

Abstract
Background and Purpose
The existence of functional Kv7 channels in thalamocortical (TC) relay neurons and the effects of the K+-current termed M-current (IM) on thalamic signal processing have long been debated. Immunocytochemical evidence suggests their presence in this brain region. Therefore, we aimed to verify their existence, pharmacological properties and function in regulating activity in neurons of the ventrobasal thalamus (VB).
Experimental Approach
Characterization of Kv7 channels was performed by combining in vitro, in vivo and in silico techniques with a pharmacological approach. Retigabine (30 μM) and XE991 (20 μM), a specific Kv7 channel enhancer and blocker, respectively, were applied in acute brain slices during electrophysiological recordings. The effects of intrathalamic injection of retigabine (3 mM, 300 nL) and/or XE991 (2 mM, 300 nL) were investigated in freely moving animals during hot-plate tests by recording behaviour and neuronal activity.
Key Results
Kv7.2 and Kv7.3 subunits were found to be abundantly expressed in TC neurons of mouse VB. A slow K+-current with properties of IM was activated by retigabine and inhibited by XE991. Kv7 channel activation evoked membrane hyperpolarization, a reduction in tonic action potential firing, and increased burst firing in vitro and in computational models. Single-unit recordings and pharmacological intervention demonstrated a specific burst-firing increase upon IM activation in vivo. A Kv7 channel-mediated increase in pain threshold was associated with fewer VB units responding to noxious stimuli, and increased burst firing in responsive neurons.
Conclusions and Implications
Kv7 channel enhancement alters somatosensory activity and may reflect an anti-nociceptive mechanism during acute pain processing.
Tables of Links
| LIGANDS | |
|---|---|
| 4-aminopyridine | Oxotremorine-M |
| 4-DAMP | Pirenzepine |
| Cd2+ | Retigabine |
| Carbachol (CCh) | Tetraethylammonium |
| Carprofen | TTX |
| Diclofenac | XE991 |
| Mibefradil | Xylocaine |
| Nifedipine | ZD7288 |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,cAlexander et al., 2013a,b,c,,).
Introduction
Sensory information processing requires signal transfer from the periphery to the cortex (Llinás and Steriade, 2006). Most sensory signals reach the thalamus where initial processing steps are performed. Due to inputs arising from the brainstem, the reticular thalamic nucleus and reciprocal connections with the cortex, the thalamus also regulates sensory gating (Weyand et al., 2001) and arousal (Coulon et al., 2012). The principle cells in the thalamus, the thalamocortical (TC) neurons, communicate with the cortex through two distinct activity patterns: tonic series of action potentials (APs) and oscillatory burst firing. The latter is based on the activation of low-threshold Ca2+ spikes (LTS). The tonic firing of TC neurons is considered fundamental for relaying sensory stimuli to the cortex while the occurrence of bursting is considered as a mechanism that interferes with tonic AP generation (Weyand et al., 2001). In this respect, the consequences of burst firing intermingled with tonic firing during the relay mode could underlie gating features of the thalamus, such as anti-nociceptive effects of burst stimulation during a painful stimulus (Huh et al., 2012).
Various voltage-gated ion channels expressed by TC neurons shape their firing patterns (Llinás and Steriade, 2006). The biophysical properties and functional relevance of a great number of thalamic K+ channels have been formerly analysed (Bista et al., 2014). However, a contribution of the channels belonging to the Kv7 family (Alexander et al., 2013a), also called KCNQ channels, was considered of minor importance (Huguenard and McCormick, 1992; McCormick and Huguenard, 1992). This general belief is mainly based on the fact that TC neurons do not show spike frequency adaptation, a typical effect of Kv7 channel activation in extrathalamic neurons (Yue and Yaari, 2004; Rivera-Arconada and Lopez-Garcia, 2005). The Kv7.1–Kv7.5 channel subunits assemble to form functional channels and four of these subunits (Kv7.2–Kv7.5) are expressed in the nervous system as homo- and heteromeric channels (Tatulian and Brown, 2003; Brown and Passmore, 2009). Activation of these channels leads to the generation of a membrane K+ current termed M-current (IM), which dampens cell excitability (Yue and Yaari, 2004; Rivera-Arconada and Lopez-Garcia, 2005). Indeed, mutations of different Kv7 channel subunits can induce alterations in cell excitability, leading to specific forms of epilepsy and neuropathic pain (Jentsch, 2000; Passmore et al., 2003). Recent evidence shows that Kv7 channels are expressed in the thalamus and are involved in regulating cell excitability (Cooper et al., 2001; Geiger et al., 2006; Kasten et al., 2007). For that reason, the presence of Kv7 channels in the thalamus might be crucial for regulating physiological and pathological processes that are triggered by sensory stimulation.
Therefore, we thought it is timely to thoroughly assess the contribution of Kv7 channels to the modulation of firing modes of TC neurons and their possible function in the processing of the somatosensory information within the thalamus. In view of the documented presence of Kv7.2 and Kv7.3 subunits in the ventrobasal thalamus (VB) and the involvement of this nucleus in the processing of thermal and noxious signals (Passmore et al., 2003), our experimental strategy was: (i) to use a model of acute thermal pain in mice; (ii) to focus on the VB, which represents the prototypical thalamic nucleus for the relay of noxious signals; (iii) to record electrophysiological activity of single VB units in mice during specific behaviour combined with pharmacological approaches probing Kv7 involvement in pain responses; (iv) to identify the relevant membrane current (IM) in VB TC neurons using patch-clamp techniques in vitro; (v) to complement the physiological findings with a mathematical model of TC activity, implementing IM; and (vi) to re-address the expression of Kv7 subunits in the VB at the mRNA and protein level.
Methods
Animals
All animal care and experimental work was approved by local authorities (LANUV NRW; approval IDs: 8.87-51.05.20.10.117, 84-02.04.2011.A177, 87-51.04.2010.A322) and was performed in accordance with Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the protection of animals used for scientific purposes, the 3Rs law, and the ARRIVE guidelines for reporting experiments involving living animals with respect to anaesthesia and animal handling (Kilkenny et al., 2010). A total of 46 mice were used for the experiments performed in vivo. Animals were singly caged and kept in a 12 h light/dark cycle with food and water available ad libitum.
Tissue preparation
C57Bl/6J mice (15–30 days old) were deeply anaesthetized using isoflurane (7% in O2), decapitated and used for electrophysiological and expression analyses. Brains were rapidly removed and placed in chilled (2–4°C) oxygenated slicing solution (see Supporting Information). Coronal thalamic slices (300 μM) were cut on a vibratome and were kept submerged in standard artificial CSF until the recordings began (see Supporting Information).
Patch-clamp recordings
Whole-cell recordings were performed on TC neurons of the VB at 21°C (voltage-clamp recordings) or 32°C (current-clamp recordings) in a standard bath solution (see Supporting Information). Electrical activity was measured with glass electrodes pulled from borosilicate glass (GC150TF-10; Clark Electromedical Instruments, Pangbourne, UK), connected to an EPC-9 amplifier (HEKA Elektronik, Lambrecht, Germany) and filled with a K-gluconate-based solution (see Supporting Information). Typical electrode resistance was 2–3 MΩ, with a series resistance in the range of 5–15 MΩ (compensation ≥30%). All recordings were governed by Pulse software (HEKA Elektronik) and corrected for liquid junction potential.
Voltage-clamp analysis
Changes in current amplitude were measured in bath solution containing mibefradil (2 μM), nifedipine (1 μM), ZD7288 (30 μM) and TTX (0.5 μM). To follow the effects of substances over time, a voltage step from −65 to −45 mV was chosen to mimic a typical depolarization from the resting membrane potential (RMP = −65.8 ± 0.99 mV; n = 52) and to maintain the voltage below the voltage of maximal IM activation (Tatulian and Brown, 2003). Thereafter, the neuron was repolarized to −60 mV. Each voltage step was preceded by a short depolarizing step (from −65 to −45 mV, 80 ms, ‘pre-pulse’) to inactivate transient Ca2+ and K+ conductances. Current amplitudes under various conditions were analysed at the furthermost 500 ms of the depolarizing voltage step to −45 mV. Retigabine (Ret)- and XE991-sensitive currents were calculated as (Ret − control) and (Ret − (Ret + XE991)) respectively.
A ramp protocol was used to establish the current–voltage (I–V) relationship in the presence of ZD7288, TTX and Cd2+ (150 μM). Neurons were held at −35 mV to open Kv7 channels, subsequently ramped to −135 mV (1 mV·ms−1, 100 ms) and repolarized back to −35 mV. The reversal potential of the sensitive current was obtained empirically and compared with the calculated Nernst potential for K+ (EK; temperature = 21°C; K+ concentrations of intracellular and extracellular solutions).
Current-clamp analysis
The current-clamp experiments were performed using the bath solution without blockers. The RMP was adjusted to about −55 mV by (DC) current injection, and burst and tonic firing was induced by alternating (1 s interval) hyperpolarizing (−200 pA, 250 ms) and depolarizing current steps (200 pA, 250 ms) respectively (Budde et al., 2008). The number of APs was calculated using a custom script written in MATLAB (MathWorks, Natick, MA, USA).
Drugs
The following drugs were added to the bath solution or injected into the VB: ZD7288 and mibefradil (Abcam Biochemicals, Cambridge, UK); retigabine (AWD, Dresden, Germany and LGM Pharma, Boca Raton, FL, USA); XE991 and TTX (Biotrend, Cologne, Germany); Alexa488 (Life Technologies GmbH, Darmstadt, Germany); nifedipine (Sigma-Aldrich, Taufkirchen, Germany); linopirdine, oxotremorine-M, pirenzepine, 4-diphenylacetoxy-N-methylpiperidine methiodide (4-DAMP) and carbachol (CCh; Tocris Bioscience, Bristol, UK). When substances were dissolved in DMSO, solvent concentration did not exceed 2‰ and had no effect on the TC neurons (not shown).
