

Abstract
Background and Purpose
Calcitonin gene-related peptide (CGRP) plays an important role in the pathology of migraine, and recent clinical trials suggest the inhibition of CGRP-mediated processes as a new therapeutic option in migraine. In this study, we describe the generation of NOX-L41, a CGRP-neutralizing mirror-image (l-)aptamer (Spiegelmer) and investigate its in vitro and in vivo function.
Experimental Approach
A CGRP-binding Spiegelmer was identified by in vitro selection. Binding studies were performed using surface plasmon resonance (SPR), and the inhibitory activity was determined in cell-based assays. The pharmacokinetic profile comparing i.v. and s.c. dosing was analysed in rats. Intravital two-photon microscopy was employed to follow extravasation from meningeal vessels. Finally, in vivo efficacy was tested in a model of electrically evoked meningeal plasma protein extravasation (PPE) in rats.
Key Results
We identified NOX-L41, a novel CGRP-neutralizing Spiegelmer. SPR studies showed that NOX-L41 binds to human and rat/mouse CGRP with sub-nanomolar affinities and is highly selective against related peptides such as amylin. In vitro, NOX-L41 effectively inhibited CGRP-induced cAMP formation in SK-N-MC cells. In rats, NOX-L41 had a plasma half-life of 8 h. Pharmacodynamic studies showed that NOX-L41 extravasates from blood vessels in the dura mater and inhibits neurogenic meningeal PPE for at least 18 h after single dosing.
Conclusions and Implications
This is the first description of the CGRP-neutralizing Spiegelmer NOX-L41. Preclinical studies confirmed a role for CGRP in neurogenic PPE and provided proof-of-concept for the potential use of this new drug candidate for the treatment or prevention of migraine.
Tables of Links
| RECEPTOR |
|---|
| CGRP receptor |
| LIGANDS | ||
|---|---|---|
| α-CGRP (human) | β-CGRP (rat) | BIBN4096 (olcegepant) |
| α-CGRP (mouse, rat) | Amylin (human) | Calcitonin |
| α-CGRP-(8-37) (human) | Amylin (mouse, rat) | cAMP |
| α-CGRP-(8-37) (rat) | Adrenomedullin (human) | Intermedin (human) |
| β-CGRP (human) | Adrenomedullin (mouse) | Intermedin (rat) |
| β-CGRP (mouse) | Adrenomedullin (rat) | Pramlintide |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
Migraine affects more than 10% of the population in Western countries and severely influences the quality of life of the individuals concerned (Lipton et al., 2007). A substantial number of patients are not effectively treated by the currently approved drugs (non-steroidal anti-inflammatory drugs, triptans, ergotamines, etc.) and a range of adverse effects may occur (Bigal et al., 2013). Thus, there is a high medical need for alternative therapies in migraine (Lipton et al., 2013). One of the most promising targets in migraine is the neuropeptide (α-) calcitonin gene-related peptide (CGRP). CGRP receptor (a complex of the GPCR calcitonin receptor-like receptor, CLR, and the receptor activity-modifying protein 1, RAMP1) is expressed at peripheral sites (e.g. trigeminal neurons and the smooth muscle layer of cranial blood vessels) as well as central brain regions (cerebellum, brainstem; Eftekhari and Edvinsson, 2010; Alexander et al., 2013). Engagement of the receptor by CGRP results in the activation of the Gs pathway and cAMP formation. Elevated levels of CGRP have been reported in the external jugular blood of patients during migraine attacks (Goadsby et al., 1990) and infusion of CGRP provokes actual migraine-like attacks in individuals with a past history of migraine (Lassen et al., 2002). Furthermore, pain relief upon triptan (serotonin receptor agonist) treatment is associated with reduced CGRP secretion (Juhasz et al., 2005). In animal models, neutralization of CGRP by monoclonal antibodies or antagonism of its receptor inhibited meningeal blood flow evoked by either CGRP infusion or electrical stimulation (Petersen et al., 2004; Juhl et al., 2007; Troltzsch et al., 2007; Zeller et al., 2008). Similarly, plasma protein extravasation (PPE) in the dura mater of guinea pigs induced by electrical stimulation of the trigeminal ganglion is blocked by the CGRP antagonist CGRP(8-37) (O'Shaughnessy and Connor, 1994). A major role of CGRP in migraine was ultimately established by phase II and III clinical trials showing that CGRP receptor antagonists BIBN4096 (olcegepant; Olesen et al., 2004), telcagepant (MK-0974; Ho et al., 2008), MK-3207 (Hewitt et al., 2011), BI 44370 TA (Diener et al., 2011) and rimegepant (BMS-927711; Marcus et al., 2014) effectively reduced acute migraine pain.
In the present study, we describe a novel CGRP-neutralizing modality that is based on a mirror-image (l)-aptamer, a so-called Spiegelmer (Spiegelmer is a registered trademark of NOXXON Pharma AG; Klussmann et al., 1996). Aptamers are synthetic oligonucleotides that adopt a three-dimensional structure and bind to a target molecule. They are identified in vitro by subjecting combinatorial oligonucleotide libraries to repeated steps of alternating selection for target binding and enzymatic amplification of the bound sequences known as ‘systematic evolution of ligands by exponential enrichment’ (SELEX; Ellington and Szostak, 1990; Tuerk et al., 1992). Unlike conventional aptamers, Spiegelmers are built from nucleotides of non-natural chirality and are therefore resistant to nuclease attack and do not interact with toll-like receptors (Klussmann et al., 1996; Kulkarni et al., 2007; and Buchner et al., unpublished data). Spiegelmers have proven to be well suited for therapeutic use and can provide a chemically synthesized alternative to monoclonal antibodies (Vater and Klussmann, 2015). In a previous report, we have described the generation of a rat/mouse CGRP-targeting Spiegelmer (STAR-F12-Δ43-48, later called NOX-C89; Vater et al., 2003) that inhibited electrically evoked medial meningeal artery blood flow after topical and systemic administration (Denekas et al., 2006). Similarly, polyethylene glycol (PEG)-conjugated NOX-C89 prevented dural and pial artery dilation provoked by CGRP infusion in an ex vivo model (Juhl et al., 2007). In a behavioural model, NOX-C89 abolished strain-related hypersensitivity to noxious heat (Mogil et al., 2005). Since NOX-C89 is cross-reactive to amylin and only shows moderate inhibition of human CGRP, it was considered insufficient for further (clinical) development. The present paper describes the novel 40 kDa PEGylated Spiegelmer NOX-L41 that binds human CGRP with high affinity and selectivity. In preclinical proof-of-concept studies, we showed that NOX-L41 extravasates from the dural circulation and prevents experimentally induced meningeal PPE. These results may warrant the further development of NOX-L41 for the treatment or prevention of migraine.
Methods
Peptides and nucleic acids
Peptides were obtained from Bachem (Bubendorf, Switzerland) with the exception of pramlintide, which was purchased from Biotrend (Köln, Germany). Oligonucleotides were synthesized using standard solid-phase phosphoramidite chemistry as previously described (Hoffmann et al., 2011). The nucleotide sequence of the Spiegelmer l-226-F2-001 is 5'-CCGUG CUGUC GGAGA CUACU CGUCG AGUAG AAAUA GGUCC CCUCC CACGG-3’. NOX-L41 is 40 kDa Y-shaped PEG-5'-CCGUG CUGUC GGAGA CUACU CGUCG AGUAG AAAUA GGUCC dCCUCC CACGG-3’ whereby dC is l-2'-deoxycytidine. All other nucleotides are l-ribonucleotides. The web-based mfold programme was used for secondary structure prediction (Zuker, 2003). NOX-C89 is 40 kDa Y-shaped PEG-5'-GGACU GAUGG CGCGG UCCUA UUACG CCGAU AGGGU GAGGG GA-3’. The PEG moiety (JenKem, Allen, TX, USA) was conjugated to the 5'-end of the oligonucleotides via an aminohexyl linker (American International Chemical Inc., Framingham, MA, USA). For intravital microscopy, fluorescently labelled NOX-L41-Alexa488 was synthesized by covalent conjugation of Alexa Fluor 488 (Alexa488)-C5-maleimide (Invitrogen, Karlsruhe, Germany) to a thiol-C3 linker at the Spiegelmer's 3'-end. Doses always refer to the oligonucleotide part of the molecules as anhydrous free acid.
