

Abstract
Background and Purpose
Toll-like receptor 4 (TLR4) signalling contributes to inflammatory cardiovascular diseases, but its role in hypertension and the associated vascular damage is not known. We investigated whether TLR4 activation contributed to angiotensin II (AngII)-induced hypertension and the associated vascular structural, mechanical and functional alterations.
Experimental Approach
AngII was infused (1.44 mg·kg−1·day−1, s.c.) for 2 weeks in C57BL6 mice, treated with a neutralizing anti-TLR4 antibody or IgG (1 μg·day−1); systolic BP (SBP) and aortic cytokine levels were measured. Structural, mechanical and contractile properties of aortic and mesenteric arterial segments were measured with myography and histology. RT-PCR and Western blotting were used to analyse these tissues and cultured vascular smooth muscle cells (VSMC) from hypertensive rats (SHR).
Key Results
Aortic TLR4 mRNA levels were raised by AngII infusion. Anti-TLR4 antibody treatment of AngII-treated mice normalised: (i) increased SBP and TNF-α, IL-6 and CCL2 levels; (ii) vascular structural and mechanical changes; (iii) altered aortic phenylephrine- and ACh-induced responses; (iv) increased NOX-1 mRNA levels, superoxide anion production and NAD(P)H oxidase activity and effects of catalase, apocynin, ML-171 and Mito-TEMPO on vascular responses; and (v) reduced NO release and effects of L-NAME on phenylephrine-induced contraction. In VSMC, the MyD88 inhibitor ST-2825 reduced AngII-induced NAD(P)H oxidase activity. The TLR4 inhibitor CLI-095 reduced AngII-induced increased phospho-JNK1/2 and p65 NF-κB subunit nuclear protein expression.
Conclusions and Implications
TLR4 up-regulation by AngII contributed to the inflammation, endothelial dysfunction, vascular remodelling and stiffness associated with hypertension by mechanisms involving oxidative stress. MyD88-dependent activation and JNK/NF-κB signalling pathways participated in these alterations.
Tables of Links
| TARGETS |
|---|
| Catalytic receptorsa |
| TLR4, Toll-like receptor 4 |
| GPCRsb |
| AT1 receptor |
| Enzymesc |
| JNK1/2 |
| LIGANDS |
|---|
| AII, angiotensin II |
| CCL2 |
| IL-6 |
| L-NAME |
| NO |
| TNF-α |
These Tables list key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 2014) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (a,b,cAlexander et al., 2013a,b,c,).
Introduction
Structural alterations or vascular remodelling, increased stiffness and endothelial dysfunction are key features of hypertension and are associated to vascular smooth muscle cells (VSMC) reorganization, increased extracellular matrix (ECM) deposition and altered contractility or impaired endothelium-dependent relaxation (Mulvany, 2008; Briones et al., 2010; Rizzoni and Agabiti-Rosei, 2012; Ponticos and Smith, 2014). Both endothelial dysfunction and remodelling of small vessels are relevant prognostic factors of cardiovascular events in high-risk patients (de Ciuceis et al., 2007; Mathiassen et al., 2007; Münzel et al., 2008). Therefore, elucidating the mechanisms responsible for these alterations is of considerable interest.
It is recognized that chronic low-grade inflammation has a crucial role in the pathogenesis of hypertension (Savoia and Schiffrin, 2006) and the role of adaptive immunity in the pathogenesis of hypertension has been recently reviewed (see de Ciuceis et al., 2014; Harrison, 2014; Schiffrin, 2014). Angiotensin II (AngII) is now considered a proinflammatory mediator in hypertension through mechanisms involving production of reactive oxygen species (ROS) that contribute to the observed vascular alterations. (Touyz, 2005; Briones et al., 2009; Briones and Touyz, 2010). However, how the inflammation associated with this pathology is initiated has not been fully elucidated.
The Toll-like receptor 4 (TLR4) is a member of the family of pattern recognition receptors and plays an important role mediating inflammatory response (Carpenter and O'Neill, 2009; McCarthy et al., 2014). Apart from its expression in immune cells, TLR4 is expressed in cells of the cardiovascular system and signalling through TLR4 has been proposed to contribute to vascular inflammatory pathologies such as atherosclerosis, diabetes or the metabolic syndrome (den Dekker et al., 2010; Jialal et al., 2014). Moreover, we and others have recently described an emerging role for TLR4 in hypertension. Thus, TLR4 expression is increased in hypertension and this expression seems to be associated with the increased BP (Eissler et al., 2011; Bomfim et al., 2012; Marketou et al., 2012; de Batista et al., 2014; Sollinger et al., 2014). TLR4 is activated by lipopolysaccharide as well as by non-infectious host-endogenous compounds (damage-associated molecular pattern molecules, DAMPs), such as heat shock proteins (HSP), fibrinogen and tenascin-C, many of which are present in hypertension as a consequence of chronic cell injury and death (Tsan and Gao, 2004; McCarthy et al., 2014). Furthermore, AngII up-regulates the expression of TLR4, an effect that contributes to the proinflammatory effects of this factor (Wolf et al., 2006; Ji et al., 2009; Yuen et al., 2012). In this context, we have already described that, through AT1 receptors, AngII contributes to the up-regulation of vascular TLR4 in spontaneously hypertensive rats (SHR; de Batista et al., 2014). After activation, TLR4 signalling might occur via MyD88 (myeloid differentiation factor 88)-dependent or -independent pathways that ultimately activate NF-κB, a transcription factor for several proinflammatory genes (Carpenter and O'Neill, 2009; Kawai and Akira, 2010).
Recent studies have demonstrated that TLR4 is involved in the vascular dysfunction observed in SHR and L-NAME-induced hypertension (de Batista et al., 2014; Sollinger et al., 2014). While the imbalance between Th1 effector and the anti-inflammatory Treg lymphocytes in the progression of vascular remodelling in hypertension has been proposed (de Ciuceis et al., 2014; Harrison, 2014; Schiffrin, 2014), to the best of our knowledge, the involvement of TLR4 in the structural and mechanical alterations observed in this pathology has not still been explored. Therefore, we investigated here, if TLR4 contributed to AngII-induced hypertension and the associated vascular structural, mechanical and functional alterations, focusing on the role of oxidative stress in these alterations. Our findings have provided new evidence that TLR4 mediated AngII-induced hypertension and inflammation as well as the vascular functional alterations and remodelling associated with this pathology. Our results also suggested that the JNK/NF-κB signalling pathways and increased oxidative stress from NAD(P)H oxidase and mitochondria may be involved in these vascular effects, through MyD88-dependent activation.
Methods
Animal models
All animal care and experimental procedures complied with the guidelines for ethical care of experimental animals of the European Community, the current Spanish and European laws (RD 223/88 MAPA and 609/86) and were approved by the Ethical Committee of Research of the Universidad Autónoma de Madrid, Spain (CEI-UAM 31–759). The ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010) have been followed in this paper. A total of 65 mice and 9 SHR were used in the experiments described here.
Male C57BL6 mice (26.8 ± 0.3 g; obtained from the Animal Facility of the Facultad de Medicina of the Universidad Autónoma of Madrid, reg. n. EX021U) were allocated randomly to the following experimental groups: (i) control mice treated with a non-specific IgG (IgG2a, 1 μg·day−1, saline-diluted, i.p., Santa Cruz Biotechnology Inc., Dallas, TX, USA); (ii) treated with AngII (1.44 mg·kg−1·day−1 for 2 weeks, s.c., by Alzet osmotic minipumps, Alza Corp., Cupertino, CA, USA) and treated with IgG; (iii) treated with AngII and treated with the neutralizing anti-TLR4 antibody (rat monoclonal IgG2a, 1 μg·day−1 saline-diluted, i.p., Santa Cruz Biotechnology Inc.). For some experiments, control mice treated with anti-TLR4 antibody were also studied. Such treatment did not modify either systolic BP (SPB) or structural and mechanical parameters of mesenteric arteries or aortic vascular responses (results not shown). Treatments started 24 h prior to AngII infusion. SBP was measured by tail-cuff plethysmography. In addition, VSMC obtained from adult (5 month old) SHR (329 ± 8 g; from the same Animal Facility as the mice) were also used.
