

Abstract
Background and Purpose
Receptor tyrosine kinase inhibitors (RTKIs) targeted at VEGF receptor 2 (VEGFR2) have proved to be attractive approaches to cancer therapy based on their ability to reduce angiogenesis. Here we have undertaken a quantitative analysis of the interaction of RTKIs and two VEGF splice variants, VEGF165a and VEGF165b, with VEGFR2 by studying nuclear factor of activated T-cells (NFAT) reporter gene activity in live HEK-293 cells.
Experimental Approach
HEK-293 cells expressing the human VEGFR2 and a firefly luciferase reporter gene regulated by an NFAT response element were used for quantitative analysis of the effect of RTKIs on VEGF165a- and VEGF165b-stimulated luciferase gene expression.
Key Results
VEGF165a produced a concentration-dependent activation of the NFAT-luciferase reporter gene in living cells that was inhibited in a non-competitive fashion by four different RTKIs (cediranib, pazopanib, sorafenib and vandetanib). The potency obtained for each RTKI from this analysis was similar to those obtained in binding studies using purified VEGFR2 kinase domains. VEGF165b was a lower-efficacy agonist of the NFAT-luciferase response when compared with VEGF165a. Analysis of the concentration–response data using the operational model of agonism indicated that both VEGF165 isoforms had similar affinity for VEGFR2.
Conclusions and Implications
Quantitative pharmacological analysis of the interaction of VEGF165 isoforms and RTKIs with VEGFR2 in intact living cells has provided important insights into the relative affinity and efficacy of VEGF165a and VEGF165b for activation of the calcineurin- NFAT signalling pathway by this tyrosine kinase receptor.
Tables of Links
| TARGETS |
|---|
| Catalytic receptorsa |
| EGFR, EGF receptor |
| VEGFR1, VEGF receptor 1 |
| VEGFR2, VEGF receptor 2 |
| VEGFR3, VEGF receptor 3 |
| Enzymesb |
| Akt |
| Abl1 |
| MEK5 |
| PKC |
| PLCγ |
| LIGANDS |
|---|
| Cediranib |
| IP3, inositol-1,4,5-trisphosphate |
| Pazopanib |
| Sorafenib |
| Vandetanib |
| VEGF-A |
| VEGF-B |
| VEGF-C |
| VEGF-D |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,bAlexander et al., 2013a,b,).
Introduction
VEGF is an important mediator of cell survival, proliferation and angiogenesis (Ferrara, 2009; Shibuya, 2011; Musumeci et al., 2012). It constitutes a family of mammalian homodimeric glycoproteins, comprising VEGF-A, VEGF-B, VEGF-C, VEGF-D and placenta growth factor. VEGF-A is an important and potent mediator of tumour-induced angiogenesis (Ferrara, 2004; 2009,). VEGF family members bind to three different VEGF receptors (VEGFR1, VEGFR2 and VEGFR3) with differing selectivity profiles (Ferrara, 2009; Shibuya, 2011). VEGFR2 is the major regulator of VEGF-driven responses in vascular endothelial cells including permeability, proliferation, invasion and migration. It is also considered to be a crucial mediator of angiogenesis (Ferrara, 2009; Shibuya, 2011). Its signalling pathways are relatively well understood with tyrosine residues Y1175 and Y1214 in the human VEGFR2 being the main auto-phosphorylation sites activated by VEGF binding and tyrosine kinase activation. This creates binding sites for key intracellular signalling proteins such as Grb2, PLCγ and Shc1 (Matsumoto and Mugishima, 2006; Rososki, 2008; Koch et al., 2011).
The transmembrane glycoprotein neuropilin 1 forms a complex with VEGFR2 and acts as a co-receptor to enhance VEGF-A binding, mediate focal adhesion kinase phosphorylation and increase cell migration (Herzog et al., 2011). It has also been shown to promote VEGFR2 internalization and endosomal trafficking, leading to the regulation of ERK signalling and cell proliferation (Lanahan et al., 2013). Neuropilin 1 is engaged by specific VEGF isoforms and has recently been the target of drug discovery efforts to design low MW inhibitors of neuropilin 1 (Djordjevic and Driscoll, 2013). Multiple isoforms of VEGF-A, ranging from 121 to 206 amino acids, can be generated by alternative exon splicing that differ in their ability to bind heparin (affecting bioavailability) or neuropilin 1 and they appear to play distinctive roles in angiogenesis (Woolard et al., 2004; 2009,; Ferrara, 2009). For example, alternative splicing in exon 8 of the VEGF gene can generate VEGFxxxa and VEGFxxxb (where xxx is the amino acid length) isoforms that have been reported to have pro-angiogenic and anti-angiogenic activities respectively (Woolard et al., 2004; 2009,). In keeping with this, VEGF165b has been reported to be a weak partial agonist at VEGFR2, able to bind weakly to heparin and does not interact with neuropilin-1 (Cebe Suarez et al., 2006; Catena et al., 2010).
