

Abstract
Background and Purpose
We aimed to characterize the pharmacology and electrophysiology of N-[3-(1H-benzimidazol-2-yl)-4-chloro-phenyl]pyridine-3-carboxamide (AZSMO-23), an activator of the human ether-a-go-go-related gene (hERG)-encoded K+ channel (Kv11.1).
Experimental Approach
Automated electrophysiology was used to study the pharmacology of AZSMO-23 on wild-type (WT), Y652A, F656T or G628C/S631C hERG, and on other cardiac ion channels. Its mechanism of action was characterized with conventional electrophysiology.
Key Results
AZSMO-23 activated WT hERG pre-pulse and tail current with EC50 values of 28.6 and 11.2 μM respectively. At 100 μM, pre-pulse current at +40 mV was increased by 952 ± 41% and tail current at −30 mV by 238 ± 13% compared with vehicle values. The primary mechanism for this effect was a 74.5 mV depolarizing shift in the voltage dependence of inactivation, without any shift in the voltage dependence of activation. Structure–activity relationships for this effect were remarkably subtle, with close analogues of AZSMO-23 acting as hERG inhibitors. AZSMO-23 blocked the mutant channel, hERG Y652A, but against another mutant channel, hERG F656T, its activator activity was enhanced. It inhibited activity of the G628C/S631C non-inactivating hERG mutant channel. AZSMO-23 was not hERG selective, as it blocked hKv4.3-hKChIP2.2, hCav3.2 and hKv1.5 and activated hCav1.2/β2/α2δ channels.
Conclusion and Implications
The activity of AZSMO-23 and those of its close analogues suggest these compounds may be of value to elucidate the mechanism of type 2 hERG activators to better understand the pharmacology of this area from both a safety perspective and in relation to treatment of congenital long QT syndrome.
Tables of Links
| TARGETS |
|---|
| Ion channels |
| Cav1.2/β2/α2δ channel |
| Cav3.2 channel |
| HCN4 channel |
| hERG, Kv11.1 channel |
| Kv1.5 channel |
| Kv4.3-hKChIP2.2 channel |
| Kv7.1-hKCNE1 channel |
| Nav1.5 channel |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (Alexander et al., 2013).
Introduction
There has been considerable interest over the last decade in drugs that, as a side effect, increase or decrease the duration of the QT interval on the surface electrocardiogram. Such effects have been termed drug-induced long and short QT syndrome respectively. Focus on drug-induced long QT syndrome has been particularly intense because it indicates delayed repolarization of ventricular action potentials and is associated with a potentially fatal cardiac arrhythmia (Torsades de Pointes). Also, drug-induced long QT syndrome has been a significant contributor to attrition of drug candidates in the pharmaceutical industry (Shah, 2006). Although there are relatively little clinical data about the risks of drug-induced short QT syndrome, the safety implications of shortening ventricular action potentials are sufficiently uncertain for it to be considered a side effect best avoided (Malik, 2010; Shah, 2010).
In terms of molecular mechanisms underlying delayed ventricular repolarization, inhibition of the cardiac K+ channel encoded by the human ether-a-go-go-related gene (hERG; Kv11.1) is widely reported as the most common cause. Not only does this channel carry a key repolarizing current, but also it is sensitive to block by large numbers of structurally diverse drugs (Mitcheson, 2008). This has led to large-scale screening operations to detect and reject hERG blockers early in the drug discovery process. In the course of this, a small number of compounds have been identified that, instead, enhance hERG current. Such compounds, by increasing outward potassium current during the ventricular action potential, could reduce its duration, leading to drug-induced short QT syndrome and associated safety concerns. An understanding of the pharmacology of these hERG activators is not only important from a safety perspective however, as their enhancement of hERG current has been proposed as a means of normalizing the ventricular action potential duration of patients with congenital forms of long QT syndrome (see Zhou et al., 2011; Sanguinetti, 2014).
Low molecular weight hERG activators appear to fall into two categories based on their principal mechanism of action (Perry et al., 2010). Type 1 activators work mainly by slowing the deactivation rate; while type 2 activators shift the voltage dependence of inactivation to more depolarized levels. Here we report the characteristics of AZSMO-23, a type 2 hERG activator that induces an atypically large shift in the voltage dependence of inactivation and whose activity is significantly modified by mutation of amino acids in the hERG channel usually associated with effects on the potency of hERG blockers. Some of the data have already appeared in preliminary form (Bridgland-Taylor et al., 2008).
Methods
IonWorks™ electrophysiology overview
Apart from the conventional patch clamping used to investigate the effect of AZSMO-23 on hERG channel electrophysiology (Figures 4 and 5), the rest of the data were generated using IonWorks (Molecular Devices, Sunnyvale, CA, USA) automated electrophysiology. The original principles and operation of this device have been described by Schroeder et al. (2003). Briefly, the technology is based on a 384-well plate (PatchPlate™, Molecular Devices) in which a recording is attempted in each well by using suction to position and hold a single cell on a small hole separating two isolated, fluid-filled chambers. Once sealing has taken place, the solution on the underside of the PatchPlate is changed to one containing amphotericin B. This permeabilizes the patch of cell membrane covering the hole in each well and in effect allows a perforated, whole-cell patch clamp recording to be made. Once optimized, successful recordings can be made before and after drug or vehicle addition from around 300 of the 384 wells in a single ‘Run’ of the device. Although only a single concentration of drug is added to each well, the high number of recordings means that non-cumulative concentration–effect curves can be generated very rapidly compared with the time taken using conventional patch clamping. However, since the invention of the original device (known as IonWorks HT), an additional recording mode has been developed: IonWorks® Quattro™. In this mode, each well of the PatchPlate has 64 rather than a single hole, such that each recording is an ensemble whole-cell current from up to 64 cells; this is often referred to as population patch clamp (Finkel et al., 2006). The key benefit to this mode of operation is that all of the 384 wells yield useable data.
Figure 4.
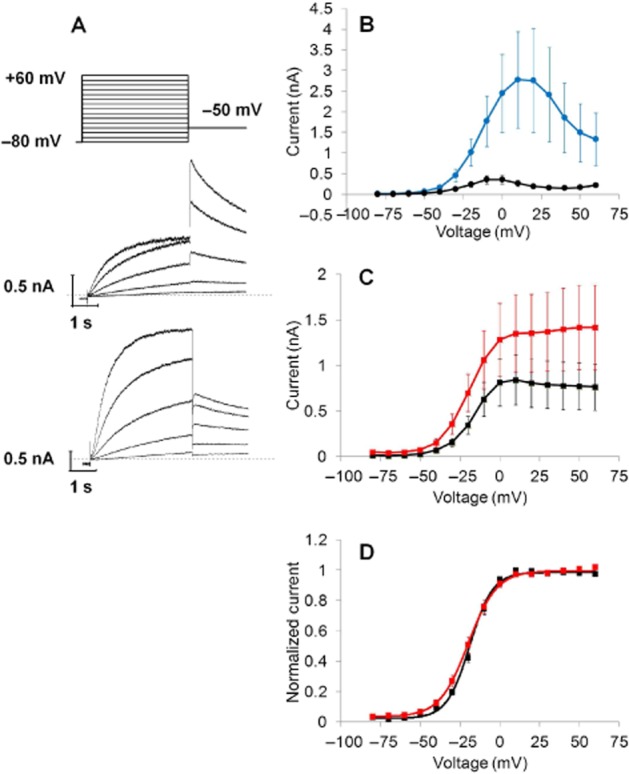
Effect of 30 μM AZSMO-23 on hERG pre-pulse and tail current activation. All data are from conventional whole-cell electrophysiology. (A) The voltage protocol used (top) to evoke typical current responses after 5 min exposure to vehicle (middle) or AZSMO-23 (bottom). Note that only the current traces to Vpre-pulse steps of −40, −30, −20, −10 and 0 mV are shown. The dashed line represents the zero current level. (B) Pre-pulse current–voltage relationship in cells after 5 min exposure to vehicle or AZSMO-23. (C) Tail current–voltage relationship in cells after 5 min exposure to vehicle or AZSMO-23. (D) Normalized data from C. For the graphs in B, C and D, each data point is mean ± SEM; n = 6 (vehicle cells); n = 7 (AZSMO-23 cells).
Figure 5.