Identification of Spiegelmers by in vitro selection of aptamers binding to human mirror-image (d-)CGRP
Aptamers (d-oligonucleotides) binding to human mirror-image (d-)CGRP were identified by in vitro selection as previously described (Purschke et al., 2006; Hoehlig et al., 2013). Briefly, a 32P-labelled RNA library composed of 40 random positions and flanking primer binding sites was incubated with biotinylated human d-α-CGRP (Bachem) in selection buffer with physiological ion concentrations and pH (20 mM Tris, pH 7.4; 150 mM NaCl; 5 mM KCl; 1 mM MgCl2; 1 mM CaCl2; 0.1% Tween 20; 50 μg·mL−1 BSA; 10 μg·mL−1 yeast RNA; 4 U·mL−1 RNaseOut, Invitrogen) at 37°C for 16 h. Complexes of RNA aptamers and the biotinylated target were immobilized on streptavidin or neutravidin-coupled agarose or ultralink plus beads (Pierce, Thermo Scientific, Nidderau, Germany). After washing, the percentage of target-bound aptamers was measured in a scintillation counter (LS6500; Beckman Coulter, Fullerton, CA, USA). Target-bound aptamers were reverse transcribed (SCRIPT reverse transcriptase; Jena Bioscience, Jena, Germany), PCR amplified (Vent (exo-) DNA polymerase; New England BioLabs, Frankfurt, Germany), and transcribed back to single-stranded RNA (T7 RNA polymerase; Stratagene, Waldbronn, Germany). In subsequent rounds, the stringency of in vitro selection was continuously increased by reducing incubation times and the concentrations of both biotinylated human d-CGRP and the enriched RNA library. Human d-amylin(1-17) was added to counter-select against amylin-binding aptamers. Pre-selection steps with beads in the absence of target were performed to avoid enrichment of bead-binding aptamers. DNA from round 11 was cloned and sequenced (LGC Genomics, Berlin, Germany).
Surface plasmon resonance (SPR)
SPR measurements were performed on a Biacore 2000 instrument (GE Healthcare, Munich, Germany). Peptides were immobilized by amine coupling to an NHS/EDC-activated carboxy-dextran-coated CM5 sensor chip (GE Healthcare). Spiegelmers were injected in twofold dilution series ranging from 2 μM to 120 pM at a flow of 30 μL·min−1. Measurements were conducted at 37°C in physiological buffer (20 mM Tris-HCl pH 7.4, 150 mM NaCl, 5 mM KCl, 1 mM MgCl2 and 1 mM CaCl2). Regeneration was performed by injection of 60 μL of 5 M NaCl. Measurements on peptide-loaded flow-cells were referenced to an NHS/EDC-activated and ethanolamine-blocked control flow cell. Kinetic rate constants ka and kd were determined with the BIAevaluation 3.1.1 software (BIACORE AB, Uppsala, Sweden) using a Langmuir 1:1 stoichiometric fitting algorithm. The equilibrium dissociation constant (Kd) is the ratio of the kinetic rate constants (Kd = kd/ka). Each measurement was done at least three times. For competitive binding assays, NOX-L41 (15.6 nM) was pre-incubated with related peptides (100 nM), namely human α-CGRP, β-CGRP, adrenomedullin, amylin, calcitonin and intermedin (adrenomedullin 2). Binding to immobilized human α-CGRP was subsequently measured by duplicate injections. Response units (RU) after 240 s of injection were recorded. For statistical analysis, Student's unpaired t-test was performed using Prism 5 software (GraphPad Software, San Diego, CA, USA).
In vitro inhibition of cAMP production
SK-N-MC human neuroblastoma cells (DSMZ, Braunschweig, Germany), which express the CGRP receptor (CLR/RAMP1) and show CGRP-stimulated cAMP formation (Choksi et al., 2002), were cultured in DMEM, low glucose (Invitrogen) supplemented with 10% heat-inactivated fetal calf serum, 4 mM GlutaMAX (Invitrogen), 50 U·mL−1 penicillin and 50 μg·mL−1 streptomycin. For stimulation of receptor signalling, 1 nM human or rat/mouse CGRP (Bachem) was pre-incubated with Spiegelmer at the indicated concentrations (triplicates) in HBSS (Fisher Scientific, Nidderau, Germany) supplemented with 1 mg·mL−1 BSA and 20 mM HEPES (HBSS/BSA) at 37°C for a total of 60 min. After 40 min of pre-incubation, 1 mM IBMX (Sigma-Aldrich, Taufkirchen, Germany) was added to the stimulation solutions as well as to the cells for 20 min. Cell culture medium was replaced by the stimulation solutions. After 30 min at 37°C stimulation, solutions were removed and the cells were lysed by adding assay/lysis buffer (supplied with Tropix cAMP-ScreenTM System kit, Applied Biosystems, Darmstadt, Germany) for 30 min at 37°C. The amount of cAMP in cell lysates was measured using the Tropix cAMP-Screen ELISA System kit (Applied Biosystems) according to the manufacturer's instructions. Data were normalized to the amount of cAMP produced upon stimulation in the absence of Spiegelmer (maximal response) and the values were plotted against the respective Spiegelmer concentration. IC50 values, that is, the concentration of Spiegelmer required for 50% inhibition of maximum cAMP production, were determined with non-linear regression (4-parameter fit) using Prism 5 software (GraphPad Software). The inhibitory effect on amylin, pramlintide and adrenomedullin signalling was determined likewise using MCF-7 human breast cancer cells (DMSZ), which show cAMP formation after stimulation with amylin (Zimmermann et al., 1997) and adrenomedullin (Miller et al., 1996), whereas SK-N-MC only respond poorly to amylin (Zimmermann et al., 1995). MCF-7 cells were cultured in a 1:1 mixture of Ham's F12 nutrient mix (Fisher Scientific) and DMEM, low glucose (Invitrogen) with final addition of 10% fetal calf serum and 50 U·mL−1 penicillin and 50 μg·mL−1 streptomycin. To prevent aggregation, amylin (Bachem) was dissolved in hexafluoroisopropanol (Sigma-Aldrich) at 100 μM stock concentration. Immediately before use, the stock solution was diluted first in hexafluoroisopropanol to 3 μM and then in HBSS/BSA to a final concentration of 3 nM, which was used for Spiegelmer pre-incubation and stimulation. For statistical analysis, Student's unpaired t-test was performed using Prism 5 software.
Animals
All animal care and experimental procedures complied with national guidelines and legislation (as outline in detail below) and are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). Animals were kept under standard climate controlled housing conditions with 12 h light/dark cycle and ad libitum access to food and water. In total, 46 animals were used in the study.
Pharmacokinetics of NOX-L41 in rat
Male Wistar rats (220–280 g; Janvier, Le Genest St Isle, France) received a single i.v. or s.c. injection of 10 mg·kg−1 NOX-L41 (n = 3 per group) and blood was collected for lithium-heparin plasma preparation 10 min, 1, 3, 6, 24, 48, 72 and 96 h after i.v. dosing and 1, 3, 6, 12, 24, 48, 72 and 96 h after s.c. dosing. Dosing and blood sampling were performed by Heidelberg Pharma (Ladenburg, Germany). All procedures were approved by the local ethical committee and performed in accordance with the German guidelines for the care and use of animals in biomedical research. Plasma concentrations of NOX-L41 were quantified with a sandwich hybridization assay. A 5'-biotinylated l-RNA detect probe (5'-CCGUG GGAGG-3’) complementary to the 3'-end of NOX-L41 was added to the plasma sample. For hybridization, samples were then heated to 95°C and slowly cooled to 40°C before being transferred to a 96-well plate (Nunc Immobilizer™ Amino plate, Thermo Scientific™) coated with a 3’ amino modified l-RNA capture probe (5'-CCGAC AGCAC GG-3’) complementary to the 5'-end of NOX-L41. After incubation for 2 h at 40°C and washing, bound Spiegelmer was quantified with streptavidin/alkaline phosphatase (Promega, Mannheim, Germany) and a chemiluminescence substrate (CSPD Ready-To-Use with Sapphire II, Applied Biosystems). Lower limit of quantification was 2 nM. Plasma half-lives were calculated from linear regressions of log-transformed Spiegelmer plasma concentrations 3–96 h after i.v. and 48–96 h after s.c. dosing (t1/2 = −log 2/slope). AUC values between the time of dosing (0 h) and 96 h (AUC0–96 h) were calculated using the trapezoidal rule. Based on AUC0–96 h, bioavailability after s.c. dosing relative to i.v. dosing was calculated.