Receptor tyrosine kinases inhibitors (RTKIs) targeted at VEGFR2 have proved to be attractive approaches to cancer therapy based on their ability to reduce angiogenesis and/or lymph-angiogenesis (Musumeci et al., 2012). There are three known classes of RKTIs. Class I RTKIs, such as cediranib, vandetanib and pazopanib, are able to bind to the active conformation of the receptor and compete for the intracellular ATP-binding site within the catalytic domain of VEGFR2 (Gotink and Verheul, 2010; Davis et al., 2011; Blasi et al., 2012). Class II RTKIs, such as sorafenib, bind to the non-active conformation of the receptor at the hydrophobic pocket of the activation loop and inhibit kinase activity by indirectly preventing the binding of ATP (Gotink and Verheul, 2010; Davis et al., 2011). Finally, class III RTKIs such as neratinib, covalently bind to cysteine residues within the intracellular ATP-binding region of the receptor (Gotink and Verheul, 2010; Davis et al., 2011). Most of these small molecule RTKIs interact with multiple members of the PK family (Davis et al., 2011). For example, binding studies with purified kinase domains have shown that vandetanib is a more potent inhibitor of Abl1 (16 nM), EGFR (9.5 nM), MEK5 (49 nM) than VEGFR2 (820 nM) (Davis et al., 2011).
Quantitative evaluation of the interactions of RTKIs with VEGFR2 in living cells has, however, been largely lacking. This is important as, by definition, all RTKIs need to access the intracellular regions of VEGFR2 in order to elicit their pharmacological action. It is therefore vital to understand how the different RTKIs affect VEGF165a- and VEGF165b-mediated signalling in intact cells. The aim of the present study was to undertake a quantitative pharmacological analysis of the effect of VEGF165 isoforms and RTKIs on VEGFR2-mediated signalling in living cells. An important signalling pathway for VEGFR2 is the calcineurin-nuclear factor of activated T-cells (NFAT) system that, following activation by VEGF, leads to nuclear translocation of the NFAT transcription factor and expression of pro-angiogenic and pro-inflammatory genes (Suehiro et al., 2014; Yang et al., 2014). Reporter gene systems have been used extensively to study GPCRs and provide an alternative to biochemical assays for following signal transduction pathways from receptors at the cell surface to nuclear gene transcription in living cells (Hill et al., 2001). Here we have used an NFAT-luciferase reporter gene to investigate the impact of four representative RTKIs on VEGF165a- and VEGF165b-stimulated NFAT-luciferase activity in HEK-293 cells expressing human VEGFR2.
Methods
Cell lines
HEK-293 cells expressing the human VEGFR2 and an NFAT reporter gene were provided by Promega Corporation. The NFAT reporter gene contained an NFAT response element linked via a minimal promoter to the firefly luciferase gene luc2P containing a human sequence enriched in proline (P), glutamic acid (E), serine (S) and threonine (T) protein destabilization sequence (Voon et al., 2005). VEGFR2 NFAT cells were maintained in DMEM media supplemented with 10% FCS and 0.5% G418 in a humidified 5% CO2/95% air atmosphere at 37°C.
Measurement of VEGFR2-stimulated NFAT-reporter gene activity in HEK-293 cells
VEGFR2 NFAT cells were seeded in a T75 flask at 5 × 106 cells per flask using DMEM +10%FCS and incubated at 37°C in a 5% CO2/95% air atmosphere for 3 days until the cells were 100% confluent. On the fourth day, cells were washed with PBS and detached using 3 mL Versene® (ETDA 0.02% in PBS). Once cells had detached, 6 mL of DMEM +0.1%BSA was added and the cells were counted using a haemocytometer. Cells were centrifuged at 200× g for 5 min, resuspended in DMEM +0.1%BSA and seeded at a density of 4 × 104 cells per well in 80 μL DMEM +0.1%BSA in white-sided, clear flat-bottomed 96-well plates (Greiner, Stonehouse, UK), which had been coated with 0.01 mg·mL−1 poly-D-lysine in PBS for 30 min and washed with DMEM. Cells were then incubated for 1 h in a humidified 5% CO2/95% air atmosphere at 37°C. RTKIs or vehicle control were added in 10 μL DMEM +0.1%BSA for 1 h prior to addition of VEGF165a or VEGF165b in 10 μL DMEM +0.1%BSA and the incubation was continued for a further 5 h (in a humidified 5% CO2/95% air atmosphere at 37°C). After the 5 h incubation, 100 μL ONE-Glo Luciferase Assay reagent was added to each well and luminescence was measured according to the manufacturer's instructions on a Topcount platereader (Perkin Elmer, Llantrisant, UK).
Data analysis
All data were fitted using non-linear regression in Prism 6 (GraphPad Software, San Diego, CA, USA). VEGF165a and VEGF165b concentration–response curves were fitted to the following equation:
| 1 |
Where Emax is the maximal response, and the EC50 is the molar concentration of agonist required to generate 50% of the Emax. When investigating the effect of different concentrations of RTKI on concentration–response curves for VEGF165a, the data were also fitted to Equation 2013 with parameters for either EC50 of Emax shared between all curves. A comparison of the extra sum of squares that resulted from the analysis with separate EC50 or Emax values (over that with one of the parameters shared) using the F-test (Prism 6) then allowed for statistical analysis of the difference between EC50 or Emax values.
Inhibition curves obtained with RTKIs in the presence of a fixed concentration of VEGF165a or VEGF165b were fitted to the following equation:
| 2 |
Where [A] is the concentration of RTKI and the IC50 is the molar concentration of ligand required to inhibit 50% of the response to VEGF.
Partial agonist concentration–response curves to VEGF165b were also fitted to the operational model of Black and Leff (1983) using the following equation:
| 3 |
Where Emax is the maximal response of the system, [A] is the concentration of VEGF165b, n is the slope parameter, KA is the dissociation constant of the agonist VEGF165b and τ is the transducer constant, which is a practical measure of efficacy. τ is the inverse of the fraction of receptors that must be occupied by agonist to obtain the half-maximal response. Emax was determined by simultaneously fitting Equation 2013 to the concentration–response data for VEGF165a that were obtained in the same experiments as those for VEGF165b. Emax was shared between the two simultaneous fits.