Statistical analyses
All results are presented as mean ± SEM. Statistical significance of the data with a Gaussian distribution was evaluated by Student's t-test. Multiple comparisons were made by anova or repeated-measures anova. OriginPro software (version 8G; OriginLab, Friedrichsdorf, Germany), Statistica (Statsoft, Hamburg, Germany), Prism (version 5; GraphPad software, San Diego, CA, USA) and Corel-Draw (version 14; Corel software, Ottawa, ON, Canada) were used for data analysis and figure preparation.
Surgical procedures
Anaesthesia was induced with isoflurane (3% in O2; Abbot GmbH & Co. KG, Wiesbaden, Germany), maintained with i.p. injection of pentobarbital (50 mg·kg−1), and additional doses were given if necessary (10–15% of the initial dose). The depth of anaesthesia was controlled by assessing withdrawal (light pinching of a hind paw) and ocular (carefully touching the cornea) reflexes and continuously monitoring the respiratory rhythm. All pressure points were covered with 2% xylocaine gel (Astra Zeneca GmbH, Wedel, Germany) and tissue to be incised was injected with 2% xylocaine solution. Corneas were protected with a dexpanthenol-containing gel (Bepanthen®; Bayer, Leverkusen, Germany). Postsurgical analgesia was ensured with s.c. injection of carprofen (5 mg·kg−1; Pfizer GmbH, Berlin, Germany).
The head was mounted in a stereotaxic apparatus (ASI Instruments, Inc., Warren, MI, USA) via ear bars and the levels of bregma and lambda were equalized. Craniotomies were performed bilaterally, and the dura mater was removed. Stereotaxic placement of guide cannula or electrode-guide cannula assembly (see Supporting Information) was conducted for the ventral posterior thalamic nuclei, with bregma as a reference: 1.7 mm posterior, 1.9 mm lateral and 3.4 mm ventral to the cortical surface (Paxinos and Franklin, 2001). Additional craniotomy was performed above the cerebellum and an epidural grounding electrode was implanted.
Recording procedures
Extracellular unit activities were recorded in freely moving mice. Signals were pre-amplified, bandpass-filtered (0.1–13 kHz) and processed (sampling rate 40 kHz) using the Multichannel Acquisition Processor system (Plexon Inc., Dallas, TX, USA). AP waveforms with a signal-to-noise ratio ≥2 were extracted offline using Spike2 software (Cambridge Electronic Design Inc., Cambridge, UK). Timestamps of neural spiking were further analysed using NeuroExplorer (Nex Technologies, Littleton, CO, USA) or MATLAB (MathWorks). DMSO (1%; vehicle), Ret (3 mM) and XE991 (2 mM) were diluted in saline containing 100 μM Alexa488 which was added for marking the injection site. Upon completion of experiments, the position of the implants was histologically verified (see Supporting Information).
Pain-response measurements
After 5 days of recovery from surgery, thermal nociception was assessed on a modified hot plate (55 ± 0.1°C). A Plexiglas cylinder (13 cm height × 15 cm diameter) with a removable sliding floor was used as a testing chamber (custom made using material from Evonik Industries, Essen, Germany). The latency time was measured from the moment the animal touched the heated plate with all paws until it showed the first signs of pain: jumping, licking, kicking and withdrawal (Crockett et al., 1977). Immediately thereafter or after 20 s on the plate, animals were removed from the plate.
Recordings during pain-response measurements
To begin the electrophysiological recordings, the animal was lightly anaesthetized with isoflurane and the implanted electrodes were connected to the headstage. The injection cannulas were inserted into the guide cannulas, the animal was transferred to the test chamber and the headstage was connected to a motorized swivel commutator. Hamilton syringes coupled to injection cannulas were attached to the swivel with a piece of adhesive tape. After recovery from anaesthesia (15 min) baseline neuronal activity was recorded, the chamber was placed on the hot plate and the floor was removed. When the animal showed first signs of pain, the floor was slid back and the chamber was removed from the hot plate. Subsequently, animals received local intrathalamic injections (300 nL, ∼10 nL·s−1) of the drug or vehicle solution and were tested again 20–60 min later. Experiments were recorded with a digital video camera and stored on a computer hard disk for offline analysis.
Open field test
The test was carried out under light conditions and the open field test apparatus consisted of a square arena (60 cm × 60 cm) with 25 cm wall height. One hour after intrathalamic Ret or vehicle administration, mice were examined separately without previous habituation. The animal was placed in the middle of the arena and allowed to move freely for 10 min. Behaviour was recorded with VideoMot2 software (TSE Systems GmbH, Bad Homburg, Germany) and stored on a computer for offline analysis. The exploratory activity, defined as the distance travelled during 10 min of the test, was measured. To estimate the level of anxiety, the time spent in the central sector and the number of visits to the centre of the apparatus during the first 5 min of the test were measured.
Mathematical modelling
All computer simulations were conducted using the NEURON Simulation Environment (Carnevale and Hines, 2006) and were based on a modified version of a previously published single compartment TC neuron template (Destexhe et al., 1996; available at http://senselab.med.yale.edu/ModelDB/, accession number: 3343, TC.tem). Modifications included the expansion of the original template file by two additional potassium conductances, implementing an inactivating IA (Wang et al., 1996; ModelDB accession number: 3648, kamt.mod), as well as a non-inactivating IM (McCormick et al., 1991; ModelDB accession number: 3817, IM.mod). Both current modules were expanded by one or two additional range variables in order to shift activation (m) and inactivation (h) parameters. Network inputs to the single-cell model were mimicked by the addition of AMPA, GABAA and GABAB receptors (Destexhe et al., 1996; ModelDB accession number: 3343, ampa.mod, gabaa.mod, gabab.mod), which were randomly triggered. White background noise was realized via an IClamp object, directly attached to the model cell. Simulations assumed a temperature of 32°C and lasted either 4 s (step stimulation) or 60 s (network inputs). All model parameters that were modified are summarized in Supporting Information Table S1.
Nomenclature
Nomenclatures of ion channels and receptors conform to BJP's Concise Guide to Pharmacology (Alexander et al., 2013a,b,c,,).
Results
Kv7-mediated alteration of activity modes in VB TC neurons in vivo
Single units (identified by principal component analysis; Figure 1A–C) were recorded in the VB of freely moving mice showing wake-state related behaviours (exploration, grooming, rearing, etc.). Single unit baseline spike patterns were analysed. Burst spikes related to LTS activation were defined by the presence of a preceding silent period (>50 ms), at least four high-frequency spikes (125–500 Hz) with a maximal first interspike interval (ISI) of 4 ms, and last ISI with a maximum of 8 ms (Figure 1D–F; Huh et al., 2012). Spontaneous spikes occurred at rates between 1 and 70 spikes/s (average: 16.3 ± 1.4 Hz, n = 149; Figure 1H) and tonic firing with frequencies up to ∼100 Hz (Figure 1D) was predominant. In order to test the effect of Ret, which is known to activate IM and Kv7.2–5 channels (Wickenden et al., 2000; Xu et al., 2010), a total number of 99 single units were recorded. Local application of Ret resulted in the disappearance of 19 units immediately with drug injection (Tremere et al., 2010) and a nominal decrease in the total number of spikes s−1 (control, 15.4 ± 1.9 Hz, n = 99; Ret, 12.1 ± 1.1 Hz, n = 80; Figure 1G, H) accompanied by a significant increase in the number of burst-associated spikes (control, 5.5 ± 0.4%, n = 86; Ret, 8.5 ± 1.9%, n = 65; t-test: two samples assuming unequal variances, two-tailed P = 0.018, t = −2.40; Figure 1I). In 21 units randomly chosen for analysis, burst-associated spikes were fired at higher frequency (decrease in ISI 2; control, 3.9 ± 0.3 ms; Ret, 1.9 ± 0.1 ms; anova: ISI no. vs. treatment: F = 3.23; P = 0.004; n = 21; Newman–Keuls post hoc test; Figure 1J, K). Baseline activity was unaltered in the presence of the Kv7-blocker XE991 (control, 18.0 ± 1.8 Hz, n = 50; XE991, 18.2 ± 2.0 Hz, n = 51; Figure 1H, I). These findings indicate that Kv7 channel activation alters the activity pattern of TC neurons by increasing the number of fast bursts.
Figure 1.
