused with saline or with AngII. WT and mPGES-1−/− mice were maintained in a DBA/1 genetic background and were generated by corresponding heterozygous breeders derived from mPGES-1+/− embryos that were a kind gift from Pfizer. The total number of mice used were 45.
Immunofluorescence microscopy
Cells or frozen transverse aortic sections (14 μm) were rinsed with PBS, fixed with 2% or 4% paraformaldehyde (Sigma-Aldrich), respectively, permeabilized with 0.2% Triton-PBS, blocked with 3% IgG-free BSA in PBS and incubated overnight at 4°C with anti-HuR monoclonal primary antibody diluted 1:200 in blocking solution. Detection was performed using fluorescein isothiocyanate-conjugated secondary IgG (1:200; Jackson ImmunoResearch, West Grove, PA, USA) diluted in blocking buffer and incubated for 1 h at room temperature, after which the cells were counterstained with Hoechst dye 33342 (Life Technologies Inc.). Slides were examined using an Evos XL Cell Imaging System (Life Technologies, Inc.) or a Leica TCS SP2 confocal microscope with a 40× objective (Leica Microsystems, Wetzlar, Germany). Images were processed using Adobe Photoshop CS3 software (Adobe Systems Inc.).
Histological analysis
Histological analysis was performed as described previously (Avendaño et al., 2014). Briefly, cross-sections from fixed aortas were stained with haematoxylin and eosin. Morphometric determinations of the lumen and vessel areas were performed using Metamorph image analysis software (Universal Imaging, Molecular Devices Corp., Downingtown, PA, USA).
Immunohistochemistry
Immunohistochemistry was performed as described previously (Orriols et al., 2014). Briefly, mouse paraffin-fixed carotids were prepared in cross-sections with a microtome (Jung RM 2055, Leica). After deparaffinization, rehydration and hydrogen peroxide treatment, sections were blocked and incubated with anti-HuR (1:100), anti-COX-2 (1:100), anti-mPGES-1 (1:100) and anti-TXAS (1:50) antibodies. After that, sections were incubated with a biotinylated secondary antibody (Vector Laboratories, Burlingame, CA, USA). Colour was developed using 3,3′-diaminobenzidine and sections were counterstained with haematoxylin. Negative controls in which the primary antibody was omitted were included to test for non-specific binding.
Measurement of PGE2 production
The levels of PGE2 metabolites were determined in plasma of mice using an enzyme immunoassay commercial kit (Cayman Chemical). The plasma was kept at −70°C until analysis and processed following the manufacturer's instructions.
Data analysis and statistical procedures
All values are expressed as mean ± SEM of the number animals used in each experiment or independent cell culture-based experiments. Statistical analysis was performed using Student's t-test, Mann–Whitney test or by one-way anova followed by a Bonferroni test. Values were considered to be significant when P < 0.05.
Results
AngII and IL-1β synergistically induce COX-2 expression in VSMCs through the ERK1/2 pathway
In basal conditions, rat VSMCs express low-to-undetectable COX-2 mRNA and protein levels. After AngII stimulation, the levels of expression of COX-2 mRNA and protein increased reaching a maximum at 1 h, and at 24 h had returned to the basal levels. The exposure of cells to IL-1β increased COX-2 mRNA and protein, levels reached a plateau at 2 and 8 h, respectively; however, co-stimulation of rat VSMCs with AngII + IL-1β led to a synergistic increase in COX-2 mRNA and protein. This increased induction of COX-2 was observed to occur as early as 1 h after stimulation and persisted for up to 24 h (Figure 1A,B). Consistently, as shown in Figure 1C,D 24 h after stimulation of rat VSMCs, IL-1β increased COX-2, mRNA and protein levels, whereas AngII only slightly increased mRNA levels. However, at this time, AngII strongly enhanced both COX-2 mRNA and protein levels induced by IL-1β. Similar results were obtained in human VSMCs after 24 h stimulation (Supporting Information Fig. S1A,B) indicating that this effect is conserved and further support the importance of this finding.
Figure 1.

AngII potentiates IL-1β-induced COX-2 expression in VSMCs through ERK1/2. Time course of COX-2 mRNA (A) and protein (B) expression in rat VSMCs. Representative Western blots of five independent experiments are shown. Effects of AngII, IL-1β and AngII + IL-1β (24 h) on COX-2 mRNA (C) and protein expression (D) in rat VSMCs. (E) Time course of ERK1/2, JNK, p38 and Akt activation by AngII + IL-1β. (F) Effect of inhibitors of ERK1/2 (U0126, U), JNK (SP600125, SP), p38 MAPK (SB203580, SB) and PI3K (LY294002, LY) on COX-2 mRNA levels induced by AngII + IL-1β. Data are expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 versus control; †P < 0.05, ††P < 0.01 versus AngII and IL-1β; ‡‡‡P < 0.001 versus AngII + IL-1β. n = 4–9.
AngII + IL-1β promoted rapid phosphorylation of ERK1/2, JNK, p38 MAPK and Akt as soon as 5 min after treatment (Figure 1E). Interestingly, only the ERK1/2 inhibitor U0126 (10 μM) was able to decrease COX-2 mRNA expression observed with AngII + IL-1β treatment, whereas the JNK (SP600125, 20 μM), p38 MAPK (SB203580, 10 μM) and PI3K (LY294002, 10 μM) inhibitors did not (Figure 1F). These results suggest that ERK1/2 is the main pathway involved in AngII + IL-1β-induced COX-2 expression.
HuR is involved in the stabilization of COX-2 mRNA induced by AngII in VSMCs
COX-2 expression is regulated by both transcriptional and post-transcriptional mechanisms (Harper and Tyson-Capper, 2008). To determine whether the effect of AngII on IL-1β-induced COX-2 expression was due to changes in transcription, COX-2 transcriptional activity was measured in rat VSMCs transfected with a reporter plasmid containing the human COX-2 promoter fused to luciferase cDNA. IL-1β increased COX-2 promoter activity and the combination of AngII + IL-1β resulted in a similar increase in transcriptional activity (Figure 2A). These results suggested that post-transcriptional regulatory mechanisms are the primary effector of the synergistic induction of COX-2 expression induced by AngII + IL-1β. To test this hypothesis, we performed mRNA stability assays. Figure 2B shows that AngII stabilized COX-2 mRNA in IL-1β-stimulated VSMCs. We also performed transfections of VSMCs using a luciferase reporter plasmid containing a 150 bp 3′UTR from COX-2 mRNA, which included the binding site for HuR. Figure 2C shows that both stimuli were needed to increase COX-2 3′UTR luciferase activity.
Figure 2.