Intravital two-photon microscopy
For dura mater imaging, a cranial window was implanted in C57BL/6 mice. 10 mg·kg−1 of NOX-L41 conjugated to Alexa488 at its 3'-end dissolved in 5% glucose and 100 μL Qdots705 (Invitrogen) in PBS according to manufacturer's instructions were i.v. injected. Spatial and temporal dynamics of NOX-L41-Alexa488 (band pass 515–560 nm) and Qdots705 (band pass 660–740 nm) fluorescence were recorded for 180 min post-injection and after 48 h. Stacks of images were collected with vertical steps of 2 μm (total depth of stack 60–80 μm) and a temporal interval of approximately 15–20 min. Imaging fields covered 500 × 500 μm2 of dural space in XY coordinates. After background subtraction, volume reconstructions were done for each individual stack using ‘Volume Viewer’ function in Fiji/ImageJ software (Schindelin et al., 2012). Regions of interest (ROI) were drawn in several intravascular regions (separately for arterioles and venules) and tissue regions using the ‘ROI Manager’ function. Z-axis fluorescence profiles of vascular ROI in different layers 5 min after Spiegelmer injection were used to define anatomical borders between dura mater and cortex. Z-axis profiles of tissue ROI were recorded at 180 min. Mean fluorescence intensity in dural ROIs was normalized to the mean venous intensity. All experiments on animals were performed with accordance to local guidance for animal care [The Finnish Act on Animal Experimentation (62/2006)]. Animal license from local authority (ELÄINKOELAUTAKUNTA-ELLA) was obtained and included all procedures used in this study.
Dural neurogenic PPE
Male Sprague-Dawley rats (250–350 g, Harlan, Indianapolis, IN, USA) were dosed with NOX-L41 at various time points via s.c. (10 mg·kg−1 or 30 mg·kg−1) or i.v. (10 mg·kg−1) injection before the initiation of the surgical procedure. Vehicle (5% glucose in deionized, distilled water) was used as a control. The CGRP antagonist BIBN4096 (1 mg·kg−1, i.v.) was used as a positive control in these studies either 10 min or 1 h post-dose. The dose of BIBN4096 was selected based on a full dose-response curve generated following the 10 min pretreatment time (Supporting Information Table S1). The animals were anaesthetized with Nembutal (50 mg·kg−1, i.p.; Oak Pharmaceuticals, Lake Forest, IL, USA) and placed in a stereotaxic frame (David Kopf Instruments, Tujunga, CA, USA) with the incisor bar set at −2.5 mm. Depth of anaesthesia was evaluated using the paw withdrawal reflex. Following a midline sagittal scalp incision, two pairs of bilateral holes were drilled through the skull (3.2 mm posterior, 1.8 and 3.8 mm laterally, all coordinates referenced to bregma). Pairs of stainless steel stimulating electrodes (Rhodes Medical Instruments, Summerland, CA, USA) were lowered through the holes in both hemispheres to a depth of 9.2 mm below the dura. Fluorescently labelled BSA (FITC-BSA, 20 mg·kg−1, i.v.; Sigma) was injected into the femoral vein 2 min prior to stimulation as a marker for protein extravasation. The left trigeminal ganglion was stimulated for 5 min at a current intensity of 1.0 mA (5 Hz, 5 ms pulse duration) with a Model S48 Grass Instrument Stimulator with PSIU6 photoelectric isolation unit (Grass-Telefactor; Grass Technologies, Warwick, RI, USA). Five minutes following stimulation, the rats were killed by exsanguination with 40 mL of saline. Dural membrane samples were removed from both hemispheres, rinsed with water and spread flat on microscope slides. A fluorescence microscope (Zeiss, Thornwood, NY, USA) equipped with a grating monochromator and a spectrophotometer was used to quantify the amount of FITC-BSA dye in each dural sample. The extravasation induced by electrical stimulation of the trigeminal ganglion was an ipsilateral effect (i.e. occurs only on the side of the dura in which the trigeminal ganglion was stimulated). This allowed the other (unstimulated) half of the dura to be used as the control. The ratio of the amount of extravasation in the dura from the stimulated side compared with the unstimulated side was calculated. Saline controls yielded a ratio of approximately 2.0. In contrast, a compound that effectively prevented the extravasation in the dura from the stimulated side would have a ratio of approximately 1.0. Data in main body are reported as % inhibition, calculated based on maximal inhibition of extravasation set to an extravasation ratio of one and no inhibition based on results from the vehicle control group. These studies conformed to protocols approved by the Eli Lilly and Company Institutional Animal Care and Use Committee.
Results
Identification of a high-affinity CGRP-binding Spiegelmer
We have previously demonstrated that the rat/mouse CGRP-targeting Spiegelmer NOX-C89 binds to human CGRP with a mean equilibrium binding constant (Kd) of 89 nM (Figure 1A, B). Here, we sought to generate a Spiegelmer with increased affinity and selectivity to human CGRP. In a first step, RNA aptamers binding to human biotinylated mirror-image (d-)CGRP were identified by in vitro selection. Within this process, non-biotinylated d-amylin(1-17) showing high homology to CGRP's N-terminal sequence and structure was used to prevent the enrichment of cross-reactive aptamers. The aptamer binding with the highest affinity to human d-CGRP was then synthesized in the mirror-image (l-)configuration, resulting in the Spiegelmer l-226-F2-001 (Figure 1A), which bound to human l-CGRP (natural configuration) with low-nanomolar affinity (Kd = 2.4 ± 0.4 nM; P < 0.01 compared with NOX-C89; Figure 1C). In previous studies, we discovered that directed modifications of the Spiegelmer backbone by ribo-to-deoxyribonucleotide exchanges (or vice versa) can result in improved affinity (Hoehlig et al., 2013; Vater et al., 2013; Purschke et al., 2014). Using this approach, we identified a single cytidine at position 41 (l-226-F2-001-D41) at which substitution of the 2'-hydroxyl group by 2'-hydrogen, that is, converting it to deoxycytidine, resulted in a sixfold increase in affinity (Kd = 0.4 ± 0.2 nM; P < 0.05 compared with l-226-F2-001; Figure 1A, D). This improvement in binding was driven by both faster association and slower dissociation of the Spiegelmer-target complex. After conjugation of 40 kDa PEG to l-226-F2-001-D41, the candidate molecule was termed NOX-L41 (Figure 1A). Diffusion limitation of PEGylated NOX-L41 influenced binding rate constants of association and dissociation but did not result in a changed equilibrium binding constant (Kd = 0.4 ± 0.3 nM, Figure 1E) compared with the unPEGylated Spiegelmer (Figure 1D). A secondary structure model of NOX-L41 is shown in Figure 1F.
Figure 1.
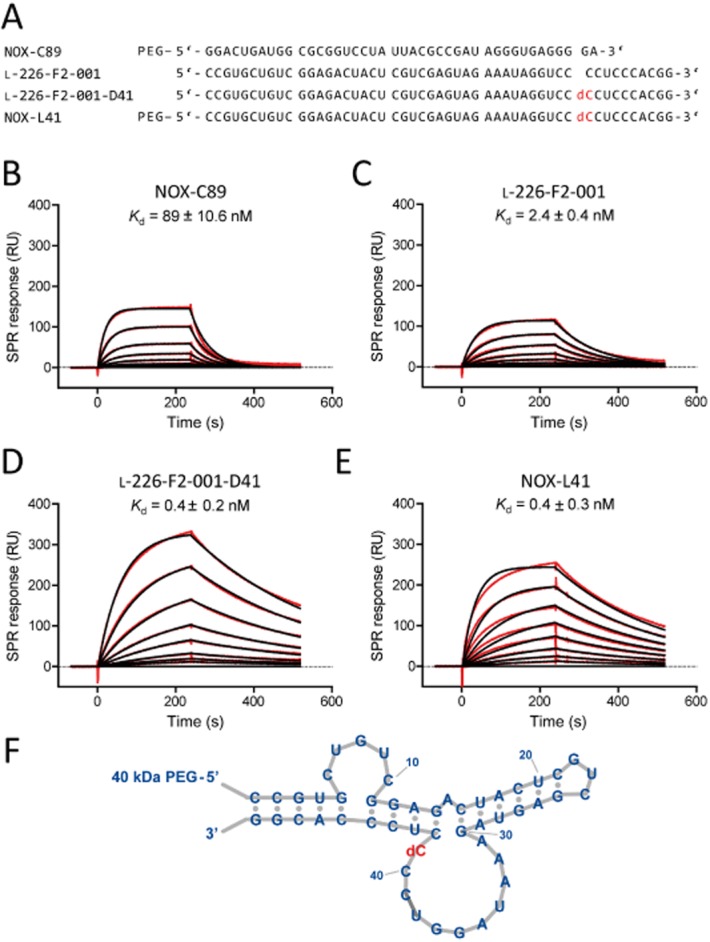
Affinity of CGRP-binding Spiegelmers to human α-CGRP. (A) Spiegelmer sequences. All nucleotides are l-ribonucleotides with the exception of dC (in red), which is l-deoxycytidine. SPR measurement for affinity determination of the Spiegelmers (B) NOX-C89, (C) l-226-F2-001, (D) l-226-F2-001-D41 (= unPEGylated NOX-L41), and (E) NOX-L41. Binding of Spiegelmers at concentrations from (B) 500 nM to 1.95 nM and (C–E) 15.6 nM to 0.12 nM to immobilized human α-CGRP in physiological buffer at 37°C is shown (red lines). Langmuir fitting is shown in black. Mean equilibrium dissociation constant (Kd) ± SEM of at least three independent experiments is given. (F) Secondary structure model of NOX-L41.