Tissue preparations
Animals were killed by exposure to CO2 and the aorta and first-order branches of the mesenteric artery were dissected free of fat and connective tissue and placed in cold (4°C) Krebs–Henseleit solution (KHS, in mM: 115 NaCl, 25 NaHCO3, 4.7 KCl, 1.2 MgSO4.7H2O, 2.5 CaCl2, 1.2 KH2PO4, 11.1 glucose and 0.01 Na2EDTA) bubbled with a 95% O2 to 5% CO2 mixture (pH = 7.4). The analyses of vascular structure and function as well as NO production were made on the same day. For assays of superoxide anion (O2−) production, segments (5 mm long) were placed in KHS containing 30% sucrose for 20 min, transferred to a cryomold containing Tissue Tek OCT (Sakura Finetek Europe B.V., Alphen aan den Rijn, The Netherlands) embedding medium and frozen in liquid nitrogen. For immunofluorescence and histological studies, arterial segments (5 mm long) were fixed with 4% paraformaldehyde (PFA; in 0.2 M phosphate buffer, pH = 7.2–7.4) for 1 h and washed in three changes of PBS. After washing, arterial segments were frozen as described above. Segments (5-7 mm long) used for gene expression studies were immediately frozen in liquid nitrogen. Tissues were kept at −70°C until the day of the experiments. The hearts were removed to assess cardiac hypertrophy. For this, the ratio between the left ventricular wet weight and the length of the tibia was calculated.
Cell culture
Primary cultures of VSMC were obtained from thoracic aortas from SHR using the explant method (Martín et al., 2012). Cells were identified as VSMC by morphological and growth characteristics and by positive immunocytochemical staining with a specific monoclonal anti-α-actin antibody (Sigma Chemical Co., St Louis, MO, USA). For experiments, cells from passages 2 to 5 were rendered quiescent by incubation in DMEM containing 0.2% FBS for 24 h. Cells (about 2x 105 per well) were stimulated with 100 nM AngII (for the times indicated in the Results section), with or without pretreatment for 1 h with the MyD88 inhibitor ST-2825 (20 μM) or the TLR4 inhibitor CLI-095 (1 μM).
Pressure myography
The structural and mechanical properties of small mesenteric arteries were studied with a pressure myograph (Danish Myo Tech, Model P100, J.P. Trading I/S, Aarhus, Denmark), as described by Briones et al., (2009). Briefly, segments of vessels (5 mm long) were placed in Ca2+-free KHS (omitting calcium and adding 1 mM EGTA) and a pressure-diameter curve was obtained by increasing intraluminal pressure in 20 mmHg steps from 3 to 120 mmHg. Internal and external diameters were continuously measured under passive conditions (Di0Ca, De0Ca) for 3 min at each intraluminal pressure. Finally, arteries were set to 45 mmHg in Ca2+-free KHS and then pressure-fixed with 4% phosphate-buffered PFA at 37°C for 60 min and kept at 4°C for confocal microscopy studies.
From internal and external diameter measurements in passive conditions, the following structural and mechanical parameters were calculated:
Incremental distensibility represents the percentage of change in the arterial internal diameter for each mmHg change in intraluminal pressure, and was calculated according to the formula:
Circumferential wall strain (ε) = (Di0Ca − D00Ca)/D00Ca, where D00Ca is the internal diameter at 3 mmHg and Di0Ca is the observed internal diameter for a given intravascular pressure, both measured in 0 Ca2+ medium.
Circumferential wall stress (σ) = (P × Di0Ca)/(2WT), where P is the intraluminal pressure (1 mmHg = 1.334 × 103 dynes·cm−2) and WT is the wall thickness at each intraluminal pressure measured in 0 Ca2+-KHS.
Arterial stiffness independent of geometry is determined by Young's elastic modulus (E = stress/strain). The stress–strain relationship is non-linear; therefore, it is more appropriate to obtain a tangential or incremental elastic modulus (Einc) by determining the slope of the stress–strain curve (Einc = δσ/δε). Einc was obtained by fitting the stress–strain data from each animal to an exponential curve using the equation: σ = σorigeβε, where σorig is the stress at the original diameter (diameter at 3 mmHg). Taking derivatives on the equation presented earlier, we see that Einc = βσ. An increase in β implies an increase in Einc, which means an increase in stiffness.
Confocal microscopy study of nuclei distribution
Quantification of nuclei distribution was analysed in pressure-fixed intact mesenteric segments, as described (Arribas et al., 1997). Briefly, arterial segments were incubated with the nuclear dye Hoechst 33342 (Sigma Chemical Co.; 0.01 mg·mL−1) and visualized with a Leica TCS SP2 confocal system (Leica Microsystems, Wetzlar, Germany; ×40 objective, zoom ×3.5). Metamorph Image Analysis Software (Universal Imaging, Molecular Devices Corp., Downingtown, PA, USA) was used for quantification.
To allow comparisons, the following calculations were performed on the basis of 1mm long segments: artery volume (mm3) [volume = wall CSA (mm2) × 1 mm]; total number of adventitial and smooth muscle cells (cell n = n of nuclei per stack × n of stacks per artery volume); total number of endothelial cells [calculated per luminal surface of 1mm long artery segments; luminal surface area = 2π × diameter (mm) × 1 mm/2].
Organization of internal elastic lamina (IEL)
The elastin organization within the IEL was studied in segments of small mesenteric arteries, using fluorescence confocal microscopy based on the autofluorescent properties of elastin (Ex 488 nm and Em 500–560 nm), as previously described (Briones et al., 2003). Briefly, intact pressure-fixed segments were visualized with a Leica TCS SP2 confocal system. From each stack of serial images, individual projections of the IEL were reconstructed, and total fenestrae number and fenestrae area were measured using Metamorph Image Analysis Software.
Collagen determination
Mesenteric arteries were cut into 5 μm sections using a cryostat. Collagen was quantified in Picrosirius Red-stained sections [0.1% (wt·vol−1), Sirius Red 3FB in saturated aqueous picric acid for 30 min with gentle agitation]. Three to four sections from each animal were analysed under microscopy transmitted light (Leica DM 2000, Leica Microsystems) using an Image System Analysis (Leica LAS Image Analysis, Leica Microsystems). The area of collagen in the media layer was identified after excluding perivascular fibrosis of the vessel as the ratio of collagen deposition to the total media area.
Histological analysis
Cross sections (14 μm) from aortas were stained with haematoxylin-eosin. All images were acquired at room temperature using a microscope (Leica DM 2000). Morphometric determinations of the lumen and vessel areas were performed by using Metamorph Image Analysis Software. All microscopic images of the sections were traced for the calculations of the areas. To determine the luminal area, the cross-sectional area enclosed by the IEL was corrected to a circle by applying the form factor l2/4π to the measurement of the IEL, where l is the length of the lamina. Vessel area was determined by the cross-sectional area enclosed by the external elastic lamina corrected to a circle, applying the same form factor (l2/4π) to the measurement of the external elastic lamina. The media area was calculated as the difference between the corrected vessel and luminal areas. Internal and external diameters were calculated from luminal and vessel areas respectively. This method avoids miscalculations of areas caused by eventual collapse of the immersion-fixed arteries.
Vascular function
Reactivity of mice aorta was studied in a wire myograph. After a 30 min equilibration period in oxygenated KHS, arterial segments (2 mm long) were stretched to their optimal lumen diameter for active tension development. Segment contractility was then tested by an initial exposure to a high-K+ solution (K+-KHS, 120 mM). The presence of endothelium was determined by the ability of 10 μM ACh to relax arteries precontracted with phenylephrine at approximately 50% the K+-KHS contraction. Afterwards, a single concentration-response curve to ACh (1 nM-10 μM), phenylephrine (1 nM-10 μM) and diethylamine NONOate (DEA-NO, 3 nM-10 μM) was performed. The effects of the hydrogen peroxide metabolising enzyme, catalase (1000 U·mL−1), the NAD(P)H oxidase inhibitor apocynin (0.3 mM), the selective NOX-1 inhibitor ML-171 (2-acetylphenothiazine, 0.5 μM), the mitochondrion-targeted SOD2 mimetic Mito-TEMPO (0.5 μM) or the non-selective inhibitor of endothelial NOS, L-NAME (100 μM) on phenylephrine- or ACh-induced responses were analysed. These drugs were incubated with the arterial segment 30 min prior to addition of phenylephrine.