Equation 1983 was also used to simultaneously fit concentration–response curves to VEGF165a in the presence and absence of increasing concentrations of a define RTKI. In this case, Emax, n and KA (which in this case is the dissociation constant of VEGF165a) were shared between the simultaneous fits.
All data are presented as mean ± SEM. The n in the text refers to the number of separate experiments. Statistical significance was determined by Student's unpaired t-test or by one or two-way anova with Dunnett's post hoc analysis and P < 0.05 was considered statistically significant.
Materials
VEGF165a and VEGF165b were obtained from R&D systems (Abingdon, UK). Vandetanib, pazopanib, cediranib and sorafenib were supplied by Sequoia Research Products (Pangbourne, UK). The ONE-Glo™ Luciferase Assay System was obtained from Promega Corporation (Madison, WI, USA). Versene was obtained from Lonza (Basal, Switzerland). G418 was purchased from Life Technologies (Paisley, UK). All other chemicals and reagents were purchased from Sigma-Aldrich (Gillingham, UK).
Results
VEGF165a-stimulated NFAT-luciferase production in intact cells
Incubation with VEGF165a produced a concentration-dependent (pEC50 9.66 ± 0.05, n = 10) increase in NFAT-mediated luciferase production in HEK-293 cells expressing VEGFR2 that was 8.30 ± 0.85-fold (n = 10) over basal levels (Table 1; Figure 1A and B). The response to 1 nM VEGF165a was inhibited by the RTKI cediranib in intact HEK-293 cells in a concentration-dependent manner (Figure 1C; Table 2). The pIC50 obtained for cediranib (9.13; Figure 2A, Table 2) was in close agreement with that reported from binding studies with the purified VEGFR2 kinase domain (Davis et al., 2011). It was also noticeable that there was no marked inhibition by cediranib below basal levels at the highest concentration used (Figures 1C and 2A), suggesting that the ability of this RTKI to inhibit other tyrosine kinases (e.g. PDGFR-A, PDGFR-B and EGFR, Davis et al., 2011) did not significantly impact on the response observed. This was confirmed when the effect of cediranib was evaluated for its ability to inhibit basal NFAT-luciferase production (Figure 1D). A significant inhibition (P < 0.05; one way anova) of the small basal NFAT-luciferase response was only observed at concentrations of cediranib above 10 nM (Figure 1D). Analysis of all five repeat experiments indicated that a significant inhibition of basal signalling was only obtained at the two highest concentrations used (P < 0.05; one way anova; n = 5).
Table 1.
Concentration–response parameters for VEGF165a- and VEGF165b-stimulated NFAT-luciferase responses
| −Log EC50 | Emax (% VEGF165a max) | n | |
|---|---|---|---|
| VEGF165a | 9.66 ± 0.05 | 100 | 10 |
| VEGF165b | 9.21 ± 0.08 | 62.1 ± 1.2 | 5 |
Values are mean ± SEM of n separate experiments. Each individual experiment was performed in quadruplicate.
Figure 1.
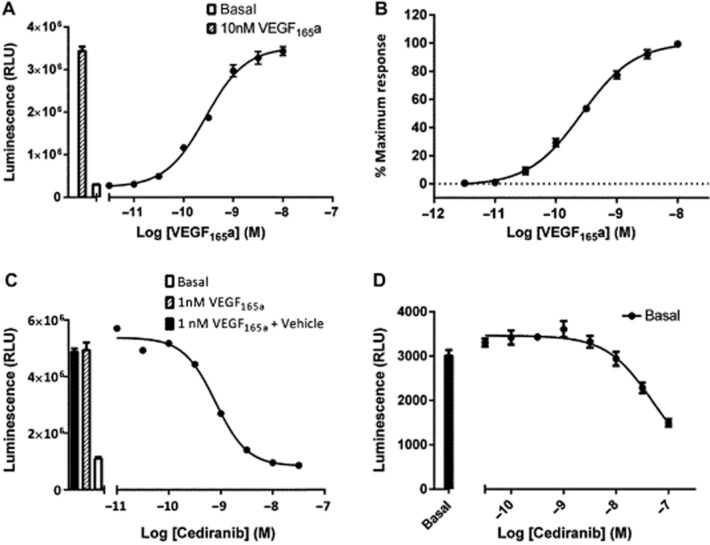
The effect VEGF165a on NFAT-mediated gene transcription in VEGFR2 NFAT cells. VEGFR2 NFAT cells were treated with VEGF165a (A and B) or cediranib +1 nM VEGF165a (C). Data are mean ± SEM from quadruplicate determinations in a single representative experiment that was repeated on five separate occasions (A and C). Normalized data from five repeat experiments expressed as a percentage of the response to 10 nM VEGF165a in each experiment (B). Effect of cediranib on basal NFAT-luciferase activity (D). Data are mean ± SEM from quadruplicate determinations in a single representative experiment that was repeated on five separate occasions. The histogram in (A) and (C) show the control response to 1 nM VEGF165a (A and C) and that to VEGF165a in the presence of the vehicle (containing DMSO) for the highest concentration of cediranib used in the competition experiment shown in (C).
Table 2.