NOX-L41 selectively binds and inhibits CGRP
The calcitonin family consists of six structurally related peptides, some of them sharing high sequence homology (Roh et al., 2004). The selectivity of NOX-L41 for these peptides was analysed by a competitive SPR-based binding assay. Pre-incubation of NOX-L41 with human α- or β-CGRP blocked subsequent binding of NOX-L41 to immobilized α-CGRP (Figure 2A). Detailed kinetic analysis confirmed that NOX-L41 binds to human α-CGRP and β-CGRP with similar affinity (α-CGRP: Kd = 0.4 ± 0.3 nM; β-CGRP: Kd = 0.3 ± 0.2 nM; P = 0.80; Figure 1E and Supporting Information Figure S1A). Rat/mouse α-CGRP was bound with comparable affinity (Kd = 0.5 ± 0.3 nM; P = 0.83 compared with human α-CGRP; Supporting Information Figure S1B). In contrast, no binding of NOX-L41 to the related human peptides amylin (islet amyloid polypeptide), calcitonin, adrenomedullin or intermedin (adrenomedullin 2) was detected (Figure 2A). Selectivity against amylin was confirmed by direct binding assays. NOX-L41 did not bind to immobilized human amylin or its non-aggregating analogue pramlintide (Supporting Information Figure S1C and S1D). In contrast, NOX-C89 bound to both peptides with comparable affinity (Supporting Information Figure S1E and S1F; P = 0.68).
Figure 2.
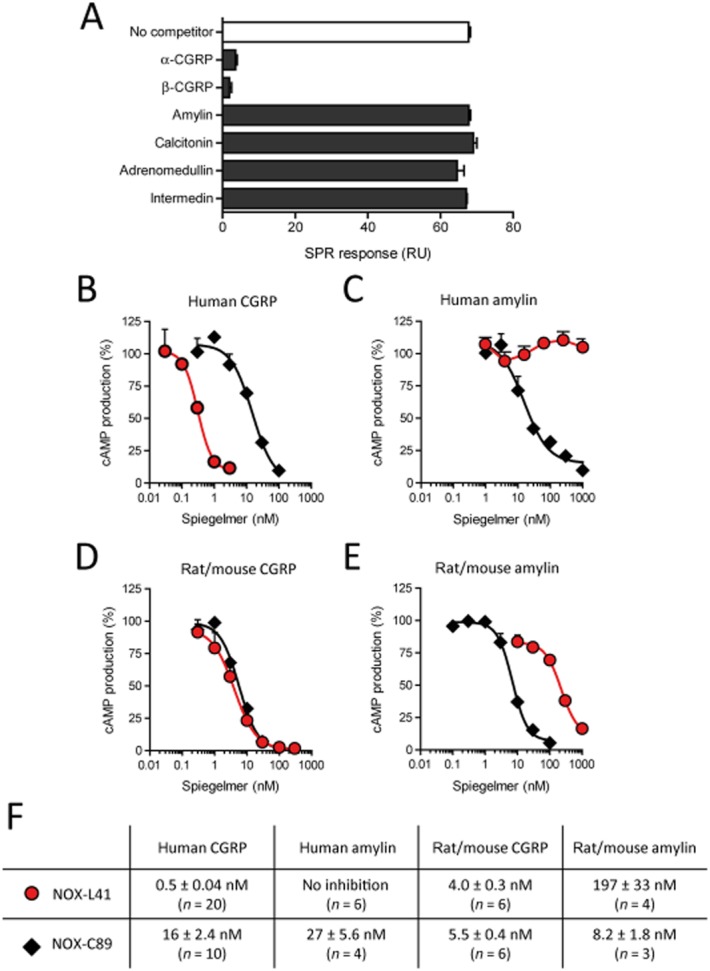
NOX-L41 selectively binds to CGRP and inhibits CGRP-induced cAMP production. (A) Selectivity of NOX-L41 was analysed by competitive SPR binding assay. NOX-L41 (15.6 nM) was pre-incubated with the respective competitor peptides (100 nM) and the binding of the remaining free NOX-L41 to immobilized human α-CGRP was subsequently measured by SPR. Mean data ± SEM of duplicate injections are shown. Data are representative of three independent experiments. Inhibition by NOX-L41 (red circles) and NOX-C89 (black diamonds) of cAMP production induced by (B) human α-CGRP (1 nM), (C) human amylin (3 nM), (D) rat/mouse α-CGRP (1 nM) and (E) rat/mouse amylin (3 nM) in SK-N-MC cells (B, D) or MCF-7 cells (C, E). Normalized data (% of maximal response, mean ± SEM of triplicates) of representative experiments are shown. (F) Summary of mean IC50 values (± SEM).
NOX-L41 inhibits CGRP-induced cAMP production
In functional in vitro assays, NOX-L41 inhibited human CGRP-induced cAMP production in human neuroblastoma SK-N-MC cells with a mean IC50 of 0.5 nM, which corresponds to a > 30-fold improvement compared with previously described NOX-C89 (mean IC50 = 16 nM; P < 0.0001; Figure 2B, F). PEGylated NOX-L41 and unPEGylated l-226-F2-001-D41 had similar inhibitory potency (Supporting Information Figure S2). In agreement with the SPR binding data, NOX-L41 showed high selectivity for CGRP and did not inhibit human amylin-induced cAMP-production by MCF-7 cells at concentrations up to 1 μM (Figure 2C, F). This was confirmed by experiments showing that NOX-L41 did not inhibit cAMP production induced by pramlintide (Supporting Figure S3A). In contrast, NOX-C89 inhibited human amylin- and pramlintide-induced cAMP production with a mean IC50 of 27 and 69 nM respectively (Figure 2C, F and Supporting Information Figure S3A). Neither NOX-C89 nor NOX-L41 showed an inhibitory effect for adrenomedullin (Supporting Information Figure S3B). Discrimination by NOX-L41 between CGRP and amylin was less pronounced for rodent peptides. Rat/mouse CGRP-induced cAMP production was inhibited by NOX-L41 with a mean IC50 of 4.0 nM (Figure 2D, F) and a substantial but 50-fold weaker inhibition of rat/mouse amylin was observed (mean IC50 = 197 nM, Figure 2E, F; P < 0.0001). Figure 2F summarizes the inhibitory constants of NOX-L41 and NOX-C89.
Pharmacokinetic profile of NOX-L41 in rats
We compared the pharmacokinetic profile of NOX-L41 after i.v. and s.c. injection of a single 10 mg·kg−1 dose. After i.v. administration, a maximum plasma concentration (cmax) of 13.7 ± 1.1 μM (mean ± SEM) was reached after 10 min (Figure 3), and the elimination half-life (t1/2) was 8.1 h. Subcutaneous injection resulted in plasma peak levels of approximately 1 μM 24 h post-dosing and a comparable elimination half-life of t1/2 = 8.2 h (Figure 3). Bioavailability after s.c. dosing was 7.2% (i.v.: AUC0–96 h = 226 μM·h; s.c.: AUC0–96 h = 16.2 μM·h).
Figure 3.
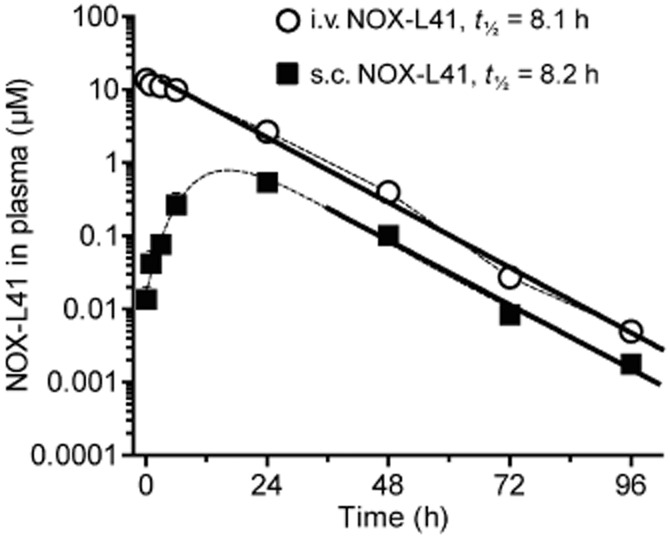
Pharmacokinetics of NOX-L41 after i.v. and s.c. administration. Rats received a single i.v. or s.c. dose of 10 mg·kg−1 NOX-L41. Lithium heparin plasma was collected at the indicated time points and NOX-L41 concentrations were determined by sandwich hybridization assay. Mean ± SEM of three animals per group and time point is shown. Linear regression of log-transformed plasma NOX-L41 concentrations at 3–96 h after i.v. and 48–96 h after s.c. injection is shown (bold lines).