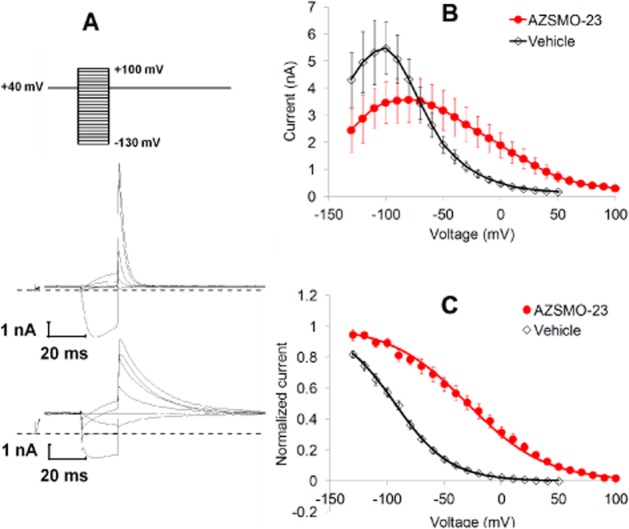
Effect of 30 μM AZSMO-23 on hERG voltage dependence of inactivation. All data are from conventional whole-cell electrophysiology. (A) The voltage protocol used (top) to evoke typical current responses after 5 min exposure to vehicle (middle) or AZSMO-23 (bottom). Note that only the current traces to Vpre-pulse steps of +80, +40, 0, −40, −80 and −120 mV are shown. Furthermore, only the initial current trace after stepping from −80 to +40 mV, and the 20 ms of current data preceding the Vpre-pulse onwards are shown. The dashed line represents the zero current level. (B) The current–voltage relationship after 5 min exposure to vehicle or AZSMO-23. (C) Normalized data from B. Each data point is mean ± SEM; n = 6 (vehicle cells); n = 7 (AZSMO-23 cells).
A disadvantage of IonWorks HT and IonWorks Quattro is that they apply a voltage clamp protocol under control conditions and repeat this after a user-defined period of exposure to vehicle/test compound; in between there is no voltage clamping, so the time course of any effects cannot be observed. This constraint has been removed by the development of IonWorks Barracuda®. Like IonWorks Quattro, it uses population patch clamp but is a step change in technology since it provides continuous voltage clamp throughout the experiment.
All the hERG data from automated electrophysiology in these experiments, came from IonWorks HT, except for the time course data in Figure 3, which were generated using IonWorks Barracuda. All the ion channel selectivity data in Figure 1 were from IonWorks Quattro except for those from hKv1.5 and hCav3.2 where IonWorks HT was used.
Figure 3.
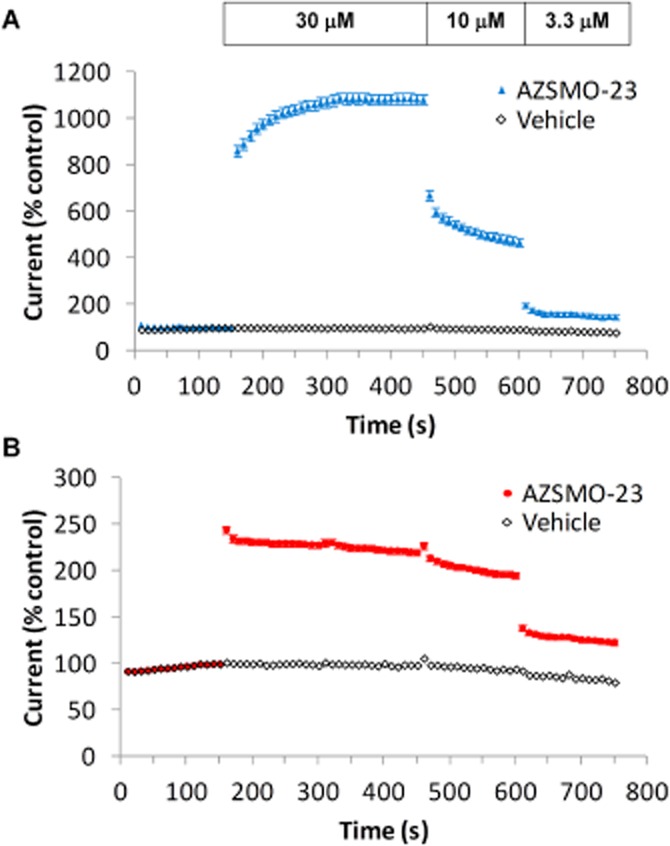
AZSMO-23 time course of effect. Using data from IonWorks Barracuda, (A) shows the onset of, and recovery from, the effect of AZSMO-23 (30 μM) on pre-pulse current compared with the effects of vehicle. (B) As for A, but measuring the effect on tail current compared with vehicle. Current amplitudes are expressed relative to the pre-pulse or tail current amplitude evoked at 150 s (i.e. immediately before addition of AZSMO-23 or vehicle). Each data point is mean ± SEM; n = 8.
Figure 1.
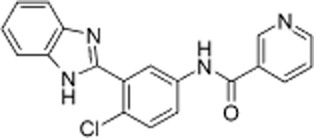
Chemical structure of AZSMO-23: (N-[3-(1H-benzimidazol-2-yl)-4-chloro-phenyl]pyridine-3-carboxamide).
IonWorks electrophysiology – hERG
As described by Bridgland-Taylor et al. (2006), for each experimental ‘Run’ of IonWorks HT, the device was operated at ∼21°C. The extracellular solution was Dulbecco's PBS (Invitrogen™, Paisley, UK), which contained (in mM): NaCl 137, KCl 2.7, Na2HPO4 8, KH2PO4 1.5, and to which was added CaCl2 and MgCl2 to give final concentrations of 0.9 and 0.5 mM respectively. The ‘pipette’ solution was (in mM): KCl 140, EGTA 1, MgCl2 1 and HEPES 20 (pH 7.25–7.3 using 10 M KOH) plus 100 μg·mL−1 amphotericin B. A pre-compound hERG current was evoked in each cell in the presence of PBS by the following voltage protocol: a 20 s holding potential (VH) of −70 mV, a 160 ms step to −60 mV (to obtain an estimate of leak), a 100 ms step back to −70 mV, a 1 s step to +40 mV (Vpre-pulse), a 2 s step to −30 mV (Vtail) and finally a 500 ms step to −70 mV. A test compound or vehicle was then added to each well and after 3 min the voltage pulse was re-applied to generate a post-compound hERG current.
In the IonWorks experiments, vehicle or test compounds were prepared and added as follows. A 96-well ‘compound plate’ was prepared where each well contained a single concentration of test compound at three times the final concentration required, dissolved in 1% dimethyl sulfoxide (DMSO) in PBS. Some wells contained just vehicle (1% DMSO in PBS). The robotics of IonWorks was set up such that 3 μL of the contents of each well of the compound plate was added to each of four wells of the 384-well PatchPlate in which the recordings were made. Each well of a PatchPlate already contained 3 μL of PBS and 3 μL of PBS containing cell suspension at a concentration of 0.25 × 106 cells/mL. Therefore, when 3 μL of test compound or vehicle was added to the PatchPlate the cells were exposed to the final test compound concentration or final vehicle concentration (0.33% DMSO).
For each experimental ‘Run’ of IonWorks Barracuda, all the experimental conditions were the same as for IonWorks HT, with the exception of the voltage protocol. As explained above, IonWorks Barracuda can perform continuous voltage clamp, so VH was maintained at −70 mV throughout, with the same hERG voltage protocol as IonWorks HT being delivered every 10 s.
Data analysis – hERG
In IonWorks HT, each current response was analysed, as follows, using the IonWorks software. Leak subtraction was first applied based on a modified P/N subtraction method described by Bezanilla and Armstrong (1977). Three measurements were then taken: the mean baseline current at VH over a 40 ms time period; the pre-pulse current at the end of Vpre-pulse and the peak tail current during Vtail. The baseline current was then subtracted from the pre-pulse and tail current values to define their amplitudes. The pre-pulse and tail current amplitudes following addition of test compound or vehicle were then expressed as a percentage of their corresponding pre-compound/vehicle values. Finally, the percentage change seen in each cell following test compound was scaled relative to the mean percentage change seen in cells in the same PatchPlate that received vehicle. In short, the post-compound currents in each cell were expressed as a percentage of the mean, time-matched vehicle control data collected in the same PatchPlate. These values were then used to construct non-cumulative concentration–effect curves with each data point represented as mean ± SEM. Current responses for the non-inactivating hERG mutant (G628C/S631C) were analysed, as for wild-type (WT) hERG, by measuring the pre-pulse current at the end of Vpre-pulse. No tail current measurements were made.
The resulting concentration–effect curves were fitted using a custom-written Origin package (OriginLab Corp., Northampton, MA, USA) to replicate the data using non-linear regression by means of a four-parameter logistic (curve bottom, curve top, nH and IC50/EC50). The lower and upper 95% confidence limit around the IC50/EC50 value was used to indicate variability in that parameter. Because curves were non-cumulative and the number of successful recordings at each test concentration varied, the range of n (the number of cells at each test concentration) is quoted.
Current traces from the IonWorks Barracuda experiments shown in Figure 3 were analysed in the same way as for IonWorks HT and current amplitude data from each well were normalized to the current values immediately before addition of AZSMO-23. Note that since IonWorks Barracuda is a population patch clamp device, each well produces a recording from up to 64 cells; hence, n for these experiments refers to the number of wells that make up the dataset.