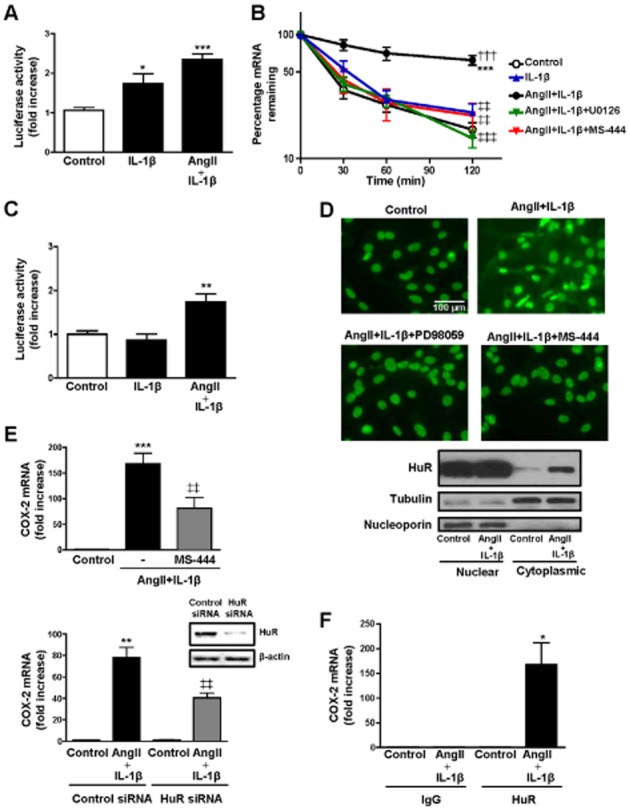
The synergistic effect of AngII and IL-1β on COX-2 expression in VSMCs is due to an increase in COX-2 mRNA stability mediated by HuR. (A) Luciferase activity in VSMCs transfected with a luciferase reporter construct containing the COX-2 promoter and incubated with vehicle (control), IL-1β or AngII + IL-1β for 4 h. (B) Effect of AngII + IL-1β on COX-2 mRNA stability (24 h after stimulation) and the inhibition of this effect by ERK1/2 (U0126) and HuR (MS-444) inhibitors. Actinomycin D, a transcriptional inhibitor, was incubated during the indicated times, after which COX-2 mRNA levels were measured by qPCR. (C) Luciferase activity in VSMCs transfected with a luciferase reporter construct containing COX-2 3′-UTR and incubated with vehicle (control), IL-1β or AngII + IL-1β for 4 h. (D) Effect of PD98059 (ERK1/2 inhibitor) and MS-444 on HuR subcellular localization assayed by HuR immunofluorescence and Western blotting of cellular fractions from VSMCs stimulated with AngII + IL-1β for 24 h. (E) Effects of MS-444 and HuR siRNA on COX-2 mRNA levels in VSMCs stimulated with AngII + IL-1β for 24 h. Representative blots of six independent experiments of HuR expression in cells transfected with control siRNA or HuR siRNA are included. (F) Ribonucleoprotein immunoprecipitation of HuR or control IgG was performed to isolate mRNA bound by HuR in VSMCs stimulated with AngII + IL-1β for 24 h. Data are expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 versus control; †††P < 0.001 versus IL-1β; ‡‡P < 0.01, ‡‡‡P < 0.001 versus AngII + IL-1β. n = 4–8.
In unstressed cells, HuR is predominantly localized to the nucleus and can be exported to the cytoplasm to stabilize different mRNAs including COX-2 mRNA (Dixon et al., 2001; Young et al., 2012). As shown in Figure 2D, treatment of VSMCs with AngII + IL-1β increased the HuR located in the cytoplasm and this effect was abolished by ERK1/2 inhibition (+PD98059, 20 μM). As expected, treatment of AngII + IL-1β-stimulated VSMCs with MS-444 (8 μM), a low-molecular weight inhibitor of HuR that prevents cytoplasmic trafficking of HuR by interfering with its homodimerization (Meisner and Filipowicz, 2011), yielded similar effects. Consistent with these observations on HuR trafficking, another ERK1/2 inhibitor (U0126, 10 μM) and MS-444 also abolished the increased stability of COX-2 mRNA observed in AngII + IL-1β-stimulated VSMCs (Figure 2B). Moreover, MS-444 or HuR knockdown by siRNA reduced the increased COX-2 mRNA levels induced by AngII + IL-1β (Figure 2E). We then determined if AngII + IL-1β signalling promotes increased cytoplasmic HuR binding to COX-2 mRNA by ribonucleoprotein immunoprecipitation using an antibody against HuR or control IgG. We observed an enrichment in COX-2 mRNA in anti-HuR precipitates from cells treated with AngII + IL-1β, but not in control cells or in anti-IgG precipitates (Figure 2F). GAPDH mRNA co-precipitation was used as a control to assess the non-specific background (data not shown).
Taken together, these findings suggest that AngII + IL-1β induce an ERK1/2-dependent activation of HuR, which is responsible for increased COX-2 mRNA stability and COX-2 levels in VSMCs.
Effect of AngII and IL-1β on mPGES-1, PGIS and TXAS expression in rat and human VSMCs
To assess whether prostanoid synthases were also regulated by AngII, IL-1β or the combination of both, we measured mPGES-1, PGIS and TXAS mRNA and protein levels.
IL-1β treatment led to a time-dependent increase in mPGES-1 mRNA levels, whereas AngII treatment decreased mPGES-1 mRNA (Supporting Information Fig. S2A). Accordingly, AngII + IL-1β increased mPGES-1 mRNA levels less than IL-1β alone. Similar results were observed when mPGES-1 mRNA and protein levels were only examined at 24 h stimulation (Figure 3A,B). In contrast to mPGES-1 expression, PGIS mRNA and protein levels were significantly increased by AngII and not affected by IL-1β (Figure 3C,D and Supporting Information Fig. S2B); the effects of AngII + IL-1β on PGIS expression were similar to those produced by AngII alone. TXAS mRNA levels were increased in a time-dependent manner by IL-1β, AngII and AngII + IL-1β (Figure 3E and Supporting Information Fig. S2C). Similar results were observed on the protein levels (Figure 3F). Human VSMCs showed a pattern similar to rat VSMCs as regards the changes observed in mPGES-1, PGIS and TXAS mRNA levels when cells were exposed for 24 h to AngII and/or IL-1β, except that PGIS that was increased by IL-1β in human cells, and human mPGES-1 protein expression was similarly regulated by these stimuli (Supporting Information Fig S1C–F).
Figure 3.
