

Abstract
Objective:
To analyze clinical features and myopathology changes in muscle fibers, connective tissue, and vessels in 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR) antibody–associated myopathies.
Methods:
Retrospective review of records and myopathologic features of 49 consecutive patients with myopathies and serum HMGCR antibodies.
Results:
Clinical features included onset age from 12 to 83 years, female predominance (67%), proximal, symmetric weakness (84%), muscle discomfort (78%), dysphagia (35%), systemic features, including skin rash and interstitial lung disease (37%), statin use (38%), and a high serum creatine kinase (83%). Myopathology included muscle fiber necrosis or regeneration (66%), myonuclear pathology (43%), perimysial connective tissue damage (61%), and lymphocytic foci (27%).
Conclusions:
Patients with HMGCR antibody–associated myopathies present with weakness and muscle discomfort and often have damage to both perimysial connective tissue and muscle fibers, with necrosis and myonuclear pathology. Only a minority of patients with HMGCR antibody–associated myopathies have a history of statin exposure.
Serum autoantibodies and myopathology features are useful for characterization and classification of acquired immune and inflammatory myopathy (IIM) syndromes.1–3 Serum IgG binding to 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR) has been identified in patients with presumed immune myopathies that usually have no lymphocytic cell foci.4,5 Myopathies with HMGCR antibodies have been suggested to be statin-induced, but statin histories are not universal and the strength of association with statin use varies among reports.5–10
Previous pathologic descriptions of myopathies with HMGCR antibodies have noted active necrosis and regeneration of scattered muscle fibers without inflammation,5 but some series mainly screened for HMGCR antibodies in groups of patients with “necrotizing myopathies.”6 Involvement of other components of muscle tissue has not been systematically evaluated. We have found that IIM syndromes often have characteristic pathology that involves several tissues in muscle, including muscle fibers, connective tissue, and vessels, and patterns of humoral and cellular immunity.3 In this study we evaluated clinical and pathologic features of 49 patients with acquired myopathies associated with serum IgG binding to HMGCR. Clinically, our patients had symmetric, proximal weakness and often had a high serum creatine kinase (CK). Only a minority had a history of statin use. Pathologic features in HMGCR antibody–associated myopathies often include damage to both perimysial connective tissue and muscle fibers.
METHODS
Patients.
We retrospectively analyzed charts, laboratory records, and muscle biopsies from 49 patients with high levels of serum HMGCR antibodies (titers > 2,800). Muscle biopsies of 44 patients were evaluated at Washington University School of Medicine in Saint Louis. The presence and degree of myonuclear pathology were evaluated later by 2 different pathologists (A.P. and A.A.) in 40 available muscle biopsies. We tested 37 additional sera from patients with a definite diagnosis of inclusion body myositis.11
Standard protocol approvals, registrations, and patient consents.
The Human Studies Committee of Washington University in Saint Louis approved all procedures.
Histochemical and immunohistochemical evaluation.
Cryostat sections of rapidly frozen muscle were processed using the same methodology described in our previous studies.12,13 Immunohistochemical stains for antibodies were performed on consecutive sections using muscle from patients and paired controls on the same glass slide. Primary antibodies used in this study were directed against HAM-56 (macrophages), CD8, CD4, CD20, C5b-9 complement (membrane attack complex) (Sigma-Aldrich, St. Louis, MO), and class I human major histocompatibility complex (MHC) (US Biological, Swampscott, MA). Ulex Europaeus Agglutinin I lectin (Sigma-Aldrich) was used to visualize endothelium in vessels.
Anti-HMGCR ELISA.