The effect of selected RTKIs on VEGF-stimulated firefly luciferase production in VEGFR2 NFAT cells
| Inhibition of 1 nM VEGF165a pIC50 | n | Inhibition of 3 nM VEGF165b pIC50 | n | Reported binding pKD for purified kinase domain* | |
|---|---|---|---|---|---|
| Cediranib | 9.13 ± 0.01 | 5 | 9.38 ± 0.07 | 5 | 8.96 |
| Pazopanib | 8.25 ± 0.03 | 5 | 8.29 ± 0.10 | 8 | 7.85 |
| Sorafenib | 8.01 ± 0.06 | 5 | 7.96 ± 0.04 | 5 | 7.23 |
| Vandetanib | 6.72 ± 0.03 | 5 | 7.00 ± 0.04 | 6 | 6.08 |
VEGFR2 NFAT cells were treated with each RTKI and either 1 nM VEGF165a or 3 nM VEGF165b. Data are mean ± SEM of n separate experiments. Each individual experiment was performed in quadruplicate. Individual fitted values for pIC50 values were obtained in each individual experiment and then analysed to provide mean ± SEM data provided here.
Values taken from Davis et al., 2011.
Figure 2.
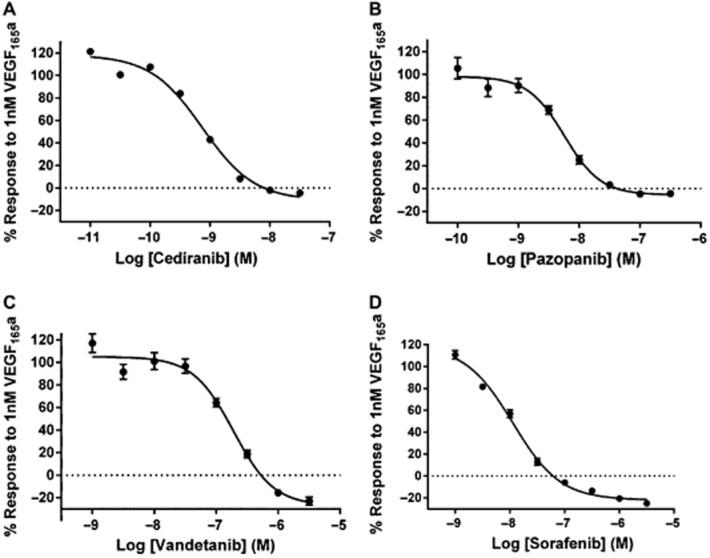
The effect of selected RTKIs on NFAT gene transcription stimulated by 1 nM VEGF165a. VEGFR2 NFAT cells were treated with (A) cediranib, (B) pazopanib, (C) vandetanib or (D) sorafenib. Data are mean ± SEM of five separate experiments. Data are expressed as a percentage of the response to 1 nM VEGF165a in the absence of RTKIs. Each individual experiment was performed in quadruplicate.
Inhibition of 1 nM VEGF165a-stimulated NFAT-luciferase activity was also obtained with a second-class I RTKI (pazopanib, which has a different inhibitor selectivity profile compared with cediranib, e.g. FGFR1-3, PDGFRA/B, VEGFR1 and EGF; Davis et al., 2011) and with sorafenib and vandetanib (Figure 2, Table 2). All RTKIs tested produced pIC50 values, which were in agreement with those reported previously in binding studies on purified VEGFR2 kinase domains (Table 2). As with cediranib, there was no marked inhibition below basal responses (Figure 2B) with pazopanib (which is also a potent PDGFR inhibitor, Davis et al., 2011). In contrast, both sorafenib (−25.0 ± 2.6%, n = 5) and vandetanib (−23.0 ± 3.7%, n = 5) produced a small significant inhibition (P < 0.05, paired t-test) below basal levels, which may reflect some interference with other tyrosine kinases at the higher concentrations required to inhibit VEGFR2 with these inhibitors.
VEGF binding to VEGFR2 requires Ig-like domains D2 and D3 in the extracellular portion of the receptor (Dosch and Ballmer-Hofer, 2010; Leppänen et al., 2010). In contrast, the kinase domain, which is the target for RTKIs lies within the intracellular portion of the receptor. As a consequence, the interaction between VEGF and RTKI in intact cells should show classical non-competitive interactions when concentration–response curves to VEGF165a are analysed in the presence of increasing concentrations of RTKIs. These data for VEGF-stimulated NFAT-luciferase production are shown in Figure 3 and Table 3. All four inhibitors produced a significant (P < 0.05) concentration-dependent reduction in the maximal response to VEGF165a (Table 3). Analysis of all the individual experiments indicated that there was a small, but significant change (P < 0.05) in EC50 at the highest concentrations of RTKIs used (Table 3). However, global analysis of the combined data presented in Figure 3 indicated that there was only a significant difference in the EC50 values for cediranib (P < 0.05). In contrast, there was a significant decrease in Emax with all four RTKIs (P < 0.001; extra sum of squares F-test; Figure 3).
Figure 3.

The effect of RTKIs on VEGF165a concentration–response curves. VEGFR2 NFAT cells were treated with (A) pazopanib, (B) vandetanib, (C) cediranib or (D) sorafenib for 1 h prior to the addition of increasing concentrations of VEGF165a. Data are mean ± SEM of five (A and B), six (C) or seven (D) replicate experiments. Each individual experiment was performed in quadruplicate. Global analysis of the combined data presented for each RTKI (A–D; extra sum of squares F-test) indicated that there was only a significant difference in the EC50 values for cetiranib (P < 0.05). In contrast, there was a significant decrease in Emax with all four RTKIs (P < 0001; Figure 3; extra sum of squares F-test).
Table 3.