Immunofluorescence
TLR4 in aorta was immunolocalized as described (de Batista et al., 2014). Briefly, frozen transverse sections (14 μm) were incubated with a polyclonal antibody against TLR4 (1:100, Santa Cruz Biotechnology Inc.) in PBS containing 2% BSA for 1 h at 37°C in a humidified chamber. After washing, rings were incubated with the secondary antibody goat anti-rat (1:200), IgG labelled with Alexa fluor-546 dye (Molecular Probes Inc., Eugene, OR, USA), for 1 h at 37°C in a humid box. Nuclei were stained with 0.01 mg·mL−1 Hoechst 33342. Immunofluorescent signals were viewed using an inverted Leica TCS SP2 confocal laser-scanning microscope with oil immersion lens. The specificity of the immunostaining was evaluated by omission of the primary antibody and processing as described above. Under these conditions, no staining was observed in the vessel wall.
Immunofluorescence was also used to detect p65 NF-κB subunit nuclear protein expression in cultured VSMC stimulated with AngII (100 nM, 45 min) after treatment or not with CLI-095 (1 μM, 1h), as described (Pérez-Girón et al., 2014). Cells were incubated overnight at 4°C with a rabbit polyclonal primary antibody against p65 (1:250, Santa Cruz Biotechnology). After washing, cells were incubated with a FITC-conjugated goat anti-rabbit secondary antibody (1:200, Molecular Probes, Life Technologies Ltd, Paisley, UK). Then, cells were counterstained for 15 min with DAPI (1:10 000, Invitrogen Life Technologies) and visualized under a fluorescence microscope (Zeiss, Axioplan 2, Carl Zeiss Microscopy, LLC, Thornwood, NY, USA). At least three independent experiments were performed.
Quantitative PCR real-time (qRT-PCR) assay
mRNA levels were determined in homogenates of mouse aorta. Total RNA was obtained using TRI Reagent (Sigma Chemical Co.) according to the manufacturer's recommendations. A total of 1 μg of DNAse I treated RNA was reverse-transcribed using the High Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA, USA) in a final volume of 10 μL. qPCR was performed using the fluorescent dye SyBRGreen (iTaq FAST SyBRGreen Supermix with ROX, Bio-Rad, Hercules, CA, USA). β2-microglobulin (Rn00560865_m1, Applied Biosystems) or 36B4 were used as internal controls. All qPCRs were performed in duplicate. Primer sequences are: for TLR4, FW: TGTGCCTTCAAAACATGACTGG, RV: CTCCCAAGATCAACCGATGG; p22phox, FW: ATCTGTCTGCTGGAGTAT, RV: CGTAGTAATTCCTGGTGAG; NOX-1, FW: CAGCGTGCCGACAACAAG; RV: CCAGCCAGTGAGGAAGAGAC; TNF-α, FW: CCACGCTCTTCTGTCTACTG, RV: TGAGGGTCTGGGCCATAGA; CCL2, FW: CATCCACGTGTTGGCTCA, RV: GATCATCTTGCTGGTGAATGAGT; IL-6, FW: TGATGGATGCTACCAAACTGG, RV: TTCATGTACTCCAGGTAGCTATGG; 36B4, FW: AGATGCAGCAGATCCGCAT, RV: GTTCTTGCCCATCAGCACC. qRT-PCR was carried out in an ABI PRISM 7000 Sequence Detection System (Applied Biosystems) using the following conditions: 2 min 50°C, 10 min 95°C and 40 cycles: 15 s 95°C, 1 min 60°C. At the end of the PCR, melting curve analysis was performed to show PCR product specificity. To calculate the relative index of gene expression, we employed the 2−ΔΔCt method using untreated samples as calibrator.
Measurement of O2− production
The oxidative fluorescent dye dihydroethidium (DHE) was used to evaluate production of O2− in situ, as previously described (Martínez-Revelles et al., 2013). Briefly, aortic sections were incubated in buffer containing DHE (2 μM, 30 min, 37°C) and then viewed by a fluorescent laser-scanning confocal microscope (Leica TCS SP2; Ex 561 and Em 610 nm), using the same imaging settings in each case. As a negative control, parallel sections were pre-incubated with tempol (100 μM). In these conditions, no staining was observed in the vascular wall. O2− production was quantitatively analysed with MetaMorph Image Analysis Software.
NAD(P)H oxidase activity
The lucigenin-enhanced chemiluminescence assay was used to determine the superoxide anion generated by NAD(P)H oxidase activity, as described (Pérez-Girón et al., 2014). Arterial samples (5 mm long) were homogenized in 125 μL of a lysis buffer (in mM: 50 KH2PO4, 1 EGTA, 150 sucrose, pH = 7.4). The reaction was started by the addition of NAD(P)H (0.1 mM) to the suspension-containing sample, lucigenin (5 μM) and assay phosphate buffer. The luminescence was measured in a plate luminometer (Auto-Lumat LB 953, Berthold Technologies GmbH & Co. KG, Bad Wildbad, Germany). Buffer blank was subtracted from each reading. Activity was expressed as relative light units per mg protein. Samples from control+IgG-treated mice or untreated cells were used as controls, and variations of activity were calculated relative to control values.
NO release
NO production was determined in aortic segments with the fluorescent probe 4,5-diaminofluorescein (DAF-2), as described (Martínez-Revelles et al., 2013). Briefly, aortic segments (5 mm long) were incubated with 2 μM DAF-2 (in 300 μL HEPES buffer, pH 7.4) for 45 min. Afterwards, basal and ACh-stimulated NO release were measured using a spectrofluorimeter (FLUOstar OPTIMA, BMG Labtech, Ortenberg, Germany). To avoid variations in fluorescence among the different days, all experimental conditions were measured each day. The amount of NO released was expressed as arbitrary units per mg tissue.
Western blot analysis
Protein expression was determined in whole cell lysates (20 μg protein) or nuclear extracts (15 μg protein) by Western blot, as described (Pérez-Girón et al., 2014). Proteins were separated by 15% SDS-PAGE and then transferred to polyvinyl difluoride membranes overnight. Membranes were incubated with rabbit polyclonal antibodies for p-JNK1/2 (1:1000; Cell Signaling Technology Inc, Danvers, MA, USA) or p65 (1:1000). Immunoreactive bands were visualized using fluorescent secondary antibodies from rabbit (1:5000; Bio-Rad Laboratories) and ECF Plus Western blot analysis system (GE Healthcare, Little Chalfont, UK). Signals on the immunoblot were quantified using the Typhoon 9210 quantification software (GE Healthcare). The same membrane was used to determine JNK (monoclonal antibody anti-JNK, 1:1000; Cell Signaling Technology Inc.) in cellular lysates and histone-3 (rabbit polyclonal antibody anti-histone-3, 1:500; Santa Cruz Biotechnology) in nuclear extracts. Results are expressed as the ratio between signals on the immunoblot corresponding to the different proteins and total JNK or histone-3 in the same preparation. To compare results for protein expression within the same experiment and with others, we assigned a value of 1 in each gel to the expression of unstimulated control cells and used that value to calculate the relative density of other bands from the same gel.
Data analysis
Vasoconstrictor responses were expressed as a percentage of the tone generated by K+-KHS. Vasodilator responses were expressed as the percentage of the previous tone induced by phenylephrine in each case. The maximum response (Emax) and pD2 values were calculated by non-linear regression analysis of each individual concentration-response curve using GraphPad Prism Software (San Diego, CA, USA). To compare the effect of L-NAME on the response to phenylephrine in segments from the three groups, results were expressed as the differences of areas under the concentration-response curves in the control and experimental situations. AUCs were calculated from the individual concentration-response curve plots using GraphPad Prism Software. Differences between mean AUC values were expressed as the percentage of the AUC of the corresponding control situation. All values are expressed as mean ± SEM of the number of animals or different cultures (each obtained from three different animals) used. Results were analysed by using unpaired Student's t-test or two-way anova followed by Bonferroni's post hoc test or Mann–Whitney's non-parametric test, using GraphPad Prism Software. P < 0.05 was considered significant.