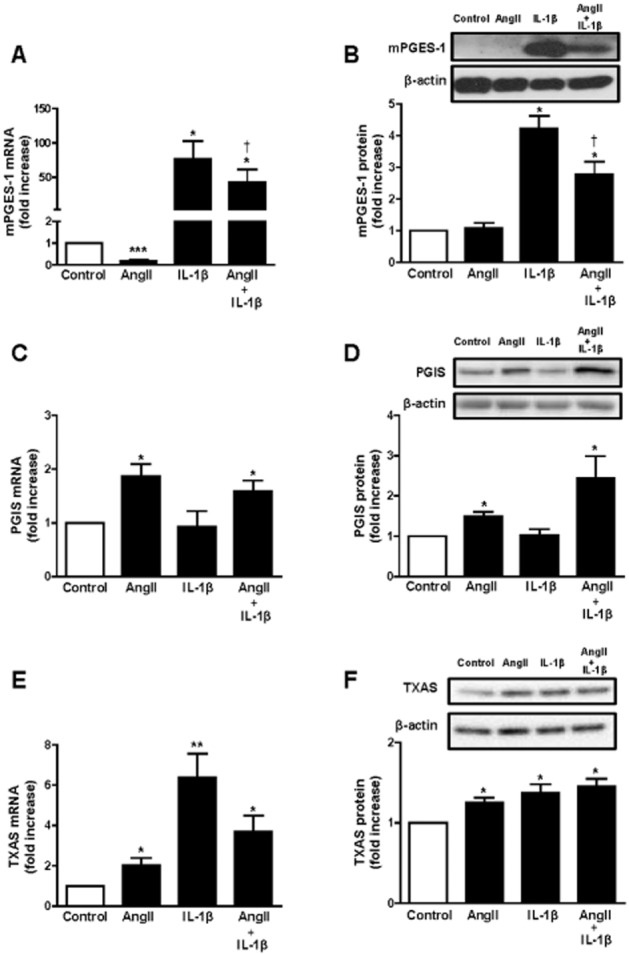
AngII and IL-1β differentially regulate mPGES-1, PGIS and TXAS expressions. Effect of AngII, IL-1β or AngII + IL-1β (24 h) on mPGES-1, PGIS and TXAS mRNA levels (A, C, E) and protein expression (B, D, F) in rat VSMC. Data are expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 versus control; †P < 0.05 versus AngII and IL-1β. n = 5–8.
The increased mPGES-1 mRNA levels observed after 24 h stimulation with AngII + IL-1β were blocked by ERK1/2 (U0126), JNK (SP600125), p38 MAPK (SB203580) and PI3K (LY294002) inhibitors (Supporting Information Fig. S3A). However, PGIS mRNA levels were diminished only by the ERK1/2 inhibitor (Supporting Information Fig. S3B). TXAS mRNA levels were reduced by ERK1/2, p38 MAPK and PI3K inhibitors (Supporting Information Fig. S3C). These results indicate that different signalling pathways are involved in the regulation in the expressions of mPGES-1, PGIS and TXAS.
COX-2-derived prostanoids mediate the synergistic effect of AngII and IL-1β on cell migration
Wound healing and transwell assays were used to study cell migration in rat VSMCs. Both AngII and IL-1β induced VSMC migration that was synergistically increased in the presence of both stimuli (Figure 4A). The addition of inhibitors against ERK1/2 (U0126, 10 μM), COX-2 (celecoxib, 10 μM), TXAS (furegrelate, 10 μM and ozagrel, 10 μM), TP receptor (SQ29548, 3 μM), EP1 receptor (SC19220, 10 μM), EP3 receptor (L798106, 1 μM), HuR (MS-444) and HuR siRNA reduced the ability of AngII + IL-1β to promote VSMC cell migration (Figure 4B,C). These results indicate that PGE2 and TXA2 derived from COX-2, along with ERK1/2 and HuR activities are involved in cell migration induced by AngII + IL-1β. In agreement with this, 16,16-dimethyl PGE2 (0.1–10 μM) or the TP agonist U46619 (0.01–1 μM) increased VSMC migration (Figure 5A–C), and these effects were abolished by their respective receptor antagonists SQ29548, SC19220 and L798106 (Figures 5D,E).
Figure 4.
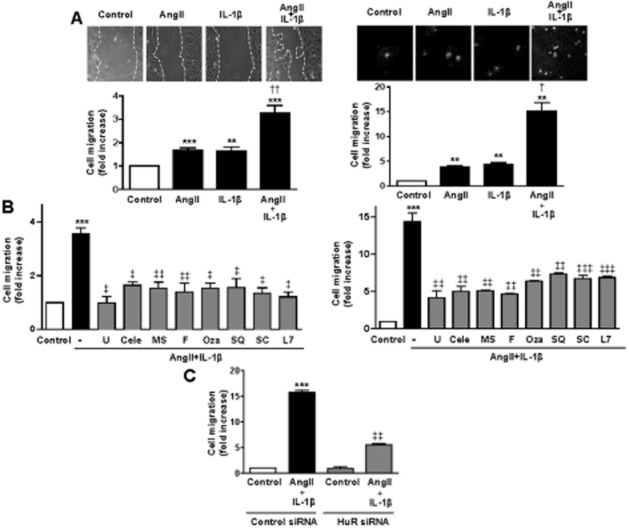
AngII + IL-1β potentiate cell migration through COX-2-derived PGE2 and TXA2. (A) Wound healing (left) and transwell (right) migration assays performed in rat VSMCs unstimulated (Control) and stimulated with AngII, IL-1β or AngII + IL-1β. (B) Wound healing (left) and transwell (right) assays of rat VSMC stimulated with AngII + IL-1β with and without inhibitors of ERK1/2 (U0126, U), COX-2 (celecoxib, Cele), HuR (MS-444, MS), TXAS (furegrelate, F; ozagrel, Oza), TP receptor (SQ29548, SQ), EP1 receptor (SC19220, SC) or EP3 receptor (L798106, L7). (C) Transwell assays of rat VSMC stimulated with AngII + IL-1β with HuR siRNA or control siRNA. Data are expressed as mean ± SEM. **P < 0.01, ***P < 0.001 versus control; †P < 0.05, ††P < 0.01 versus AngII and IL-1β. ‡P < 0.05, ‡‡P < 0.01, ‡‡‡P < 0.001 versus AngII + IL-1β. n = 3–15.
Figure 5.

PGE2 and TXA2 induce cell migration. (A,B) Wound healing assays performed in rat VSMCs unstimulated and stimulated with different concentrations of U46619 or PGE2. (C) Transwell migration assay of VSMCs unstimulated and stimulated with U46619 or PGE2. (D,E) Wound healing assays performed in rat VSMCs unstimulated and stimulated with U46619 or PGE2 with or without inhibitors of TP receptor (SQ29548, SQ), EP1 receptor (SC19220, SC), EP3 receptor (L798106, L7), ERK1/2 (U0126, U), JNK (SP600125, SP), p38 MAPK (SB203580, SB), PI3K (LY294002, LY), PKC (chelerythrine, Ch), CaM (W7, W12), pCaMKII (KN93, KN) or EGFR (AG1478, AG). Data are expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 versus control; †P < 0.05, ††P < 0.01, †††P < 0.001 versus AngII + IL-1β. n = 3–6.