IonWorks electrophysiology – other cardiac ion channels
IonWorks was also used to test the selectivity of AZSMO-23 versus heterologously expressed human cardiac ion channels. Two channel types (hKv1.5 and hCav3.2) were recorded in IonWorks HT mode but hNav1.5, hCav1.2/β2/α2δ, hKv4.3-hKChIP2.2, hKv7.1-hKCNE1 and hHCN4 were recorded in IonWorks Quattro. The detailed method for hNav1.5 has been described by Harmer et al. (2008). However, methods for this and the other channels were in general terms the same as for hERG except in terms of the voltage protocol and, for some channels, the solutions used, as described below.
Extracellular and ‘pipette’ solutions were the same as for hERG, except for hHCN4 where the extracellular solution was (in mM): Na gluconate 104, NaCl 10, KCl 30, MgCl2 1, CaCl2 1.8, HEPES 10, glucose 5 (pH 7.3 using NaOH). The ‘pipette’ solution was (in mM): K gluconate 130, NaCl 10, MgCl2 1, EGTA 1, HEPES 10, pH 7.2 using KOH. The hCav1.2/β2/α2δ ‘pipette’ solution was as for hERG but also contained 2 mM Na2ATP and 0.3 mM Na2GTP; 4 mM aescin was used as the permeabilizing agent instead of amphotericin.
Each voltage protocol consisted of a VH that was briefly interrupted by a +10 or −10 mV, non-current-activating voltage step to define the leak current, followed by a step to a test voltage (Vtest) of an amplitude and duration that would activate each channel type. Specific details are as follows: hCav1.2/β2/α2δ VH −60 mV for 10 s, Vtest 0 mV for 100 ms; hCav3.2 VH −100 mV for 20 s, Vtest −20 mV for 200 ms; hKv1.5VH −70 mV for 15 s, Vtest 0 mV for 200 ms; hKv4.3-hKChIP2.2 VH −80 mV for 20 s, Vtest +20 mV for 1000 ms; hKv7.1-hKCNE1 VH −80 mV for 5 s, Vtest +40 mV for 4 s; hHCN4 VH −30 mV for 5 s, Vtest −110 mV for 4 s. After VH −90 mV for 15 s, the hNav1.5 protocol consisted of an eight-pulse train with each pulse being Vtest 0 mV for 50 ms followed by a return to VH for 300 ms.
The cell number (×106 cells mL−1) in the suspension added to each well was: hKv1.5, 0.25; hCav3.2, 0.6; for all other channels, 1–1.5; although these suspensions were ultimately diluted threefold in the PatchPlate.
Data analysis – other cardiac ion channels
Currents were all leak subtracted using the same method as for hERG and their amplitudes defined as the difference between the baseline current at VH (calculated as a mean value over a brief period immediately before current activation) and the Vtest current. For sustained currents this was the mean value over a brief period of time at the end of Vtest (hKv1.5, hKv7.1-hKCNE1, hHCN4 channels), and for transient currents it was the peak current during Vtest (hCav1.2/β2/α2δ, hCav3.2, hKv4.3-hKChIP2.2, hNav1.5 channels). For hNav1.5 channels, the data for the eighth pulse in the train were used. For each IonWorks run, the current amplitude in each cell in the presence of PBS was compared with that in the presence of test compound for the same cell. Subsequent analysis was as described for hERG.
Conventional electrophysiology experiments – hERG
This has been described fully by Bridgland-Taylor et al. (2006). Briefly, glass coverslips seeded with hERG-expressing CHO-K1 cells were placed at the bottom of a custom-made Perspex chamber containing bath solution at room temperature (∼21°C). The bath solution contained (in mM): NaCl 137, KCl 4, MgCl2 1, CaCl2 1.8, HEPES 10 and glucose 10 (pH 7.4 using 1 M NaOH). This chamber was fixed to the stage of an inverted, phase-contrast microscope and conventional ‘gigaseal’ whole-cell patch clamp recordings were made. The pipette solution was (in mM): KCl 130, MgCl2 1, HEPES 10, Na2ATP 5, EGTA 5 (pH 7.2 using 1 M KOH). Pipette resistance was typically 1.5–4 ΜΩ. Following ‘break-in’ and appropriate adjustment of series resistance and capacitance controls, Clampex software (Molecular Devices, Sunnyvale, CA, USA) was used to set a holding potential and to deliver voltage protocols required to characterize electrophysiological parameters. Series resistance compensation of at least 70% was applied. The junction-potential error measured using the flowing 3 M KCl electrode method was found to be 4 mV but was not corrected for. The holding voltage for every protocol was −80 mV and for each protocol used, voltage pulses were delivered every 10 s. Currents were low-pass filtered by the amplifier at 5 kHz. The filtered signal was then acquired, online, by digitizing this analogue signal from the amplifier with a Digidata 1200 analogue to digital converter (Molecular Devices) at a sampling frequency of 10 kHz. The digitized signal was then captured on a computer running Clampex software.
We studied the effect of AZSMO-23 on the voltage dependence of activation and inactivation. In both cases, after achieving a whole-cell recording, we first delivered a family of voltage steps (F1) to study activation or inactivation while perfusing the recording chamber with bath solution containing vehicle (0.1% DMSO). We then either kept perfusing with vehicle or switched to perfusing with 30 μM AZSMO-23. Five minutes later, we repeated the voltage protocol (F2) to again define activation or inactivation. The data from cells where both F1 and F2 were performed in the presence of vehicle indicated that there was a time-dependent shift in the voltage dependence of activation (data not shown). For this reason, for both activation and inactivation, we defined the effect of AZSMO-23 by comparing to data from F2 in the presence of vehicle (i.e. the time-matched vehicle control data).
The voltage dependence of activation was measured as described by Mannikko et al. (2010). As shown in Figure 4A, membrane potential (Vm) was stepped from VH of −80 mV to vpre-pulse voltages ranging from −80 to +60 mV for 4 s followed by a 4 s step to −50 mV (Vtail) to evoke a tail current. The current amplitude at the end of Vpre-pulse and the peak tail current during Vtail were plotted against Vpre-pulse. Using Clampfit (Molecular Devices), a Boltzmann curve was then fitted to normalized data to derive V½ and Vslope values for activation.
Voltage dependence of inactivation was estimated using the ‘triple pulse’ protocol described by Vandenberg et al. (2012), as shown in Figure 5A. In this protocol, at the end of a 4 s step to +40 mV, 20 ms Vpre-pulse voltages ranging from −130 to +100 mV (depending on the conditions) were applied before stepping to a Vtail of +40 mV. The tail current 1 ms after stepping to Vtail gives an estimate of the extent to which channels recover from inactivation at each value of Vpre-pulse. However, tail current amplitude is underestimated owing to closure (deactivation) of the channels, especially at the most negative Vpre-pulse voltages. This effect was adjusted for by multiplying the tail current amplitude by a closing factor. This was derived from separate recordings where the time course of channel closure was studied as follows. The voltage protocol used for the ‘extrapolation’ method of defining the voltage dependence of inactivation (Vandenberg et al., 2012) was applied to cells in the presence of 30 μM AZSMO-23 or to time-matched vehicle control cells. Specifically, following a 4 s Vpre-pulse to +40 mV, the voltage was stepped to Vtail values ranging from −130 to +40 mV. A two-exponential curve was then fitted to the decaying phase of the current at Vtail and extrapolated back to the time point at the start of Vtail. The closing factor was calculated by dividing the extrapolated current amplitude at the start of Vtail by the current amplitude at the end of Vtail. This gives a measure of the extent of channel closure during the 20 ms step to Vpre-pulse in the ‘triple pulse’ protocol. Values for V½ and Vslope in AZSMO-23 and time-matched vehicle control cell were then derived via a Boltzmann fit to corrected, normalized data. Note that currents in the presence of AZSMO-23 were not fully inactivated by the initial 4 s step to +40 mV. However, attempts to use an initial step and Vtail of +80 mV or greater, invariably led to loss of the recording.
Cell lines
All cell lines were generated at AstraZeneca except: hCav1.2/β2/α2δ (ChanTest, Cleveland, OH, USA), hHCN4 and hKv7.1-hKCNE1 (Millipore, Billerica, MA, USA). WT hERG and the other channel types were stably expressed in CHO-K1 cells. The only exception was hCav3.2, which was expressed in HEK-293 cells. The Y652A and F656T hERG-binding site mutants were expressed in doxycycline-inducible CHO-K1 cells as described by Shamovsky et al. (2009). The same method was used to generate a cell line expressing a non-inactivating hERG mutant (G628C/S631C). The cell lines were maintained using standard tissue culture techniques.
Data analysis
Because all IonWorks HT hERG data comprised non-cumulative concentration–effect curves, any comparisons were made using an unpaired, two-tailed Student's t-test. Conventional electrophysiology experiments involved comparing data from AZSMO-23-treated cells with those from time-matched vehicle control cells; hence, an unpaired, two-tailed Student's t-test was also used for these experiments.
Materials
AZSMO-23 and its analogues were synthesized by or on behalf of AstraZeneca (Macclesfield, Cheshire, UK). Other chemicals were sourced from Sigma-Aldrich (St. Louis, MO, USA) or Apin Chemicals (Abingdon, Oxfordshire, UK).