HMGCR antigen (Sigma-H7039) (Sigma-Aldrich) was diluted to 2 μg/mL in 0.01 M phosphate-buffered saline (PBS). HMGCR 0.1 μg was placed in each well of Immulon 2 HB ELISA plates (Thermo Fisher Scientific, Waltham, MA) and incubated overnight at 4°C. After washing 3 times with PBS-0.05% Tween 20, residual nonspecific binding sites in ELISA wells were blocked with 1% normal goat serum in PBS (PBS-NGS) (100 μL per well) for 4 hours at room temperature and washed 3 times with PBS-NGS. Subsequent steps were performed at 4°C. Between steps, washing was performed 5 times using PBS-NGS without detergent. Patient sera were tested in duplicate at 1:3,000 dilution with overnight incubation in ELISA wells at 4°C. Binding of serum IgG to HMGCR was measured using a 4-hour exposure to goat anti-human IgG linked to horseradish peroxidase (Organon Teknika–Cappel, West Chester, PA) in PBS with 1% bovine serum albumin (1:20,000). Color was developed with 100 μL substrate buffer (0.1 M citrate buffer, pH 4.5 with 0.004% H2O2 and 0.1% phenylenediamine) for 30 minutes. Optical density was measured at 450 nm. Final anti-HMGCR antibody titers were calculated after subtracting levels of nonspecific serum IgG binding to sulfatide. A normal range of values was determined by analysis of sera from 85 adult patients with other immune or inflammatory neuromuscular disorders.
Statistical analysis.
Fisher exact test was used to compare categorical variables. Independent 2-tailed t test and mean ± standard error were used to compare and report quantitative variables. A p value < 0.05 was considered a significant difference between groups.
RESULTS
Case report.
A 26-year-old woman noted proximal weakness with onset shortly after the delivery of her first child that progressed over a few months to difficulty walking up and down stairs. There was no history of statin use or dyspnea. Examination showed symmetric, mild proximal weakness but no skin rash. Laboratory testing showed a myopathic EMG, a serum CK of 6,270 IU/L, and an antinuclear antibody titer of 1:320. Muscle biopsies (figure 1) at presentation and 8 years later showed increased endomysial connective tissue, many small- and moderate-sized basophilic muscle fibers, scattered necrotic fibers, and no foci of lymphocytic inflammation. Alkaline phosphatase diffusely stained the perimysium (figure 1C). Dystrophy-related protein staining and blots were normal for dystrophin, sarcoglycans, α-dystroglycan, dysferlin, desmin, emerin, calpain-3, caveolin-3, and laminin-α2. Gene testing was not performed. Oral treatments with high-dose prednisone, methotrexate, cyclosporine, and azathioprine produced no functional benefit. Myositis-specific antibody panels were negative on several occasions. Anti-HMGCR antibody titers were 34,000 in a serum sample obtained in 1991 and 48,000 in 2013. Weakness was slowly progressive. Twenty-three years after disease onset she required a scooter for mobility and could only walk 3 m with help. Quantitative testing showed strength that was 50%–60% of normal in proximal muscles in the arms and legs and normal in distal muscles.
Figure 1. Muscle biopsy from a patient with a slowly progressive (24 years) HMGCR antibody–associated myopathy syndrome.

(A) Hematoxylin & eosin stain, (B) Verhoeff-van Gieson stain. Muscle fiber size varies from very small to hypertrophied. Some small muscle fibers have mildly basophilic, or darkly stained, cytoplasm (white arrows). A few muscle fibers have internal nuclei. Endomysial connective tissue between muscle fibers is increased. (C) Alkaline phosphatase stains perimysial connective tissue and the cytoplasm of scattered small muscle fibers (white arrow). (D) Acid phosphatase stains scattered cells in the perimysium and cells associated with a necrotic muscle fiber (black arrow). Scale bar = 50 μM for A, B, and D and 100 μM for C. HMGCR = 3-hydroxy-3-methylglutaryl-coenzyme A reductase.
HMGCR antibodies.
HMGCR antibody titers in our initial 49 patients ranged from 2,800 to 80,000. In the few patients with serial sera available, as in our case report, antibody titers showed no clear changes over long periods of time. A second patient evaluated over a very long period had titers of 7,000 at presentation, which included rapidly progressive severe weakness with respiratory failure, and 12,000 7 years later after chronic corticosteroid treatment that was associated with improvement to near-normal strength.
Clinical characteristics.
