

Abstract
Neuritic plaque (NP) formation can be dated in vivo. This analysis attempts to “date” the progression of neurofibrillary tangles (NFT) using the spatial distribution of NP as a reference. Autopsy data from 471 participants in the Honolulu-Asia Aging Study (HAAS) were combined into latent factor measures of NFT and NP counts. The variance in “early” and “late” NP pathology was used to estimate the spatial distribution of “early” and “late” NFT formation. A third latent factor representing “non-NP-related NFT” was also constructed. “Early”NP and ‘Late”NP correlated significantly with objectively early and later cognitive performance, respectively. In contrast to our expectations, neocortical NFT correlated best with “early” NP pathology, while NFT in allocortical structures correlated best with “late” NP pathology. Therefore, the NP-related fraction of NFT appears to be co-localized spatially with NP. However, since the latter evolve corticofugally in time, this suggests that NP-related NFT do so as well. Corticotropic NFT formation must therefore be either un-related to NP formation, a temporally distinct process, or both.
Keywords: Alzheimer's Disease, dementia, MCI, Neuropathology
Introduction
Alzheimer's disease (AD) is associated with neurodegenerative lesions including the neurofibrillary tangles (NFT) and neuritic plaques (NP). Their propagation in space and time is not well understood. Autopsy data suggest that NFT spread corticotropically from an origin in the allocortex. This has been implicitly incorporated into a series of “Braak Stages” describing the pre-clinical appearance of NFT in structures such as the hippocampus and entorhinal cortex, followed by neocortical involvement in AD's clinical phases [1].
Unfortunately, autopsy studies are necessarily cross-sectional. It is difficult to confidently estimate longitudinal processes from cross-sectional data. We recently tried to overcome this limitation by fitting autopsy data to a latent growth curve (LGC) [2]. LGC models were developed to estimate change over time in a cohort's serially obtained measurements. We applied LGC techniques to the spatial distribution of AD lesions using autopsy data from 435 participants in the Honolulu-Asia Aging Study (HAAS). The result was a quantification of inter-individual variation in inter-regional vulnerability to AD lesions.
Surprisingly, we found that NFT and NP were distributed across differently ordered sets of anatomical regions. The gradient of spatial differences in NP (dNP), was significantly associated with that of NFT (dNFT), but weakly and inversely (r = -0.12, p <0.001). Only dNFT was significantly associated with longitudinal change in cognition. Braak's staging system is referenced to NFT, in deference to NP's relatively weak association with cognitive impairment.
In contrast to NFT, NP achieve their highest densities in the neocortex. Moreover, the relative vulnerability of individual structures to NP formation suggests a corticofugal progression [2]. Corticofugal NP development is consistent with the longitudinal evolution of β-amyloid in vivo [3-5] and with cross-sectional evidence of neocortical β-amyloid deposition in non-demented persons, years before the onset of cognitive impairment [6]. Less is known about the evolution of NFT in time, as those lesions cannot be imaged in vivo. However, corticofugal NP formation violates the expected propagation of NFT described by Braak.
If we assume Braak is correct about NFT formation, then allocortical NFT necessarily precede neocortical NFT. Since the appearance of NFT in neocortical regions at autopsy is usually associated with dementia [and only rarely with normal cognition [7], and since cortical β-amyloid has been shown to precede both allocortical β-amyloid formation and dementia [3], then allocortical NFT formation has to develop in isolation (i.e., in advance of NP formation in allocortical structures) [8].
That possibility conflicts with the “amyloid cascade hypothesis” [9] which posits that β-amyloid formation locally precedes NFT formation in the same structure. However, it is consistent with our observation that 89% of the brains available in HAAS have evidence of NFT formation in CA1, although <50% of participants are unambiguously cognitively impaired [10].
In a follow up to our 2012 paper, we related neocortical neuropathology to a LGC of observed cognitive change. That analysis allowed us to date the time frame within which NFT and NP formation were most likely to have occurred. We found that neocortical NFT and NP develop concurrently [11]. Therefore, NFT and NP are both likely to follow neocortical β-amyloid formation, consistent with the cascade hypothesis.