Results
Initial observations
In the process of screening for hERG blockers using IonWorks HT, a 3-min exposure to AZSMO-23 (Figure 1) was found to cause a concentration-dependent increase in both the pre-pulse and tail current. As shown in Figure 2, the proportionate increase was greater for the pre-pulse current. In the presence of 100 μM AZSMO-23, the current at the end of the pre-pulse to +40 mV increased by 952 ± 41% (n = 16) and the tail current at −30 mV by 238 ± 13% (n = 16). In terms of absolute current, the pre-pulse current in the presence of vehicle was 0.36 ± 0.05 nA (n = 16) and in the presence of 100 μM AZSMO-23 was 2.76 ± 0.23 nA (n = 16); P < 0.001. In the experiment shown in Figure 2B, the EC50 for the effect on pre-pulse current was 28.6 μM and for the effect on tail current was 11.2 μM (n = 14–20).
Figure 2.
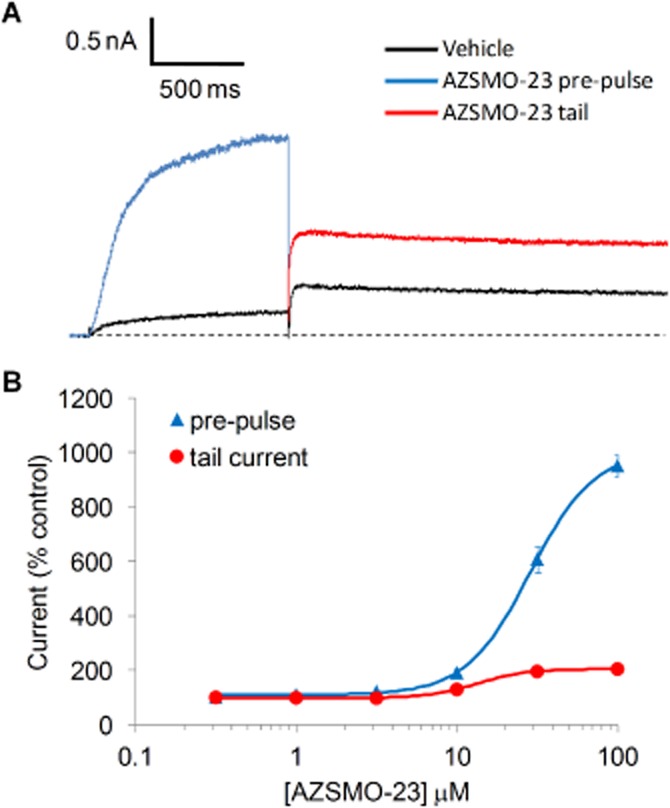
Activation of hERG current by AZSMO-23. (A) Typical leak-corrected current trace from IonWorks HT before and 3 min after exposure to 100 μM AZSMO-23. The dashed line represents the zero current level after leak subtraction. (B) Concentration–effect relationship for AZSMO-23 versus the pre-pulse current and tail current. Current amplitudes in the presence of a given concentration of AZSMO-23 were normalized by expressing them as a percentage of the mean, time-matched vehicle control data collected in the same PatchPlate. The dashed line at 100% therefore represents the vehicle control level for the pre-pulse or tail current. Each data point is mean ± SEM; n = 14–20. For most data points, the error bar is covered by the symbol. The concentration–effect curve parameters for the pre-pulse data are: EC50 28.6 μM [lower and upper 95% confidence limit (CL) 25.9, 32.6 μM respectively]; nH 2.2. The corresponding tail current data are: EC50 11.2 μM (lower and upper 95% CL 6.8, 18.4 μM respectively); nH 3.6.
Time course of effect
As we could not follow the time course of the AZSMO-23 effect using IonWorks HT, we used the next generation of IonWorks, known as IonWorks Barracuda, to determine onset and recovery of its activity. Although the same in terms of the way whole-cell recordings are made, the key attribute of this device is its ability to make additions of vehicle or test compounds while maintaining a continuous voltage clamp (see Methods). As shown in Figure 3, using this capability, we evoked hERG currents every 10 s. Baseline data were collected for 150 s and 30 μM AZSMO-23 was then added for 300 s before its concentration was diluted to 10 μM for 150 s and then 3.3 μM for a further 150 s. Note that complete washout cannot be achieved, as total buffer removal would result in loss of the recording. Hence washout can only be achieved in steps, with each step decreasing by two-thirds the drug concentration in each well. On addition of AZSMO-23, the effect on pre-pulse current reached steady state after around 180 s with a 10.8-fold increase (Figure 3A), but the effect on tail current was an immediate 2.4-fold increase (Figure 3B). When the concentration of AZSMO-23 was reduced from 30 to 10 μM, then 3.3 μM, its effects were reversed, but the offset of the effect was more rapid for the tail current.
Mechanism of action studies
To explore the mechanistic basis of the effect of AZSMO-23, we used conventional, whole-cell patch clamping to assess the effect of 30 μM AZSMO-23 on the voltage dependence of activation and inactivation compared with data from time-matched vehicle control cells. Data in the presence of AZSMO-23 were generated after 5 min incubation with the compound. A protocol to define the voltage dependence of activation (Figure 4A) revealed a 7.7-fold enhancement of the peak, pre-pulse current (Figure 4B)]. The amplitude of the maximum tail current evoked by this voltage protocol in the presence of AZSMO-23 was not statistically different (P > 0.05) from that for time-matched vehicle control data (Figure 4C). Boltzmann fits to the normalized tail current data (Figure 4D) showed that the voltage dependence of activation was not affected by AZSMO-23 [control V½ and Vslope −17.7 ± 0.9 and 7.2 ± 0.5 mV respectively (n = 6); AZSMO-23 V½ and Vslope 19.5 ± 1.7 and 8.5 ± 0.6 mV respectively (n = 7); P > 0.05 for both parameters].
In contrast, AZSMO-23 caused a depolarizing shift in the voltage dependence of inactivation (Figure 5). To quantify the effect, we used a ‘triple pulse’ protocol as described by Vandenberg et al. (2012). This involves a first step to a depolarized membrane potential that aims to ensure that all channels are inactivated. The channels are then allowed to relax to a steady state of inactivation via a second short step to different potentials, before a third step back to the initial depolarized potential to assess the extent to which channels are inactivated during the second step (Figure 5A). However, during initial experiments using this protocol, we found that in the presence of AZSMO-23, its effect on the voltage dependence of inactivation was such that we had to depolarize the cells to at least +80 mV to achieve full inactivation. This invariably led to a loss of the recording, such that we were only able to obtain data using a protocol based around a membrane potential of +40 mV. Using this voltage protocol, AZSMO-23 had a significant effect on inactivation [control V½ and Vslope −94.5 ± 3.4 and 24.5 ± 0.7 mV respectively (n = 6); AZSMO-23 V½ and Vslope −20.0 ± 6.9 and 36.0 ± 1.5 mV respectively (n = 7); P < 0.05 for both parameters]. The effect of AZSMO-23 on inactivation is also apparent by a qualitative assessment of the rate of inactivation [compare tail current decay in a vehicle-treated cell (Figure 5A, middle) with the slower rate seen in an AZSMO-23-treated cell (Figure 5A, bottom)].
Following on from these mechanistic observations, we used IonWorks HT to test the effect of AZSMO-23 on a mutant form of hERG (G628C/S631C) reported to lack the normal inactivation process (Smith et al., 1996). This was on the basis that AZSMO-23 should not increase G628C/S631C hERG current if removal of inactivation is the principal basis for its effect on WT hERG current. As shown in Figure 6, in the presence of vehicle, G628C/S631C hERG current had a time course that is characteristic of a non-inactivating potassium current (large outward current at Vpre-pulse but lacking a large outward tail current at Vtail). After a 3 min exposure to 100 μM AZSMO-23, rather than significant enhancement of pre-pulse current, as seen with WT hERG, there was a 22.9 ± 2.3% (n = 7) inhibition of the current measured at the end of Vpre-pulse.
Figure 6.
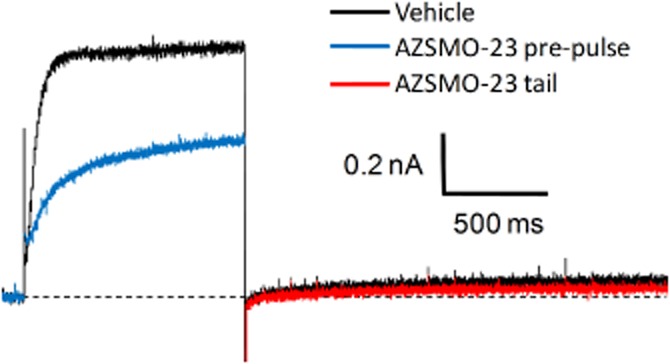
Effect of AZSMO-23 on a non-inactivating hERG mutant channel (G628C/S631C). Typical leak-corrected current trace, using IonWorks HT, before and after 100 μM AZSMO-23. The dashed line represents the zero current level after leak subtraction.