Materials
l-Phenylephrine hydrochloride, ACh chloride, catalase, apocynin, L-NAME and ML-171 were purchased from Sigma Chemical Co, mito-TEMPO from Santa Cruz Biotechnology Inc., DHE from Invitrogen, CLI-095 from Invivogen (San Diego, CA, USA) and ST-2825 from MedChem Express (Princeton, NJ, USA). All drugs were dissolved in distilled water, except for CLI-095 and ST-2825, which were dissolved in DMSO; 10 μL of DMSO did not have any effect on VSMC.
Results
TLR4 mediates AngII-induced hypertension and inflammation
TLR4 mRNA levels were higher in aortic segments from AngII-infused mice, compared with controls (Figure 1A); TLR4 expression was increased in all layers of the vascular wall (Figure 1A). Treatment with the anti-TLR4 antibody partly prevented the increased SBP observed in AngII-treated mice (Figure 1B), although neither body weight (data not shown) nor left ventricular hypertrophy were affected (Figure 1C). Aortic segments from AngII-treated mice showed increased levels of mRNA for TNF-α, IL-6 and CCL2 and these changes were prevented by treatment with anti-TLR4 antibody (Figure 1D).
Figure 1.

Increased TLR4 contributes to hypertension and inflammation in AngII-treated mice. (A) TLR4 mRNA levels and representative fluorescent confocal photomicrographs (×40 objective) of TLR4 immunolocalization in aortic segments from mice treated with a non-specific IgG and from mice treated with AngII plus a non-specific IgG. Image size: 375 × 375 μm. Positive immunostaining is indicated by arrows and the insets are magnified images of indicated areas. (B) SBP, (C) left ventricular hypertrophy and (D) levels of mRNA for TNF-α, IL-6 and CCL2 in mice treated with a non-specific IgG, with AngII plus a non-specific IgG or with AngII plus anti-TLR4 antibody. *P < 0.05 versus control + IgG, #P < 0.05 versus AngII + IgG. n = 6–16.
TLR4 mediates vascular remodelling and mechanical alterations
In small mesenteric arteries from AngII-treated mice, vessel and lumen diameters were reduced, while wall thickness and wall : lumen ratio were increased when compared with controls (Figure 2A-D). Treatment of AngII-treated mice with anti-TLR4 antibody improved these structural parameters (Figure 2A-D); the antibody treatment also improved the reduced number of smooth muscle and endothelial cells observed in mesenteric arteries from AngII-treated mice (Figure 2E).
Figure 2.

TLR4 inhibition improves structural alterations in mesenteric resistance arteries from AngII-treated mice. Vessel and lumen diameter (A,B), wall thickness (C), wall : lumen ratio (D) and total number of adventitial, smooth muscle and endothelial cells (E) in small mesenteric arteries from mice treated with a non-specific IgG, with AngII plus a non-specific IgG or with AngII plus anti-TLR4 antibody. *P < 0.05 versus control + IgG, #P < 0.05 versus AngII + IgG. n = 6–14.
Infusion of AngII reduced incremental distensibility (Figure 3A) and increased vessel stiffness of mesenteric arteries, as shown by the leftward shift of the stress–strain relationship and the larger β-value (Figure 3B), effects that were prevented by anti-TLR4 antibody treatment (Figure 3A,B). Infusion of AngII also reduced the number and size of fenestrae in the IEL (Figure 3C) and increased collagen deposition (Figure 3D). Anti-TLR4 treatment improved fenestrae size and decreased collagen deposition (Figure 3C,D). In aortae from AngII-treated mice, the media thickness, media : lumen ratio and cross-sectional area were increased, compared with controls. Treatment with anti-TLR4 antibody prevented these structural alterations (Figure 4).
Figure 3.

TLR4 inhibition improves mechanical alterations in mesenteric resistance arteries from AngII-treated mice. Incremental distensibility (A) and stress–strain relationship and β values (B) in mesenteric arteries from mice treated with a non-specific IgG, with AngII plus a non-specific IgG or with AngII plus anti-TLR4 antibody. (C) Representative confocal projections and quantification of fenestra area and total number of fenestra of the internal elastic lamina of mesenteric arteries from the three groups of mice. Projections were obtained from serial optical sections captured with a fluorescence confocal microscope (×40 objective, zoom ×7.10); image size: 52.8 × 52.8 μm. (D) Representative images and quantitative analysis of collagen content in the media layer from transverse sections of mesenteric arteries. Images were captured with a light microscope (×40 objective); image size: 311 × 233 μm. *P < 0.05 versus control + IgG, #P < 0.05 versus AngII + IgG. n = 6–14.
Figure 4.
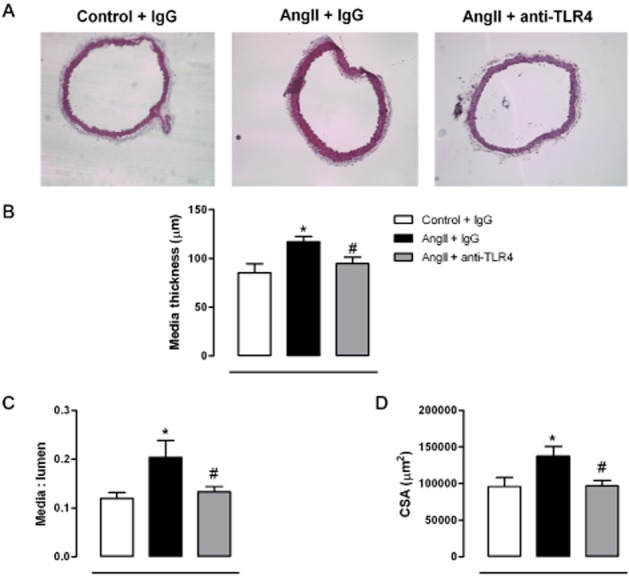
TLR4 inhibition improves structural alterations in aorta from AngII-treated mice. (A) Representative photographs of sections of aorta stained with haematoxylin-eosin, from mice treated with a non-specific IgG, with AngII plus a non-specific IgG or with AngII plus anti-TLR4 antibody. Images were captured with a light microscope (×10 objective); image size: 2.37 × 1.88 mm. (B) Media thickness, (C) media : lumen ratio and (D) cross-sectional area (CSA) in aorta from the three groups of mice. *P < 0.05 versus control + IgG, #P < 0.05 versus AngII + IgG. n = 7–8.
TLR4 mediates alterations on vascular function
Phenylephrine induced contractile responses in aortic segments and these contractions were greater in segments from AngII-treated mice than those from control mice. These responses were reduced by anti-TLR4 antibody treatment (Figure 5A). Antibody treatment also improved the reduced ACh-induced endothelium-dependent vasodilatation observed in aorta from AngII-treated mice (Figure 5B). However, endothelium-independent relaxation induced by DEA-NO was similar in all three groups (Figure 5C).
Figure 5.
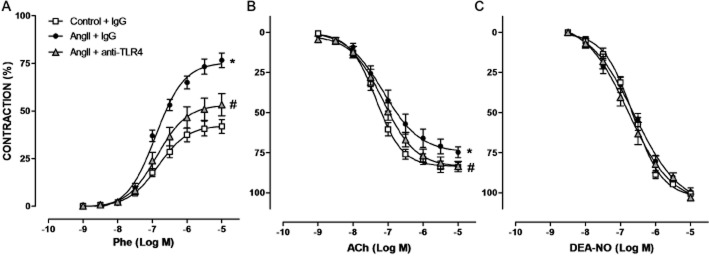
TLR4 inhibition improves vasoconstrictor and vasodilator responses in aorta from AngII-treated mice. Concentration-response curves to phenylephrine (Phe) (A), ACh (B) and diethylamine NONOate (DEA-NO) (C) in aorta from mice treated with a non-specific IgG, with AngII plus a non-specific IgG or with AngII plus anti-TLR4 antibody. *P < 0.05 versus control + IgG, #P < 0.05 versus AngII + IgG. n = 12–17.