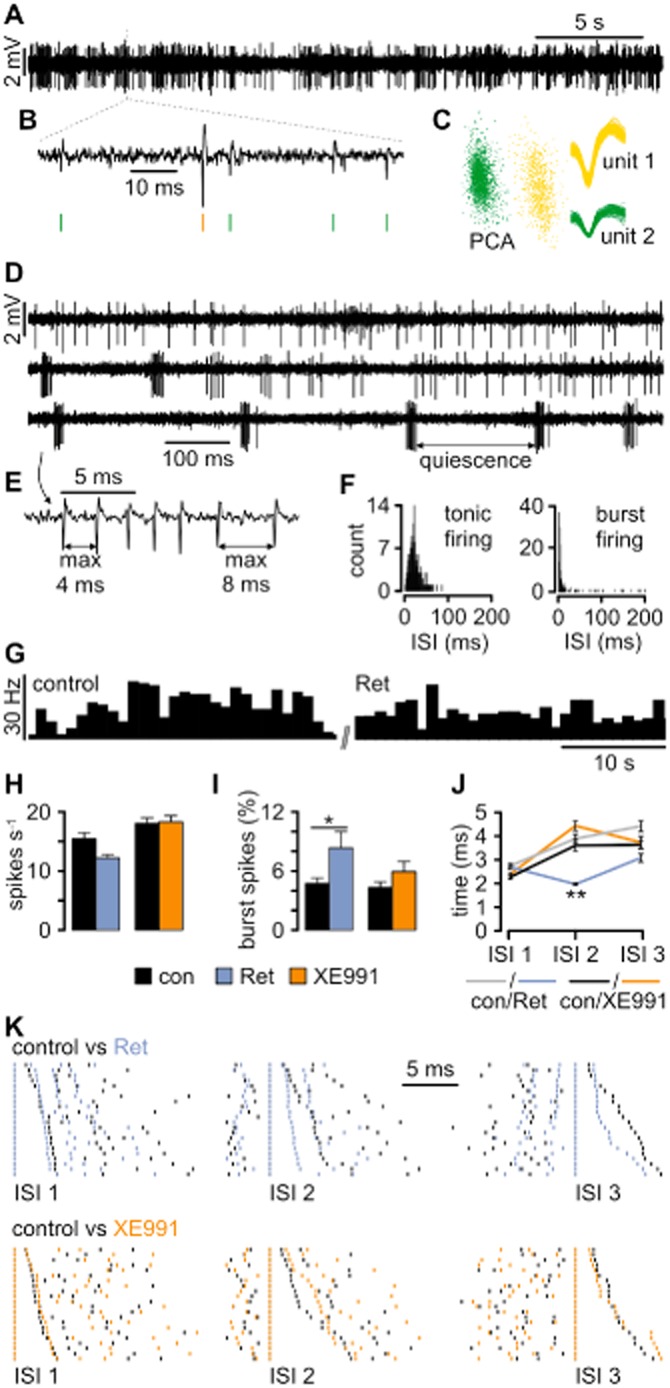
Kv7 channels influence the firing behaviour of TC neurons in vivo. (A) Representative traces recorded in VB revealing spontaneous spiking. (B) The typical waveforms of extracellularly recorded APs are visible on an expanded timescale, thereby allowing spike sorting (green and yellow vertical lines). (C) After spike extraction, waveforms were clustered and sorted into units based on principal component analysis (PCA). (D) Extracellular recording revealing the two distinct TC firing patterns: tonic firing (upper trace) characterized by single APs occurring independently of each other and burst firing (bottom trace) characterized by grouped APs and high-frequency spiking. Bursts are separated by a quiet period of at least 50 ms duration. The two firing patterns can occur concurrently (middle trace). (E) Definition of burst-firing characteristics at higher temporal resolution. (F) Distribution histograms of interspike intervals (ISIs) for tonic and burst firing calculated from adequate activity periods selected manually. Note the narrow peak (reflecting high-frequency firing) and long tail (corresponding to quiescence periods) when the recorded unit is in bursting mode. (G) An example of a firing rate histogram (bin size: 1 s) for a single unit recorded under control conditions and after local Ret injection. (H) Local Ret injection caused a slight, although not significant, decrease in the firing rate, while the injection of XE991 had no effect. (I) Burst analysis revealed that Ret application facilitated burst firing in VB neurons. The application of XE991 caused a smaller and insignificant increase. (J) Analysis of ISI within bursts (intraburst ISI) revealed that Ret application caused a highly significant shortening of the second intraburst ISI (ISI2). (K) Examples of burst ISI distribution during control conditions versus periods after drug application. Each row represents a single burst and every vertical line corresponds to a single AP recorded during the control period (black lines) and after drug injection (coloured lines). To demonstrate the difference between the ISI values, corresponding spikes within each burst were time aligned. The bursts were sorted in length, with the shortest ISI displayed in the first row and the longest in the last row. Ret considerably shortened ISI 2. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01.
Contribution of Kv7 channels in VB to the modulation of pain responsiveness
Unit activity in the VB was recorded and related to nociceptive responses during the hot-plate test. After bringing an animal in contact with the surface, the latency to the first sign of aversive behaviour was determined. Application of Ret in the VB significantly increased the latency (control, 6.7 ± 0.3 s; Ret, 11.5 ± 0.3 s; P < 0.001; n = 7) while XE991 decreased it (control, 6.4 ± 0.4 s; XE991, 4.0 ± 0.3 s; P < 0.05; n = 7). Simultaneous application of Ret and XE991, as well as of DMSO, had no significant effect (n = 7; Figure 2A). Recording and application sites were histologically verified and confined to the VB (Figure 2B, C). Naïve Ret-injected animals displayed a similar increase in the pain threshold as animals that had underwent a control hot-plate trial before (control, 6.71 ± 0.32 s; n = 7, naïve Ret, 9.6 ± 0.82 s; n = 4; t-test for independent samples, naïve Ret vs. control t = −3.91, P = 0.003; Figure 2D). Furthermore, the effect of Ret was long lasting (24 h, 10.36 ± 0.61 s; n = 6; t-test for independent samples: t = −5.47, P = 0.002; 24 h after Ret vs. control). Importantly, Ret infusion did not alter locomotor activity and anxiety level (Figure 2E–G). The average distance travelled by control animals was 44.74 ± 10.29 m (n = 4) and 35.54 ± 3.70 m in Ret-injected animals (n = 5; t-test for independent samples t = 0.92, P = 0.38, Ret vs. control; Figure 2F). The level of anxiety was measured as the time spent in the centre of an open field arena and was not different (t-test for independent samples: t = −0.2, P = 0.84, Ret vs. control) between control (15.19 ± 8.81 s; n = 4) and animals injected with Ret (17.2 ± 5.86 s; n = 5; Figure 2G). The number of visits to the centre was not different between groups (control, 16.75 ± 5.35; n = 4; Ret, 17.20 ± 1.98; n = 5; t-test for independent samples: t = −0.08, P = 0.93), suggesting that Ret injection did not affect the anxiety level.
Figure 2.
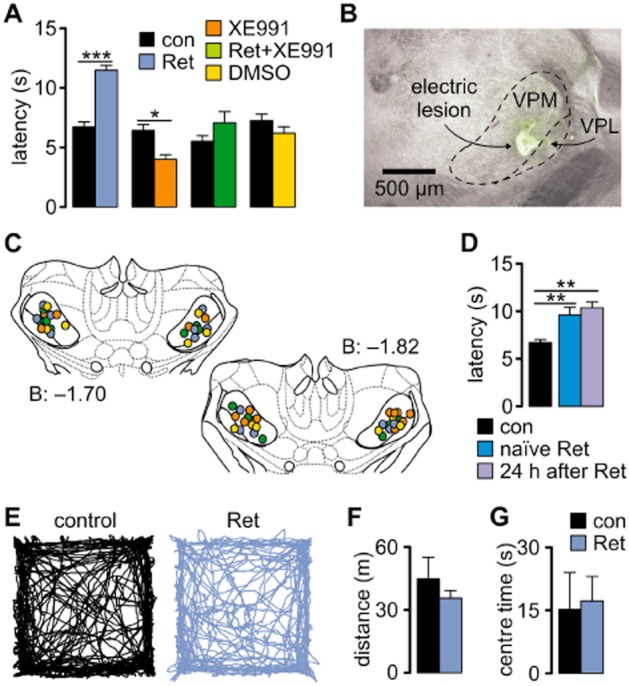
Kv7 channels modulate pain perception. (A) The hot-plate test showed that the latency to the appearance of pain-related behaviour was significantly increased after Ret injection, and this effect was diminished when Ret was co-applied with the Kv7-blocker XE991. The blocker alone caused a significant shortening of the latency. Specificity of drug action was tested by injecting a vehicle solution (DMSO), which did not significantly change the latency (repeated-measures anova with Newman–Keuls post hoc test). (B) Photomicrograph of a coronal section of the mouse brain, indicating the co-localization of the recording (lesion) and injection site marked by Alexa488 (green staining around the lesion; VPL, ventral posterior lateral nucleus; VPM, ventral posterior medial nucleus). (C) Schematic coronal sections through the thalamus, marking the injection/recording sites (Paxinos and Franklin, 2001). The numbers indicate the posterior distance from bregma in mm. The colour of circles corresponds to the colour of the bars and legend in A. (D) Increased latencies observed in Ret-injected animals were not a result of learning, since naïve Ret-injected animals also displayed increased latencies which differed significantly from the control group shown in A (t-test for independent samples), and the effect of Ret was long lasting (t-test for independent samples). (E) Examples of open field test traces obtained from a control and a Ret-injected animal. (F) Bar graph showing the average distance travelled by the tested animals during 10 min of open field test. (G) The level of anxiety was not different between control and Ret-injected animals (t-test for independent samples) and was measured as time spent in the centre of an open field arena during the first 5 min of the test. Data are presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
In order to determine the changes in neuronal firing during noxious stimulation, firing rates of individual units were analysed relative to baseline activity in 1 s segments, 10 s before (pre), during (test), and 10 s after (post) exposing the mice to the thermal stimulus. The recorded units were classified as responsive or non-responsive based on their firing rate. Neurons were considered responsive when the calculated z-score values for the firing frequency during the pain response were higher than 2.5 times the standard deviation of the baseline pre-test activity, indicating a significant change in the firing rate (P < 0.01; Figure 3A). A response to pain-related behaviour was observed in a subpopulation of VB neurons. The activity of roughly 35% of the recorded units was associated with an aversive response upon exposure to the hot-plate test, irrespective of injection of vehicle solution (Figure 3B). Local application of Ret in the VB reduced the number of responsive units to 25%, while application of XE991 increased it to 49% (Figure 3B). On average, the peak activity of responsive cells, measured as the time from the start of the test to the first peak above the z-score of 2.5, occurred ∼7 s after exposure to the hot plate under control conditions and injection of vehicle solution, while Ret increased this to ∼14 s and XE991 decreased it to ∼3 s (Figure 3C). Moreover, Ret significantly increased the relative mean number of burst spikes during the post-test period (control, 1.0 ± 0.19; n = 36; Ret, 1.7 ± 0.28; n = 21; t-test for independent samples: t = −2.08, P = 0.04), while tonic spike activity was not altered in pain-responsive neurons (Figure 3D). These findings indicate that Kv7 activation in VB promotes burst firing and decreases the overall number of units responding to pain, which is associated with a delay in behavioural responses to noxious stimuli.