Effect of RTKIs on VEGF165a concentration–response parameters
| Vandetanib | Pazopanib | Cediranib | Sorafenib | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| nM | pEC50 | % Emax | nM | pEC50 | % Emax | nM | pEC50 | % Emax | nM | pEC50 | % Emax |
| 0 | 9.90 ± 0.14 | 100.0 | 0 | 9.66 ± 0.11 | 100.0 | 0 | 9.68 ± 0.09 | 100.0 | 0 | 9.72 ± 0.06 | 100.0 |
| 30 | 9.77 ± 0.10 | 81.0 ± 4.1 | 1 | 9.50 ± 0.09 | 85.2 ± 7.0 | 0.3 | 9.40 ± 0.10 | 64.8 ± 10.4* | 3 | 9.50 ± 0.05 | 70.1 ± 6.9* |
| 100 | 9.75 ± 0.12 | 58.5 ± 5.7* | 3 | 9.43 ± 0.13 | 71.4 ± 5.2* | 1 | 9.28 ± 0.12 | 31.1 ± 6.6* | 10 | 9.33 ± 0.06* | 52.9 ± 5.1* |
| 300 | 9.41 ± 0.14* | 29.5 ± 7.9* | 10 | 9.14 ± 0.16* | 32.8 ± 2.0* | 3 | 9.13 ± 0.18* | 8.8 ± 1.9* | 30 | 9.00 ± 0.13* | 18.9 ± 8.9* |
pEC50 and Emax values for VEGF165a obtained in the presence of increasing concentrations of four RTKIs.
P < 0.05 compared with corresponding control in the absence of RTKI (one-way anova). Values are mean ± SEM from six (cediranib), five (pazopanib), seven (sorafenib) and five (vandetanib) separate experiments.
Pharmacological characteristics of the splice variant, VEGF165b, in HEK-293 cells
In the present study, VEGF165b produced a robust NFAT-luciferase response in HEK-293 cells expressing human VEGFR2 that accounted for 62.1% (Table 1, Figure 4) of the maximum response obtained with VEGF165a in the same experiments. The EC50 values were, however, very similar (Table 1, Figure 4A). Analysis of the concentration–response curves using the operational model of Black and Leff (1983) for partial agonists indicated that the log KA for VEGF165b was −8.83 ± 0.13 (n = 5) and the transducer constant τ was 1.65 ± 0.23 (n = 5). τ is a measure of agonist efficacy and represents the inverse of the fraction of receptors (60.1%) that must be occupied by agonist to obtain the half-maximal response (Black and Leff, 1983). The response to 3 nM VEGF165b was sensitive to inhibition by RTKIs with similar potencies to those obtained when VEGF165a was used as agonist (Figure 4B; Table 2).
Figure 4.
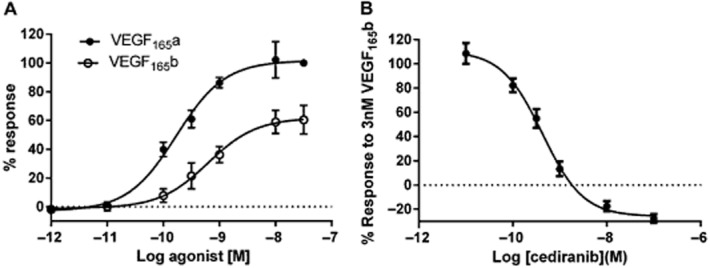
Characterization of the effect of VEGF165b on NFAT-luciferase repsonses. (A) A comparison of VEGF165a and VEGF165b concentration–response curves. (B) Inhibition of VEGF165b-stimulated NFAT-luciferase responses by cediranib. In (B), the concentration of VEGF165b was 3 nM. Data are mean ± SEM of five separate experiments. Four replicates were made for each condition in each individual experiment.
Discussion
VEGF receptors have been shown to activate several intracellular signalling pathways including PKC, PLCγ, MAPK and calcium-calcineurin (Suehiro et al., 2014). Calcineurin signalling activates NFAT transcription factors leading to the stimulation of gene transcription (Hill et al., 2001; Suehiro et al., 2014; Yang et al., 2014). Stimulation of PLCγ increases levels of inositol-1,4,5-trisphosphate (IP3) and diacyglycerol. IP3 then stimulates the release of intracellular calcium while diacylglycerol activates PKC. Increased intracellular calcium concentration stimulates calcineurin leading to the dephosphorylation of cytoplasmic NFAT transcription factors allowing them to translocate to the nucleus. In parallel, PKC activation results in the production of the AP-1 immediate early genes c-fos and c-jun. Once in the nucleus, NFAT binds with c-fos and c-jun to form a transcriptional complex capable of synergistically activating both the NFAT and the AP-1 response elements to stimulate gene expression (Masuda et al., 1998; Macian et al., 2000; Hill et al., 2001). In endothelial cells, VEGF treatment leads to NFAT nuclear localization and the expression of pro-angiogenic and pro-inflammatory genes (Suehiro et al., 2014; Yang et al., 2014). Furthermore, the calcineurin-NFAT pathway appears to be an important route for VEGF-mediated signalling (Suehiro et al., 2014; Yang et al., 2014). Here we have used a reporter gene containing the NFAT promoter coupled to the expression of firefly luciferase (Hill et al., 2001; Voon et al., 2005) to investigate in living cells the pharmacological characteristics of VEGF165a- and VEGF165b-induced gene expression in HEK-293 cell transfected with human VEGFR2.