To understand the signalling by which TXA2 and PGE2 mediate cell migration, VSMCs were pretreated with inhibitors targeting various kinase pathways: ERK1/2 (U0126), JNK (SP600125), p38 MAPK (SB203580), PI3K (LY294002), PKC (chelerythrine, 20 μM), calmodulin (CaM) (W7, 10 μM), pCaMKII (KN93, 20 μM) and EGFR (AG1478, 10 μM). Wound healing assays were then performed in the presence of the TP receptor agonist U46619 (0.1 μM) and 16,16-dimethyl PGE2 (1 μM). As a negative control, the less potent CaM inhibitor W12 (10 μM) was used. As shown in Figure 5D and E, only U0126, SP600125, W7, KN93 and AG1478 inhibited 16,16-dimethyl PGE2- and U46619-induced cell migration indicating the participation of ERK1/2, JNK, CaM, PKC and EGFR in such effects.
TN-C is expressed predominantly in pathological conditions and it is involved in cell migration (Wang et al., 2011; Yu et al., 2013). In addition, PGE2 induces its expression in the mouse model of femoral artery wire injury (Wang et al., 2011). We observed that TN-C mRNA levels were increased by AngII or IL-1β and this effect was potentiated in the presence of both stimuli (Figure 6A). The effect of AngII + IL-1β on TN-C mRNA levels was abolished by celecoxib, U0126, furegrelate, SC19220 and L798106 (Figure 6B).
Figure 6.
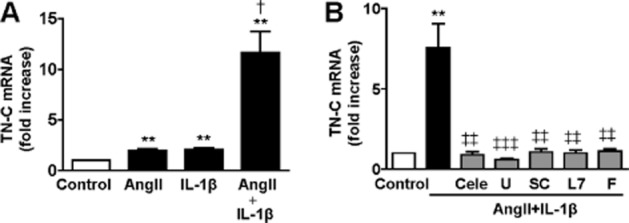
AngII + IL-1β potentiate TN-C mRNA levels in VSMCs through COX-2-derived PGE2 and TXA2. (A) Effect of AngII, IL-1β or AngII + IL-1β (24 h) on TN-C mRNA levels in rat VSMCs. (B) Effect of inhibitors of COX-2 (celecoxib, Cele), ERK1/2 (U0126, U), EP1 receptor (SC19220, SC), EP3 receptor (L798106, L7) or TXAS (furegrelate, F) on TN-C mRNA levels in rat VSMC stimulated with AngII + IL-1β (24 h). Data are expressed as mean ± SEM. **P < 0.01 versus control; †P < 0.05 versus AngII or IL-1β; ‡‡P < 0.01, ‡‡‡P < 0.001 versus AngII + IL-1β. n = 6.
Taken together, our results demonstrate that PGE2 and TXA2 derived from the COX-2 pathway participate in the effects induced by AngII and IL-1β on cell migration by mechanisms involving ERK1/2, HuR and TN-C.
Involvement of COX-2-derived prostanoids in vascular remodelling
To study the potential implications of the results obtained in cultured VSMCs, we used two models of vascular remodelling characterized by thickening of the vascular wall influenced by VSMC migration and inflammation. Increased expression of COX-2, mPGES-1, TXAS and HuR was observed in ligated carotid arteries compared with non-ligated controls (Figure 7A). Similar results were obtained in aorta from AngII-infused mice (Figure 7B). In agreement with these findings, plasma PGE2 levels were increased in the carotid ligation and AngII-infused mouse models (Supporting Information Fig. S4). Moreover, AngII-infused mice showed increased aortic TN-C expression that was reduced by pharmacological blockade of COX-2 with celecoxib and by mPGES-1 deletion (Figure 7C). Aorta from AngII-infused mice also showed increased media : lumen ratio and cross-sectional area (markers of hypertrophic remodelling) that was reduced by celecoxib treatment and mPGES-1 deletion (Figure 7D,E). These results suggest that COX-2- and mPGES-1-derived TXA2 and PGE2 have a role in TN-C expression and vascular hypertrophic remodelling.
Figure 7.
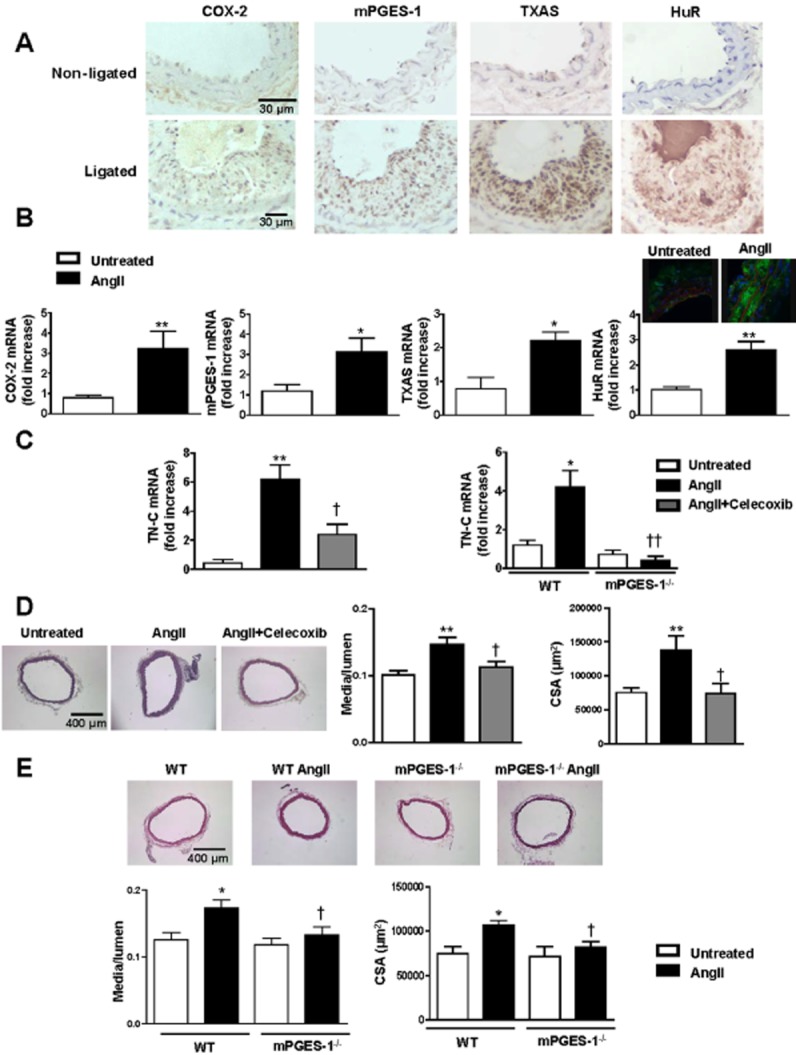
COX-2-derived PGE2 participates in vascular remodelling. (A) Representative immunohistochemical staining of three independent experiments of COX-2, mPGES-1, TXAS and HuR expression in non-ligated and ligated carotid arteries. (B) COX-2, mPGES-1, TXAS and HuR mRNA levels and HuR immunofluorescence in aorta from untreated or AngII-treated mice. (C) TN-C mRNA levels in aorta from mice untreated or treated with AngII or AngII plus celecoxib and mPGES-1+/+ (wild type, WT) and mPGES-1−/− mice infused or not with AngII. (D,E) Representative photographs of haematoxylin and eosin stained aortic sections, media/lumen ratio and cross-sectional area (CSA) from untreated or treated with AngII or AngII plus celecoxib-treated and from WT and mPGES-1−/− mice infused or not with AngII. Data are expressed as mean ± SEM. *P < 0.05, **P < 0.01 versus untreated; †P < 0.05, ††P < 0.01 versus AngII. n = 3–7.