Figure 3.
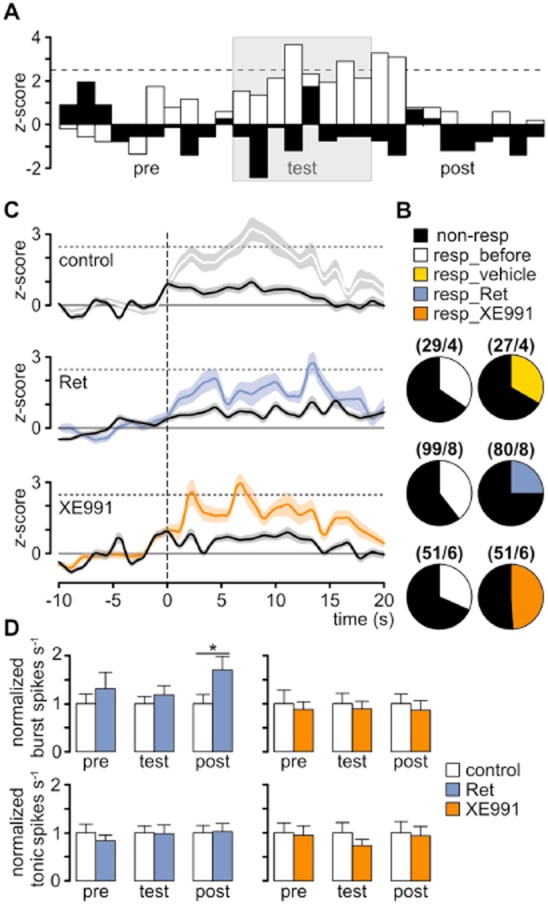
Kv7 channels modulate pain responses of thalamic neurons in freely moving mice. (A) In order to determine the changes in neuronal firing during aversive behaviour, firing rates of individual units were analysed relative to baseline activity in 1 s segments, 10 s before (pre), during (test), and 10 s after (post) exposing the mice to the thermal stimulus. The recorded units were classified as responsive or non-responsive based on their firing rate. Neurons were considered responsive when the calculated z-score values for the firing frequency during the pain response were higher than 2.5 times the SD of the baseline activity 10 s before the pain stimulus. A response to pain-related behaviour was observed in a subpopulation of the VB neurons. Examples of normalized time histograms illustrating the changes in the firing rate of responsive (white) and non-responsive (black) units under pre-injection conditions (bin size: 1 s). The duration of the hot-plate test is indicated by a grey background. (B) The ratio of responsive (resp) to non-responsive neurons before drug injection was ∼1:2 and was not changed after injection of vehicle. After Ret injection, this decreased to ∼1:3, while after XE991 injection it increased to ∼1:1. For every group, the total number of recorded cells/number of animals is indicated in parentheses. (C) Mean population z-scores for responsive (coloured lines) and non-responsive cells (black lines) with SEM indicated by shading. After Ret injection (middle panel), the peak of the z-score was delayed compared with control (upper panel), indicating a later response to the painful stimulus. After XE991 injection (bottom panel), this latency was reduced, indicating an earlier response to the painful stimulus. The vertical dashed line marks the start of a hot-plate test. (D) The propensity of responsive neurons to fire APs in the burst mode, but not in the tonic mode, was increased after Ret application, while XE991 did not change the firing pattern. For the testing periods, the 10 s preceding (pre) and following (post) the exposure to the hot plate was analysed. The time of exposure to the hot plate never exceeded 20 s. Data are presented as mean ± SEM. *P < 0.05.
Identification of IM in VB TC neurons in vitro
We identified IM under whole cell voltage-clamp conditions in TC neurons of the VB in the presence of Ca2+ and pacemaker current blockers (see Methods). A depolarizing voltage step from a holding potential of −65 to −45 mV was used to evoke an outward current (inset in Figure 4; for families of currents see Supporting Information Fig. S1A–E) that was increased in amplitude by Ret (30 μM, Figure 4A). Graphical subtraction (Ret − control) yielded a membrane current with a slow time course of activation which displayed no inactivation during the depolarizing steps (Figure 4B; Supporting Information Fig. S1D). Upon return to −60 mV, slow outward tail currents indicated deactivation of involved channels (Figure 4B; Supporting Information Fig. S1D). Ret application increased the outward current amplitude by 71.2 ± 23.6% (n = 8, P < 0.05 vs. control; Figure 4G). Subsequent co-application of Ret and the Kv7 channel blockers linopirdine (20 μM) and XE991 (20 μM; Aiken et al., 1995; Wladyka and Kunze, 2006) reversed the observed effect (XE991, 2.9 ± 16.4% of control, n = 8, P > 0.05 vs. Ret; linopirdine, 7.3 ± 8%, n = 8, P > 0.05 vs. Ret; Figure 4G; Supporting Information Fig. S1C). Furthermore, the current blocked by XE991 (Ret − (Ret + XE991)) displayed similar kinetics as the Ret-induced current (Figure 4C; Supporting Information Fig. S1D, E). Application of XE991 alone reduced the membrane current at −45 mV (by 26.6 ± 18.9% compared with control, n = 8, P > 0.05 vs. Ret; Figure 4G), suggesting some basal channel activity. When XE991 was applied first, subsequent wash-in of Ret had no effect on the outward current, thereby suggesting specific effects of the two current modulators (Figure 4G). Current versus time plots of the Ret effect revealed a long lasting modulation of outward current amplitudes (Supporting Information Fig. S2).
Figure 4.
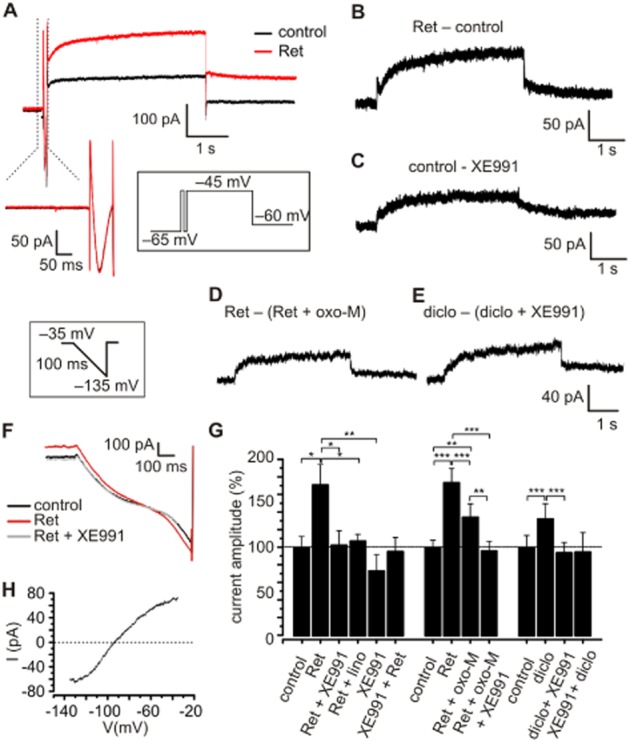
Characterization of IM in TC neurons of the VB. (A) Representative current traces evoked by a depolarizing voltage step from −65 to −45 mV (duration 4 s) recorded under control conditions and in the presence of Ret. Each test pulse was preceded by a short depolarization (pre-pulse; −45 mV, 80 ms) in order to inactivate fast-transient Ca2+ currents which were not affected by Ret application (see magnification of the pre-pulse). (B, C) Currents activated by Ret (B) or inhibited by XE991 (C) were isolated by graphical subtraction (Ret − control; control − XE991). Subtracted currents showed a slow activation and deactivation, typical for IM. (D, E) The Ret-activated current blocked by oxo-M and the diclofenac-sensitive current revealed typical IM kinetics. (F, H) Current–voltage relationship of the Ret-sensitive current. A fast hyperpolarizing ramp, affecting only constitutively open channels, was applied under control conditions in the presence of Ret and Ret + XE991. The Ret-sensitive current was obtained by subtracting control currents from the current activated by Ret and reversed at −94.04 ± 2.8 mV (n = 9), close to the calculated EK of −103 mV. (G) Bar graph showing the changes in current amplitude (voltage steps from a holding potential of −65 to −45 mV were analysed). Ret application significantly increased the current amplitude compared with control conditions. Adding Kv7 channel blockers [XE991 or linopirdine (lino)] reversed this effect. XE991 alone (i.e. without Ret) induced a pronounced current reduction; however, this was not significant. Wash-in of XE991 before Ret application prevented current activation by Ret. The presence of the muscarinic agonist oxo-M decreased the Ret-evoked current. Diclofenac (diclo) significantly increased the current amplitude, which was abolished by adding XE991 before or after drug application. Data are presented as mean ± SEM. For Ret: repeated-measures anova: F = 4.57; P = 0.005; for oxo-M: repeated-measures anova: F = 12.29; P < 0.0001; for diclo: repeated-measures anova: F = 14.85; P = 0.0002; Newman–Keuls post hoc test: *P < 0.05, **P < 0.01, ***P < 0.001 versus respective control as suggested by the horizontal bars.