Both VEGF165a and VEGF165b were able to produce a robust and potent stimulation of NFAT-mediated luciferase gene expression after 5 h of incubation. Both isoforms had very similar EC50 values that were in the nanomolar range (Table 1). This is in keeping with previous reports that VEGF165b has a lower efficacy than VEGF165a for VEGFR2-mediated responses (Woolard et al., 2004; Cebe Suarez et al., 2006; Kawamura et al., 2008; Catena et al., 2010). The alternatively spliced variant VEGF165b was a partial agonist of NFAT-luciferase production eliciting a maximal response that was only 62% of that achieved by VEGF165a. Analysis of the VEGF165b concentration–response data using the operational model of Black and Leff (1983) provides a means by which both the dissociation binding constant (KA) and the efficacy (in terms of the τ constant) of VEGF165b can be estimated. This produced an estimate for pKA of VEGF165b at VEGFR2 (8.83) and a value of 1.65 for the efficacy parameter τ (indicating that 60.1% of receptors need to be occupied by VEGF165b in order to achieve 50% of the maximal cellular response). The similarity of EC50 values for the two VEGF isoforms, however, suggests that the relatively large response to VEGF165b compared with that seen in other studies (Cebe Suarez et al., 2006; Kawamura et al., 2008; Catena et al., 2010) is not a consequence of a large amplification of the signalling pathways to NFAT-mediated gene expression in these cells. Furthermore, previous work has suggested that VEGF165a and VEGF165b have the same binding affinities for VEGFR2 (Woolard et al., 2004; Cebe Suarez et al., 2006).
Interestingly, previous work has indicated that the extent to which VEGF165b can elicit responses may depend on the cellular context and the signalling cascade measured. Thus, while VEGF165b was a very weak agonist of MAPK and Akt phosphorylation in VEGFR2 transfected CHO cells, it was able to stimulate a robust MAPK and Akt phosphorylation in human microvascular endothelial cells (Woolard et al., 2004). Furthermore, signalling pathway differences in the relative efficacy of VEGF-A isoforms have been observed for the activation of VEGFR2-mediated responses (ERK1/2, p38 MAPK, Akt) in HUVECs by VEGF165a and VEGF121a (Fearnley et al., 2014). Previous studies, however, have largely been based on Western blot analysis, and it is clear that the NFAT-luciferase system reported here provides a powerful system for the quantitative evaluation of concentration–response relationships for drugs interacting with human VEGFR2 in living cells.
Four representative RTKIs (cediranib, pazopanib, vandetanib and sorafenib) were able to inhibit both VEGF165a- and VEGF165b-mediated NFAT-luciferase expression with similar potency, and yielded IC50 values that were similar to the KD values reported from binding studies with purified VEGFR2 kinase domains (Davis et al., 2011). This similarity suggests that all four compounds readily cross the cell membrane in intact living cells. The target for VEGF binding within VEGFR2 is to domains D2 and D3 of the extracellular portion of the receptor (Dosch and Ballmer-Hofer, 2010; Leppänen et al., 2010). In contrast, RTKIs interact in various ways (depending on RTKI class) with the intracellular kinase domain of the receptor. As a consequence, the interaction between VEGF and RTKI in intact cells would be expected to show classical non-competitive interactions and lead to a marked change in the maximum response to VEGF165a with little impact on the EC50 value for the agonist. This is what was observed in the present study (Figure 3). All four RTKIs produced a marked reduction in the Emax values for VEGF165a with only a small change in the pEC50 value. At the highest concentrations of RTKIs used, the pEC50 was generally between 9.00 and 9.14, which provides an indication of the PKA for VEGF165a. Global analysis of the combined data shown in Figure 3 for each inhibitor (using the extra sum of squares F-test) indicated that it was only cediranib that had a significant difference in EC50 between the four sets of VEGF165a concentration–responses curves.
The non-competitive nature of the inhibition produced by RTKIs via the intracellular kinase domain, however, provided an opportunity to estimate the binding affinity of VEGF165a by utilizing the operational model of Black and Leff (1983). In this model, the transducer ratio tau (τ) is a measure of efficacy and reflects the ratio (total receptor number)/KE where KE describes the hyperbolic relationship in the system between the response and the concentration of agonist-receptor complexes. Receptor alkylation experiments have been used previously to reduce the number of binding sites as a way to obtain concentration–response relationships with different τ values, but with common values for KE, Emax and KA. Use of an RTKI targeted against VEGFR2 kinase activity is also a way of reducing τ values without changing the maximal capacity of the NFAT reporter gene system in the cells. In this case, the inhibition of the VEGFR2 kinase activity will interfere with signal transduction at the level of the receptor. This will effectively alter the KE value and increase the concentration of agonist–receptor complexes needed to produce a function response. However, this analysis assumes that the RTKI has no allosteric effect on the binding affinity of VEGF165a. Thus, increasing concentrations of RTKIs should decrease the apparent efficacy of VEGF165a in this cellular system and reduce the transducer constant τ. All other parameters in terms of KA, Emax and should, however, remain unchanged.
If the concentration–response curves obtained with a given RTKI presented in Figure 3 are simultaneously analysed on this basis (with these assumptions), the following estimates of the pKA value for VEGF165a are obtained: 8.9, 8.9, 9.4 and 9.0 (for the data sets obtained with cediranib, sorafenib, vandetanib and pazopanib respectively). The mean value obtained from this analysis for VEGF165a (8.9) is almost identical to that obtained for the partial agonist VEGF165b (8.8) in keeping with previous reports that their affinities are identical (Woolard et al., 2004; Cebe Suarez et al., 2006). Interestingly, vandetanib produced a higher estimate for the pKA of VEGF165a than that obtained with the other RTKIs. This suggests that the nature of the interaction of vandetanib with VEGFR2 has produced an allosteric conformational change in the ligand binding site for VEGF165a and altered its affinity for VEGF165a.