NOX-L41 extravasates in the dura mater
During neurogenic inflammation, CGRP promotes vasodilation and PPE by acting on abluminal vascular smooth muscle cells and mast cells. Therefore, we analysed the ability for fluorescently-labelled NOX-L41-Alexa488 to extravasate from meningeal blood vessels using two-photon microscopy. The functionality of NOX-L41 was not compromised by conjugation to the fluorophore (Supporting Information Figure S4). Upon i.v. injection, NOX-L41-Alexa488 was promptly detectable in meningeal vessels (Figure 4A). For the following 3 h, the fluorescence intensity within meningeal arterioles and venules was unchanged suggesting a constant concentration of Spiegelmer during the length of the experiment (Figure 4B). In the surrounding tissue, fluorescence could be detected as early as 5 min after Spiegelmer injection and intensity continuously increased thereafter (Figure 4B). A plateau was reached after about 2 h. The maximum fluorescence intensity in the surrounding tissue was 15–25% of that inside the vessels with a higher intensity near venules (Figure 4B). After 48 h, fluorescence intensities had fallen below 10% in vessels and tissue. Qdots705 co-injected with NOX-L41-Alexa488 as a control for vascular integrity was not detected outside the vessels (Supporting Information Figure S5). Analyses of vertical (Z-axis) image stacks showed that the fluorescence in extravascular tissue ROI (Figure 4C, lower panel) corresponded closely to the profile of dural vessels (0–40 μm) (Figure 4C, upper panel), whereas no extravascular fluorescence was detected in deeper layers (below 55 μm; Figure 4A, right panel) showing that NOX-L41-Alexa488 extravasates from dural but not from cortical vessels.
Figure 4.

NOX-L41 extravasates from the circulation in dura mater but not in the cortex. Fluorescently labelled NOX-L41-Alexa488 (10 mg·kg−1) was injected i.v. and distribution in the dura mater with cortex were visualized over somatosensory cortex by two-photon microscopy. (A) Representative images taken from the dura mater before (baseline), 5 min and 3 h after NOX-L41-Alexa488 injection and from the cortex after 3 h. (B) Quantification of intravascular and tissue fluorescence intensity. Data are normalized to mean venous intensity. Mean ± SEM of fluorescence intensities measured in at least five ROI is shown. (C) Z-axis profiles of vascular versus tissue fluorescence. For identification of the anatomical borders between dural and cortical layers, Z-axis profiles of fluorescence in the vascular ROI 5 min after dosing were analysed (upper panel). Zero level corresponds to the surface of the dura mater. Dural layer constitutes the first 30–40 μm, followed by the subdural space (SDS), and cortical layers that are located below 55–60 μm of depth. AU, arbitrary units.
NOX-L41 inhibits neurogenic PPE in the dura mater
We evaluated the ability of NOX-L41 to reduce PPE in the dura of rats induced by unilateral electrical stimulation of the trigeminal ganglia. Based on maximal exposure of NOX-L41 being achieved 12–24 h following s.c. injection (Figure 3), we evaluated the inhibition of PPE in the dura of rats 18 h following s.c. injection. Doses of 10 and 30 mg·kg−1 significantly reduced PPE (46 and 74% respectively; Figure 5A). After 72 h, inhibition of PPE by the 10 mg·kg−1 dose was not significant (9% inhibition; Figure 5A). Based on the rapid distribution of NOX-L41 into the dural tissue (Figure 4B) after i.v. administration, we also characterized inhibition of PPE at various time points following i.v. dosing. Significant inhibition of PPE by NOX-L41 was observed 1 and 18 h (23 and 46%, respectively) after i.v. administration, but not at 10 min (Figure 5B). For control, the CGRP receptor antagonist BIBN4096 was used. BIBN4096 significantly inhibited PPE 10 min and 1 h after i.v. dosing (93 and 91%, respectively), illustrating that the model is capable of showing efficacy following relatively short pretreatment times with the test compound (Figure 5B). Dose and pretreatment times for BIBN4096 were selected based on preliminary experiments (Supporting Information Table S1). In these experiments, no inhibition of PPE by BIBN4096 was observed 18 h post-dose (Supporting Information Table S1). The actual extravasation ratio data (non-normalized) are included in Supporting Information Table S2.
Figure 5.
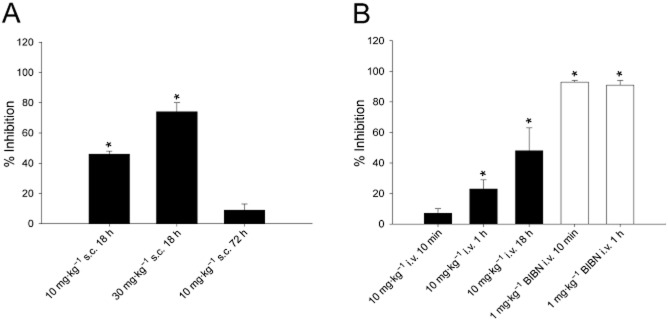
NOX-L41 inhibits PPE in dura of rats induced by electrical stimulation of the trigeminal ganglia. (A) Inhibition of PPE in rats following s.c. administration of NOX-L41 (10 mg·kg−1 or 30 mg·kg−1) evaluated 18 or 72 h post-dose. (B) Inhibition of PPE in rats following i.v. administration of either NOX-L41 (10 mg·kg−1) or BIBN4096 (1 mg·kg−1) at various time points post-dose. Data are presented as mean ± SEM (n = 3–4) of % inhibition data calculated based on maximal inhibition of extravasation set to an extravasation ratio of one and no inhibition based on results from vehicle control group. *P < 0.05 vs. vehicle control group (by anova and Dunnett's post-test).
Discussion
In the present study, we describe the Spiegelmer NOX-L41, a novel CGRP-neutralizing l-aptamer, and demonstrate proof for its in vivo functionality in a preclinical model of neurogenic inflammation. The Spiegelmer acts by binding to the neuropeptide CGRP, thus inhibiting it from signalling at its receptor. NOX-L41 differs from the previously described Spiegelmer NOX-C89 by enhanced affinity and selectivity for human CGRP. High affinity was a result of an in vitro selection process against the human target (NOX-C89 was raised against rat/mouse CGRP) and subsequent optimization by directed modification of the oligonucleotide backbone (cytidine to deoxy-cytidine exchange at position 41). This is in line with previous studies in which we showed that directed exchanges of ribo- to corresponding deoxyribonucleotides in RNA Spiegelmers and vice versa in DNA Spiegelmers can significantly improve affinity to the target (Hoehlig et al., 2013; Vater et al., 2013; Purschke et al., 2014). These modifications may improve the stability of the three-dimensional structure of the Spiegelmer or may directly impact on the binding interface. However, detailed structural analyses will be required for each individual Spiegelmer to understand the molecular details of functional improvements.
High selectivity is an important premise for prospective therapeutic use. NOX-L41 has similar affinity for α- and β-CGRP but does not show binding to other members of the human calcitonin peptide family. Due to high sequence homology (34 out of 37 residues identical) and engagement of the same receptor (CLR/RAMP1), α- and β-CGRP will likewise be blocked by CGRP receptor antagonists (Petersen et al., 2004) and presumably anti-CGRP antibodies. More importantly, NOX-L41 does not cross-react with human amylin (as the result of counter-selection for amylin(1-17) during the in vitro selection process), eliminating the risk for potential metabolic side effects due to amylin's involvement in food intake and blood glucose regulation (Roth et al., 2012). Selectivity was validated by showing that NOX-L41 does not bind and inhibit pramlintide, a synthetic non-aggregating amylin mimetic used in diabetes therapy. In contrast to human, NOX-L41 showed some cross-reactivity to amylin from rat/mouse, which may reflect a slightly higher homology of CGRP and amylin in rat/mouse than in human (19 vs. 16 out of 37 residues identical). The other members of the calcitonin family of peptides, namely calcitonin, adrenomedullin and intermedin, have only structural (conserved disulfide bridge and amidated C-terminus) but no sequence similarities to CGRP and are not recognized by NOX-L41.
Choosing appropriate animal models in migraine research is challenging as the clinical pathophysiology is still not completely understood and preclinical models only mimic particular aspects of the disease. Among the pathological events that have most intensively been investigated and that are frequently addressed in animal models is neurogenic inflammation (Erdener and Dalkara, 2014). Activation of trigeminal neurons results in the perivascular release of mediators such as substance P and CGRP that promote meningeal vasodilation, PPE and the release of pro-inflammatory substances, which, in turn, causes hypersensitivity and pain. Within this process, CGRP acts on abluminal smooth muscle cells and mast cells resulting in the need for CGRP antagonizing compounds to extravasate from dural circulation into the surrounding tissue in order to interfere with neurogenic inflammation (Edvinsson et al., 2007). Using intravital two-photon microscopy, we show that in the dura mater where vessels are not blood–brain barrier protected, NOX-L41 extravasates gradually leading to stable extravascular levels about 2 h after i.v. dosing. This correlates with a delayed onset of action following i.v. administration of NOX-L41 in the PPE model and supports an abluminal site of action in this model. Fluorescence intensities in the dural tissue were about 15–25% of those observed in blood. From pharmacokinetic analyses showing plasma levels ≥10 μM after i.v. dosing of 10 mg·kg−1 NOX-L41, we anticipate that NOX-L41 will also be present in the dural tissue at low micromolar concentrations.