I–V relationships were determined by using ramp protocols (bottom inset in Figure 4 and Methods). Graphical subtraction (Ret − control) revealed a current that reversed at −94.0 ± 2.8 mV (n = 9), close to the calculated EK (−103 mV; Figure 4H). Currents obtained in the presence of both Ret and XE991 were indistinguishable from control currents (Figure 4F). Furthermore, Ret had no effect when K+ conductances were blocked (Supporting Information Fig. S1F) by using a Cs+-based electrode solution containing 4-aminopyridine (45 mM) and tetraethylammonium (10 mM).
The suppression of IM by muscarinic ACh receptor (mAChR) stimulation (Brown and Adams, 1980) was tested using oxotremorine methiodide (oxo-M) and CCh. Application of oxo-M significantly reduced the Ret-induced current (relative current amplitude change compared with control; Ret: 173.7 ± 16.6%, n = 11, P < 0.001 vs. control; Ret + oxo-M: 134.4 ± 15.5%, n = 11, P < 0.01 vs. control and P < 0.001 vs. Ret; Figure 4G). The oxo-M-sensitive current revealed the typical properties of IM (Figure 4D). Additional application of XE991 further reduced the outward current (96.1 ± 11.3% of control, n = 11, P < 0.01 vs. Ret + oxo-M and P < 0.001 vs. Ret; Figure 4G). Similar results were obtained using CCh (112 ± 15.9% of control, n = 8, P < 0.01 vs. Ret; Supporting Information Fig. S1G, H). Finally, the M1 receptor antagonist pirenzepine (10 μM) and the M3 receptor antagonist 4-DAMP (10 μM) prevented the effects of oxo-M on the Ret-induced current (Supporting Information Fig. S1G).
The anti-inflammatory drug diclofenac, a known positive enhancer of Kv7.2–4 (Peretz et al., 2005; Brueggemann et al., 2011), increased the outward current by 32.6 ± 17.5% (n = 9, P < 0.001 vs. control; Figure 4G). The diclofenac-sensitive current revealed the typical kinetics of IM (Figure 4E) and was significantly inhibited by co-application of XE991 (Figure 4G). When XE991 was applied first, subsequent wash-in of diclofenac had no effect on the outward current (Figure 4G).
Taken together, these findings indicate that Ret induces an outward current in TC neurons displaying electrophysiological and pharmacological features typical of IM.
Contribution of IM to the firing properties of VB TC neurons
TC neurons were held at about −55 mV and subjected to alternating hyperpolarizing and depolarizing current steps under current-clamp conditions (Figure 5A). A delayed onset of firing and early high-frequency firing upon depolarization was regarded as tonic activity and LTS mediated, respectively (Deleuze et al., 2012). Application of Ret (n = 28) hyperpolarized the membrane potential by −4.2 ± 0.28 mV (P < 0.001; Figure 5A, B) and the overall number of APs triggered by depolarizing current steps decreased to a normalized value of 0.53 ± 0.11 (P < 0.01) compared with controls (control = 1.0; Figure 5G). The overall number of LTS-mediated APs elicited by a depolarizing step in the presence of Ret increased to a normalized value of 2.73 ± 0.05 (P < 0.001; Figure 5H). While under control conditions, 64.3% of the tested neurons showed LTS-mediated APs at the beginning of a depolarizing current step, presence of Ret increased it to 100%. Upon return from a hyperpolarizing current step, rebound LTS bursts were elicited (Figure 5C–F). The number of APs crowning this Ca2+ spike in the presence of Ret decreased to a normalized value of 0.81 ± 0.08 (P < 0.05; Figure 5I). Application of XE991 (n = 24) depolarized the membrane by +1.5 ± 0.38 mV (P < 0.001), induced an increase in the number of depolarization-induced APs (normalized value of 2.06 ± 0.31 of control; P < 0.01; Figure 5G), reduced the overall number of LTS-induced APs elicited by a depolarizing step (normalized value of 0.71 ± 0.12 of control; P < 0.05; Figure 5H), decreased the number of neurons presenting LTS-induced APs from 70.8 to 58.3%, and increased the number of APs on a rebound LTS (normalized value of 1.12 ± 0.07 of control; P < 0.05; Figure 5I).
Figure 5.
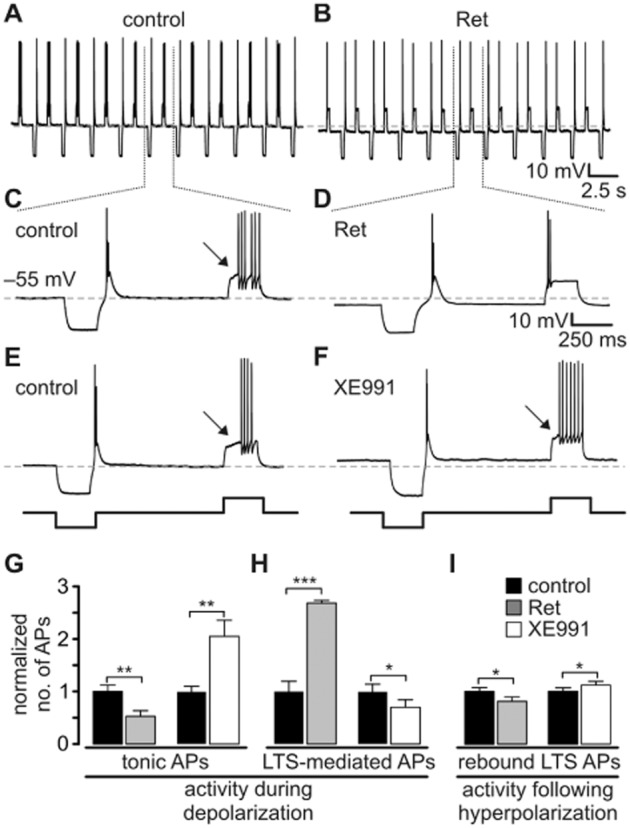
IM modulates thalamic activity modes. (A–F) TC neurons were subjected to a series of hyperpolarizing and depolarizing current steps under current-clamp conditions (±200 pA, 250 ms duration). Recordings show that Ret hyperpolarized the TC neurons, thereby reducing the number of tonic APs and favouring burst-like activity (A–D). Application of XE991 induced opposite effects on the firing pattern (E, F). (G–I) Normalized numbers of APs induced by depolarizing pulses (G, H) and subsequent to hyperpolarizing pulses (rebound burst triggered by a LTS, I). AP sequences typically following a delayed onset of firing (indicated by arrow on C, E and F) were regarded as tonic firing (G). Early firing (see D) was regarded as LTS mediated (H). Data are presented as mean ± SEM. Paired t-test, *P < 0.05, **P < 0.01, ***P < 0.001 versus respective control.
Next, a modified single compartment TC neuron model (Budde et al., 2008) containing an IM module with a basal conductance of gkbar_im = 0.69e−5 S·cm−2 (30% of maximal current) was set to a membrane potential of −57 mV by DC current injection. A depolarizing current step (+500 pA, 250 ms) induced a series of tonic APs, while upon release from a hyperpolarizing step (−500 pA, 250 ms) a burst of APs was generated (Figure 6B). Increasing IM (gkbar_im = 2.30e−5 S·cm−2) resulted in a membrane hyperpolarization and induction of burst firing in response to depolarizing current steps (Figure 6C). To verify that bursting was based on de-inactivation of T-type Ca2+ channels (Tscherter et al., 2011) and subsequent LTS generation, the Hodgkin–Huxley formalism was removed from the computer model (Figure 6D). In addition, removal of IM (gkbar_im = 0) depolarized the membrane potential and increased the number of tonic APs (Figure 6A).
Figure 6.