It should be noted that all four RTKIs produced a small inhibition of the responses to VEGF165a and VEGF165b below basal levels (Figure 2; Figure 4B). A similar effect has been seen with other RTKs (Forsell et al., 2012) and has been ascribed to constitutive activity of the receptor. However, in the present study, the effect is likely to be due to inhibition of other tyrosine kinases within this cell line. The effect of cediranib on basal NFAT signalling seen in Figure 1D is consistent with this, particularly at the higher concentration used. Thus, the potency of cediranib for inhibition of VEGF-stimulated NFAT signalling and basal NFAT signalling, respectively, are quite different (Figure 1C and D). Thus, although the major effects of RTKIs reported here are consequences of an interaction with VEGFR2, it must be remembered that interference with other tyrosine kinase signalling cascades is possible at higher concentrations of these inhibitors.
In summary, the present study has shown that the VEGFR2 NFAT-luciferase reporter gene system provides a robust way to investigate, in a quantitative manner, the interaction of drugs (both agonists and RTKIs) with VEGFR2 in an intact cellular environment. Quantitative pharmacological analysis of the interaction of these drugs with VEGFR2 in living cells has provided important insights into the relative affinity and efficacy of VEGF165a and VEGF165b for activation of the calcineurin–NFAT signalling pathway by this tyrosine kinase receptor. This opens the way for similar quantitative approaches to be used to evaluate affinity and efficacy measures for different VEGF isoforms in mediating responses via other signalling cascades. This should shed light on the potential for VEGFR2 agonists to bias signalling to particular intracellular pathways.
Acknowledgments
We thank Matt Robers (Promega Corporation) for helpful discussions and the Medical Research Council (G0800006), BBSRC (BB/L19418) and Promega Corporation for financial support.
Glossary
- NFAT
nuclear factor of activated T-cells
- RTKIs
receptor tyrosine kinase inhibitors
- VEGFR1
VEGF receptor 1
- VEGFR2
VEGF receptor 2
- VEGFR3
VEGF receptor 3
Author contributions
J. J. C., A. J. W., S. J. H. and J. W. participated in the research design. J. J. C. conducted experiments. J. J. C. and S. J. H. performed data analysis. J. J. C., S. J. H. and J. W. wrote or contributed to the writing of the paper.
Conflict of interest
The authors declare no conflict of interest.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Catalytic Receptors. Br J Pharmacol. 2013;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black JW, Leff P. Operational models of pharmacological agonism. Proc R Soc Lond B Biol Sci. 1983;220:141–162. doi: 10.1098/rspb.1983.0093. [DOI] [PubMed] [Google Scholar]
- Blasi E, Heyen J, Patyna S, Hemkens M, Ramirez D, John-Baptiste A, et al. Sunitinib, a receptor tyrosine kinase inhibitor, increases blood pressure in rats without associated changes in cardiac structure and function. Cardiovasc Ther. 2012;30:287–294. doi: 10.1111/j.1755-5922.2011.00278.x. [DOI] [PubMed] [Google Scholar]
- Catena R, Larzabal L, Larrayoz M, Molina E, Hermida J, Agorreta J, et al. VEGF121b and VEGF165b are weakly angiogenic isoforms of VEGF-A. Mol Cancer. 2010;9:320. doi: 10.1186/1476-4598-9-320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cebe Suarez S, Pieren M, Cariolato L, Arn S, Hoffmann U, Bogucki A, et al. A VEGF-A splice variant defective for heparan sulfate and neuropilin-1 binding shows attenuated signaling through VEGFR-2. Cell Mol Life Sci. 2006;63:2067–2077. doi: 10.1007/s00018-006-6254-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MI, Hunt JP, Herrgard S, Ciceri P, Wodicka LM, Pallares G, et al. Comprehensive analysis of kinase inhibitor selectivity. Nat Biotechnol. 2011;29:1046–1051. doi: 10.1038/nbt.1990. [DOI] [PubMed] [Google Scholar]
- Djordjevic S, Driscoll PC. Targeting VEGF signalling via the neuropilin co-receptor. Drug Discov Today. 2013;18:447–455. doi: 10.1016/j.drudis.2012.11.013. [DOI] [PubMed] [Google Scholar]
- Dosch DD, Ballmer-Hofer K. Transmembrane domain-mediated orientation of receptor monomers in active VEGFR-2 dimers. FASEB J. 2010;24:32–38. doi: 10.1096/fj.09-132670. [DOI] [PubMed] [Google Scholar]
- Fearnley GW, Odell AF, Latham AM, Mughal NA, Bruns AF, Burgoyne NJ, et al. VEGF-A isoforms differentially regulate ATF-2-dependent VCAM-1 gene expression and endothelial-leukocyte interactions. Mol Biol Cell. 2014;25:2509–2521. doi: 10.1091/mbc.E14-05-0962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev. 2004;25:581–611. doi: 10.1210/er.2003-0027. [DOI] [PubMed] [Google Scholar]
- Ferrara N. VEGF-A: a critical regulator of blood vessel growth. Eur Cytokine Netw. 2009;20:158–163. doi: 10.1684/ecn.2009.0170. [DOI] [PubMed] [Google Scholar]