TLR4 mediates vascular ROS production
The increased ROS production induced by AngII may contribute to the vascular alterations occurring in hypertension (Briones and Touyz, 2010; Virdis et al., 2011). As expected, O2− production was greater in the aortic wall from AngII-treated mice than control mice and this difference was particularly evident in adventitia and media layers (Figure 6A). Aortic NAD(P)H oxidase activity (Figure 6B) and p22phox and NOX-1 mRNA levels (Figure 6C) were also greater in samples from AngII-treated mice than in those from control mice. Anti-TLR4 antibody treatment prevented the increased O2− production, NAD(P)H oxidase activity and NOX-1 mRNA levels, without modifying p22phox levels (Figure 6).
Figure 6.

TLR4 inhibition reduces the increased oxidative stress in aorta from AngII-treated mice. (A) Representative fluorescent confocal photomicrographs and quantification in the media and in the adventitia of vascular superoxide anion production in aortic segments from mice treated with a non-specific IgG, with AngII plus a non-specific IgG or with AngII plus anti-TLR4 antibody. Vessels were labelled with the oxidative dye DHE and viewed by using a fluorescence confocal microscope (×40 objective). Image size: 375 × 375 μm. (B) Basal NAD(P)H oxidase activity and (C) NOX-1 and p22phox mRNA levels in aorta from the three groups of mice. *P < 0.05 versus control + IgG, #P < 0.05 versus AngII + IgG. n = 5–8.
We have previously shown that AngII increased oxidative stress in VSMC through TLR4 activation (de Batista et al., 2014). Here, we observed the NAD(P)H oxidase activity in VSMC induced by exposure to AngII for 1h was reduced by the MyD88 inhibitor ST-2825 (Figure 7A). We have also reported that AngII increases oxidative stress in VSMC through JNK1/2 MAPK and NF-κB (Pérez-Girón et al., 2014). The phosphorylation of JNK1/2, induced by exposure to AngII for 10 min, was reduced by the TLR4 inhibitor CLI-095 (Figure 7B). In addition, the increased nuclear expression and cytosol to the nucleus translocation of the p65 NF-κB subunit induced by AngII (45 min incubation) was also reduced by CLI-095 (Figure 7C,D).
Figure 7.

AngII activates MyD88, JNK and NF-κB in VSMCs. (A) Effect of ST-2825 (20 μM, 1 h) on AngII (100 nM, 1 h)-induced NADPH oxidase activity. (B) Effect of CLI-095 (1 μM, 1 h) on AngII (10 min)-induced p-JNK1/2 protein expression; a representative blot is shown above. (C) Effect of CLI-095 on nuclear p65 NF-κB protein expression induced by AngII (45 min); representative blots of the cytosolic (Cy) and nuclear (Nu) expression are shown above; Histone-3 (H3) expressions are also shown to validate the cellular fractioning. (D) Representative photomicrographs of p65 NF-κB immunofluorescence (red) in VSMC in control and after incubation with AngII in the absence and the presence of CLI-095. The insets are magnified images of indicated areas. Images were captured with a fluorescence microscope (×20 objective); image size: 444.16 × 332.8 μm. *P < 0.05 versus control, #P < 0.05 versus AngII. n = 4–5.
TLR4 mediates the contribution of oxidative stress to altered vascular function
We then assessed whether the increased ROS production associated with TLR4 up-regulation contributed to the hypertension-associated vascular dysfunction. Catalase, apocynin, ML-171 and Mito-TEMPO did not affect phenylephrine-induced contraction in arterial segments taken from control mice, but they reduced those responses in samples from AngII-treated mice. Treatment with the anti-TLR4 antibody abolished the inhibitory effect of the four antioxidants, in AngII-treated mice (Figure 8). Similarly, catalase, apocynin, ML-171 and Mito-TEMPO increased ACh-induced relaxation only in AngII-treated mice and anti-TLR4 antibody treatment abolished these effects (Figure 9). Altogether, these results suggest that TLR4 increased oxidative stress and that this increase contributed to the impaired vascular function observed in hypertensive mice.
Figure 8.

TLR4 inhibition reduces the involvement of oxidative stress on vasoconstrictor responses in aorta from AngII-treated mice. Effect of catalase (1000 U·mL−1), apocynin (0.3 mM), ML-171 (0.5 μM) and Mito-TEMPO (0.5 μM) on the concentration-response curve to phenylephrine (Phe) in aortic segments from mice treated with a non-specific IgG, with AngII plus a non-specific IgG or with AngII plus anti-TLR4 antibody. *P < 0.05. n = 5–11.
Figure 9.

TLR4 inhibition reduces the involvement of oxidative stress on vasodilator responses in aorta from AngII-treated mice. Effect of catalase (1000 U·mL−1), apocynin (0.3 mM), ML-171 (0.5 μM) and Mito-TEMPO (0.5 μM) on the concentration-response curve to ACh in aortic segments from mice treated with a non-specific IgG, with AngII plus a non-specific IgG or with AngII plus anti-TLR4 antibody. *P < 0.05. n = 5–11.
TLR4 reduces NO release and its participation in vasoconstriction
Then, we evaluated the effects of the anti-TLR4 antibody on NO release and its contribution to constrictor responses in vascular tissue. In aortae from AngII-treated mice, we observed a reduced NO release that was normalized by anti-TLR4 treatment (Figure 10A). Furthermore, the NOS inhibitor L-NAME shifted the concentration-response curve to phenylephrine to the left and this shift was greater in segments from control than in those from AngII-treated mice. Treatment with the anti-TLR4 antibody reversed this effect (Figure 10B and Table 1), indicating that the diminished involvement of NO in the contractile response to phenylephrine after AngII infusion was restored by TLR4 blockade.
Figure 10.

TLR4 inhibition increases NO production and its participation on vasoconstrictor responses in AngII-treated mice. (A) Quantification of NO release in aortic segments from mice treated with a non-specific IgG, with AngII plus a non-specific IgG or with AngII plus anti-TLR4 antibody. (B) Effect of L-NAME (100 μM) on the concentration-response curve to phenylephrine (Phe) in aortic segments from the three groups of mice. Differences between the AUCs (dAUC) of the concentration-response curves to Phe in the presence and the absence of L-NAME are also shown. *P < 0.05 versus control or versus control + IgG, #P < 0.05 versus AngII + IgG. n = 5–9.
Table 1.
Effect of L-NAME on maximum response (Emax) and pD2 values of phenylephrine responses in aortic segments from mice treated with a non-specific IgG, with AngII plus a non-specific IgG or with AngII plus anti-TLR4 antibody
| Control | L-NAME | Fold increase | ||
|---|---|---|---|---|
| Control + IgG | Emax (%) | 30.1 ± 2.6 | 85.7 ± 6.4* | 2.84 ± 0.21 |
| pD2 | 6.53 ± 0.17 | 7.12 ± 0.06* | – | |
| AngII + IgG | Emax (%) | 89.6 ± 4.2# | 148.2 ± 17.7*# | 1.65 ± 0.20# |
| pD2 | 7.02 ± 0.04# | 7.30 ± 0.07* | – | |
| AngII + anti-TLR4 | Emax (%) | 44.8 ± 8.7$ | 103.7 ± 9.8*$ | 2.32 ± 0.22$ |
| pD2 | 6.72 ± 0.08$ | 7.02 ± 0.07*$ | – |
The enhancing effect of L-NAME in Emax is expressed as fold increase over control values.
P < 0.05 versus control.
P < 0.05 versus control + IgG mice.
P < 0.05 versus AngII + IgG mice; n = 5–9.
Discussion
The results of the present study demonstrated, for the first time, that up-regulation of TLR4 by AngII participates in the pathogenesis of hypertension by affecting, not only the function, but also the structure and the mechanical properties of the vasculature by mechanisms likely to involve oxidative stress. MyD88-dependent mechanisms and the JNK/NF-κB signalling pathways might also contribute to these effects.