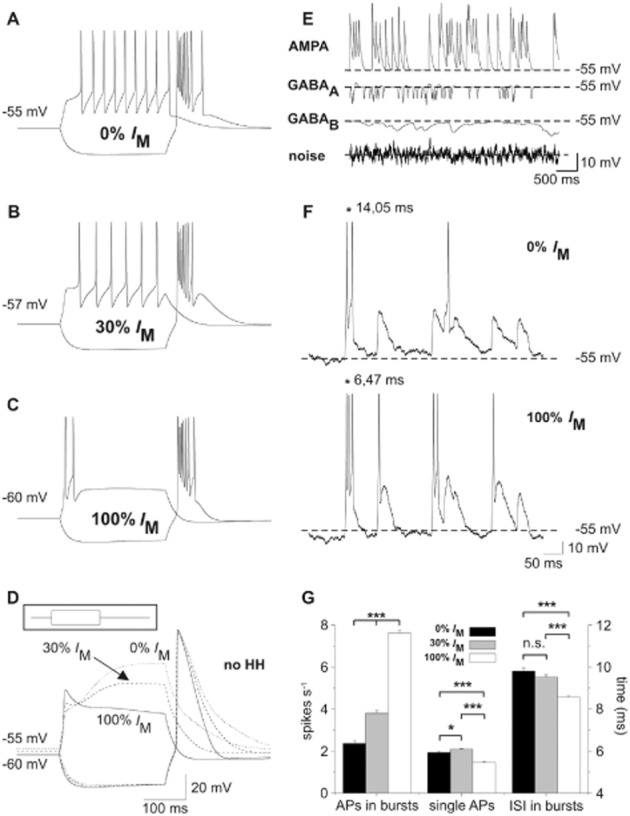
Effect of IM on the firing behaviour of a thalamocortical (TC) relay cell model. (A–C) Hyperpolarizing and depolarizing current pulses were applied in a TC neuron model (±500 pA, 250 ms, see inset in D). (B) Setting the conductance of IM to 30% of the maximal value and RMP to −57 mV using DC current injection was regarded as control condition. A depolarizing current step induced six APs. (C) An increase of IM to 100% hyperpolarized the RMP to −60 mV and shifted the mode of activity elicited by the depolarizing current pulse from tonic towards burst firing. (A) Removing IM from the computational model depolarized the RMP of the model cell to −55 mV and promoted tonic firing. (D) Hodgkin–Huxley (HH)-like conductances were set to zero (no HH-like conductances) thereby preventing AP generation. At 100% IM, a LTS is discernible at the beginning of the depolarizing step. (E) Network inputs were mimicked by adding randomly triggered excitatory and inhibitory synaptic activity, as well as by including a white noise source to the model cell. The individual effect of each of these parameters on the membrane potential is shown when all other external inputs, AP generation and IM are deactivated. Dashed lines mark the potential of −55 mV. (F) Application of all random network inputs (as shown in E) to the model cell with 0 and 100% IM revealed increased burst activity for the latter condition. While bursts of APs were triggered more frequently, single APs appeared less often. Moreover, APs within a burst were observed with reduced ISI. The asterisks in the upper and lower panel exemplarily mark the first ISI. Dashed lines mark the RMP of −55 mV. (G) The bar graph shows the rate (spikes per second) of burst-associated APs and single APs as well as the duration of the ISI (ms) calculated for the burst-associated APs only. Repeating the random network stimulation (60 s duration) 10 times revealed increased burst firing (defined as at least two APs ≤ 25 ms apart), reduced single-spike activity and reduced ISI with 100% IM. Adding 30% IM resulted in intermediate effects. *P < 0.05, ***P < 0.001.
Synaptic background activity in a TC neuron was mimicked by randomly triggering GABAA, GABAB and AMPA receptors of the computational model and adding white background noise (Figure 6E). The TC neuron was given a DC current to maintain a potential of −55 mV and different levels of IM conductance (0, 30 and 100%), thereby mimicking the XE991 effect, control condition and the Ret effect respectively. Addition of IM increased the number of burst-associated APs (0% IM: 2.35 ± 0.13; 100% IM: 7.61 ± 0.13; P < 0.001) together with a reduction of their mean ISI (0% IM: 9.79 ± 0.15 ms; 100% IM: 8.56 ± 0.05 ms; P < 0.001; Figure 6F, G) and decreased the number of single APs (0% IM: 1.93 ± 0.05; 100% IM: 1.46 ± 0.04; P < 0.001). These findings indicate that IM activation and the associated hyperpolarization limit tonic firing, but increase bursting. The latter is due to an increased availability of IT.
Expression of Kv7 subtypes in VB
Using a RT-PCR assay of mouse VB tissue, PCR fragments from Kv7.2–7.4, but not Kv7.1 and Kv7.5, were detected (Supporting Information Fig. S3A). The use of antibodies specific for various Kv7 channel subunits showed no Kv7.4 proteins, but revealed clear soma staining for Kv7.2 and diffuse neuropil staining for Kv7.3 (Supporting Information Fig. S3B). For both subtypes, staining of cell membranes was overlapping. These findings were corroborated by Western blots revealing bands close to the expected size of ∼97 kDa for Kv7.2 and Kv7.3 (Supporting Information Fig. S3C).
Discussion
Characteristic features of TC neurons, largely accounting for their unique physiological functions, are the two modes of activity: tonic and burst firing (Llinás and Steriade, 2006; Sherman and Guillery, 2006). Tonic firing is characterized by a delayed onset of AP generation and a lack of pronounced spike frequency adaptation (McCormick and Pape, 1988). It is thought to underlie the faithful relay of sensory information from the periphery to the cortex, including the discriminative and somatotopic aspects of pain sensation, which have been shown to be governed by information processing in the VB (Willis and Westlund, 1997; Sherman and Guillery, 2006). In contrast, burst firing was traditionally thought to hinder information transfer and is defined as rapid AP generation followed by a prolonged quiescence period that occurs mostly during slow-wave sleep (Llinás and Steriade, 2006). However, there is increasing evidence for a role of thalamic burst activity during wakefulness (Fanselow et al., 2001; Bezdudnaya et al., 2006; Ohara et al., 2007; Hughes et al., 2008). On the cellular level, it is important to take into consideration that the principal component of thalamic burst activity is the activation of IT. Subtle modifications of the membrane potential around −60 mV impact T-type Ca2+ channels, and this has profound consequences for stimulus-induced AP generation (Tscherter et al., 2011; Deleuze et al., 2012). Even the small hyperpolarization conveyed by IM activation will de-inactivate T-type Ca2+ channels, thereby allowing the generation of an LTS, which in turn triggers a burst of APs. Our mathematical modelling confirmed that this effect was based on the IM-dependent enhancement of IT and also occurred when firing dynamics were evoked by synaptic currents, indicating that activation of IM influences the membrane potential and, in consequence, modifies IT.
Convergent evidence obtained from studies in various types of neurons and preparations indicates that XE991 and Ret affect Kv7 channels with high specificity (Brown and Passmore, 2009). Here we confirmed their preserved specificity, as the Ret effect was reversible and blocked by pre-application of XE991 in thalamic slices. The lack of Ret effects on other TC neuron properties, like the T-type Ca2+ current (Figure 1A), and the reversal of Ret effects by linopirdine, another Kv7 channel blocker, support this conclusion. Following intraperitoneal injection, Ret plasma concentrations were found to be low after 24 h; however, half-life was longer and the concentration was higher in the brain (Rostock et al., 1996). Therefore, the lasting effect of Ret on behavioural responses following central administration in different pain models is in agreement with pharmacokinetic data (Xu et al., 2010). Sole application of XE991 exerted relatively small effects on thalamic neuronal activity and significant XE991 effects were thus detectable through in vitro recordings rather than in vivo electrophysiology in the present study. Furthermore, the strong temporal non-linearity of acute thermal pain perception by the entire pain processing system of the brain (vs. unit activity of a single neuron) may be the basis for the detection of XE991 effects in behavioural experiments (Cecchi et al., 2012).
The effects of XE991 application on firing pattern observed in the present study in vitro indicated that AP generation during both activity modes is limited by basal Kv7 channel activity. This conclusion is corroborated by recordings obtained from lateral dorsal thalamic slices, where XE991 moderately enhanced the firing rate in mice TC neurons (Kasten et al., 2007). Therefore, enhancement of these channels should strongly limit tonic firing. Indeed, application of Ret hyperpolarized the RMP of the TC neurons, reduced the firing rate during depolarizing current steps, and decreased the number of APs triggered by a rebound LTS in vitro. These findings indicate that the activation of IM hyperpolarizes the membrane potential and, in consequence, strongly affects the output of TC neurons and thus may influence TC information processing.
In thalamic synaptic circuits, release of ACh from cholinergic terminals (Llinás and Steriade, 2006) is an important mechanism for shifting firing modes from burst to tonic AP generation. Activation of Gαq protein-coupled mAChR inhibits K2P channels and inwardly rectifying K+ channels, causing a depolarizing shift of the membrane potential in TC neurons and results in the appearance of tonic activity (Coulon et al., 2010; Bista et al., 2012; 2015,). Therefore, the mAChR-dependent inhibition of IM (Brown and Passmore, 2009) seems to contribute to the enhancement of tonic firing in TC neurons (Kasten et al., 2007; Bista et al., 2014).
On the functional level, thalamic burst firing interferes with information relay, and its role in sensory perception has been a matter of discussion in recent years. In the context of nociception, studies performed in humans and rats found that, under conditions of neuropathic pain, the incidence of burst firing in VB is increased (Lenz et al., 1989; Jeanmonod et al., 1993; Hains et al., 2006; Wang and Thompson, 2008). Other models showed that under anaesthesia, the absence of bursting relates to increased pain responses, while excessive bursting decreased them (Kim et al., 2003; Liao et al., 2011). Recordings in awake and normal behaving animals confirmed that tonic firing of VB neurons reliably reflects the behavioural nociceptive responses, while increased burst firing corresponds to decreased behavioural nociceptive responses (Huh et al., 2012). Accordingly, our results demonstrated that upon local thalamic Kv7 channel activation, the incidence of burst generation is increased, while behavioural and electrophysiological correlates of pain response are delayed. Specifically during burst firing, activation of IM results in a decrease in ISIs within a burst, thereby shaping early phases of activity (Storm, 1989). This is important here, as the dynamic properties of bursting are crucial for interfering with the faithful relay of the information, and therefore should reduce pain responses (Huh et al., 2012; Huh and Cho, 2013a). In particular, it has been shown that formalin-induced pain altered the intraburst ISI1 and ISI2 of VB TC neurons in a subtle time-dependent manner which was associated with changes in pain-related behaviour (Huh et al., 2012). Electrical stimulation of the VB with train stimuli of various intratrain intervals that mimicked physiologically occurring bursts revealed that only high-frequency stimulation (intratrain interval ≤3 ms) with at least three pulses per train was able to reliably ease pain responses induced by formalin (Huh and Cho, 2013a). Along this line, Ret-facilitated bursting and associated shortening of intraburst ISI2 observed in VB TC neurons has the potential to delay responses to incoming sensory signals, such as acute heat stimuli. Indeed, the behavioural pain response was delayed in Ret-treated animals while XE991 application advanced it. Additionally, Kv7 channel activation reduced the number of pain-responsive units, supporting the conclusion of an anti-nociceptive function that depended on the local thalamic action of retigabine, and thus differed in its mechanism of action from μ-opioid receptor agonists in the VB, which were found to exert strong anti-nociceptive effects in acute, tonic and chronic pain models in rats (Pozza et al., 2010). The exact mechanisms underlying the alteration of specific intraburst ISI are not clear yet, but seem to depend on a complex interplay between intrinsic ion channels (Broicher et al., 2007; Tscherter et al., 2011; Ehling et al., 2013) and cortical feedback (Huh and Cho, 2013b).