Patients with HMGCR antibodies included 33 females (67%) and 16 males (33%) (table). Mean age at disease onset was 50 years (range 12–83 years). Ethnicities were Caucasian (76%), African American (22%), and Asian (2%). Neuromuscular symptoms included myalgia (78%) and dysphagia (35%). Weakness was present on examination in 84%. The 8 patients (16%) with no weakness reported muscle discomfort, cramps, or fasciculations. On examination, weakness was generally symmetric (98%) and proximal-greater-than-distal (100%). The course of disease progression ranged from subacute (progression to respiratory failure over 2 months) to chronic (slow progression over decades mimicking limb-girdle muscular dystrophies; see case report). Systemic features (37%) included skin rash affecting the face, chest, and dorsum of the hands (22%); interstitial lung disease (by chest imaging) (20%); and associated malignancy (10%) (pancreatic [2], colon, prostate, and hematologic). Statin use was reported in 38% of patients. Statin users were older (p = 0.005; mean 58 years) than nonusers (mean 44 years). The frequency of a history of statin use in our cohort was somewhat higher than, but not statistically different from, patients in an age-matched concurrent group with other immune myopathies (38% vs 23%; p = 0.2). There were no differences in any clinical features between patients with or without statin exposure (table).
Table.
Clinical features of HMGCR myopathy patients
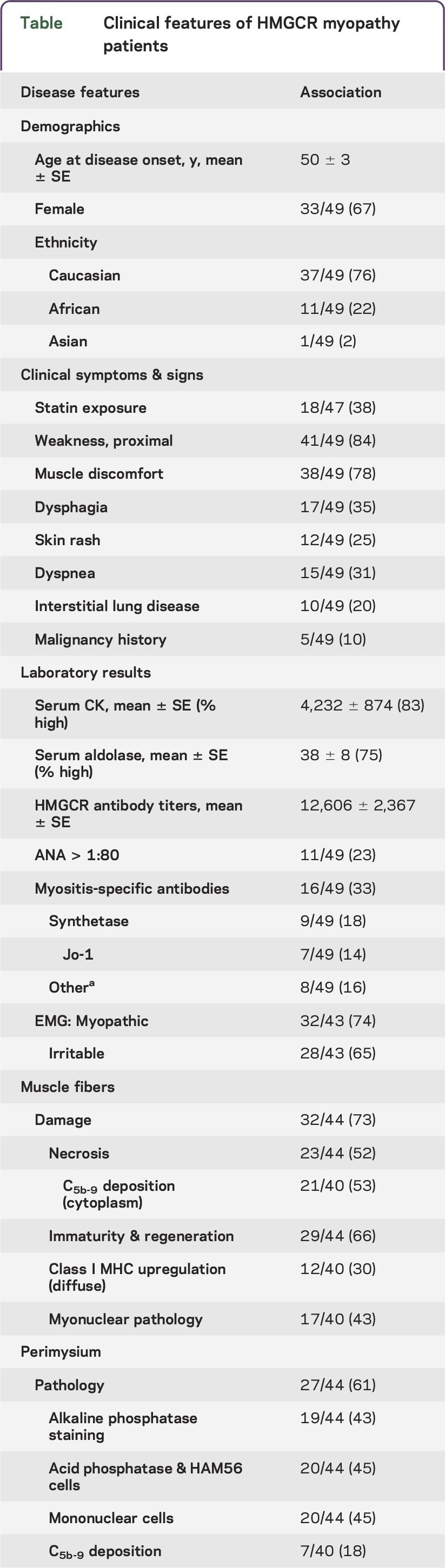
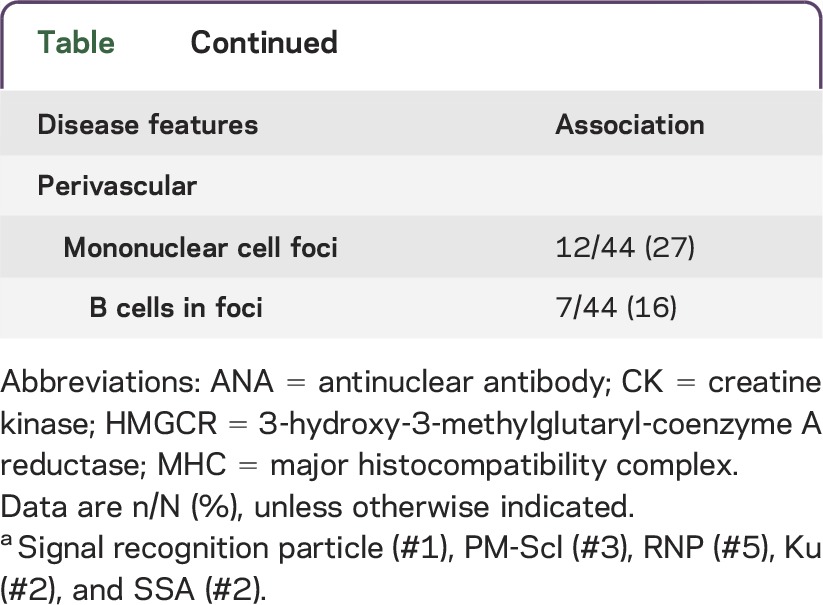
Laboratory data.