Discussion and conclusions
In this study, we present evidence that the combination of two inflammatory stimuli, AngII and IL-1β, synergistically induce COX-2 expression and cell migration in VSMCs by a HuR-mediated increase in mRNA stability and this mechanism is involved in the vascular remodelling associated with cardiovascular diseases.
AngII and cytokines have been implicated in several cardiovascular pathologies through their inflammatory actions in the vascular wall (Marchesi et al., 2008). COX-2 is an early response gene overexpressed in pathological conditions associated with inflammatory processes, such as cancer (Dixon et al., 2001; Young et al., 2012), atherosclerosis (Cipollone and Fazia, 2006) and hypertension (Martínez-Revelles et al., 2013). In this study, we found that AngII and IL-1β synergistically increased COX-2 protein levels in VSMCs, as we previously demonstrated in vascular fibroblasts (Galán et al., 2011). More importantly, the effects of AngII + IL-1β on COX-2 expression in VSMCs had a functional consequence as the combination of both stimuli synergistically induced VSMC migration by a mechanism dependent on COX-2-derived prostanoids. The participation of COX-2-derived prostanoids in cell migration has been observed previously in VSMCs stimulated with AngII (Zhang et al., 2013).
COX-2 expression is tightly regulated by both transcriptional and post-transcriptional mechanisms (Dixon et al., 2000; Harper and Tyson-Capper, 2008; Young et al., 2012). Pham et al. (2008) demonstrated that the synergistic up-regulation of COX-2 induced by AngII and EGF in intestinal epithelial cells was due to an increase in its transcriptional rate and its mRNA stability. Our results in VSMCs demonstrate that the effect of AngII on IL-1β-induced COX-2 expression was mainly due to a mechanism involving mRNA stabilization, as previously described in adventitial fibroblasts (Galán et al., 2011). RNA stability is regulated by different mRNA-binding proteins such as HuR, which is most noted to play a role in cancer through stabilization of various target genes such as COX-2 (Dixon et al., 2001; Abdelmohsen and Gorospe, 2010). HuR is mainly located in the nucleus and shuttles between the nucleus and the cytoplasm. The presence of cytoplasmic HuR is known to contribute in promoting cell growth, proliferation, angiogenesis and survival (Abdelmohsen and Gorospe, 2010), and in this capacity, HuR stabilizes ARE-containing mRNAs such as cyclins, TNF-α, VEGF and COX-2 (Abdelmohsen and Gorospe, 2010). This is evident in colorectal and other tumour types, where HuR is overexpressed and present within the cytoplasm leading to ARE-mRNA stabilization and increased expression of COX-2 (Dixon et al., 2001; Young et al., 2012). Post-translational modifications of HuR such as serine and threonine phosphorylation by several kinases can modify the subcellular location of HuR (Meisner and Filipowicz, 2011). Specifically, ERK1/2 can participate in HuR phosphorylation, modifying its activity in a lung cancer cell line (Yang et al., 2004,2004) or HuR cytoplasmic location in hepatic stellate cells (Woodhoo et al., 2012). Herein, we demonstrated, for the first time in VSMCs, that ERK1/2-mediated HuR activation is involved in the synergistic effect of AngII and IL-1β on COX-2 expression and cell migration. This is supported by the following findings: (i) AngII + IL-1β interfered with the rapid decay of COX-2 mRNA resulting in increased 3′UTR reporter activity in VSMCs. This effect was accompanied by HuR translocation to the cytoplasm and its binding to COX-2 mRNA; (ii) HuR silencing and/or treatment with the HuR inhibitor MS-444 reduced COX-2 mRNA stability, AngII + IL-1β-induced COX-2 mRNA levels and cell migration; and (iii) ERK1/2 phosphorylation was increased by AngII + IL-1β and ERK1/2 inhibition reduced COX-2 levels, COX-2 mRNA stability, HuR cytoplasmic translocation and cell migration. All together, these results suggest that the cytoplasmic trafficking of HuR, by modulating COX-2 expression and cell migration, could be a central node involved in the vascular remodelling associated with cardiovascular pathologies. In agreement with this hypthesis, we observed increased vascular expression of HuR in AngII-infused mice and in the carotid ligation mouse model, as demonstrated in different vascular injuries such as intimal hyperplasia, arterialized saphenous vein or atherosclerotic plaque (Pullmann et al., 2005).
During the last few years, prostanoids downstream of COX-2 involved in vascular remodelling have received an increasing amount of attention (Wang et al., 2011; Sparks et al., 2013; Zhang et al., 2013). Our results demonstrated that COX-2-derived PGE2 and TXA2 are involved in cell migration induced by AngII + IL-1β. Thus, we found that not only COX-2 but also mPGES-1 and TXAS expressions were increased in VSMCs treated with AngII + IL-1β. In addition, cell migration induced by AngII + IL-1β was blocked by pharmacological inhibitors of COX-2 and the TXA2 and PGE2 pathways. Finally, a PGE2 analogue and a TP agonist mimicked AngII + IL-1β effects on cell migration, effects that were blocked by EP1 and EP3 and TP receptor blockade respectively. Furthermore, PGE2 and TXA2 have been shown to induce cell migration in VSMCs through EP3 receptors and in endothelial cells through TP receptors (Nie et al., 2000; Zhang et al., 2013). The effects of prostanoids on cell migration might be mediated by TN-C, an ECM protein which participates in cell proliferation and migration (Wang et al., 2011; Yu et al., 2013) and is regulated by PGI2 or PGE2 (Wang et al., 2011). Our results demonstrated that not only EP1 and EP3 receptors but also TXA2 participate in the AngII + IL-1β-mediated induction of TN-C. More importantly, the effects of AngII + IL-1β on COX-2, mPGES-1, TXAS and TN-C expression were replicated in two animal models of vascular remodelling associated with inflammation and increased cell migration/proliferation (the carotid artery ligation model and the AngII infusion model). Accordingly, heightened plasma PGE2 levels were observed in both models of vascular damage. Moreover, the hypertrophic vascular remodelling and the increased TN-C expression induced by AngII were reversed by celecoxib treatment and genetic mPGES-1 deletion, demonstrating that COX-2/mPGES-1/TXAS-derived prostanoids are key mediators in vascular damage/remodelling. Supporting these findings, other authors have demonstrated a role for COX-2, PGE2 and TXA2 in animal models of restenosis and hypertension (Wang et al., 2011; Sparks et al., 2013; Zhang et al., 2013). The effect of celecoxib treatment and mPGES-1 deletion in vascular remodelling might depend on changes in BP. However, while celecoxib prevented the increase in BP induced by AngII (Martínez-Revelles et al., 2013), mPGES-1 deletion did not (Facemire et al., 2010). The effect of AngII on the hypertrophic vascular remodelling may depend on the generation of pro-inflammatory cytokines, as observed with increased levels of IL-1β in hearts of AngII-infused rats (Dange et al., 2014). In addition, we cannot exclude the possibility that a synergistic effect of AngII and IL-1β also occurs in vivo and thus modulates vascular remodelling.