Hypertension has been described as an inflammatory disease in which an inadequate activation of the immune system may contribute to its pathophysiology (Savoia and Schiffrin, 2006; de Ciuceis et al., 2014; Harrison, 2014; McCarthy et al., 2014; Schiffrin, 2014; Virdis et al., 2014) and the role of adaptive immunity as contributor to hypertension has been widely reported. For instance, subsets of T-lymphocytes are involved in the development of hypertension and vascular remodelling in different hypertension models (de Ciuceis et al., 2014; Harrison, 2014; Schiffrin, 2014; Virdis et al., 2014). Moreover, in the last few years, activation of TLR4 has become a new research focus because this receptor was up-regulated in hypertension, contributing to its occurrence (Eissler et al., 2011; Bomfim et al., 2012; de Batista et al., 2014; Sollinger et al., 2014). An increased expression of TLR4 induced by AngII is one of the proposed mechanisms explaining the involvement of this receptor in hypertension. Thus, AngII would induce inflammatory responses via increased TLR4 activation. Consistent with this proposal, in VSMC AngII, through AT1 receptors, activated TLR4, leading to ERK1/2 and NF-κB activation and subsequent inflammatory responses (Ji et al., 2009). Moreover, osteocalcin-induced AngII release activated the PKCδ/TLR4/ROS/COX-2 signalling cascade that contributed to the transformation of vascular fibroblasts to myofibroblasts (Yuen et al., 2012). Recently, we found that treatment with an AT1 receptor antagonist reduced the increased TLR4 levels in SHR (de Batista et al., 2014).
In the present set of experiments, AngII infusion increased the expression of TLR4 in the aorta. More importantly, anti-TLR4 antibody treatment prevented the increased SBP and proinflammatory cytokine levels in aortic tissue, confirming the role of TLR4 in the AngII-induced hypertension and associated inflammation. In SHR, such treatment had already been shown to reduce SBP (Bomfim et al., 2012; de Batista et al., 2014) and the increased vascular proinflammatory cytokine levels (Bomfim et al., 2015). Further experiments are necessary to ascertain the exact mechanisms by which AngII up-regulates the TLR4 signalling pathway in our hypertension model.
The role of TLR4 in hypertension might be expressed through its effects on vascular structure and mechanical properties. For instance, TLR4 is involved in the outward arterial remodelling after atherosclerotic plaque formation (Hollestelle et al., 2004) and TLRs, through MyD88-dependent mechanisms, contribute to the flow-mediated inward remodelling of conduit arteries (Tang et al., 2008). Furthermore, the TLR4 gene Asp299Gly polymorphism is associated with increased carotid artery compliance in young adults (Hernesniemi et al., 2008). Our present study provided evidence that TLR4 blockade prevented AngII-induced vascular remodelling and mechanical alterations, these effects contributing to the anti-hypertensive effects of the anti-TLR4 antibody treatment. Of note, these effects on vessel structure were observed in both conductance and resistance arteries, as demonstrated by the decreased wall : lumen ratio and wall thickness, which are well-known predictors of cardiovascular risk (Rizzoni and Agabiti-Rosei, 2012). Normalization of vessel structure by anti-TLR4 antibody treatment was achieved by increasing the number of cells, which was reduced after AngII (Briones et al., 2009). Moreover, TLR4 blockade prevented the increased collagen deposition and altered elastin structure induced by AngII, probably accounting for the restored wall : lumen ratio and the diminished vessel stiffness observed. Similarly, in human VSMC, the endogenous ligand for TLR4, HSP70, increased fibronectin and collagen type-I (González-Ramos et al., 2013). Besides improving vascular structure and mechanics, anti-TLR4 treatment normalized contractile responses and endothelial function in aortic segments from AngII-treated mice, in agreement with earlier results from SHR (Bomfim et al., 2012; de Batista et al., 2014). We also showed, here, that the TLR4 antibody prevented the reduction in endothelial cell number induced by AngII, thus providing a novel mechanism for improvement of endothelial function after TLR4 blockade.
One of the mechanisms by which TLR4 blockade might prevent the development of hypertension and improve the vascular alterations found in AngII-treated mice is by reducing oxidative stress. Earlier studies had described the prevention, by antioxidants, of hypertension and structural alterations of both resistance and conductance arteries in AngII-treated mice (Virdis et al., 2004; Martínez-Revelles et al., 2013). The mechanisms responsible for the vascular alterations induced by increased ROS production include VSMC proliferation and migration, oxidation of matrix metalloproteinases and generation of matrix proteins such as collagen (Virdis et al., 2004; Briones et al., 2009; Drummond et al., 2011). Furthermore, the increase of ROS has been causally linked to endothelial dysfunction in hypertension (Briones and Touyz, 2010; Virdis et al., 2011). In addition, increased ROS production by the TLR4 pathway contributes to the effects of AngII on apoptosis in mesangial cells and the differentiation of fibroblasts (Lv et al., 2009; Yuen et al., 2012). In the work presented here, treatment with anti-TLR4 antibody reduced the increased NOX-1 expression, NAD(P)H oxidase activity and superoxide anion production observed in aorta from AngII-treated mice. Such treatment also abolished the effect of different antioxidants on both vasoconstrictor and vasodilator responses, confirming that TLR4 contributed to the endothelial dysfunction in AngII-induced hypertension by increasing oxidative stress, as suggested recently in other hypertension models (de Batista et al., 2014; Sollinger et al., 2014). In obese and diabetic mice, TLR4 activation had been shown to increase ROS levels leading to a reduced NO production and bioavailability (Liang et al., 2013). Our present results pointed to both NAD(P)H oxidase and mitochondria as sources of the TLR4-induced oxidative stress. Following oxidative stress reduction by anti-TLR4 antibody treatment, bioavailability of NO could be increased. We found that TLR4 blockade increased release of NO and its contribution to the phenylephrine-induced vasoconstriction, in aortic samples from AngII-treated mice. The increased output of NO would also be related to the increased number of endothelial cells following TLR4 antibody treatment, as discussed above. Altogether, our results suggest that oxidative stress reduction by TLR4 blockade might account for the beneficial effects not only on the functional, but also on the structural and mechanical, alterations in the vasculature. Although the role of oxidative stress after TLR4 activation on vascular remodelling in hypertension has not been previously explored, superoxide-initiated inflammation is necessary for MyD88-dependent inward remodelling of conduit arteries induced by flow (Tang et al., 2008). Interestingly, TLR4 activation contributes to the vascular dysfunction associated with hypertension in SHR by COX-dependent mechanisms (Bomfim et al., 2012). Contractile prostanoids from COX-2 contribute to hypertension and also to the functional alterations observed in AngII-treated mice and this is in part dependent on ROS (Martínez-Revelles et al., 2013). Therefore, it is possible that by affecting the level of oxidative stress, TLR4 also modulates the COX pathway and its contribution to the vascular alterations, associated with our hypertension model.
TLR4 signals through different adaptor proteins, mainly MyD88, ultimately activating NF-κB, which controls transcription of different proinflammatory genes (Kawai and Akira, 2010). Another signalling pathway recruited by TLR4 activation, via the MyD88-dependent pathway, is the MAPK cascade (Kawai and Akira, 2010). Previously, we have shown that JNK and NF-κB contributed to the oxidative stress induced by AngII (Pérez-Girón et al., 2014). Here we showed, in VSMC, that AngII-induced NAD(P)H oxidase activity was reduced by a specific MyD88 inhibitor. Additionally, the TLR4 antagonist CLI-095 reduced the AngII-induced increased p65 nuclear expression and JNK1/2 phosphorylation. Altogether, our results suggest that after TLR4 up-regulation, through the MyD88-dependent signalling pathway, AngII activates the NF-κB/JNK pathways, which would contribute to the increased oxidative stress and inflammation occurring in hypertension. This suggestion would be compatible with the recent finding that, in SHR mesenteric arteries, TLR4 signalling was activated, leading to activation of MyD88/NF-κB pathway (Bomfim et al. 2015).