Anti-HMGCR antibody titers ranged from 2,800 to 80,000 (mean 12,606) and were similar in patients with and without statin exposure (table). Serum CK was high in 83% of our patients (mean 4,232 IU/L; range 26 to 27,000). Fifty percent of patients with normal strength had high serum CK (mean 447 IU/L; range 48–1,846). Patients with very high titers of anti-HMGCR antibodies (≥6,000) had higher levels of serum CK (p = 0.006). Serum aldolase was high in 75% (mean 38 U/L; range 3.9–291). Three patients with normal serum CK levels had a high aldolase. Other serum antibodies included antinuclear (22%), anti-synthetase (18% [Jo-1, 14%]), signal recognition particle (2%), and connective tissue disease overlap antibodies (PM-Scl, SSA, or RNP) (16%). EMG was often myopathic (74%), with fibrillation potentials and/or positive sharp waves in many (65%).
Myopathology.
Common myopathic features involved muscle fibers and perimysial connective tissue (table; figures 1–3). Muscle fiber pathology included evidence of ongoing damage, with fibers in varied stages of necrosis (52%) and regeneration or immaturity (66%) (figure 1). C5b-9 complement stained the cytoplasm of some necrotic muscle fibers but was not typically deposited on the surface of non-necrotic muscle fibers. Large clusters of contiguous damaged fibers were not present. In some muscles, immature fibers and necrosis were most prominent at the edge of fascicles near damaged or cellular perimysial connective tissue (figure 3, C and F). Focal invasion of muscle fibers by cells was present in a few patients (7%). Myonuclear pathology (43%) (figure 2) included enlargement, irregular shapes, empty central regions, and clusters. Perimysial connective tissue pathology was present in 61% of biopsies, with diffuse alkaline phosphatase staining (figure 1C), histiocytic (acid phosphatase- and esterase-positive) cells (figure 3, B and E), or damaged structure (figure 3D). The most common general pathologic pattern (57%; 25/44) was for both muscle fiber and perimysial pathology to be present. Muscles with neither muscle fiber nor perimysial pathology occurred in 25% (11/44). Weakness was more common (p = 0.017) in patients with perimysial damage (100%) than in those without perimysial damage (77%). Anti-HMGCR antibody titers were higher in patients with muscle biopsies showing muscle fiber necrosis (19,748 ± 4,509 vs 6,538 ± 1,249; p = 0.001) and connective tissue damage (17,841 ± 3,998 vs 6,459 ± 1,027; p = 0.03). Some biopsies contained lymphocytic cell foci (27%), generally surrounding intermediate-sized perimysial vessels. MHC class I was diffusely upregulated by all muscle fibers in some biopsies (30%). In some cases MHC class I expression was more prominent on fibers at the edge of fascicles near perimysium. There were no differences in biopsy features between patients with or without statin use. Endomysial connective tissue was increased in a few biopsies (8%), mostly in patients with longer histories of disease (figure 1). Vacuoles in muscle fibers were not observed.
Figure 2. Abnormal myofiber nuclei in HMGCR antibody–associated myopathy.

Myonuclei are often enlarged (dark arrow) with clear centers (dark arrowhead) or irregular shapes (light arrowhead). Internal nuclei are common. Some myonuclei are in abnormal clusters or clumps (light arrows). Scale bar = 18 μM. HMGCR = 3-hydroxy-3-methylglutaryl-coenzyme A reductase.