An intriguing finding that deserves further consideration is the fact that COX-2-selective non-steroidal anti-inflammatory drugs seem to be associated with increased cardiovascular risk mainly through the reduced biosynthesis of endothelial COX-2-dependent PGI2 leading to an increase in thrombotic events (reviewed in Patrignani and Patrono, 2015). Care is needed in drawing direct comparisons between results obtained from animals- and cell-based studies, similar to the ones presented here, to clinical trials. Various factors such as the well-controlled nature of animal models, the duration or type of the pathology studied and the particular inhibitor or gene regulatory pathway assayed in these studies are factors that might explain differences between experimental and clinical studies. Our results are part of a proof of concept that builds on information available on the effects of prostanoids on vascular structure. Whether these effects have a translational consequence remains to be identified.
Different intracellular signalling pathways are responsible for cell migration. Among the most extensively studied are ERK1/2 and JNK. Both are able to phosphorylate different proteins leading to cytoskeleton reorganization and cell migration (Huang et al., 2004). CaM and pCaMKII are also important mediators of VSMC migration (Scott et al., 2012). CaM/pCaMKII is activated by Gq proteins which are associated with EP1, EP3 and TP receptors (Bos et al., 2004; Yamaoka et al., 2009). The participation of EGFR in VSMC migration has been suggested in response to AngII (Mugabe et al., 2010). Interestingly, the EGFR can be activated directly by TN-C (Huang et al., 2004) or transactivated by EP and TP receptors (Han et al., 2006; Uchiyama et al., 2009). Herein, we demonstrated that ERK1/2, JNK, CaM and pCaMKII and the EGFR are key mediators responsible for PGE2- and TXA2-dependent VSMC migration.
In summary, we demonstrated that the synergistic effect of AngII and IL-1β upon COX-2 expression in VSMCs occurs through post-transcriptional regulation, mediated by ERK1/2-dependent effects, on the activity and localization of HuR. COX-2-derived TXA2 and PGE2 acting on TP and EP1 and EP3 receptors, respectively, then induce TN-C expression and promote cell migration (Figure 8). Increased COX-2, mPGES-1, TXAS, TN-C, HuR and PGE2 levels were observed in animal models of vascular damage. Furthermore, blockade of COX-2 and mPGES-1 protected the vessel against TN-C expression and vascular remodelling. In conclusion, the results of this study suggest that HuR and the COX-2/mPGES-1/TXAS pathway could be pharmacological targets for vascular remodelling in cardiovascular diseases.
Figure 8.
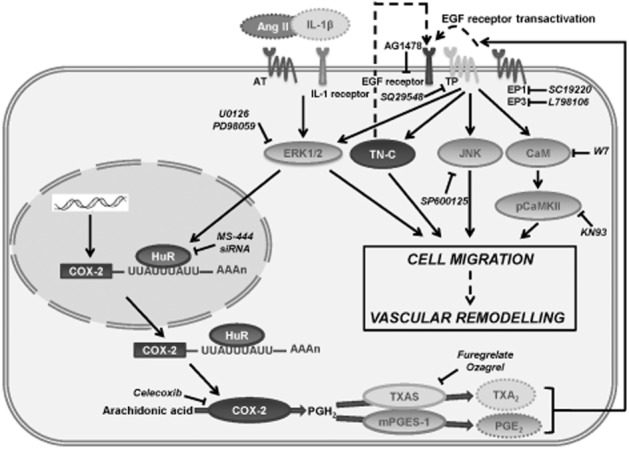
Scheme showing the role of HuR in AngII and IL-1β effects on vascular COX-2 expression and cell migration. In VSMCs, AngII and IL-1β synergistically induce COX-2 expression through the ERK1/2 pathway. ERK1/2 activation leads to HuR binding to COX-2 3′UTR mRNA and exportation to the cytoplasm where HuR increases COX-2 mRNA stability. COX-2 is translated to protein and converts arachidonic acid in PGH2 and TXAS and mPGES-1 will transform it into PGE2 and TXA2. Both prostanoids, through TP and EP1/EP3 receptors, increase TN-C expression and would activate EGFR (EGFR transactivation) that will lead to ERK1/2, JNK, CaM/pCaMKII activation resulting in cell migration and vascular remodelling. Inhibitors used in the experiments are shown in the scheme with their protein targets marked with bar-headed lines. Dashed lines indicate non-investigated mechanisms.
Acknowledgments
We thank Laura García-Redondo for her excellent technical assistance. This study was supported by MINECO (SAF2012-36400 and SAF2012-40127), ISCIII (RD12/0042/0024, RD12/0042/0053, PI13/01488, PI12/01952), Fundación Mutua Madrileña, UAM-Grupo Santander and NIH (R01CA134609). The study was cofunded by Fondo Europeo de Desarrollo Regional (FEDER ‘a way to make Europe’). A. A. and A. M. B. were supported by a FPI fellowship and the Ramón y Cajal programme (RYC-2010–06473) respectively.
Glossary
- 3′UTR
3′ untranslated region
- AngII
angiotensin II
- ARE
adenylate- and uridylate-rich element
- ECM
extracellular matrix
- PGS
prostaglandin synthases
- PGIS
prostacyclin synthase
- TN-C
tenascin-C
- TXAS
thromboxane A2 synthase
- VSMC
vascular smooth muscle cells
Author contributions
A. A., C. R., S. M. R., M. S. A., O. Z. and M. O. performed the experiments. C. R., J. M.-G., A. M. B., D. A. D. and M. S. designed the research study. C. R., J. M.-G. and D. A. D. contributed essential reagents or tools. A. A., S. M. R., M. S. A., M. J. A. analysed the data. A. A., A. M. B. and M. S. wrote the manuscript. A. A., C. R., S. M. R., M. S. A., O. Z., M. O., J. M.-G., M. J. A., A. M. B., D. A. D. and M. S. approved the manuscript.
Conflict of interest
None declared.
Supporting Information
Figure S1 AngII and IL-1β regulate differentially COX-2, mPGES-1, PGIS and TXAS expressions in human VSMCs.
Figure S2 Effect of AngII, IL-1β and AngII + IL-1β on mPGES-1, PGIS and TXAS expressions.
Figure S3 Different signalling pathways are involved in mPGES-1, PGIS and TXAS expression.