Summarizing, we present evidence that Kv7 channels located in thalamic VB neurons are capable of affecting pain sensation, as assessed by using a hot-plate test. This modulation occurs through Kv7 channel-induced moderate hyperpolarization of the TC neurons, resulting in a facilitation of T-type Ca2+ current-dependent high-frequency bursting that leads to decreased transmission of pain information. Therefore, the contribution of Kv7 channels to the understanding of physiological and pathological conditions associated with alterations in sensory gating, neuropsychiatric disorders such as thalamic dysrhythmia syndromes, or epilepsy may be of paramount importance.
Acknowledgments
The authors wish to thank Elke Nass, Svetlana Kiesling, Birgit Herrenpoth and Elisabeth Boening for excellent technical assistance; Manfred Daweke and Andreas Kolkmann for designing and building the equipment for acute pain tests; Professor Dr Erwin-Josef Speckmann for his support of the project; Dr Michael Doengi for actively helping many times; and Dr Kay Jüngling for helpful discussions on experimental paradigms. This project was supported by grants from IZKF Münster (Bud3/010/10) and DFG (BU1019/9-2 BU1019/11-1, SFB-TR128/B6), and a Max-Planck-Research Award 2007 (to H. C. P).
Glossary
- 4-DAMP
4-diphenylacetoxy-N-methylpiperidine methiodide
- AP
action potential
- IM
M-current
- IT
T-type calcium current
- ISI
interspike interval
- mAChR
muscarinic ACh receptor
- Ret
retigabine
- RMP
resting membrane potential
- TC
thalamocortical
- VB
ventrobasal thalamus
Author contributions
All authors commented on the final version and approved it. M. C. and H. J. S. conducted and designed the experiments, analysed the data, and prepared the figures. T. B., P. C. and S. G. M. initiated the project and designed the experiments. P. C. made the initial in vitro findings and conducted the experiments together with X. V. N; P. M. designed, performed and analysed the computational modelling. T. K. designed, performed and analysed the immunohistochemical and RT-PCR experiments. K. G. designed, performed and analysed the Western blot experiments. T. S. conducted initial in vivo experiments. H. C. P. designed the experiments. T. B., P. C., S. G. M., M. C., H. J. S. and H. C. P. wrote the paper. T. B., P. C., S. G. M. and H. C. P. supervised the project.
Conflict of interest
The authors declare that they have no conflicts of interest.
Supporting Information
Figure S1 Pharmacological characterization of Kv7 channels in VB.
Figure S2 Time course of IM modulation by Ret.
Figure S3 Expression of Kv7 channels in the ventrobasal thalamus (VB).
Table S1 List of modelled parameters and their default values.
References
- Aiken SP, Lampe BJ, Murphy PA, Brown BS. Reduction of spike frequency adaptation and blockade of M-current in rat CA1 pyramidal neurones by linopirdine (DuP 996), a neurotransmitter release enhancer. Br J Pharmacol. 1995;115:1163–1168. doi: 10.1111/j.1476-5381.1995.tb15019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The Concise Guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The Concise Guide to PHARMACOLOGY 2013/14: ligand-gated ion channels. Br J Pharmacol. 2013b;170:1582–1606. doi: 10.1111/bph.12446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: ion channels. Br J Pharmacol. 2013c;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezdudnaya T, Cano M, Bereshpolova Y, Stoelzel CR, Alonso J-M, Swadlow HA. Thalamic burst mode and inattention in the awake LGNd. Neuron. 2006;49:421–432. doi: 10.1016/j.neuron.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Bista P, Meuth SG, Kanyshkova T, Cerina M, Pawlowski M, Ehling P, et al. Identification of the muscarinic pathway underlying cessation of sleep-related burst activity in rat thalamocortical relay neurons. Pflügers Arch. 2012;463:89–102. doi: 10.1007/s00424-011-1056-9. [DOI] [PubMed] [Google Scholar]
- Bista P, Cerina M, Ehling P, Leist M, Pape H-C, Meuth SG, et al. The role of two-pore-domain background K+ (K2P) channels in the thalamus. Pflügers Arch. 2014 doi: 10.1007/s00424-014-1632-x. doi: 10.1007/s00424-014-1632-x [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Bista P, Pawlowski M, Cerina M, Ehling P, Leist M, Meuth P, et al. Differential phospholipase C-dependent modulation of TASK and TREK two-pore domain K(+) channels in rat thalamocortical relay neurons. J Physiol. 2015;593:127–144. doi: 10.1113/jphysiol.2014.276527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broicher T, Seidenbecher T, Meuth P, Munsch T, Meuth SG, Kanyshkova T, et al. T-current related effects of antiepileptic drugs and a Ca(2+) channel antagonist on thalamic relay and local circuit interneurons in a rat model of absence epilepsy. Neuropharmacol. 2007;53:431–446. doi: 10.1016/j.neuropharm.2007.05.030. [DOI] [PubMed] [Google Scholar]
- Brown DA, Adams PR. Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature. 1980;283:673–676. doi: 10.1038/283673a0. [DOI] [PubMed] [Google Scholar]
- Brown DA, Passmore GM. Neural KCNQ (Kv7) channels. Br J Pharmacol. 2009;156:1185–1195. doi: 10.1111/j.1476-5381.2009.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brueggemann LI, Mackie AR, Martin JL, Cribbs LL, Byron KL. Diclofenac distinguishes among homomeric and heteromeric potassium channels composed of KCNQ4 and KCNQ5 subunits. Mol Pharmacol. 2011;79:10–23. doi: 10.1124/mol.110.067496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budde T, Coulon P, Pawlowski M, Meuth P, Kanyshkova T, Japes A, et al. Reciprocal modulation of I (h) and I (TASK) in thalamocortical relay neurons by halothane. Pflügers Arch. 2008;456:1061–1073. doi: 10.1007/s00424-008-0482-9. [DOI] [PubMed] [Google Scholar]
- Carnevale N, Hines M. The NEURON Book. Cambridge: Cambridge University Press; 2006. [Google Scholar]
- Cecchi GA, Huang L, Hashmi JA, Baliki M, Centeno MV, Rish I, et al. Predictive dynamics of human pain perception. PLoS Comput Biol. 2012;8:e1002719. doi: 10.1371/journal.pcbi.1002719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper EC, Harrington E, Jan YN, Jan LY. M channel KCNQ2 subunits are localized to key sites for control of neuronal network oscillations and synchronization in mouse brain. J Neurosci. 2001;21:9529–9540. doi: 10.1523/JNEUROSCI.21-24-09529.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulon P, Kanyshkova T, Broicher T, Munsch T, Wettschureck N, Seidenbecher T, et al. Activity modes in thalamocortical relay neurons are modulated by G(q)/G(11) family G-proteins – serotonergic and glutamatergic signaling. Front Cell Neurosci. 2010;4:1–10. doi: 10.3389/fncel.2010.00132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulon P, Budde T, Pape H-C. The sleep relay – the role of the thalamus in central and decentral sleep regulation. Pflügers Arch. 2012;463:53–71. doi: 10.1007/s00424-011-1014-6. [DOI] [PubMed] [Google Scholar]
- Crockett RS, Bornschein RL, Smith RP. Diurnal variation in response to thermal stimulation: mouse-hotplate test. Physiol Behav. 1977;18:193–196. doi: 10.1016/0031-9384(77)90120-2. [DOI] [PubMed] [Google Scholar]
- Deleuze C, David F, Béhuret S, Sadoc G, Shin H-S, Uebele VN, et al. T-type calcium channels consolidate tonic action potential output of thalamic neurons to neocortex. J Neurosci. 2012;32:12228–12236. doi: 10.1523/JNEUROSCI.1362-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Destexhe A, Bal T, McCormick DA, Sejnowski TJ. Ionic mechanisms underlying synchronized oscillations and propagating waves in a model of ferret thalamic slices. J Neurophysiol. 1996;76:2049–2070. doi: 10.1152/jn.1996.76.3.2049. [DOI] [PubMed] [Google Scholar]
- Ehling P, Cerina M, Meuth P, Kanyshkova T, Bista P, Coulon P, et al. Ca(2+)-dependent large conductance K(+) currents in thalamocortical relay neurons of different rat strains. Pflügers Arch. 2013;465:469–480. doi: 10.1007/s00424-012-1188-6. [DOI] [PubMed] [Google Scholar]
- Fanselow EE, Sameshima K, Baccala LA, Nicolelis MA. Thalamic bursting in rats during different awake behavioural states. Proc Natl Acad Sci U S A. 2001;98:15330–15335. doi: 10.1073/pnas.261273898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger J, Weber YG, Landwehrmeyer B, Sommer C, Lerche H. Immunohistochemical analysis of KCNQ3 potassium channels in mouse brain. Neurosci Lett. 2006;400:101–104. doi: 10.1016/j.neulet.2006.02.017. [DOI] [PubMed] [Google Scholar]
- Hains BC, Saab CY, Waxman SG. Alterations in burst firing of thalamic VPL neurons and reversal by Na(v)1.3 antisense after spinal cord injury. J Neurophysiol. 2006;95:3343–3352. doi: 10.1152/jn.01009.2005. [DOI] [PubMed] [Google Scholar]