Figure 3. Perimysial and muscle fiber pathology in HMGCR antibody–associated myopathy.

(A) Perimysial pathology with histiocytic cells and widening (hematoxylin & eosin stain). (B) Most cells in the perimysium are histiocytic and stain for acid phosphatase. (C) Muscle fibers at the edge of some fascicles are small and may be dark-stained (immature) (nicotinamide adenine dinucleotide stain). (D) Fragmentation of perimysial connective tissue (Gomori trichrome stain). (E) Cells in perimysial connective tissue are scattered, large, and irregularly shaped (nonspecific esterase stain). (F) Necrotic (dark) muscle fibers, with C5b-9 complement staining of cytoplasm, are scattered through the muscle but may have increased frequency at the edge of fascicles, a feature common in immune myopathies with perimysial pathology. C5b-9 complement is also deposited in perimysial connective tissue, occasionally extending into the endomysium. Scale bar = 100 μM for A, B, and D and 50 μM for C, E, and F. HMGCR = 3-hydroxy-3-methylglutaryl-coenzyme A reductase.
HMGCR antibodies in patients with inclusion body myositis.
To evaluate the specificity of HMGCR antibodies for the myopathologic features identified in this study, we tested sera from 37 patients with definite inclusion body myositis, a group in whom we did not expect to detect positive results. We found 2 sera with high HMGCR antibody titers (2,900 and 8,400). Both patients also had serum IgG binding to NT5C1A antigen. The muscle pathology in these 2 patients showed changes consistent with inclusion body myositis, including vacuoles containing basophilic debris, endomysial foci of CD4 and CD8 lymphocytes with focal invasion of muscle fibers, and diffuse upregulation of MHC class I by muscle fibers. However, there were also scattered necrotic muscle fibers and perimysial connective tissue pathology (alkaline phosphatase staining and acid phosphatase–positive cells), as noted above in other HMGCR antibody–positive patients. Similar changes were found in only 2 of the 35 HMGCR antibody–negative inclusion body myositis patients (p = 0.008).
DISCUSSION
We retrospectively reviewed clinical and myopathology features of 49 patients with serum anti-HMGCR antibodies. Some clinical features in our patients with serum IgG binding to HMGCR were similar to those in previous studies.5,6 We observed a moderate female predominance. Onset ages varied widely from childhood to older adulthood. Presenting features often included proximal, symmetric weakness, myalgia, and a high serum CK. The disease course varied from acute to very chronic. Three patients with slowly progressive proximal weakness were initially thought to have limb-girdle muscular dystrophies but had no histologic or molecular diagnosis despite extensive testing. Patients with previous exposure to statin drugs were older, but clinical and laboratory findings were similar to those in patients without statin exposure.
Differences in our cohort from prior studies included a small group of HMGCR antibody patients who presented with myalgia, cramps, and elevated CK but no weakness.14,15 Systemic manifestations, including skin rash and interstitial lung disease, were more frequent than previously reported. Interstitial lung disease, ascertained by chest imaging, was found in 20% of our cohort. Other myositis-associated antibodies were identified in 33% of our patients. The wider spectrum of clinical and laboratory features seen in our series likely reflects our inclusive pattern of serum testing (i.e., most patients with acquired myopathies). The initial series of HMGCR antibody patients excluded patients with inflammation in the muscle biopsy and selectively studied patients referred to a myositis clinic.4 Another series analyzed only patients who had noninflammatory biopsies with active and prominent muscle fiber necrosis and regeneration.6
Our data suggest that myopathies associated with HMGCR antibodies should not be called “statin myopathies.” Statin-intolerant patients with a clear temporal association between statin intake and symptoms have a very low frequency of HMGCR antibodies.14 In all but one study, a substantial proportion of HMGCR antibody–positive patients (37% to 62%) have been statin-naive.5,6,16 In some HMGCR antibody–positive patients, the statin exposure was years prior to the onset of myopathy. An Australian report is an outlier, finding that 94% of HMGCR antibody–positive patients had statin exposure.9 In this study the clinical correlations, including myopathy type and sex predominance, also differed from other reports.