In preclinical models, neurogenic inflammation is often addressed by monitoring changes in the meningeal circulation (vasodilation and increased blood flow) that are experimentally induced by CGRP infusion or electrical stimulation of trigeminal afferents. In these models, efficacy was shown for small-molecule CGRP receptor antagonists (Petersen et al., 2004; Troltzsch et al., 2007), CGRP-neutralizing monoclonal antibodies (Juhl et al., 2007; Zeller et al., 2008) and the Spiegelmer NOX-C89 (Denekas et al., 2006; Juhl et al., 2007). Although for BIBN4096, this translated into clinical efficacy (Olesen et al., 2004), there appears to be no general correlation between cerebral blood flow and migraine pain as it has been reported that CGRP-induced dural vasodilation is not sufficient to activate or sensitize meningeal nociceptors (Levy et al., 2005) and dilation of cranial arteries by vasoactive intestinal peptide does not trigger headache attacks in migraineurs (Rahmann et al., 2008).
In the present study, we chose a related model that analyses the extravasation of plasma proteins in the meninges upon electrical stimulation of the trigeminal ganglion. It has been suggested that meningeal PPE is triggered by the activation of neurokinin receptors (e.g. by substance P) and that this process is potentiated by CGRP (Gamse and Saria, 1985; Lee et al., 1994). In a prior study, O'Shaughnessy and Connor reported that blockade of CGRP receptor by CGRP(8-37) attenuated PPE in the dura mater of guinea pigs but not rats (O'Shaughnessy and Connor, 1994). Here we show that neutralization of CGRP by NOX-L41 or blockade of its receptors by BIBN4096 does also significantly reduce neurogenic PPE in rats. Similarly, inhibition of PPE has been reported for anti-migraine drugs ergotamine and triptans (Markowitz et al., 1988; Johnson et al., 1997; Williamson et al., 1997) and has been used as a model to select several novel, non-vasoconstrictive molecules that were efficacious in the clinic for the treatment of migraine (5-HT1F receptor agonists, ionotropic glutamate receptor 5 antagonists; Johnson et al., 1997; 2008,; Goldstein et al., 2001; Weiss et al., 2006). On the other hand, there have been a number of substances (e.g. neurokinin 1 receptor antagonists and endothelin receptor antagonists) that inhibited PPE but failed in clinical trials (Lee et al., 1994; May et al., 1996; Williamson et al., 1997; Diener, 2003). Thus, although neither vasodilatation nor PPE per se appear sufficient to fully explain migraine pathology (Ho et al., 2010), both models have successfully been used to provide predictive preclinical in vivo proof for CGRP-targeting compounds. Nonetheless, future studies will be required to better understand the precise mode and site of action for CGRP and its antagonists in migraine pathology and therapy.
The clinical development of CGRP-targeting drugs experienced a setback after orally available, small-molecule CGRP receptor antagonists for the abortive treatment of acute migraine attacks [including BIBN4096 (olcepegant) and follow-up BI-44370 TA, telcagepant and follow-up MK-3207] were discontinued despite encouraging late-stage clinical trial results, with two (telcagepant, MK-3207) being stopped due to concerns regarding liver toxicity (Bigal et al., 2013). The development status of two other small-molecule CGRP receptor antagonists, BMS-927711 and MK-1602, is currently unclear. Lately, CGRP antagonizing monoclonal antibodies have come into focus for the preventive treatment of episodic/chronic migraine. Three anti-CGRP antibodies (LY2951742, ALD-403 and LBR-101) and one anti-CGRP receptor antibody (AMG-334) are currently being tested in early-stage clinical trials (Dodick et al., 2014a,b,). The use of antibodies for preventive rather than acute treatments arises from prolonged in vivo half-lives, a need for parenteral administration and a slower onset of action compared with small-molecule CGRP receptor antagonists as suggested by preclinical studies (Zeller et al., 2008). Engagement by CGRP of receptors other than CLR/RAMP1 may furthermore discriminate the mode of action of receptor antagonists and ligand-neutralizing compounds (Walker and Hay, 2013). Similarly, PEGylated Spiegelmers are injectable drugs and NOX-L41 showed slow onset but long-lasting efficacy in the PPE model. In comparison with mice and rats, in which 40 kDa PEG-conjugated Spiegelmers are eliminated from circulation with half-lives of 6–8 h (Hoehlig et al., 2013; Vater et al., 2013), extended plasma half-lives of 24–48 h and a higher bioavailability upon s.c. dosing are observed in humans (Vater and Klussmann, 2015). Thus, it is expected that the pharmacological effect of NOX-L41 will be available for a significantly longer period of time in patients and that NOX-L41 will most appropriately be used in patients with severe episodic migraine or other recurring pain disorders. Despite some similarities with antibodies, there are important differences between Spiegelmers and monoclonal antibodies: Spiegelmers are not biologics but are synthesized chemically. They are manufactured by solid-phase synthesis, which allows for controlled and scalable production processes. Aggregation of the highly hydrophilic Spiegelmers is not an issue. Likewise, immunogenicity problems that may occur for antibodies over time and can be responsible for a loss of efficacy and adverse events have not been observed for Spiegelmers. Moreover, the different chemical properties of both substance groups are likely to provide Spiegelmers with a unique pharmacological profile, although comparative studies will be required to analyse those in detail.
The present studies introduce a novel CGRP-neutralizing Spiegelmer, NOX-L41, with improved affinity and a favourable selectivity profile. Proof of efficacy is provided in an in vivo model of neurogenic inflammation, which moreover confirms a role for CGRP in PPE. This establishes NOX-L41 as a new drug candidate that may serve as a chemically synthesized alternative to monoclonal antibodies for the treatment or prevention of migraine and potentially for other CGRP-mediated pain disorders.
Acknowledgments
The authors would like to thank Simone Schülzchen for technical assistance in the in vitro selection group; John Turner, Lucas Bethge, Gabriele Anlauf and Tino Struck for synthesizing aptamers and Spiegelmers; Lisa Bauer and Katrin Schindele for technical assistance with cell-based assays; Seike Gericke and Hanna Davideit for technical assistance in bioanalytics; and Christian Mihm for editing the figures.
Glossary
- CGRP
(α-) calcitonin gene-related peptide
- PEG
polyethylene glycol
- PPE
plasma protein extravasation
- ROI
region of interest
- SPR
surface plasmon resonance
Author contributions
K. H. performed the in vitro pharmacology experiments and wrote the paper. K. W. J. and A. C.-S. designed and performed the dural PPE experiments. E. P. and L. K. designed and performed the intravital two-photon microscopy experiments. C. M. designed and performed SPR binding experiments. W. G. P. performed the in vitro selection and identified NOX-L41. S. Vauléon designed and performed bioanalytics for pharmacokinetic studies. K. B. designed the in vitro pharmacology experiments. F. J., A. V., S. K. designed and supervised the study. A. V., W. G. P., K. B. and K. W. J. revised the manuscript.
Conflict of interest
K. H., C. M., W. G. P., S. V., K. B., F. J., A. V. and S.K. are full-time employees of NOXXON Pharma AG, which has filed patents on the CGRP-inhibiting Spiegelmers. K. W. J. and A. C.-S. are full-time employees of Eli Lilly and Company. E. P. and L. K. are full-time employees of Neurotar Ltd. which acted as a CRO on behalf of NOXXON.
Supporting Information
Figure S1 Binding selectivity of NOX-L41.
Figure S2 PEGylation does not affect the inhibitory activity of NOX-L41.
Figure S3 NOX-L41 does not inhibit pramlintide and adrenomedullin-induced cAMP production.
Figure S4 NOX-L41-Alexa488 inhibits CGRP-induced cAMP production.
Figure S5 Qdots705 do not extravasate from dural vessels.
Table S1 Inhibition of dural PPE (reported as extravasation ratios) by BIBN4096 at various doses and pretreatment times following intravenous administration.
Table S2 Inhibition of dural PPE (reported as extravasation ratios) by NOX-L41 and BIBN4096 at various doses, pretreatment times and administration routes.