Structure–activity relationships
Three compounds with very similar structures to AZSMO-23 were tested to explore structure–activity relationships. After a 3 min exposure in IonWorks HT, all three compounds inhibited, rather than activated, both the pre-pulse and tail current. Even compound 1, which differed from AZSMO-23 only in the position of the nitrogen on the pyridine ring, blocked rather than activated hERG. These data and the structure of the close analogues (compounds 1, 2 and 3) are shown in Figure 7. Only data for inhibition of the tail current are shown, since the pre-pulse current is relatively small, such that the signal-to-noise ratio in the presence of an inhibitor is poor.
Figure 7.
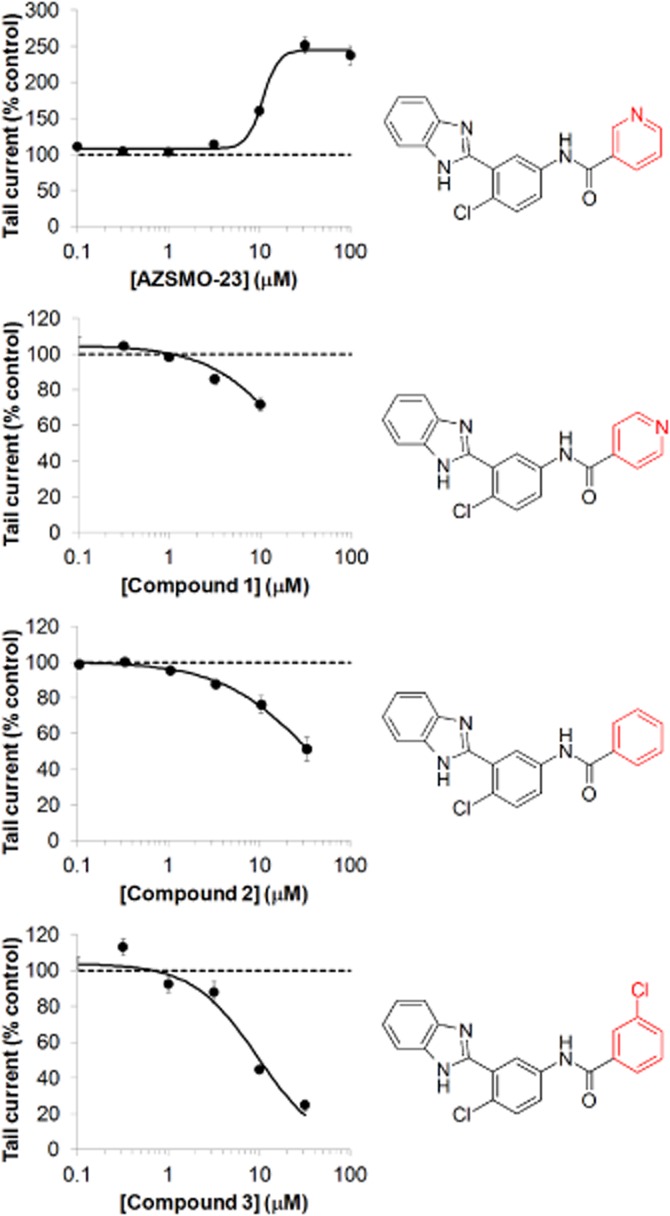
Effect of AZSMO-23 and its close analogues on hERG tail current. Concentration–effect relationship for AZSMO-23 and its close analogues, compounds 1, 2 and 3, using IonWorks HT. Current amplitudes in the presence of a given concentration of test compound were normalized by expressing them as a percentage of the mean, time-matched, vehicle control data collected in the same PatchPlate. The dashed line at 100% therefore represents the tail current vehicle control level. Each data point is mean ± SEM; n = 4–11. AZSMO-23 concentration–effect curve parameters for the tail current data shown are: EC50 12.3 μM (lower and upper 95% CL 8.7, 20.1 μM respectively); nH 3.4. Concentration–effect curve parameters were not defined for AZSMO-23 analogues since curves were incomplete.
AZSMO-23 has some structural similarities to ICA-105574, the type 2 activator reported by Gerlach et al. (2010): both compounds have an N-phenyl benzamide core structure (see Figure 8). We therefore tested, using IonWorks HT, two further compounds (compounds 4 and 5) that combined features of both molecules. After a 3 min incubation, neither compound activated hERG but instead inhibited the pre-pulse current (data not shown) and the tail current (Figure 8).
Figure 8.
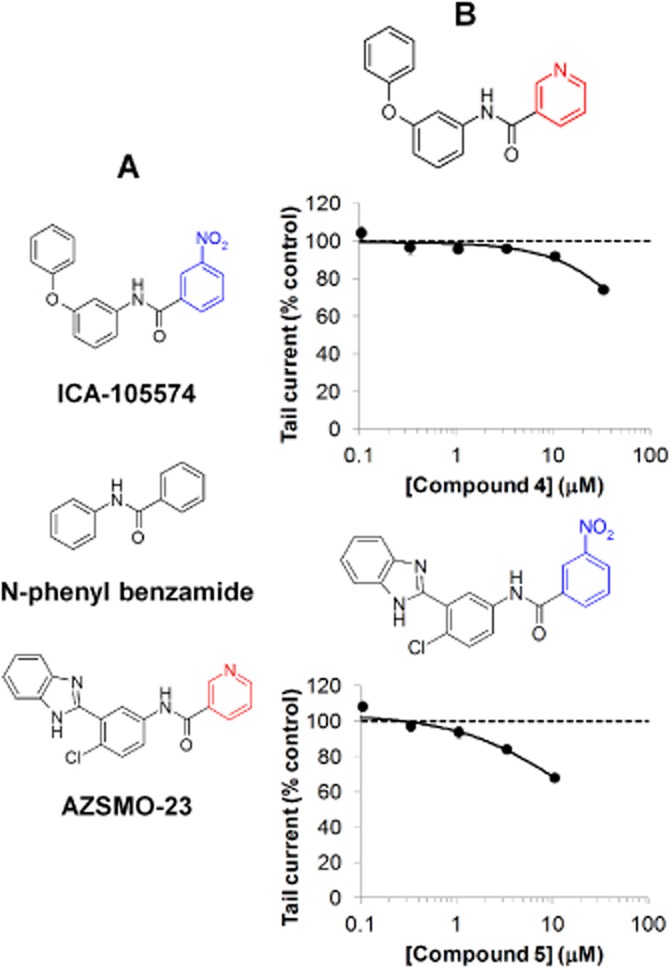
Effect of combining structural features of AZSMO-23 and ICA-105574. (A) Structures of AZSMO-23, ICA-105574 and the N-phenyl benzamide core common to both compounds. The ICA-105574 structural element is shown in blue and the AZSMO-23 structural element shown in red, were exchanged to make compounds 4 and 5. (B) Concentration–effect relationship for compounds 4 and 5, using IonWorks HT. Current amplitudes in the presence of a given concentration of test compound were normalized by expressing them as a percentage of the mean, time-matched, vehicle control data collected in the same PatchPlate. The dashed line at 100% therefore represents the tail current vehicle control level. Each data point is mean ± SEM; n = 4–8. Concentration–effect curve parameters were not defined since curves were incomplete.
Activity versus hERG-binding site mutants
Archetypal hERG blockers such as cisapride are reported to be less potent inhibitors of the mutant channels Y652A hERG, where tyrosine is changed to alanine, and F656T hERG, where phenylalanine is changed to threonine (Fernandez et al., 2004). These residues are therefore considered critical for blocker activity. However, the activator properties of ICA-105574 have been reported to be modified at Y652A and F656T hERG mutant channels (Garg et al., 2011), so we sought to determine whether this was also true for AZSMO-23. To reproduce this property of the binding site mutants in IonWorks HT, we first tested cisapride and, as expected, found a significant decrease in potency (Figure 9A–D and Table 1). However, at the Y652A mutant, AZSMO-23 inhibited rather than enhanced both the pre-pulse and tail current (Figure 9E, F and Table 1). In contrast, at the F656T mutant channel, its activating properties were enhanced (Figure 9 G, H and Table 1). Typical current traces from experiments with Y652A and F656T hERG channels are shown in Figure 10.
Figure 9.