The initial focus of our study was to analyze the myopathology features of HMGCR antibody patients. Our results show that 2 components of muscle tissue are commonly involved: muscle fibers and connective tissue. Muscle fibers showed 2 patterns of pathology. Non-necrotic muscle fibers often had nuclear pathology with irregular shapes, enlargement, and aggregation (figure 2). This pattern of myopathology of uncertain cause is not common in most other myopathies with prominent necrosis. A study of myopathies associated with Jo-1 antibodies found nuclear actin aggregates in many patients.17 The relationship between these nuclear aggregates and the myonuclear pathology seen on histologic sections in our study is unclear because ultrastructural analysis was not available. A second pattern of myofiber pathology is the scattered fibers in different stages of necrosis and regeneration that has been described in all previous studies of HMGCR antibody patients. This general pattern of scattered, ongoing muscle fiber necrosis is not specific for HMGCR antibody syndromes and is common in many immune and hereditary myopathies.18 One distinctive feature of the distribution of the muscle fiber necrosis in some of our HMGCR antibody patients was a tendency for necrotic and regenerating fibers to be more common at the edge of fascicles near the perimysium. This distribution of necrosis occurs in immune myopathies with perimysial pathology (IMPP), such as those seen with Jo-1 antibodies.12,17 The patterns of scattered necrosis differ from the clusters of muscle fibers in similar stages of necrosis seen in regional ischemic immune myopathy, which is likely due to vascular pathology.19
The second area of muscle commonly involved in HMGCR antibody patients is connective tissue, especially the perimysium. Perimysial pathology with scattered histiocytic cells, alkaline phosphatase staining, and damaged structure (figures 1 and 3) was as common as muscle fiber necrosis in our HMGCR antibody patients. HMGCR antibody–associated myopathology is similar to disorders in the IMPP pathologic group.3 Damage to both perimysial connective tissue and muscle fibers is the most common pattern of myopathology in IMPP, Jo-1 antibody–associated myopathies, and our HMGCR patients. The common types of muscle fiber damage in IMPP syndromes are necrosis and regeneration, which are more prominent in perifascicular regions near perimysium and nuclear pathology.3,12,17 There is also some clinical overlap of features between IMPP syndromes and our patients. Systemic features present in 37% of our patient series included interstitial lung disease or a skin rash.
Our myopathology results differ from previous descriptions of HMGCR patients, which have mainly emphasized the myopathy as “necrotic.”5,6,9,10 One reason for this discrepancy is that some studies have biased their results by selecting patients based on the presence of muscle fiber necrosis. Another issue reducing reporting of perimysial damage may be that our pathology laboratory routinely performs histochemical analysis of muscle biopsies that includes acid and alkaline phosphatase stains. Acid and alkaline phosphatase stains are especially sensitive for observing cellular and molecular pathology in perimysial connective tissue.3 They are not routinely performed in many laboratories and were not reported in previous HMGCR series.
The specificity of HMGCR antibodies in immune and other myopathies requires further study. One series reported that HMGCR antibodies occur in 10% of patients with an entirely different disorder, inclusion body myositis.9 Our initial results, identifying 49 antibody-positive patients, were at least partly the result of clinicians with a suspicion of an HMGCR antibody–associated syndrome ordering serum testing. Our follow-up review of patients diagnosed with inclusion body myositis suggested a prevalence of 5.4%. However, review of the muscle biopsies in the 2 positive patients showed perimysial connective tissue damage in addition to the typical inclusion body myositis pathology. It is well-known that there is an increased frequency of associated immune features in inclusion body myositis, including antinuclear antibodies, SSA, and M proteins.20,21 Clinically, the patients had features that were somewhat atypical for inclusion body myositis, including younger onset in the fifth decade and prominent proximal weakness early in the disease course. This suggests that these patients may have had 2 concurrent myopathy syndromes, inclusion body myositis and HMGCR antibody–associated myopathology. HMGCR antibodies may correlate better with specific pathology features than with general clinical diagnoses.