Figure S4 PGE2 levels are increased in two models of vascular damage.
Table S1 Primer sequences of the different mRNAs analysed.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Catalytic receptors. Br J Pharmacol. 2013b;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013c;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelmohsen K, Gorospe M. Posttranscriptional regulation of cancer traits by HuR. Wiley Interdiscip Rev RNA. 2010;1:214–229. doi: 10.1002/wrna.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguado A, Galán M, Zhenyukh O, Wiggers GA, Roque FR, Redondo S, et al. Mercury induces proliferation and reduces cell size in vascular smooth muscle cells through MAPK, oxidative stress and cyclooxygenase-2 pathways. Toxicol Appl Pharmacol. 2013;268:188–200. doi: 10.1016/j.taap.2013.01.030. [DOI] [PubMed] [Google Scholar]
- Aotani Y, Saitoh Y. Structure determination of MS-444; a new myosin light chain kinase inhibitor. J Antibiot (Tokyo) 1995;48:952–953. doi: 10.7164/antibiotics.48.952. [DOI] [PubMed] [Google Scholar]
- Avendaño MS, Lucas E, Jurado-Pueyo M, Martínez-Revelles S, Vila-Bedmar R, Mayor F, Jr, et al. Increased nitric oxide bioavailability in adult GRK2 hemizygous mice protects against angiotensin II-induced hypertension. Hypertension. 2014;63:369–375. doi: 10.1161/HYPERTENSIONAHA.113.01991. [DOI] [PubMed] [Google Scholar]
- Bos CL, Richel DJ, Ritsema T, Peppelenbosch MP, Versteeg HH. Prostanoids and prostanoid receptors in signal transduction. Int J Biochem Biol. 2004;36:1187–1205. doi: 10.1016/j.biocel.2003.08.006. [DOI] [PubMed] [Google Scholar]
- Cipollone F, Fazia ML. COX-2 and atherosclerosis. J Cardiovasc Pharmacol. 2006;47(Suppl. 1):S26–S36. doi: 10.1097/00005344-200605001-00006. [DOI] [PubMed] [Google Scholar]
- Dange RB, Agarwal D, Masson GS, Vila J, Wilson B, Nair A, et al. Central blockade of TLR4 improves cardiac function and attenuates myocardial inflammation in angiotensin II-induced hypertension. Cardiovasc Res. 2014;103:17–27. doi: 10.1093/cvr/cvu067. [DOI] [PubMed] [Google Scholar]
- Dixon DA, Kaplan CD, McIntyre TM, Zimmerman GA, Prescott SM. Post-transcriptional control of cyclooxygenase-2 gene expression. The role of the 3′-untranslated region. J Biol Chem. 2000;275:11750–11757. doi: 10.1074/jbc.275.16.11750. [DOI] [PubMed] [Google Scholar]
- Dixon DA, Tolley ND, King PH, Nabors LB, McIntyre TM, Zimmerman GA, et al. Altered expression of the mRNA stability factor HuR promotes cyclooxygenase-2 expression in colon cancer cells. J Clin Invest. 2001;108:1657–1665. doi: 10.1172/JCI12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon DA, Tolley ND, Bemis-Standoli K, Martinez ML, Weyrich AS, Morrow JD, et al. Expression of COX-2 in platelet-monocyte interactions occurs via combinatorial regulation involving adhesion and cytokine signaling. J Clin Invest. 2006;116:2727–2738. doi: 10.1172/JCI27209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doller A, Akool E-S, Huwiler A, Müller R, Radeke HH, Pfeilschifter J, et al. Posttranslational modification of the AU-rich element binding protein HuR by protein kinase Cdelta elicits angiotensin II-induced stabilization and nuclear export of cyclooxygenase 2 mRNA. Mol Cell Biol. 2008;28:2608–2625. doi: 10.1128/MCB.01530-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facemire CS, Griffiths R, Audoly LP, Koller BH, Coffman TM. The impact of microsomal prostaglandin E synthase 1 on blood pressure is determined by genetic background. Hypertension. 2010;55:531–538. doi: 10.1161/HYPERTENSIONAHA.109.145631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galán M, Miguel M, Beltrán AE, Rodríguez C, García-Redondo AB, Rodríguez-Calvo R, et al. Angiotensin II differentially modulates cyclooxygenase-2, microsomal prostaglandin E2 synthase-1 and prostaglandin I2 synthase expression in adventitial fibroblasts exposed to inflammatory stimuli. J Hypertens. 2011;29:529–536. doi: 10.1097/HJH.0b013e328342b271. [DOI] [PubMed] [Google Scholar]
- Han C, Michalopoulos GK, Wu T. Prostaglandin E2 receptor EP1 transactivates EGFR/MET receptor tyrosine kinases and enhances invasiveness in human hepatocellular carcinoma cells. J Cell Physiol. 2006;207:261–270. doi: 10.1002/jcp.20560. [DOI] [PubMed] [Google Scholar]
- Harper KA, Tyson-Capper AJ. Complexity of COX-2 gene regulation. Biochem Soc Trans. 2008;36(Pt 3):543–545. doi: 10.1042/BST0360543. [DOI] [PubMed] [Google Scholar]
- Huang C, Jacobson K, Schaller MD. MAP kinases and cell migration. J Cell Sci. 2004;117(Pt 20):4619–4628. doi: 10.1242/jcs.01481. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal A, Mazan-Mamczarz K, Kawai T, Yang X, Martindale JL, Gorospe M. Concurrent versus individual binding of HuR and AUF1 to common labile target mRNAs. EMBO J. 2004;23:3092–3102. doi: 10.1038/sj.emboj.7600305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesi C, Paradis P, Schiffrin EL. Role of the renin-angiotensin system in vascular inflammation. Trends Pharmacol Sci. 2008;29:367–374. doi: 10.1016/j.tips.2008.05.003. [DOI] [PubMed] [Google Scholar]
- Martín A, Perez-Girón JV, Hernanz R, Palacios R, Briones AM, Fortuño A, et al. Peroxisome proliferator-activated receptor-γ activation reduces cyclooxygenase-2 expression in vascular smooth muscle cells from hypertensive rats by interfering with oxidative stress. J Hypertens. 2012;30:315–326. doi: 10.1097/HJH.0b013e32834f043b. [DOI] [PubMed] [Google Scholar]
- Martínez-Revelles S, Avendaño MS, García-Redondo AB, Álvarez Y, Aguado A, Pérez-Girón JV, et al. Reciprocal relationship between reactive oxygen species and cyclooxygenase-2 and vascular dysfunction in hypertension. Antioxid Redox Signal. 2013;18:51–65. doi: 10.1089/ars.2011.4335. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisner NC, Filipowicz W. Properties of the regulatory RNA-binding protein HuR and its role in controlling miRNA repression. Adv Exp Med Biol. 2011;700:106–123. doi: 10.1007/978-1-4419-7823-3_10. [DOI] [PubMed] [Google Scholar]