Here we attempt to “date” the progression of NFT across these structures by using the spatial distribution of NP as a reference. Since beta-amyloid can be imaged in vivo, its longitudinal spatial development can be used to define “early” and “late” NP formation. That knowledge can be leveraged to reveal NFT's development through its association(s) with regional NP. If neocortical and allocortical NFT represent two distinct tangleopathies, the cascade hypothesis might be reconciled with isolated allocortical NFT formation.
Methods
The Honolulu-Asia Aging Study (HAAS): Autopsy tissue and clinical data were obtained from HAAS [12] HAAS began in 1991 as an add-on to the Honolulu Heart Program (HHP). It is a longitudinal study of heart disease and stroke established in 1965 with the examination of 8006 Japanese-American men born 1900-1919. Brain autopsy and cognitive exams have been performed continuously since 1991.
HAAS autopsy material
838 autopsies had been performed prior to May, 2010. These represent approximately 20% of HAAS deaths since 1991. Demented decedents with autopsies do not differ significantly on several demographic and clinical measures from those who have not been autopsied, and non-demented and autopsied decedents were similar to autopsied, non-demented decedents. Although a diagnosis of dementia increased the likelihood that family members would contact the HAAS and /or agree to autopsy at the time of death, previous analyses indicate a general comparability across the two groups (demented and non-demented) with regard to clinical and demographic features [13]. The current analyses are limited to autopsies obtained between 1991 and 2001. Microscopic examinations performed since 2001 have been done by a different team of neuropathologists, and have not yet been pooled for common analyses. Complete microscopic data generated by the first team are available in 493 decedents.
The gross exams include external measurements and examination of 1 cm thick coronal sections of the entire brain for lacunes and large infarcts. The microscopic exam includes multiple stains for each of 38 tissue blocks from the brainstem and left hemisphere [14]. These are typically stained using Hematoxylin and Eosin (H&E) staining, Bielschowshi, Gallyas, and anti-Aβ. Anti-α-synuclein staining was done on isocortical sections from brains in which Lewy bodies were observed in H & E stained sections from the substantia nigra and /or locus ceruleus, and on a sample of brains in which no brainstem Lewy bodies were observed.
Microscopic AD pathology data include NFT counts, neuritic and diffuse (non- neuritic) amyloid plaque densities, vascular amyloid indices, substantia nigra neuronal counts, and anti-α-synuclein assessments of isocortical Lewy bodies. Focal ischemic lesions assessed included large cortical infarctions, small grey and white matter lacunes and “microinfarcts” (i.e., ischemic lesions visible only on light microscopy).
Pathological materials
Brains were fixed by submersion in 10% neutral formalin. Tissue samples were embedded in paraffin. Slides were cut at 8 micron thicknesses and stained as mentioned. Modified Bielschowsky, Gallyas and α- synuclein-stained slides were examined to quantify neuritic plaques (NP), neurofibrillary tangles (NFT), and to determine Braak stage [1]. NP were defined as extracellular accumulations of abnormal agyrophilic and anti-amyloid staining aggregates containing a central amyloid core and identifiable neurites (abnormal dark, coarse, tangled or irregular neuritic processes). NFT were defined by intraneuronal, cytoplasmic dense accumulations of agyrophilic (Bielschowsky or Gallyas stain) filamentous material that may be globoid, circumferential or flame-shaped. Extracellular or “tombstone” neurofibrillary tangles were interpreted as indicating that the neuron in which the NFT had developed had died and deteriorated.
NP, and NFT were enumerated in 5 fields for each anatomical region, with post-assessment adjustment to produce counts standardized to areas of 1 square millimeter. Fields with the highest counts (2-dimensional densities) were selected for either the total plaque count (neuritic plus diffuse) or the total NFT count. Mean NP, and NFT counts were calculated across 20 isocortical fields, from the right frontal, parietal, temporal, and occipital lobes.
Cognitive Abilities Screening Instrument (CASI)
The CASI was developed by merging an expanded MMSE (the 3MS) with the Hasegawa dementia scale [15-16]. The resulting measure has been rescaled to 100 points (higher score is better), and contains items addressing 9 cognitive domains, including long-term and short-term memory, attention, concentration, orientation, visuospatial abilities, judgment and abstract thinking, word fluency and language. This analysis is restricted to participants with “normal” cognitive performance (CASI > 70/100) at baseline (N = 471).