HMGCR antibody–associated myopathies can be considered in the context of the common clinical classification and our proposed pathologic groups of immune myopathies.3 Polymyositis and inclusion body myositis can be ruled out because there are few lymphocytic inflammatory foci or inclusion bodies in HMGCR myopathies. Dermatomyositis is not a useful diagnosis because skin changes were present in only a minority of our patients. Necrotic myopathies are a possible class within which to group HMGCR patients, but such a classification is misleading and lacks pathologic specificity. The term “necrotic myopathy” can be applied equally well to rhabdomyolysis and a variety of hereditary, ischemic, and toxic disorders.18 Denoting the HMGCR syndrome as a necrotic myopathy implies that myofiber necrosis is a constant feature and that the disorder involves mainly damage to muscle fibers. However, connective tissue in perimysium is also commonly damaged in HMGCR myopathies. The connective tissue damage includes distinctive cellular mechanisms with histiocytic, not lymphocytic, infiltration. In muscles in which necrosis is present, the myopathic features are often nonuniform and more prominent near the perimysium. An additional non-necrotic muscle fiber pathology, abnormal myonuclear morphology, was also observed in many biopsies.
The therapeutic response of some patients with HMGCR antibodies to immunomodulation supports the idea that these myopathies are autoimmune disorders.4,6,22 The absence of lymphocytic inflammation suggests that humoral mechanisms may be important. However, despite the presence of serum autoantibodies, intracellular molecules such as HMGCR have been difficult to implicate as pathogenic target antigens of humoral or cellular immune mechanisms in immune disorders. The specific molecular antigenic targets of the immune response in muscle and connective tissue in HMGCR antibody–associated myopathy remain to be defined.
GLOSSARY
- CK
creatine kinase
- HMGCR
3-hydroxy-3-methylglutaryl-coenzyme A reductase
- IIM
immune and inflammatory myopathy
- IMPP
immune myopathies with perimysial pathology
- MHC
major histocompatibility complex
- NGS
normal goat serum
- PBS
phosphate-buffered saline
AUTHOR CONTRIBUTIONS
Ali Alshehri: obtaining, analysis, and interpretation of the data, drafting and revising the manuscript. Rati Choksi: obtaining the data. Robert Bucelli: obtaining and interpretation of the data, revising the manuscript. Alan Pestronk: study design, obtaining, analysis, and interpretation of the data, revising the manuscript.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
A. Alshehri and R. Choksi report no disclosures. R. Bucelli received a financial gift from a patient's family for Parsonage-Turner research. A. Pestronk is on the scientific advisory board for The Myositis Association; holds patents and receives payments for TS-HDS antibody, GALOP antibody, GM1 Ganglioside antibody, and Sulfatide antibody; received speaker honoraria from Athena; is on the speakers' bureau for The Myositis Association; received research support from Genzyme, Insmed, Knopp, Ultragenyx, ISIS, Sanofi, Cytokinetics, GSK, Biogen, CSL Behring, NIH, Washington University Neuromuscular Research Fund, CINRG Children's Hospital Washington DC, and Muscular Dystrophy Association; holds stock in Johnson & Johnson; and receives payments for antibody testing from Athena. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Hoogendijk J, Amato A, Lecky B, et al. 119th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10–12 October 2003, Naarden, The Netherlands. Neuromuscul Disord 2004;14:337–345. [DOI] [PubMed] [Google Scholar]
- 2.Gunawardena H, Betteridge ZE, McHugh NJ. Myositis-specific autoantibodies: their clinical and pathogenic significance in disease expression. Rheumatology (Oxford) 2009;48:607–612. [DOI] [PubMed] [Google Scholar]
- 3.Pestronk A. Acquired immune and inflammatory myopathies: pathologic classification. Curr Opin Rheumatol 2011;23:595–604. [DOI] [PubMed] [Google Scholar]