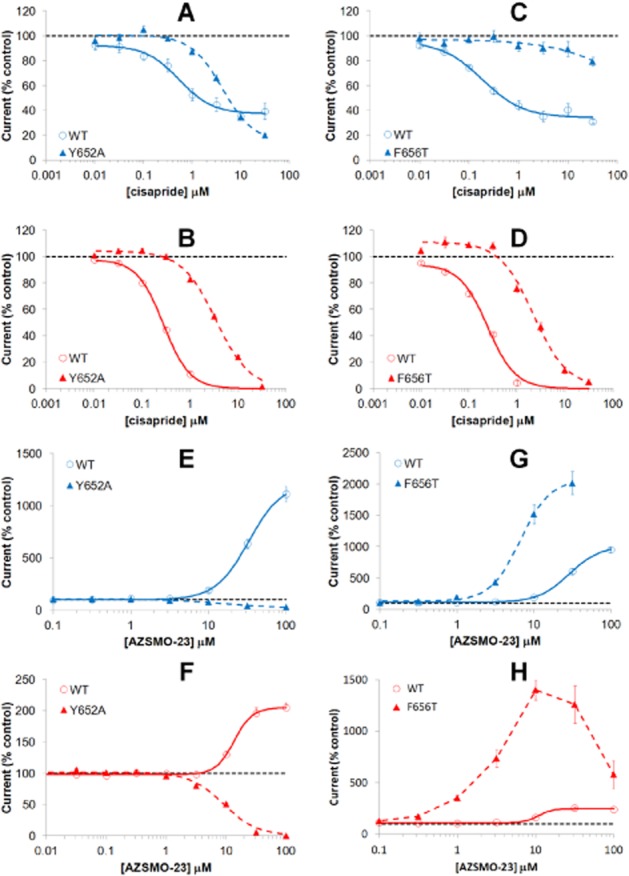
Effect of cisapride or AZSMO-23 on Y652A and F656T hERG mutant channels. Data are from experiments using IonWorks HT. Concentration–effect curves shown are for pre-pulse current (A, C, E, G) and for tail current (B, D, F, H). The top four graphs are for cisapride (A, B, C, D) and the bottom four (E, F, G, H) are for AZSMO-23. Current amplitudes in the presence of a given concentration of test compound were normalized by expressing them as a percentage of the mean, time-matched, vehicle control data collected in the same PatchPlate. The dashed line at 100% therefore represents the pre-pulse or tail current vehicle control level. Each data point is mean ± SEM (see Table 1 for n numbers). Concentration–effect curve parameters are summarized in Table 1.
Table 1.
Concentration–effect curve parameters for cisapride and AZSMO-23 with WT, Y652A and F656T hERG channels
| hERG | Pre-pulse/tail | IC50 (μM) | EC50 (μM) | LCI (μM) | UCI (μM) | nH | α | n | |
|---|---|---|---|---|---|---|---|---|---|
| Cisapride | WT | Pre-pulse | 0.5 | 0.3 | 1.0 | 1.3 | 12–17 | ||
| Y652A | Pre-pulse | 4.2 | 3.2 | 5.4 | 1.3 | 10–17 | |||
| WT | Tail | 0.3 | 0.3 | 0.3 | 1.6 | 12–17 | |||
| Y652A | Tail | 3.4 | 3.0 | 4.0 | 1.2 | 10–17 | |||
| WT | Pre-pulse | 0.2 | 0.1 | 0.3 | 1.1 | 12–20 | |||
| F656T | Pre-pulse | ND | ND | 8–13 | |||||
| WT | Tail | 0.2 | 0.2 | 0.3 | 1.5 | 12–20 | |||
| F656T | Tail | 2.1 | 1.7 | 2.6 | 1.3 | 8–13 | |||
| AZSMO-23 | WT | Pre-pulse | 30.8 | 25.9 | 36.6 | 2.1 | 1192 | 14–18 | |
| Y652A | Pre-pulse | 14.8 | 10.5 | 20.7 | 1.2 | 12–15 | |||
| WT | Tail | 12.8 | 10.0 | 16.5 | 3.4 | 205 | 14–17 | ||
| Y652A | Tail | 8.9 | 7.7 | 10.3 | 1.6 | 12–16 | |||
| WT | Pre-pulse | 28.6 | 25.0 | 32.6 | 2.2 | 1007 | 14–20 | ||
| F656T | Pre-pulse | 5.9 | 4.6 | 7.5 | 2.6 | 1904 | 13–16 | ||
| WT | Tail | 11.2 | 6.8 | 18.4 | 4.8 | 251 | 14–17 | ||
| F656T | Tail | ND | ND | ND | 4–16 |
IC50 or EC50 values are shown, depending on whether block or activation was seen. The 95% lower (LCl) and upper (UCl) confidence limit for these potency values are also shown. Where activation was observed, the estimated curve top is indicated (α). Curve parameters for the effect of cisapride on F656T hERG pre-pulse current were not determined (ND) since the concentration–effect curve was incomplete. Likewise, the effect of AZSMO-23 on F656T hERG tail current since the concentration–effect curve was bell shaped. Because the curves are non-cumulative, the number of cells making up data for each test concentration varies across a curve; hence the range of cell numbers underlying each point on curves is shown as n.
Figure 10.
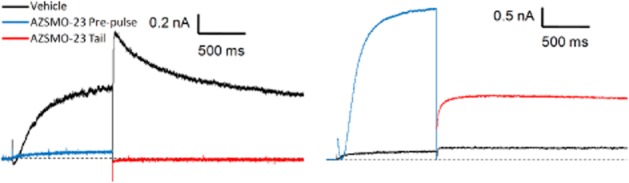
Typical current traces showing the effect of AZSMO-23 on Y652A and F656T hERG mutant channels. (A) Typical leak-corrected current traces, using IonWorks HT, for Y652A hERG channels before and after 30 μM AZSMO-23. (B) Equivalent current traces for F656T hERG. The dashed line represents the zero current level after leak subtraction.
Selectivity
We tested AZSMO-23 at several types of cardiac ion channels to see whether its selectivity was good enough to evaluate the effect of its hERG activation properties in native cardiac tissues or animals. At 30 μM, it was inactive at hHCN4 and caused only a small degree of inhibition of hNav1.5 [18.4 ± 2.0% (n = 15)] and hKv7.1-hKCNE1 channels [10.2 ± 4.1% (n = 12)]. However, as shown in Figure 11, it was more active as a blocker of hKv4.3-hKChIP2.2, hCav3.2 and hKv1.5. Of particular note was the fact that AZSMO-23 activated, rather than blocked, the hCav1.2/β2/α2δ channels. Current amplitude with these channels was doubled at 30 μM but an EC50 could not be determined since the concentration–effect curve was incomplete.
Figure 11.
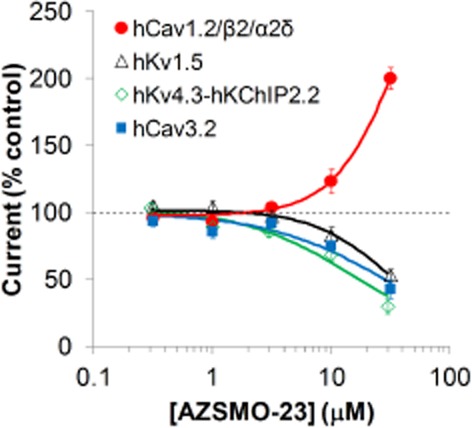
Effect of AZSMO-23 on other cardiac ion channel types. Concentration–effect curves for AZSMO-23 on hCav3.2 and hKv1.5 channels using IonWorks HT, and hKv4.3-hKChIP2.2 and hCav1.2/β2/α2δ channels, using IonWorks Quattro. Current amplitudes in the presence of a given concentration of test compound were normalized by expressing them as a percentage of the mean, time-matched, vehicle control data collected in the same PatchPlate. The dashed line at 100% therefore represents the vehicle control level. Each data point is mean ± SEM. n values were: hCav3.2 (4–8 cells); hKv1.5 (7–8 cells); hKv4.3-hKChIP2.2 (10–16 wells); hCav1.2/β2/α2δ (6–8 wells). Concentration–effect curve parameters were not defined since curves were incomplete.
Discussion and conclusions
This study characterized AZSMO-23, a hERG channel activator that acts by causing a depolarizing shift in the voltage dependence of inactivation. This mechanism was supported by the fact that, when tested against the non-inactivating hERG mutant channel (G628C/S631C) described by Smith et al. (1996), AZSMO-23 caused a small inhibition, rather than a large increase, in pre-pulse current.
Such compounds have been categorized by Perry et al. (2010) as type 2 activators and several have been described PD118057 (Zhou et al., 2005), NS1643 (Casis et al., 2006; Hansen et al., 2006a), NS3623 (Hansen et al., 2006b), PD307243 (Xu et al., 2008), A935142 (Su et al., 2009) and ICA-105574 (Gerlach et al., 2010).
A range of shifts in the voltage dependence of inactivation have been seen for type 2 activators, but AZSMO-23 is closest in profile to ICA-105574 (Gerlach et al., 2010). The latter caused a 183 mV shift whereas the effect of AZSMO-23 was 74.5 mV. However, this difference may not represent a fundamental difference in the two compounds because they were not tested at comparable concentrations relative to their EC50: ICA-105574 was tested at 2 μM (fourfold above its EC50 of 0.5 μM) while AZSMO-23 was tested at 30 μM (which is essentially at its EC50 of 28.6 μM). These very large shifts compare with relatively small changes seen with NS1643 (35 mV; Casis et al., 2006), A935142 (15 mV; Su et al., 2009), PD-118057 (19 mV; Perry et al., 2009) and NS3623 (17 mV; Hansen et al., 2006b). Although these are not truly comparable data collected in the same assay and at the same point on the concentration–effect curve, they were generated at concentrations above EC50. This suggests that AZSMO-23 and ICA-105574 are distinct from other type 2 compounds in terms of the magnitude of their effect on the voltage dependence of inactivation.