Statistical Approach
Latent Neuropathology Measures
We constructed latent neuropathology measures derived from observed regional lesion counts supplied by HAAS. The raw pathological data were submitted to confirmatory factor analyses in a structural equation modeling (SEM) framework. All observed indicators were adjusted for age at death, last CASI score available, and education. All analyses were performed using Analysis of Moment Structures (AMOS) software [17].
First, we constructed latent variables (ovals) representing “Early” (EarlyNP) and “Late” (LateNP) NP pathology (Figure 1). These are indicated by observed variables (squares) representing NP counts in regions affected early and late in AD's clinical evolution, according to longitudinal studies of in vivo amyloid formation [3-5]. The validity of these two factors was confirmed by factor analysis.
Figure 1.
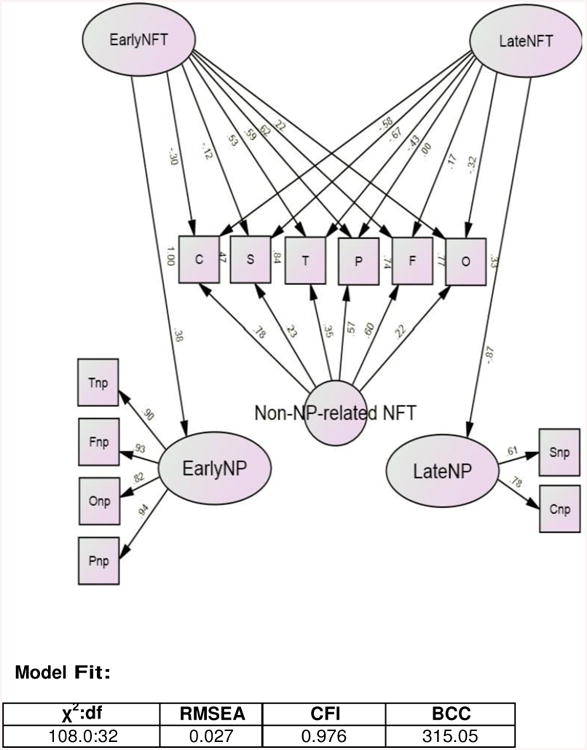
Observed NP-related NFT Counts are Most Strongly Associated with “Early” and “Late” NP lesions in the Same Regions of Interest*.
* Restricted to subjects with unimpaired CASI > 70/100 at baseline. All observed indicators are adjusted for age at death, last CASI score, and education.
χ2 = chi-square; BCC = Browne Cudeck Criteria; CASI = Cognitive Abilities Screening Instrument; CFI = comparative fit index; df = degrees of freedom; NFT = neurofibrillary tangle (C = CA1 NFT, F = frontal lobe NFT, O = occipital lobe NFT, P = parietal lobe NFT, S = subiculum NFT, T = temporal lobe NFT); NP = neuritic plaque (Cnp = CA1 NP, Fnp = frontal lobe NP, Onp = occipital lobe NP, Pnp = parietal lobe NP, Snp = subiculum NP, Tnp = temporal lobe NP); RMSEA = root mean square error of approximation.
Next we constructed latent variables representing the variance in NFT counts shared with those “early” and “late” NP lesions (i.e., “EarlyNFT” and “LateNFT” respectively). The NP-derived latent variables (EarlyNP and LateNP) were themselves used as indicators of these new latent constructs. At this stage, all the variance in regional NFT counts was effectively been parsed between: 1) that which is shared with “Early” NP lesions, 2) that which is shared with “Late” NP lesions, and 3) residual measure specific “measurement error”, including any interregional NFT variance not related to NP.
In Figure 1, the observed NFT variables have been ordered in a specific corticotropic sequence from left to right [i.e. Cornus Ammon 1 (C) > subiculum (S) > temporal (T) > parietal (P) > frontal (F)]. We have previously shown that this sequence best describes the interregional vulnerability of these structures to NFT formation in the HAAS sample [2].