- 4.Christopher-Stine L, Casciola-Rosen LA, Hong G, Chung T, Corse AM, Mammen AL. A novel autoantibody recognizing 200-kd and 100-kd proteins is associated with an immune-mediated necrotizing myopathy. Arthritis Rheum 2010;62:2757–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mammen AL, Chung T, Christopher-Stine L, et al. Autoantibodies against 3-hydroxy-3-methylglutarylcoenzyme a reductase (HMGCR) in patients with statin-associated autoimmune myopathy. Arthritis Rheum 2011;63:713–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allenbach Y, Drouot L, Rigolet A, et al. Anti-HMGCR autoantibodies in European patients with autoimmune necrotizing myopathies: inconstant exposure to statin. Medicine (Baltimore) 2014;93:150–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Albayda J, Mammen AL. Is statin-induced myositis part of the polymyositis disease spectrum? Curr Rheumatol Rep 2014;16:433. [DOI] [PubMed] [Google Scholar]
- 8.Hamann PD, Cooper RG, McHugh NJ, Chinoy H. Statin-induced necrotizing myositis—a discrete autoimmune entity within the “statin-induced myopathy spectrum”. Autoimmun Rev 2013;12:1177–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Limaye V, Bundell C, Hollingsworth P, et al. Clinical and genetic associations of autoantibodies to 3-hydroxy-3-methyl-glutaryl-coenzyme a reductase in patients with immune-mediated myositis and necrotizing myopathy. Muscle Nerve Epub 2014 Dec 18. [DOI] [PubMed]
- 10.Watanabe Y, Suzuki S, Nishimura H, et al. Statins and myotoxic effects associated with anti-3-hydroxy-3-methylglutaryl-coenzyme a reductase autoantibodies: an observational study in Japan. Medicine (Baltimore) 2015;94:e416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rose MR; ENMC IBM Working Group. 188th ENMC International Workshop: Inclusion Body Myositis, 2–4 December 2011, Naarden, The Netherlands. Neuromuscul Disord 2013;23:1044–1055. [DOI] [PubMed] [Google Scholar]
- 12.Mozaffar T, Pestronk A. Myopathy with anti-Jo-1 antibodies: pathology in perimysium and neighbouring muscle fibres. J Neurol Neurosurg Psychiatry 2000;68:472–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller T, Al-Lozi MT, Lopate G, Pestronk A. Myopathy with antibodies to the signal recognition particle: clinical and pathological features. J Neurol Neurosurg Psychiatry 2002;73:420–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mammen AL, Pak K, Williams EK, et al. Rarity of anti-3-hydroxy-3-methylglutaryl-coenzyme A reductase antibodies in statin users, including those with self-limited musculoskeletal side effects. Arthritis Care Res (Hoboken) 2012;64:269–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mohassel P, Mammen AL. The spectrum of statin myopathy. Curr Opin Rheumatol 2013;25:747–752. [DOI] [PubMed] [Google Scholar]
- 16.Drouot L, Allenbach Y, Jouen F, et al. Exploring necrotizing autoimmune myopathies with a novel immunoassay for anti-3-hydroxy-3-methyl-glutaryl-CoA reductase autoantibodies. Arthritis Res Ther 2014;16:R39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stenzel W, Preuße C, Allenbach Y, et al. Nuclear actin aggregation is a hallmark of anti-synthetase syndrome-induced dysimmune myopathy. Neurology 2015;84:1346–1354. [DOI] [PubMed] [Google Scholar]
- 18.Allenbach Y, Benveniste O. Acquired necrotizing myopathies. Curr Opin Neurol 2013;26:554–560. [DOI] [PubMed] [Google Scholar]
- 19.Cai C, Alshehri A, Choksi R, Pestronk A. Regional ischemic immune myopathy: a paraneoplastic dermatomyopathy. J Neuropathol Exp Neurol 2014;73:1126–1133. [DOI] [PubMed] [Google Scholar]
- 20.Koffman BM, Rugiero M, Dalakas MC. Immune-mediated conditions and antibodies associated with sporadic inclusion body myositis. Muscle Nerve 1998;21:115–117. [DOI] [PubMed] [Google Scholar]
- 21.Dalakas MC, Illa I, Gallardo E, Juarez C. Inclusion body myositis and paraproteinemia: incidence and immunopathologic correlations. Ann Neurol 1997;41:100–104. [DOI] [PubMed] [Google Scholar]
- 22.Werner JL, Christopher-Stine L, Ghazarian SR, et al. Antibody levels correlate with creatine kinase levels and strength in anti-HMG-CoA reductase-associated autoimmune myopathy. Arthritis Rheum 2012;64:4087–4093. [DOI] [PMC free article] [PubMed] [Google Scholar]