References
- Alexander SP, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The concise guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigal ME, Walter S, Rapoport AM. Calcitonin gene-related peptide (CGRP) and migraine current understanding and state of development. Headache. 2013;53:1230–1244. doi: 10.1111/head.12179. [DOI] [PubMed] [Google Scholar]
- Choksi T, Hay DL, Legon S, Poyner DR, Hagner S, Bloom SR, et al. Comparison of the expression of calcitonin receptor-like receptor (CRLR) and receptor activity modifying proteins (RAMPs) with CGRP and adrenomedullin binding in cell lines. Br J Pharmacol. 2002;136:784–792. doi: 10.1038/sj.bjp.0704761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denekas T, Troltzsch M, Vater A, Klussmann S, Messlinger K. Inhibition of stimulated meningeal blood flow by a calcitonin gene-related peptide binding mirror-image RNA oligonucleotide. Br J Pharmacol. 2006;148:536–543. doi: 10.1038/sj.bjp.0706742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diener HC. RPR100893, a substance-P antagonist, is not effective in the treatment of migraine attacks. Cephalalgia. 2003;23:183–185. doi: 10.1046/j.1468-2982.2003.00496.x. [DOI] [PubMed] [Google Scholar]
- Diener HC, Barbanti P, Dahlof C, Reuter U, Habeck J, Podhorna J. BI 44370 TA, an oral CGRP antagonist for the treatment of acute migraine attacks: results from a phase II study. Cephalalgia. 2011;31:573–584. doi: 10.1177/0333102410388435. [DOI] [PubMed] [Google Scholar]
- Dodick DW, Goadsby PJ, Spierings EL, Scherer JC, Sweeney SP, Grayzel DS. Safety and efficacy of LY2951742, a monoclonal antibody to calcitonin gene-related peptide, for the prevention of migraine: a phase 2, randomised, double-blind, placebo-controlled study. Lancet Neurol. 2014a;13:885–892. doi: 10.1016/S1474-4422(14)70128-0. [DOI] [PubMed] [Google Scholar]
- Dodick DW, Goadsby PJ, Silberstein SD, Lipton RB, Olesen J, Ashina M, et al. Safety and efficacy of ALD403, an antibody to calcitonin gene-related peptide, for the prevention of frequent episodic migraine: a randomised, double-blind, placebo-controlled, exploratory phase 2 trial. Lancet Neurol. 2014b;13:1100–1107. doi: 10.1016/S1474-4422(14)70209-1. [DOI] [PubMed] [Google Scholar]
- Edvinsson L, Nilsson E, Jansen-Olesen I. Inhibitory effect of BIBN4096BS, CGRP(8–37), a CGRP antibody and an RNA-Spiegelmer on CGRP induced vasodilatation in the perfused and non-perfused rat middle cerebral artery. Br J Pharmacol. 2007;150:633–640. doi: 10.1038/sj.bjp.0707134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eftekhari S, Edvinsson L. Possible sites of action of the new calcitonin gene-related peptide receptor antagonists. Ther Adv Neurol Disord. 2010;3:369–378. doi: 10.1177/1756285610388343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- Erdener SE, Dalkara T. Modelling headache and migraine and its pharmacological manipulation. Br J Pharmacol. 2014;171:4575–4594. doi: 10.1111/bph.12651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamse R, Saria A. Potentiation of tachykinin-induced plasma protein extravasation by calcitonin gene-related peptide. Eur J Pharmacol. 1985;114:61–66. doi: 10.1016/0014-2999(85)90520-5. [DOI] [PubMed] [Google Scholar]
- Goadsby PJ, Edvinsson L, Ekman R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurol. 1990;28:183–187. doi: 10.1002/ana.410280213. [DOI] [PubMed] [Google Scholar]
- Goldstein DJ, Roon KI, Offen WW, Ramadan NM, Phebus LA, Johnson KW, et al. Selective seratonin 1F (5-HT(1F)) receptor agonist LY334370 for acute migraine: a randomised controlled trial. Lancet. 2001;358:1230–1234. doi: 10.1016/s0140-6736(01)06347-4. [DOI] [PubMed] [Google Scholar]
- Hewitt DJ, Aurora SK, Dodick DW, Goadsby PJ, Ge YJ, Bachman R, et al. Randomized controlled trial of the CGRP receptor antagonist MK-3207 in the acute treatment of migraine. Cephalalgia. 2011;31:712–722. doi: 10.1177/0333102411398399. [DOI] [PubMed] [Google Scholar]
- Ho TW, Ferrari MD, Dodick DW, Galet V, Kost J, Fan X, et al. Efficacy and tolerability of MK-0974 (telcagepant), a new oral antagonist of calcitonin gene-related peptide receptor, compared with zolmitriptan for acute migraine: a randomised, placebo-controlled, parallel-treatment trial. Lancet. 2008;372:2115–2123. doi: 10.1016/S0140-6736(08)61626-8. [DOI] [PubMed] [Google Scholar]
- Ho TW, Edvinsson L, Goadsby PJ. CGRP and its receptors provide new insights into migraine pathophysiology. Nat Rev Neurol. 2010;6:573–582. doi: 10.1038/nrneurol.2010.127. [DOI] [PubMed] [Google Scholar]
- Hoehlig K, Maasch C, Shushakova N, Buchner K, Huber-Lang M, Purschke WG, et al. A novel C5a-neutralizing mirror-image (l-)aptamer prevents organ failure and improves survival in experimental sepsis. Mol Ther. 2013;21:2236–2246. doi: 10.1038/mt.2013.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann S, Hoos J, Klussmann S, Vonhoff S. RNA aptamers and Spiegelmers: synthesis, purification, and post-synthetic PEG conjugation. Curr Protoc Nucleic Acid Chem. 2011:1–30. doi: 10.1002/0471142700.nc0446s46. Chapter 4 (Unit 4.46) [DOI] [PubMed] [Google Scholar]
- Johnson KW, Schaus JM, Durkin MM, Audia JE, Kaldor SW, Flaugh ME, et al. A novel therapeutic target for migraine: 5-HT1F receptor agonists inhibit neurogenic inflammation. Neuroreport. 1997;8:2237–2240. doi: 10.1097/00001756-199707070-00029. [DOI] [PubMed] [Google Scholar]
- Johnson KW, Nisenbaum ES, Johnson MP, Dieckman DK, Clemens-Smith A, Siuda ER. Innovative drug development for headache disorders-glutamate. In: Olesen J, Ramadan NM, et al., editors. Innovative Drug Development for Headache Disorders. Vol. 16. Oxford: Oxford University Press; 2008. pp. 185–194. [Google Scholar]
- Juhasz G, Zsombok T, Jakab B, Nemeth J, Szolcsanyi J, Bagdy G. Sumatriptan causes parallel decrease in plasma calcitonin gene-related peptide (CGRP) concentration and migraine headache during nitroglycerin induced migraine attack. Cephalalgia. 2005;25:179–183. doi: 10.1111/j.1468-2982.2005.00836.x. [DOI] [PubMed] [Google Scholar]
- Juhl L, Edvinsson L, Olesen J, Jansen-Olesen I. Effect of two novel CGRP-binding compounds in a closed cranial window rat model. Eur J Pharmacol. 2007;567:117–124. doi: 10.1016/j.ejphar.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klussmann S, Nolte A, Bald R, Erdmann VA, Furste JP. Mirror-image RNA that binds D-adenosine. Nat Biotechnol. 1996;14:1112–1115. doi: 10.1038/nbt0996-1112. [DOI] [PubMed] [Google Scholar]
- Kulkarni O, Pawar RD, Purschke W, Eulberg D, Selve N, Buchner K, et al. Spiegelmer inhibition of CCL2/MCP-1 ameliorates lupus nephritis in MRL-(Fas)lpr mice. J Am Soc Nephrol. 2007;18:2350–2358. doi: 10.1681/ASN.2006121348. [DOI] [PubMed] [Google Scholar]
- Lassen LH, Haderslev PA, Jacobsen VB, Iversen HK, Sperling B, Olesen J. CGRP may play a causative role in migraine. Cephalalgia. 2002;22:54–61. doi: 10.1046/j.1468-2982.2002.00310.x. [DOI] [PubMed] [Google Scholar]
- Lee WS, Moussaoui SM, Moskowitz MA. Blockade by oral or parenteral RPR 100893 (a non-peptide NK1 receptor antagonist) of neurogenic plasma protein extravasation within guinea-pig dura mater and conjunctiva. Br J Pharmacol. 1994;112:920–924. doi: 10.1111/j.1476-5381.1994.tb13168.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, Burstein R, Strassman AM. Calcitonin gene-related peptide does not excite or sensitize meningeal nociceptors: implications for the pathophysiology of migraine. Ann Neurol. 2005;58:698–705. doi: 10.1002/ana.20619. [DOI] [PubMed] [Google Scholar]
- Lipton RB, Bigal ME, Diamond M, Freitag F, Reed ML, Stewart WF. Migraine prevalence, disease burden, and the need for preventive therapy. Neurology. 2007;68:343–349. doi: 10.1212/01.wnl.0000252808.97649.21. [DOI] [PubMed] [Google Scholar]