While the effect of AZSMO-23 on V½ was large and was also apparent using the ‘rectification’ or ‘extrapolation’ methods described by Vandenberg et al. (2012) (data not shown), the significance of the effect of AZSMO-23 on Vslope is harder to assess and would need further electrophysiological assessment.
The voltage dependence of activation was unaffected by AZSMO-23, compared with time-matched vehicle control data. This contrasts with the small hyperpolarizing shift in the voltage dependence of activation seen with other type 2 activators at the same concentrations where there were differences in the voltage dependence of inactivation [ICA-105574, 11 mV (Gerlach et al., 2010); NS1643, 9.8 mV (Xu et al., 2008); A935142, 9 mV (Su et al., 2009)]. In protocols to assess the voltage dependence of activation, the pre-pulse duration should ideally be long enough to ensure steady-state activation (Vandenberg et al., 2012). As seen in Figure 4A, a 4 s Vpre-pulse did not necessarily achieve steady-state current responses. Hence, our results are better described as 4 s isochronal activation data, but are nevertheless a like-for-like comparison of data from time-matched vehicle control and AZSMO-treated cells. Despite this, we cannot rule out the possibility that AZSMO-23 changes the rate of channel activation and therefore the error in estimating V½ differs between control and drug-treated cells.
AZSMO-23 is not a selective activator of hERG channels given that it modulated four of seven other types of cardiac ion channel over the same concentration range as it caused hERG activation. Particularly noteworthy was the fact that it activated the hCav1.2/β2/α2δ channel. This lack of selectivity precludes its use to understand the effect of its hERG activation in native in vitro or in vivo studies and contrasts with the selectivity of other type 2 compounds. Although the selectivity of ICA-105574 was not directly assessed by testing against other cardiac ion channels, an inference from the action potential data reported by Gerlach et al. (2010) is that it was a selective hERG activator, at least to a concentration of 3 μM.
Perhaps with the exception of ICA-105574, AZSMO-23 is structurally distinct from activators previously described (see Zhou et al., 2011; Sanguinetti, 2014). Our exploration of the structure–activity relationship for close analogues of AZSMO-23 suggests it is unique in terms of hERG channel activation, in that very close analogues, in particular the structural isomer (compound 1), and compound 2, in which the pyridine nitrogen has been substituted for an aromatic carbon atom, were inhibitors of hERG channels, rather than activators. This subtle structure–activity relationship suggests that highly specific molecular recognition, potentially involving a hydrogen bond donor, is required for activator activity. In addition to the mechanistic similarity between AZSMO-23 and ICA-105574, it might be argued that they are structurally related, since both contain a N-phenyl benzamide core structure (see Figure 8). Nevertheless, when the uncommon structural features were combined to give compounds 4 and 5, neither compound was an activator, suggesting that the binding modes of AZSMO-23 and ICA-105574 are not identical.
While little has been described concerning the structural features for hERG activators, structure–activity relationships from the series containing PD-307243 also suggest that relatively small changes in structure can impart this activity to otherwise inactive compounds. For example, PD-118057 and PD-117780 differ only in the nature of two aromatic substituents yet have very different effects on hERG current (Zhou et al., 2005).
Information about the molecular determinants of hERG channel activation has only recently been collated in order to form molecular models (see Sanguinetti, 2014) and try to understand the complex evolving picture for compounds such as ICA-105574 that have intrinsic inhibitory as well as activator activity (Garg et al., 2011). While we did not carry out a detailed analysis of molecular determinants for AZSMO-23 activity, we did specifically assess the effects on the mutant channels Y652A and F656T, as these mutants have been reported to affect the activity of ICA-105574. While AZSMO-23 inhibited rather than activated Y652A hERG channels, ICA-105574's activator activity, although reduced, was still apparent (Garg et al., 2011). In contrast, the data for F656T channels were clearly divergent: AZSMO-23's activator activity was enhanced while that of ICA-105574 was significantly reduced (Garg et al., 2011). The small AZSMO-23-induced inhibition of the G628C/S631C non-inactivating hERG mutant is analogous to that reported for ICA-105574, however. This observation, the inhibitory activity of AZSMO-23 at Y652A hERG channels, evidence of inhibition of WT hERG at negative voltages (see Figure 5) and the inhibitory activity of close analogues of AZSMO-23, suggests similar properties of these compounds and ICA-105574. Further work to explore the pharmacological subtleties of AZSMO-23 and its close analogues may be useful to gain further understanding of whether AZSMO-23 exerts its effects via distinct inhibitor and activator sites, or whether both properties involve a single binding site, as hypothesized by Garg et al. (2011) for ICA-105574. The inhibition of other cardiac ion channel types, but activation of the hCav1.2/β2/α2δ channels, certainly suggests a complex pharmacology for AZSMO-23.
There are some potentially interesting differences in the concentration–effect curve data for AZSMO-23, when comparing its effect on the pre-pulse and tail current. As quantified in Table 1, apart from the much greater relative increase in pre-pulse current, AZSMO-23 is more potent against the tail current but with a less steep mid-point curve slope; although both pre-pulse and tail current curves have nH values >1. It was also apparent that the rate of onset of AZSMO-23's effect is faster for the tail current than the pre-pulse current. However, this may simply reflect the law of mass action in operation. That is, the fact that AZSMO-23's effect is around threefold more potent on the tail current; hence 30 μM is threefold the EC50 for tail current but around EC50 for the pre-pulse. While these differences are worth noting, the limitations of the IonWorks device (see below) mean that more detailed experiments would be needed to confirm and interpret these differences.
The IonWorks HT methodology used for most of this work has the advantage of being able to generate concentration–effect curves on around 100 compounds per day, hence the capacity to screen enough compounds to detect AZSMO-23 in the first place. It is important, however, to consider the potential disadvantages of this type of electrophysiology in the context of our findings. The device does not enable series resistance compensation, which is particularly relevant given that the large currents observed in the presence of AZSMO-23 will result in significant voltage errors. This may distort concentration–effect curves, especially for the effect on pre-pulse current: at low concentrations of AZSMO-23, when absolute currents are relatively small, the discrepancy between the command and actual Vm will be smaller than for the large currents seen at high concentrations. It is difficult to quantify the extent of this effect without doing concentration–effect curves using conventional electrophysiology, or an automated device that can apply series resistance compensation. It is for this reason that we used conventional methodology to assess the effect of AZSMO-23 on voltage dependence of activation and inactivation. An additional limitation is that IonWorks HT applies a voltage clamp protocol before addition of vehicle/test compound and, in this study, applies the same protocol again after a 3 min exposure to the compound; in between there is no voltage clamping so an assessment of whether steady state has been achieved cannot be made. However, the time course and magnitude of effect data from IonWorks Barracuda (Figure 3), which does provide a continuous voltage clamp, suggest that results from IonWorks HT were not significantly compromised by a lack of continuous voltage clamp.
Selective activation of hERG channels with compounds such as ICA-105574 leads to a shortening of ventricular action potential duration (Gerlach et al., 2010). The safety risks of this effect in man remain a matter of debate (Holbrook et al., 2009), but hERG channel activation is an off-target effect ideally to be avoided. Fortunately, at least in our experience, hERG channel activators are rare. Of the 60 119 compounds tested to date, only 52 increased hERG tail current (0.086%) when tested at 30 or 100 μM. This is in stark contrast to the proportion of compounds identified that have some blocking activity (>20% inhibition of hERG tail current) at these concentrations (50 409 of 60 119; 84%).
The data presented here, in two respects, represent initial observations worthy of further investigation. Firstly, a more detailed electrophysiological assessment would enable an in-depth comparison with other type 2 compounds. Furthermore, further work to understand the molecular determinants of the effects of AZSMO-23 is needed in order to add to the emerging picture for hERG activators (Sanguinetti, 2014). This would increase our understanding of the structure–activity relationships for hERG activators as means of avoiding this activity from a safety perspective and potentially in order to design selective analogues as a treatment for congenital long QT syndrome.
Acknowledgments
We would like to thank Dr Jean-Pierre Valentin and Dr Tim Hammond for their support to do this work.
Glossary
- α
curve top
- AZSMO-23
N-[3-(1H-benzimidazol-2-yl)-4-chloro-phenyl]pyridine-3-carboxamide
- confidence limit
CL
- hERG
human ether-a-go-go-related gene
- nH
Hill coefficient
- V½
half maximal activation or inactivation
- Vm
membrane potential
- WT
wild type
Author contributions
R. M. and M. B. T. contributed to the acquisition of data, its interpretation and drafting, and review of the manuscript. N. A., H. P. and M. J. M. contributed to the acquisition of data and review of the manuscript. S. S. provided chemistry input to the design and interpretation of the work and reviewed the manuscript. C. E. P. drafted the article and took responsibility for the integrity of the work as a whole from inception to the finished article.