- Mugabe BE, Yaghini FA, Song CY, Buharalioglu CK, Waters CM, Malik KU. Angiotensin II-induced migration of vascular smooth muscle cells is mediated by p38 mitogen-activated protein kinase-activated c-Src through spleen tyrosine kinase and epidermal growth factor receptor transactivation. J Pharmacol Exp Ther. 2010;332:116–124. doi: 10.1124/jpet.109.157552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie D, Lamberti M, Zacharek A, Li L, Szekeres K, Tang K, et al. Thromboxane A(2) regulation of endothelial cell migration, angiogenesis, and tumor metastasis. Biochem Biophys Res Commun. 2000;267:245–251. doi: 10.1006/bbrc.1999.1840. [DOI] [PubMed] [Google Scholar]
- Ohnaka K, Numaguchi K, Yamakawa T, Inagami T. Induction of cyclooxygenase-2 by angiotensin II in cultured rat vascular smooth muscle cells. Hypertension. 2000;35:68–75. doi: 10.1161/01.hyp.35.1.68. [DOI] [PubMed] [Google Scholar]
- Orriols M, Guadall A, Galán A, Martí-Pàmies I, Varona S, Rodríguez-Calvo R, et al. Lysyl oxidase (LOX) in vascular remodelling: insight from a new animal model. Thromb Haemost. 2014;112:812–824. doi: 10.1160/TH14-01-0024. [DOI] [PubMed] [Google Scholar]
- Patrignani P, Patrono C. Cyclooxygenase inhibitors: from pharmacology to clinical read-outs. Biochim Biophys Acta. 2015;181:422–432. doi: 10.1016/j.bbalip.2014.09.016. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl. Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham H, Chong B, Vincenti R, Slice LW. Ang II and EGF synergistically induce COX-2 expression via CREB in intestinal epithelial cells. J Cell Physiol. 2008;214:96–109. doi: 10.1002/jcp.21167. [DOI] [PubMed] [Google Scholar]
- Pullmann R, Jr, Juhaszova M, López de Silanes I, Kawai T, Mazan-Mamczarz K, Halushka MK, et al. Enhanced proliferation of cultured human vascular smooth muscle cells linked to increased function of RNA-binding protein HuR. J Biol Chem. 2005;280:22819–22826. doi: 10.1074/jbc.M501106200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Calvo R, Guadall A, Calvayrac O, Navarro MA, Alonso J, Ferrán B, et al. Over-expression of neuron-derived orphan receptor-1 (NOR-1) exacerbates neointimal hyperplasia after vascular injury. Hum Mol Genet. 2013;22:1949–1959. doi: 10.1093/hmg/ddt042. [DOI] [PubMed] [Google Scholar]
- Scott JA, Xie L, Li H, Li W, He JB, Sanders PN, et al. The multifunctional Ca2+/calmodulin-dependent kinase II regulates vascular smooth muscle migration through matrix metalloproteinase 9. Am J Physiol Heart Circ Physiol. 2012;302:H1953–H1964. doi: 10.1152/ajpheart.00978.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparks MA, Makhanova NA, Griffiths RC, Snouwaert JN, Koller BH, Coffman TM. Thromboxane receptors in smooth muscle promote hypertension, vascular remodeling, and sudden death. Hypertension. 2013;6:166–173. doi: 10.1161/HYPERTENSIONAHA.112.193250. [DOI] [PubMed] [Google Scholar]
- Uchiyama K, Saito M, Sasaki M, Obara Y, Higashiyama S, Nakahata N. Thromboxane A2 receptor-mediated epidermal growth factor receptor transactivation: involvement of PKC-delta and PKC-epsilon in the shedding of epidermal growth factor receptor ligands. Eur J Pharm Sci. 2009;38:504–511. doi: 10.1016/j.ejps.2009.09.016. [DOI] [PubMed] [Google Scholar]
- Wang M, Ihida-Stansbury K, Kothapalli D, Tamby MC, Yu Z, Chen L, et al. Microsomal prostaglandin E2 synthase-1 modulates the response to vascular injury. Circulation. 2011;123:631–639. doi: 10.1161/CIRCULATIONAHA.110.973685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhoo A, Iruarrizaga-Lejarreta M, Beraza N, García-Rodríguez JL, Embade N, Fernández-Ramos D, et al. Human antigen R contributes to hepatic stellate cell activation and liver fibrosis. Hepatology. 2012;56:1870–1882. doi: 10.1002/hep.25828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaoka K, Yano A, Kuroiwa K, Morimoto K, Inazumi T, Hatae N, et al. Prostaglandin EP3 receptor superactivates adenylyl cyclase via the Gq/PLC/Ca2+ pathway in a lipid raft-dependent manner. Biochem Biophys Res Commun. 2009;389:678–682. doi: 10.1016/j.bbrc.2009.09.064. [DOI] [PubMed] [Google Scholar]
- Yang HM, Kim HS, Park KW, You HJ, Jeon SI, Youn SW, et al. Celecoxib, a cyclooxygenase-2 inhibitor, reduces neointimal hyperplasia through inhibition of Akt signaling. Circulation. 2004;110:301–308. doi: 10.1161/01.CIR.0000135467.43430.16. [DOI] [PubMed] [Google Scholar]
- Yang X, Wang W, Fan J, Lal A, Yang D, Cheng H, et al. Prostaglandin A2-mediated stabilization of p21 mRNA through an ERK-dependent pathway requiring the RNA-binding protein HuR. J Biol Chem. 2004;279:49298–49306. doi: 10.1074/jbc.M407535200. [DOI] [PubMed] [Google Scholar]
- Young LE, Moore AE, Sokol L, Meisner-Kober N, Dixon DA. The mRNA stability factor HuR inhibits microRNA-16 targeting of COX-2. Mol Cancer Res. 2012;10:167–180. doi: 10.1158/1541-7786.MCR-11-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Ricciotti E, Miwa T, Liu S, Ihida-Stansbury K, Landersberg G, et al. Myeloid cell 5-lipoxygenase activating protein modulates the response to vascular injury. Circ Res. 2013;112:432–440. doi: 10.1161/CIRCRESAHA.112.300755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zou F, Tang J, Zhang Q, Gong Y, Wang Q, et al. Cyclooxygenase-2-derived prostaglandin E2 promotes injury-induced vascular neointimal hyperplasia through the E-prostanoid 3 receptor. Circ Res. 2013;113:104–114. doi: 10.1161/CIRCRESAHA.113.301033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 AngII and IL-1β regulate differentially COX-2, mPGES-1, PGIS and TXAS expressions in human VSMCs.
Figure S2 Effect of AngII, IL-1β and AngII + IL-1β on mPGES-1, PGIS and TXAS expressions.
Figure S3 Different signalling pathways are involved in mPGES-1, PGIS and TXAS expression.
Figure S4 PGE2 levels are increased in two models of vascular damage.
Table S1 Primer sequences of the different mRNAs analysed.