In conclusion, TLR4 up-regulation by AngII contributed to the inflammation, endothelial dysfunction, vascular remodelling, ECM alterations and vascular stiffness associated with hypertension. These effects appear to be mediated by the MyD88/NF-κB/JNK pathways and by increased oxidative stress from NAD(P)H oxidase and mitochondria. Although the role of adaptive immunity and T-lymphocytes on vascular remodelling has been established (de Ciuceis et al., 2014; Harrison, 2014; Schiffrin, 2014; Virdis et al., 2014), to the best of our knowledge, this is the first evidence that the TLR4 signalling pathway contributes to the vascular structural and mechanical alterations observed in hypertension. The fact that TLRs are expressed not only in immune cells, but also in vascular cells, allows us to suggest that TLR4 might be the link between low-grade inflammation, vascular remodelling and hypertension. According to the current model, hypertension, with elevated circulating levels of several compounds, which act as DAMPs, such as AngII, after being recognized by TLRs on antigen-presenting cells, would lead to the activation of T-cells. The subsequent release of cytokines and chemokines causes inflammation and subsequently vascular damage through several mechanisms, including oxidative stress. This results in increased BP, leading to further damage, which would increase the release of DAMPs, creating a positive feedback loop, which contributes to the maintenance of hypertension (de Ciuceis et al., 2014; Harrison, 2014; Schiffrin, 2014). Therefore, our observations open new approaches to antihypertensive therapy, using agents that interfere with the TLR4 signalling pathway.
Acknowledgments
We are grateful to Dr. Ernesto Moro for his help in some experiments. This work was supported by Ministerio de Economía y Competitividad (SAF2012-36400), Instituto de Salud Carlos III (PI13/01488 and Red de Investigación Cardiovascular RD12/0042/0024 and RD12/0042/0033) and URJC (PRIN13_CS12). A. M. B. was supported by the Ramón y Cajal Program (RYC-2010-06473).
Glossary
- AngII
angiotensin II
- DAMPs
damage-associated molecule patterns
- ECM
extracellular matrix
- HSP
heat shock proteins
- IEL
internal elastic lamina
- KHS
Krebs–Henseleit solution
- ML-171
2-acetylphenothiazine
- ROS
reactive oxygen species
- SBP
systolic BP
- SHR
spontaneously hypertensive rats
- TLR4
Toll-like receptor 4
- VSMC
vascular smooth muscle cells
Author contributions
R. H., S. M.-R., R. P., A. M., V. C., A. A., L. G.-R., M. T. B. and P. R. B. performed the research. R. H., A. M. B., M. S. and M. J. A. designed the research study. R. H., S. M.-R. and A. A. analysed the data. R. H., A. M. B., M. S. and M. J. A. wrote the paper. R. H., S. M.-R., R. P., A. M., V. C., A. A., L. G.-R., M. T. B., P. R. B., A. M. B., M. S. and M. J. A. approved the MS.
Conflict of interest
None.
References
- Alexander SP, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Catalytic receptors. Br J Pharmacol. 2013a;170:1676–1705. doi: 10.1111/bph.12449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: G Protein-Coupled Receptors. Br J Pharmacol. 2013b;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013c;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arribas SM, Hillier C, González C, McGrory S, Dominiczak AF, McGrath JC. Cellular aspects of vascular remodeling in hypertension revealed by confocal microscopy. Hypertension. 1997;30:1455–1464. doi: 10.1161/01.hyp.30.6.1455. [DOI] [PubMed] [Google Scholar]
- de Batista PR, Palacios R, Martín A, Hernanz R, Médici CT, Silva MASC, et al. Toll-like receptor 4 upregulation by angiotensin II contributes to hypertension and vascular dysfunction through reactive oxygen species production. PlosOne. 2014;9:e104020. doi: 10.1371/journal.pone.0104020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomfim GF, Dos Santos RA, Oliveira MA, Giachini FR, Akamine EH, Tostes RC, et al. Toll-like receptor 4 contributes to blood pressure regulation and vascular contraction in spontaneously hypertensive rats. Clin Sci. 2012;122:535–543. doi: 10.1042/CS20110523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bomfim GF, Echem C, Martins CB, Costa TJ, Sartoretto SM, Dos Santos RA, et al. Toll-like receptor 4 inhibition reduces vascular inflammation in spontaneously hypertensive rats. Life Sci. 2015;122:1–7. doi: 10.1016/j.lfs.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briones AM, Touyz RM. Oxidative stress and hypertension: current concepts. Curr Hypertens Rep. 2010;12:135–142. doi: 10.1007/s11906-010-0100-z. [DOI] [PubMed] [Google Scholar]
- Briones AM, González JM, Somoza B, Giraldo J, Daly CJ, Vila E, et al. Role of elastin in spontaneously hypertensive rat small mesenteric artery remodelling. J Physiol. 2003;552:185–195. doi: 10.1113/jphysiol.2003.046904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briones AM, Rodríguez-Criado N, Hernanz R, García-Redondo AB, Rodrigues-Díez RR, Alonso MJ, et al. Atorvastatin prevents angiotensin II-induced vascular remodeling and oxidative stress. Hypertension. 2009;54:142–149. doi: 10.1161/HYPERTENSIONAHA.109.133710. [DOI] [PubMed] [Google Scholar]
- Briones AM, Arribas SM, Salaices M. Role of extracellular matrix in vascular remodelling of hypertension. Curr Opin Nephrol Hypertens. 2010;19:187–194. doi: 10.1097/MNH.0b013e328335eec9. [DOI] [PubMed] [Google Scholar]
- Carpenter S, O'Neill LA. Recent insights into the structure of Toll-like receptors and post-translational modifications of their associated signalling proteins. Biochem J. 2009;422:1–10. doi: 10.1042/BJ20090616. [DOI] [PubMed] [Google Scholar]
- de Ciuceis C, Porteri E, Rizzoni D, Rizzardi N, Paiardi S, Boari GE, et al. Structural alterations of subcutaneous small-resistance arteries may predict major cardiovascular events in patients with hypertension. Am J Hypertens. 2007;20:846–852. doi: 10.1016/j.amjhyper.2007.03.016. [DOI] [PubMed] [Google Scholar]
- de Ciuceis C, Rossini C, La Boria E, Porteri E, Petroboni B, Gavazzi A, et al. Immune mechanisms in hypertension. High Blood Press Cardiovasc Prev. 2014;21:227–234. doi: 10.1007/s40292-014-0040-9. [DOI] [PubMed] [Google Scholar]
- den Dekker WK, Cheng C, Pasterkamp G, Duckers HJ. Toll like receptor 4 in atherosclerosis and plaque destabilization. Atherosclerosis. 2010;209:314–320. doi: 10.1016/j.atherosclerosis.2009.09.075. [DOI] [PubMed] [Google Scholar]
- Drummond GR, Selemidis S, Griendling KK, Sobey CG. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat Rev Drug Discov. 2011;10:453–471. doi: 10.1038/nrd3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eissler R, Schmaderer C, Rusai K, Kühne L, Sollinger D, Lahmer T, et al. Hypertension augments cardiac Toll-like receptor 4 expression and activity. Hypertens Res. 2011;34:551–558. doi: 10.1038/hr.2010.270. [DOI] [PubMed] [Google Scholar]
- González-Ramos M, Calleros L, López-Ongil S, Raoch V, Griera M, Rodríguez-Puyol M, et al. HSP70 increases extracellular matrix production by human vascular smooth muscle through TGF-β1 up-regulation. Int J Biochem Cell Biol. 2013;45:232–242. doi: 10.1016/j.biocel.2012.10.001. [DOI] [PubMed] [Google Scholar]
- Harrison DG. The immune system in hypertension. Trans Am Clin Climatol Assoc. 2014;125:130–138. [PMC free article] [PubMed] [Google Scholar]