- Hughes SW, Errington A, Lorincz ML, Kékesi KA, Juhász G, Orbán G, et al. Novel modes of rhythmic burst firing at cognitively-relevant frequencies in thalamocortical neurons. Brain Res. 2008;1235:12–20. doi: 10.1016/j.brainres.2008.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguenard JR, McCormick DA. Simulation of the currents involved in rhythmic oscillations in thalamic relay neurons. J Neurophysiol. 1992;68:1373–1383. doi: 10.1152/jn.1992.68.4.1373. [DOI] [PubMed] [Google Scholar]
- Huh Y, Cho J. Discrete pattern of burst stimulation in the ventrobasal thalamus for anti-nociception. PLoS ONE. 2013a;8:e67655. doi: 10.1371/journal.pone.0067655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh Y, Cho J. Urethane anesthesia depresses activities of thalamocortical neurons and alters its response to nociception in terms of dual firing modes. Front Behav Neurosci. 2013b;7:1–9. doi: 10.3389/fnbeh.2013.00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh Y, Bhatt R, Jung D, Shin H, Cho J. Interactive responses of a thalamic neuron to formalin induced lasting pain in behaving mice. PLoS ONE. 2012;7:e30699. doi: 10.1371/journal.pone.0030699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeanmonod D, Magnin M, Morel A. Thalamus and neurogenic pain: physiological, anatomical and clinical data. Neuroreport. 1993;4:475–478. doi: 10.1097/00001756-199305000-00003. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ. Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci. 2000;1:21–30. doi: 10.1038/35036198. [DOI] [PubMed] [Google Scholar]
- Kasten MR, Rudy B, Anderson MP. Differential regulation of action potential firing in adult murine thalamocortical neurons by Kv3.2, Kv1, and SK potassium and N-type calcium channels. J Physiol. 2007;584:565–582. doi: 10.1113/jphysiol.2007.141135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Park D, Choi S, Lee S, Sun M, Kim C, et al. Thalamic control of visceral nociception mediated by T-type Ca2+ channels. Science. 2003;302:117–119. doi: 10.1126/science.1088886. [DOI] [PubMed] [Google Scholar]
- Lenz FA, Kwan HC, Dostrovsky JO, Tasker RR. Characteristics of the bursting pattern of action potentials that occurs in the thalamus of patients with central pain. Brain Res. 1989;496:357–360. doi: 10.1016/0006-8993(89)91088-3. [DOI] [PubMed] [Google Scholar]
- Liao Y-F, Tsai M-L, Chen C-C, Yen C-T. Involvement of the Cav3.2 T-type calcium channel in thalamic neuron discharge patterns. Mol Pain. 2011;7:1–10. doi: 10.1186/1744-8069-7-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás RR, Steriade M. Bursting of thalamic neurons and states of vigilance. J Neurophysiol. 2006;95:3297–3308. doi: 10.1152/jn.00166.2006. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Huguenard JR. A model of the electrophysiological properties of thalamocortical relay neurons. J Neurophysiol. 1992;68:1384–1400. doi: 10.1152/jn.1992.68.4.1384. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Pape HC. Acetylcholine inhibits identified interneurons in the cat lateral geniculate nucleus. Nature. 1988;334:246–248. doi: 10.1038/334246a0. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Wang Z, Huguenard J. Neurotransmitter control of neocortical neuronal activity and excitability. Cereb Cortex. 1991;3:387–398. doi: 10.1093/cercor/3.5.387. [DOI] [PubMed] [Google Scholar]
- Ohara S, Taghva A, Kim JH, Lenz FA. Spontaneous low threshold spike bursting in awake humans is different in different lateral thalamic nuclei. Exp Brain Res. 2007;180:281–288. doi: 10.1007/s00221-007-0856-9. [DOI] [PubMed] [Google Scholar]
- Passmore GM, Selyanko AA, Mistry M, Al-Qatari M, Marsh SJ, Matthews EA, et al. KCNQ/M currents in sensory neurons: significance for pain therapy. J Neurosci. 2003;23:7227–7236. doi: 10.1523/JNEUROSCI.23-18-07227.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledgebase of drug targets and their ligands. Nucl. Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin K. The Mouse Brain in Stereotaxic Coordinates. San Diego, CA: Academic Press; 2001. [Google Scholar]
- Peretz A, Degani N, Nachman R, Uziyel Y, Gibor G, Shabat D, et al. Meclofenamic acid and diclofenac, novel templates of KCNQ2/Q3 potassium channel openers, depress cortical neuron activity and exhibit anticonvulsant properties. Mol Pharmacol. 2005;67:1053–1066. doi: 10.1124/mol.104.007112. [DOI] [PubMed] [Google Scholar]
- Pozza DH, Potes CS, Barroso PA, Azevedo L, Castro-Lopes JM, Neto FL. Nociceptive behaviour upon modulation of mu-opioid receptors in the ventrobasal complex of the thalamus of rats. Pain. 2010;148:492–502. doi: 10.1016/j.pain.2009.12.013. [DOI] [PubMed] [Google Scholar]
- Rivera-Arconada I, Lopez-Garcia JA. Effects of M-current modulators on the excitability of immature rat spinal sensory and motor neurones. Eur J Neurosci. 2005;22:3091–3098. doi: 10.1111/j.1460-9568.2005.04507.x. [DOI] [PubMed] [Google Scholar]
- Rostock A, Tober C, Rundfeldt C, Bartsch R, Engel J, Polymeropoulos EE, et al. D-23129: a new anticonvulsant with a broad spectrum activity in animal models of epileptic seizures. Epilepsy Res. 1996;23:211–223. doi: 10.1016/0920-1211(95)00101-8. [DOI] [PubMed] [Google Scholar]
- Sherman SM, Guillery R. Exploring the Thalamus and Its Role in Cortical Function. Cambridge, London: MIT Press; 2006. [Google Scholar]
- Storm JF. An after-hyperpolarisation of medium duration in rat hippocampal pyramidal cells. J Physiol. 1989;409:171–190. doi: 10.1113/jphysiol.1989.sp017491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatulian L, Brown DA. Effect of the KCNQ potassium channel opener retigabine on single KCNQ2/3 channels expressed in CHO cells. J Physiol. 2003;549:57–63. doi: 10.1113/jphysiol.2003.039842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremere LA, Terleph TA, Jeong JK, Pinaud R. Bilateral multielectrode neurophysiological recordings coupled to local pharmacology in awake songbirds. Nat Protoc. 2010;5:191–200. doi: 10.1038/nprot.2009.224. [DOI] [PubMed] [Google Scholar]
- Tscherter A, David F, Ivanova T, Deleuze C, Renger JJ, Uebele VN, et al. Minimal alterations in T-type calcium channel gating markedly modify physiological firing dynamics. J Physiol. 2011;589:1707–1724. doi: 10.1113/jphysiol.2010.203836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Thompson SM. Maladaptive homeostatic plasticity in a rodent model of central pain syndrome: thalamic hyperexcitability after spinothalamic tract lesions. J Neurosci. 2008;28:11959–11969. doi: 10.1523/JNEUROSCI.3296-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XY, McKenzie JS, Kemm RE. Whole-cell K+ currents in identified olfactory bulb output neurones of rats. J Physiol. 1996;490:63–77. doi: 10.1113/jphysiol.1996.sp021127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyand TG, Boudreaux M, Guido W. Burst and tonic response modes in thalamic neurons during sleep and wakefulness. J Neurophysiol. 2001;85:1107–1118. doi: 10.1152/jn.2001.85.3.1107. [DOI] [PubMed] [Google Scholar]
- Wickenden AD, Yu W, Zou A, Jegla T, Wagoner PK. Retigabine, a novel anti-convulsant, enhances activation of KCNQ2/Q3 potassium channels. Mol Pharmacol. 2000;58:591–600. doi: 10.1124/mol.58.3.591. [DOI] [PubMed] [Google Scholar]
- Willis WD, Westlund KN. Neuroanatomy of the pain system and of the pathways that modulate pain. J Clin Neurophysiol. 1997;14:2–31. doi: 10.1097/00004691-199701000-00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wladyka CL, Kunze DL. KCNQ/M-currents contribute to the resting membrane potential in rat visceral sensory neurons. J Physiol. 2006;575:175–189. doi: 10.1113/jphysiol.2006.113308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Wu Y, Bi Y, Tan L, Gan Y, Wang K. Activation of voltage-gated KCNQ/Kv7 channels by anticonvulsant retigabine attenuates mechanical allodynia of inflammatory temporomandibular joint in rats. Mol Pain. 2010;6:1–10. doi: 10.1186/1744-8069-6-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue C, Yaari Y. KCNQ/M channels control spike after depolarisation and burst generation in hippocampal neurons. J Neurosci. 2004;24:4614–4624. doi: 10.1523/JNEUROSCI.0765-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Pharmacological characterization of Kv7 channels in VB.
Figure S2 Time course of IM modulation by Ret.
Figure S3 Expression of Kv7 channels in the ventrobasal thalamus (VB).
Table S1 List of modelled parameters and their default values.