Conflict of interest
The authors are employees of AstraZeneca, the company involved in the synthesis of the compounds reported in the manuscript.
References
- Alexander SP, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The Concise Guide to PHARMACOLOGY 2013/14: Ion Channels. Br J Pharmacol. 2013;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla F, Armstrong CM. Inactivation of the sodium channel. I. Sodium current experiments. J Gen Physiol. 1977;70:549–566. doi: 10.1085/jgp.70.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridgland-Taylor MH, Hargreaves AC, Easter A, Orme A, Henthorn DC, Ding M, et al. Optimisation and validation of a medium-throughput electrophysiology-based hERG assay using IonWorks HT. J Pharmacol Toxicol Methods. 2006;54:189–199. doi: 10.1016/j.vascn.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Bridgland-Taylor MH, Pye H, Mannikko R, Abi Gerges N, Easter A, Valentin J-P, et al. Abstract. Characterisation of an activator of the hERG-encoded potassium channel. J Pharmacol Toxicol Methods. 2008;58:156. [Google Scholar]
- Casis O, Olesen SP, Sanguinetti MC. Mechanism of action of a novel human ether-a-go-go-related gene channel activator. Mol Pharmacol. 2006;69:658–665. doi: 10.1124/mol.105.019943. [DOI] [PubMed] [Google Scholar]
- Fernandez D, Ghanta A, Kauffman GW, Sanguinetti MC. Physicochemical features of the HERG channel drug binding site. J Biol Chem. 2004;279:10120–10127. doi: 10.1074/jbc.M310683200. [DOI] [PubMed] [Google Scholar]
- Finkel A, Wittel A, Yang N, Handran S, Hughes J, Costantin J. Population patch clamp improves data consistency and success rates in the measurement of ionic currents. J Biomol Screen. 2006;11:488–496. doi: 10.1177/1087057106288050. [DOI] [PubMed] [Google Scholar]
- Garg V, Stary-Weinzinger A, Sachse F, Sanguinetti MC. Molecular determinants for activation of human ether-a-go-go-related gene 1 potassium channels by 3-nitro-n-(4-phenoxyphenyl) benzamide. Mol Pharmacol. 2011;80:630–637. doi: 10.1124/mol.111.073809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach AC, Stoehr SJ, Castle NA. Pharmacological removal of human ether-a-go-go-related gene potassium channel inactivation by 3-nitro-N-(4-phenoxyphenyl) benzamide (ICA-105574) Mol Pharmacol. 2010;77:58–68. doi: 10.1124/mol.109.059543. [DOI] [PubMed] [Google Scholar]
- Hansen RS, Diness TG, Christ T, Demnitz J, Ravens U, Olesen SP, et al. Activation of human ether-a-go-go-related gene potassium channels by the diphenylurea 1,3-bis-(2-hydroxy-5-trifluoromethyl-phenyl)-urea (NS1643) Mol Pharmacol. 2006a;69:266–277. doi: 10.1124/mol.105.015859. [DOI] [PubMed] [Google Scholar]
- Hansen RS, Diness TG, Christ T, Wettwer E, Ravens U, Olesen SP, et al. Biophysical characterization of the new human ether-a-go-go-related gene channel opener NS3623 [N-(4-bromo-2-(1H-tetrazol-5-yl)-phenyl)-N'-(3'-trifluoromethylphenyl)urea. Mol Pharmacol. 2006b;70:1319–1329. doi: 10.1124/mol.106.026492. [DOI] [PubMed] [Google Scholar]
- Hansen RS, Olesen SP, Ronn LC, Grunnet M. In vivo effects of the IKr agonist NS3623 on cardiac electrophysiology of the guinea pig. J Cardiovasc Pharmacol. 2008;52:35–41. doi: 10.1097/FJC.0b013e31817dd013. [DOI] [PubMed] [Google Scholar]
- Harmer AR, Abi-Gerges N, Easter A, Woods A, Lawrence CL, Small BG, et al. Optimisation and validation of a medium-throughput electrophysiology-based hNav1.5 assay using IonWorks. J Pharmacol Toxicol Methods. 2008;57:30–41. doi: 10.1016/j.vascn.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Holbrook M, Malik M, Shah RR, Valentin JP. Drug induced shortening of the QT/QTc interval: an emerging safety issue warranting further modelling and evaluation in drug research and development? J Pharmacol Toxicol Methods. 2009;59:21–28. doi: 10.1016/j.vascn.2008.09.001. [DOI] [PubMed] [Google Scholar]
- Malik M. Facts, fancies and follies of drug-induced QT/QTc interval shortening. Br J Pharmacol. 2010;159:70–76. doi: 10.1111/j.1476-5381.2009.00554.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannikko R, Overend G, Perrey C, Gavaghan CL, Valentin JP, Morten J, et al. Pharmacological and electrophysiological characterization of nine, single nucleotide polymorphisms of the hERG-encoded potassium channel. Br J Pharmacol. 2010;159:102–114. doi: 10.1111/j.1476-5381.2009.00334.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitcheson JS. hERG potassium channels and the structural basis of drug-induced arrhythmias. Chem Res Toxicol. 2008;21:1005–1010. doi: 10.1021/tx800035b. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl Acids Res. 2014;42:D1098–D1106. doi: 10.1093/nar/gkt1143. (Database Issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry M, Sachse FB, Abbruzzese J, Sanguinetti MC. PD-118057 contacts the pore helix of hERG1 channels to attenuate inactivation and enhance K+ conductance. Proc Natl Acad Sci U S A. 2009;106:20075–20080. doi: 10.1073/pnas.0906597106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry M, Sanguinetti M, Mitcheson J. Revealing the structural basis of action of hERG potassium channel activators and blockers. J Physiol. 2010;588:3157–3167. doi: 10.1113/jphysiol.2010.194670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti M. hERG1 channel agonists and cardiac arrhythmia. Curr Opin Pharmacol. 2014;15:22–27. doi: 10.1016/j.coph.2013.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder K, Neagle B, Trezise DJ, Worley J. Ionworks HT: a new high-throughput electrophysiology measurement platform. J Biomol Screen. 2003;8:50–64. doi: 10.1177/1087057102239667. [DOI] [PubMed] [Google Scholar]
- Shah RR. Can pharmacogenetics help rescue drugs withdrawn from the market? Pharmacogenomics. 2006;7:889–908. doi: 10.2217/14622416.7.6.889. [DOI] [PubMed] [Google Scholar]
- Shah RR. Drug-induced QT interval shortening: potential harbinger of proarrhythmia and regulatory perspectives. Br J Pharmacol. 2010;159:58–69. doi: 10.1111/j.1476-5381.2009.00191.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamovsky I, de Graaf C, Alderin L, Bengtsson M, Bladh H, Borjesson L, et al. Increasing selectivity of CC chemokine receptor 8 antagonists by engineering nondesolvation related interactions with the intended and off-target binding sites. J Med Chem. 2009;52:7706–7723. doi: 10.1021/jm900713y. [DOI] [PubMed] [Google Scholar]
- Smith PL, Baukrowitz T, Yellen G. The inward rectification mechanism of the HERG cardiac potassium channel. Nature. 1996;379:833–836. doi: 10.1038/379833a0. [DOI] [PubMed] [Google Scholar]
- Su Z, Limberis J, Souers A, Kym P, Mikhail A, Houseman K, et al. Electrophysiologic characterization of a novel hERG channel activator. Biochem Pharmacol. 2009;77:1383–1390. doi: 10.1016/j.bcp.2009.01.015. [DOI] [PubMed] [Google Scholar]
- Vandenberg JI, Perry MD, Perrin MJ, Mann SA, Ke Y, Hill AP. hERG K+ channels: structure, function and clinical significance. Physiol Rev. 2012;92:1393–1478. doi: 10.1152/physrev.00036.2011. [DOI] [PubMed] [Google Scholar]
- Xu X, Recanatini M, Roberti M, Tseng GN. Probing the binding sites and mechanisms of action of two human ether-a-go-go-related gene channel activators, 1,3-bis-(2-hydroxy-5-trifluoromethyl-phenyl)-urea (NS1643) and 2-[2-(3,4-dichloro-phenyl)-2,3-dihydro-1H-isoindol-5-ylamino]-nicotinic acid (PD307243) Mol Pharmacol. 2008;73:1709–1721. doi: 10.1124/mol.108.045591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Augelli-Szafran CE, Bradley JA, Chen X, Koci BJ, Volberg WA, et al. Novel potent human ether-a-go-go-related gene (hERG) potassium channel enhancers and their in vitro antiarrhythmic activity. Mol Pharmacol. 2005;68:876–884. doi: 10.1124/mol.105.014035. [DOI] [PubMed] [Google Scholar]
- Zhou PZ, Babcock J, Liu LQ, Li M, Gao ZB. Activation of human ether-a-go-go related gene (hERG) potassium channels by small molecules. Acta Pharmacol Sin. 2011;32:781–788. doi: 10.1038/aps.2011.70. [DOI] [PMC free article] [PubMed] [Google Scholar]