- Forsell P, Almqvist H, Hillertz P, Akerud T, Magdalena O, Eisele L, et al. The use of TrkA-PathHunter assay in high-throughput screening to identify compounds that affect nerve growth factor signalling. J Biomol Screen. 2012;18:659–669. doi: 10.1177/1087057113479401. [DOI] [PubMed] [Google Scholar]
- Gotink KJ, Verheul HM. Anti-angiogenic tyrosine kinase inhibitors: what is their mechanism of action? Angiogenesis. 2010;13:1–14. doi: 10.1007/s10456-009-9160-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog B, Pellet-Many C, Britton G, Hatzoulakis B, Zachery IC. VEGF binding to NRP1 is essential for VEGF stimulation of endothelial cell migration, complex formation between NRP1 and VEGFR2, and signaling via FAK Tyr407 phosphorylation. Mol Biol Cell. 2011;22:2766–2776. doi: 10.1091/mbc.E09-12-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill SJ, Baker JG, Rees S. Reporter gene systems for the study of G-protein-coupled receptors. Curr Opin Pharmacol. 2001;1:526–532. doi: 10.1016/s1471-4892(01)00091-1. [DOI] [PubMed] [Google Scholar]
- Kawamura H, Li X, Harper SJ, Bates DO, Claesson-Welsh L. Vascular endothelial growth factor (VEGF)-A165b is a weak in vitro agonist for VEGF receptor-2 due to lack of coreceptor binding and deficient regulation of kinase activity. Cancer Res. 2008;68:4683–4692. doi: 10.1158/0008-5472.CAN-07-6577. [DOI] [PubMed] [Google Scholar]
- Koch S, Tugues S, Li X, Gualandi L, Claesson-Welsh L. Signal transduction by vascular endothelial growth factor receptors. Biochem J. 2011;437:169–183. doi: 10.1042/BJ20110301. [DOI] [PubMed] [Google Scholar]
- Lanahan A, Zhang X, Fantin A, Zhuang Z, Rivera-Molnia F, Speichinger K, et al. The neuropilin 1 cytoplasmic domain is required for VEGF-A-dependent arteriogenesis. Dev Cell. 2013;25:156–168. doi: 10.1016/j.devcel.2013.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppänen VM, Prota AE, Jeltsch M, Anisimov A, Kalkkinen N, Strandin T, et al. Structural determinants of growth factor binding and specificity by VEGF receptor 2. Proc Natl Acad Sci U S A. 2010;107:2425–2430. doi: 10.1073/pnas.0914318107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macian F, Garcia-Rogriguez C, Rao A. Gene expression elicited by NFAT in the presence or absence of cooperative recruitment of fos and jun. EMBO J. 2000;19:4783–4795. doi: 10.1093/emboj/19.17.4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda ES, Imamura R, Amasaki Y, Arai K, Arai N. Signalling into the T-cell nucleus: NFAT regulation. Cell Signal. 1998;10:599–611. doi: 10.1016/s0898-6568(98)00019-9. [DOI] [PubMed] [Google Scholar]
- Matsumoto T, Mugishima H. Signal transduction via vascular endothelial growth factor (VEGF) receptors and their roles in atherogenesis. J Atheroscler Thromb. 2006;13:130–135. doi: 10.5551/jat.13.130. [DOI] [PubMed] [Google Scholar]
- Musumeci F, Radi M, Brullo C, Schenone S. Vascular endothelial growth factor (VEGF) receptors: drugs and new inhibitors. J Med Chem. 2012;55:10797–10822. doi: 10.1021/jm301085w. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rososki R. VEGF receptor protein-tyrosine kinases: structure and regulation. Biochem Biophys Res Commun. 2008;375:287–291. doi: 10.1016/j.bbrc.2008.07.121. [DOI] [PubMed] [Google Scholar]
- Shibuya M. Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: a crucial target for anti- and pro-angiogenic therapies. Genes Cancer. 2011;2:1097–1105. doi: 10.1177/1947601911423031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suehiro JI, Kanki Y, Makahira C, Schadler K, Miura M, Manabe Y, et al. Genome-wide approaches reveal functional vascular endothelial growth factor (VEGF)-inducible nuclear factor of activated T cells (NFAT) c1 binding to angiogenesis-related genes in endothelium. J Biol Chem. 2014;17:29044–29059. doi: 10.1074/jbc.M114.555235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voon DC, Subrata LS, Baltic S, Leu MP, Whiteway JM, Wong A, et al. Use of mRNA- and protein-destabilizing elements to develop a highly responsive reporter system. Nucleic Acids Res. 2005;33:e27. doi: 10.1093/nar/gni030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolard J, Wang WY, Bevan HS, Qiu Y, Morbidelli L, Pritchard-Jones RO, et al. VEGF165b, an inhibitory vascular endothelial growth factor splice variant: mechanism of action, in vivo effect on angiogenesis and endogenous protein expression. Cancer Res. 2004;64:7822–7835. doi: 10.1158/0008-5472.CAN-04-0934. [DOI] [PubMed] [Google Scholar]
- Woolard J, Bevan H, Harper S, Bates DO. Molecular diversity of VEGF-A as a regulator of its biological activity. Microcirculation. 2009;10:1–21. doi: 10.1080/10739680902997333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Guan H, He J, Zeng L, Yuan Z, Zhang W, et al. VEGF increases the proliferation capacity and eNOS/NO levels of endothelial progenitor cells through the caclineurin/NFAT signalling pathway. Cell Biol Int. 2014;36:21–27. doi: 10.1042/CBI20100670. [DOI] [PubMed] [Google Scholar]