- Lipton RB, Buse DC, Serrano D, Holland S, Reed ML. Examination of unmet treatment needs among persons with episodic migraine: results of the American Migraine Prevalence and Prevention (AMPP) Study. Headache. 2013;53:1300–1311. doi: 10.1111/head.12154. [DOI] [PubMed] [Google Scholar]
- Marcus R, Goadsby PJ, Dodick D, Stock D, Manos G, Fischer TZ. BMS-927711 for the acute treatment of migraine: a double-blind, randomized, placebo controlled, dose-ranging trial. Cephalalgia. 2014;34:114–125. doi: 10.1177/0333102413500727. [DOI] [PubMed] [Google Scholar]
- Markowitz S, Saito K, Moskowitz MA. Neurogenically mediated plasma extravasation in dura mater: effect of ergot alkaloids. A possible mechanism of action in vascular headache. Cephalalgia. 1988;8:83–91. doi: 10.1046/j.1468-2982.1988.0802083.x. [DOI] [PubMed] [Google Scholar]
- May A, Gijsman HJ, Wallnofer A, Jones R, Diener HC, Ferrari MD. Endothelin antagonist bosentan blocks neurogenic inflammation, but is not effective in aborting migraine attacks. Pain. 1996;67:375–378. doi: 10.1016/0304-3959(96)03137-5. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JN, Martinez A, Unsworth EJ, Thiele CJ, Moody TW, Elsasser T, et al. Adrenomedullin expression in human tumor cell lines. Its potential role as an autocrine growth factor. Journal of Biological Chemistry. 1996;271:23345–23351. doi: 10.1074/jbc.271.38.23345. [DOI] [PubMed] [Google Scholar]
- Mogil JS, Miermeister F, Seifert F, Strasburg K, Zimmermann K, Reinold H, et al. Variable sensitivity to noxious heat is mediated by differential expression of the CGRP gene. Proc Natl Acad Sci U S A. 2005;102:12938–12943. doi: 10.1073/pnas.0503264102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesen J, Diener HC, Husstedt IW, Goadsby PJ, Hall D, Meier U, et al. Calcitonin gene-related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N Engl J Med. 2004;350:1104–1110. doi: 10.1056/NEJMoa030505. [DOI] [PubMed] [Google Scholar]
- O'Shaughnessy CT, Connor HE. Investigation of the role of tachykinin NK1, NK2 receptors and CGRP receptors in neurogenic plasma protein extravasation in dura mater. Eur J Pharmacol. 1994;263:193–198. doi: 10.1016/0014-2999(94)90541-x. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl Acids Res. 2014;42:D1098–D1106. doi: 10.1093/nar/gkt1143. (Database Issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen KA, Birk S, Doods H, Edvinsson L, Olesen J. Inhibitory effect of BIBN4096BS on cephalic vasodilatation induced by CGRP or transcranial electrical stimulation in the rat. Br J Pharmacol. 2004;143:697–704. doi: 10.1038/sj.bjp.0705966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purschke WG, Eulberg D, Buchner K, Vonhoff S, Klussmann S. An L-RNA-based aquaretic agent that inhibits vasopressin in vivo. Proc Natl Acad Sci U S A. 2006;103:5173–5178. doi: 10.1073/pnas.0509663103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purschke WG, Hoehlig K, Buchner K, Zboralski D, Schwoebel F, Vater A, et al. Identification and characterization of a mirror-image oligonucleotide that binds and neutralizes sphingosine 1-phosphate, a central mediator of angiogenesis. Biochem J. 2014;462:153–162. doi: 10.1042/BJ20131422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahmann A, Wienecke T, Hansen JM, Fahrenkrug J, Olesen J, Ashina M. Vasoactive intestinal peptide causes marked cephalic vasodilation, but does not induce migraine. Cephalalgia. 2008;28:226–236. doi: 10.1111/j.1468-2982.2007.01497.x. [DOI] [PubMed] [Google Scholar]
- Roh J, Chang CL, Bhalla A, Klein C, Hsu SY. Intermedin is a calcitonin/calcitonin gene-related peptide family peptide acting through the calcitonin receptor-like receptor/receptor activity-modifying protein receptor complexes. J Biol Chem. 2004;279:7264–7274. doi: 10.1074/jbc.M305332200. [DOI] [PubMed] [Google Scholar]
- Roth JD, Erickson MR, Chen S, Parkes DG. GLP-1R and amylin agonism in metabolic disease: complementary mechanisms and future opportunities. Br J Pharmacol. 2012;166:121–136. doi: 10.1111/j.1476-5381.2011.01537.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troltzsch M, Denekas T, Messlinger K. The calcitonin gene-related peptide (CGRP) receptor antagonist BIBN4096BS reduces neurogenic increases in dural blood flow. Eur J Pharmacol. 2007;562:103–110. doi: 10.1016/j.ejphar.2007.01.058. [DOI] [PubMed] [Google Scholar]
- Tuerk C, MacDougal S, Gold L. RNA pseudoknots that inhibit human immunodeficiency virus type 1 reverse transcriptase. Proc Natl Acad Sci U S A. 1992;89:6988–6992. doi: 10.1073/pnas.89.15.6988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vater A, Klussmann S. Turning mirror-image oligonucleotides into drugs: the evolution of Spiegelmer therapeutics. Drug Discov Today. 2015;20:147–155. doi: 10.1016/j.drudis.2014.09.004. [DOI] [PubMed] [Google Scholar]
- Vater A, Jarosch F, Buchner K, Klussmann S. Short bioactive Spiegelmers to migraine-associated calcitonin gene-related peptide rapidly identified by a novel approach: tailored-SELEX. Nucleic Acids Res. 2003;31:e130. doi: 10.1093/nar/gng130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vater A, Sell S, Kaczmarek P, Maasch C, Buchner K, Pruszynska-Oszmalek E, et al. A mixed mirror-image DNA/RNA aptamer inhibits glucagon and acutely improves glucose tolerance in models of type 1 and type 2 diabetes. J Biol Chem. 2013;288:21136–21147. doi: 10.1074/jbc.M112.444414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker CS, Hay DL. CGRP in the trigeminovascular system: a role for CGRP, adrenomedullin and amylin receptors? Br J Pharmacol. 2013;170:1293–1307. doi: 10.1111/bph.12129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss B, Alt A, Ogden AM, Gates M, Dieckman DK, Clemens-Smith A, et al. Pharmacological characterization of the competitive GLUK5 receptor antagonist decahydroisoquinoline LY466195 in vitro and in vivo. J Pharmacol Exp Ther. 2006;318:772–781. doi: 10.1124/jpet.106.101428. [DOI] [PubMed] [Google Scholar]
- Williamson DJ, Hargreaves RJ, Hill RG, Shepheard SL. Sumatriptan inhibits neurogenic vasodilation of dural blood vessels in the anaesthetized rat – intravital microscope studies. Cephalalgia. 1997;17:525–531. doi: 10.1046/j.1468-2982.1997.1704525.x. [DOI] [PubMed] [Google Scholar]
- Zeller J, Poulsen KT, Sutton JE, Abdiche YN, Collier S, Chopra R, et al. CGRP function-blocking antibodies inhibit neurogenic vasodilatation without affecting heart rate or arterial blood pressure in the rat. Br J Pharmacol. 2008;155:1093–1103. doi: 10.1038/bjp.2008.334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann U, Fischer JA, Muff R. Adrenomedullin and calcitonin gene-related peptide interact with the same receptor in cultured human neuroblastoma SK-N-MC cells. Peptides. 1995;16:421–424. doi: 10.1016/0196-9781(94)00195-c. [DOI] [PubMed] [Google Scholar]
- Zimmermann U, Fluehmann B, Born W, Fischer JA, Muff R. Coexistence of novel amylin-binding sites with calcitonin receptors in human breast carcinoma MCF-7 cells. J Endocrinol. 1997;155:423–431. doi: 10.1677/joe.0.1550423. [DOI] [PubMed] [Google Scholar]
- Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Binding selectivity of NOX-L41.
Figure S2 PEGylation does not affect the inhibitory activity of NOX-L41.
Figure S3 NOX-L41 does not inhibit pramlintide and adrenomedullin-induced cAMP production.
Figure S4 NOX-L41-Alexa488 inhibits CGRP-induced cAMP production.
Figure S5 Qdots705 do not extravasate from dural vessels.
Table S1 Inhibition of dural PPE (reported as extravasation ratios) by BIBN4096 at various doses and pretreatment times following intravenous administration.
Table S2 Inhibition of dural PPE (reported as extravasation ratios) by NOX-L41 and BIBN4096 at various doses, pretreatment times and administration routes.