- Hernesniemi JA, Raitakari OT, Kähönen M, Juonala M, Hutri-Kähönen N, Marniemi J, et al. Toll-like receptor 4 gene (Asp299Gly) polymorphism associates with carotid artery elasticity. The cardiovascular risk in young Finns study. Atherosclerosis. 2008;198:152–159. doi: 10.1016/j.atherosclerosis.2007.09.024. [DOI] [PubMed] [Google Scholar]
- Hollestelle SC, De Vries MR, Van Keulen JK, Schoneveld AH, Vink A, Strijder CF, et al. Toll-like receptor 4 is involved in outward arterial remodeling. Circulation. 2004;109:393–398. doi: 10.1161/01.CIR.0000109140.51366.72. [DOI] [PubMed] [Google Scholar]
- Ji Y, Liu J, Wang Z, Liu N. Angiotensin II induces inflammatory response partly via toll-like receptor 4-dependent signaling pathway in vascular smooth muscle cells. Cell Physiol Biochem. 2009;23:265–276. doi: 10.1159/000218173. [DOI] [PubMed] [Google Scholar]
- Jialal I, Kaur H, Devaraj S. Toll-like receptor status in obesity and metabolic syndrome: a translational perspective. J Clin Endocrinol Metab. 2014;99:39–48. doi: 10.1210/jc.2013-3092. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: Reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang CF, Liu JT, Wang Y, Xu A, Vanhoutte PM. Toll-like receptor 4 mutation protects obese mice against endothelial dysfunction by decreasing NADPH oxidase isoforms 1 and 4. Arterioscler Thromb Vasc Biol. 2013;33:777–784. doi: 10.1161/ATVBAHA.112.301087. [DOI] [PubMed] [Google Scholar]
- Lv J, Jia R, Yang D, Zhu J, Ding G. Candesartan attenuates Angiotensin II-induced mesangial cell apoptosis via TLR4/MyD88 pathway. Biochem Biophys Res Commun. 2009;380:81–86. doi: 10.1016/j.bbrc.2009.01.035. [DOI] [PubMed] [Google Scholar]
- Marketou ME, Kontaraki JE, Zacharis EA, Kochiadakis GE, Giaouzaki A, Chlouverakis G, et al. TLR2 and TLR4 gene expression in peripheral monocytes in nondiabetic hypertensive patients: the effect of intensive blood pressure-lowering. J Clin Hypertens (Greenwich) 2012;14:330–335. doi: 10.1111/j.1751-7176.2012.00620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín A, Pérez-Girón JV, Hernanz R, Palacios R, Briones AM, Fortuño A, et al. Peroxisome proliferator-activated receptor-γ activation reduces cyclooxygenase-2 expression in vascular smooth muscle cells from hypertensive rats by interfering with oxidative stress. J Hypertens. 2012;30:315–326. doi: 10.1097/HJH.0b013e32834f043b. [DOI] [PubMed] [Google Scholar]
- Martínez-Revelles S, Avendaño MS, García-Redondo AB, Alvarez Y, Aguado A, Pérez-Girón JV, et al. Reciprocal relationship between reactive oxygen species and cyclooxygenase-2 and vascular dysfunction in hypertension. Antioxid Redox Signal. 2013;18:51–65. doi: 10.1089/ars.2011.4335. [DOI] [PubMed] [Google Scholar]
- Mathiassen ON, Buus NH, Sihm I, Thybo NK, Mørn B, Schroeder AP, et al. Small artery structure is an independent predictor of cardiovascular events in essential hypertension. J Hypertens. 2007;25:1021–1026. doi: 10.1097/HJH.0b013e32805bf8ed. [DOI] [PubMed] [Google Scholar]
- McCarthy CG, Goulopoulou S, Wenceslau CF, Spitler K, Matsumoto T, Webb RC. Toll-like receptors and damage-associated molecular patterns: novel links between inflammation and hypertension. Am J Physiol Heart Circ Physiol. 2014;306:H184–H196. doi: 10.1152/ajpheart.00328.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvany MJ. Small artery remodelling in hypertension: causes, consequences and therapeutic implications. Med Biol Eng Comput. 2008;46:461–467. doi: 10.1007/s11517-008-0305-3. [DOI] [PubMed] [Google Scholar]
- Münzel T, Sinning C, Post F, Warnholtz A, Schulz E. Pathophysiology, diagnosis and prognostic implications of endothelial dysfunction. Ann Med. 2008;40:180–196. doi: 10.1080/07853890701854702. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl. Acids Res. 2014;42:D1098–1106. doi: 10.1093/nar/gkt1143. (Database Issue): [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Girón JV, Palacios R, Martín A, Hernanz R, Aguado A, Martínez-Revelles S, et al. Pioglitazone reduces angiotensin II-induced COX-2 expression through inhibition of ROS production and ET-1 transcription in vascular cells from spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol. 2014;306:H1582–H1593. doi: 10.1152/ajpheart.00924.2013. [DOI] [PubMed] [Google Scholar]
- Ponticos M, Smith BD. Extracellular matrix synthesis in vascular disease: hypertension, and atherosclerosis. J Biomed Res. 2014;28:25–39. doi: 10.7555/JBR.27.20130064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzoni D, Agabiti-Rosei E. Structural abnormalities of small resistance arteries in essential hypertension. Intern Emerg Med. 2012;7:205–212. doi: 10.1007/s11739-011-0548-0. [DOI] [PubMed] [Google Scholar]
- Savoia C, Schiffrin EL. Inflammation in hypertension. Curr Opin Nephrol Hypertens. 2006;15:152–158. doi: 10.1097/01.mnh.0000203189.57513.76. [DOI] [PubMed] [Google Scholar]
- Schiffrin EL. Immune mechanisms in hypertension and vascular injury. Clin Sci. 2014;126:267–274. doi: 10.1042/CS20130407. [DOI] [PubMed] [Google Scholar]
- Sollinger D, Eiβler R, Lorenz S, Strand S, Chmielewski S, Aoqui C, et al. Damage-associated molecular pattern activated Toll-like receptor 4 signalling modulates blood pressure in L-NAME-induced hypertension. Cardiovasc Res. 2014;101:464–472. doi: 10.1093/cvr/cvt265. [DOI] [PubMed] [Google Scholar]
- Tang PC, Qin L, Zielonka J, Zhou J, Matte-Martone C, Bergaya S, et al. MyD88-dependent, superoxide-initiated inflammation is necessary for flow-mediated inward remodeling of conduit arteries. J Exp Med. 2008;205:3159–3171. doi: 10.1084/jem.20081298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touyz RM. Intracellular mechanisms involved in vascular remodelling of resistance arteries in hypertension: role of angiotensin II. Exp Physiol. 2005;90:449–455. doi: 10.1113/expphysiol.2005.030080. [DOI] [PubMed] [Google Scholar]
- Tsan MF, Gao B. Endogenous ligands of Toll-like receptors. J Leukoc Biol. 2004;76:514–519. doi: 10.1189/jlb.0304127. [DOI] [PubMed] [Google Scholar]
- Virdis A, Neves MF, Amiri F, Touyz RM, Schiffrin EL. Role of NAD(P)H oxidase on vascular alterations in angiotensin II-infused mice. J Hypertens. 2004;22:535–542. doi: 10.1097/00004872-200403000-00016. [DOI] [PubMed] [Google Scholar]
- Virdis A, Duranti E, Taddei S. Oxidative stress and vascular damage in hypertension: role of angiotensin II. Int J Hypertens. 2011;2011:916310. doi: 10.4061/2011/916310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virdis A, Dell'Agnello U, Taddei S. Impact of inflammation on vascular disease in hypertension. Maturitas. 2014;78:179–183. doi: 10.1016/j.maturitas.2014.04.012. [DOI] [PubMed] [Google Scholar]
- Wolf G, Bohlender J, Bondeva T, Roger T, Thaiss F, Wenzel UO. Angiotensin II upregulates toll-like receptor 4 on mesangial cells. J Am Soc Nephrol. 2006;17:1585–1593. doi: 10.1681/ASN.2005070699. [DOI] [PubMed] [Google Scholar]
- Yuen CY, Wong SL, Lau CW, Tsang SY, Xu A, Zhu Z, et al. From skeleton to cytoskeleton: osteocalcin transforms vascular fibroblasts to myofibroblasts via angiotensin II and Toll-like receptor 4. Circ Res. 2012;111:e55–e66. doi: 10.1161/CIRCRESAHA.112.271361. [DOI] [PubMed] [Google Scholar]