It was our intention that a spatial gradient should emerge from the factor loadings between each successive structure and the latent variables “EarlyNFT” and “LateNFT”. In this way, the temporal propagation of NFT across these structures might be revealed. We predicted that hippocampal NFT counts would be most strongly associated with “early” NP development in the neocortex, and that neocortical NFT will be most strongly associated with “late” NP in the hippocampus, i.e., that NFT and NP formation would originate in different locations and propagate toward each other. Furthermore, we hoped to leverage the temporal evolution of NP derived from recent imaging data in order to “date” the spatial distribution of corticotropic NFT formation, as described by Braak.
However, model fit at this stage was marginally adequate and the residuals of the latent factors' NFT indicators had to be densely intercorrelated to achieve that fit. This suggested the existence of additional factor(s) indicated solely by the NFT variables. We therefore added such a factor and labeled it “non-NP-related NFT”. The final model is presented in Figure 1.
Missing data
AMOS uses Full Information Maximum Likelihood (FIML) methods to address missing data. FIML uses the entire observed data matrix to estimate parameters with missing data. In contrast to listwise or pairwise deletion, FIML yields unbiased parameter estimates, preserves the overall power of the analysis, and is currently the accepted state-of-the-art method in addressing issues of missing data [18-19].
The final model contains only pathological variables and covariates, including age at death, and last CASI score. The analysis was limited to cases with normal baseline CASI scores. Pathological material was limited to 471 cases with normal baseline CASI scores and complete autopsy data. Last CASI score and date of death were available in 440 or those. Braak stage was available in 240.
Goodness of fit
The fit of each structural model was assessed using three common test statistics. A non-significant chi-square signifies that the data are consistent with the model [20]. However, this statistic is vulnerable to large sample sizes, and often ignored in favor of additional measures. A root mean square error of approximation (RMSEA) of 0.05 or less indicates a close fit to the data, with models below 0.05 considered “good” fit, and up to 0.08 as “acceptable” [21]. The comparative fit index (CFI) compares the specified model with a model of no change [22]. CFI values below 0.95 suggest model misspecification. Values >0.95 indicate adequate to excellent fit. The Browne Cudeck Criteria (BCC) [23] address the issue of parsimony and is useful for comparing two models that are not necessarily nested, with lower BCC values indicating better fit.
Results
Sample characteristics are presented in Table 1. Mean lesion counts by anatomical region have been reported elsewhere [2]. Model fit was excellent (χ2:df = 108.0:32, p <0.001; RMSEA = 0.027; CFI= 0.976; BCC = 315.05).
Table 1. Demographic Features of Autopsied Decedents*.
| N | Mean | SD | |
|---|---|---|---|
| Baseline Age (yrs) | 471 | 77.8 | 4.4 |
| Education (yrs) | 471 | 11.1 | 3.3 |
| Baseline CASI Score | 471 | 86.8 | 6.6 |
| Last CASI Score Before Death | 440 | 72.1 | 22.4 |
| Age at Death (yrs) | 440 | 86.3 | 5.1 |
| Braak Stage | 240 | 3.6 | 1.3 |
| Brain Weight (g) | 471 | 1233.7 | 123.5 |
Restricted to subjects with unimpaired CASI > 70/100 at baseline.
The NP data load strongly on “EarlyNP” and “LateNP” (Figure 1). This suggests that they are robust factors. Their validity is further supported by their relative associations with pre-morbid CASI scores (Table 2). Only EarlyNP is significantly associated with Exam 5 CASI performance (circa 1994-1996) (partial r = -0.19, p = 0.003, adjusted for age at baseline, education, and LateNP). Only EarlyNP is again significantly associated with Exam 6 CASI performance (circa 1997-1999) (partial r = -0.16, p = 0.037). However, LateNP's association increases in strength and approaches significance. In contrast, only LateNP is significantly associated with Exam 7 CASI performance (circa 1999-2000) (partial r = -0.29, p = 0.023, adjusted for age at baseline, education, and EarlyNP). This suggests that from a non-demented baseline, neocortical NP can propagate into the allocortex over a period of 2-3 years, while neocotical NP formation slows or remains stable. Both findings are consistent with longitudinal β-amyloid neuroimaging [6].
Table 2. Partial Correlations from Multivariate Regression Models*.
| Predictors | |||
|---|---|---|---|
| Outcomes | “Early” NP | “Late” NP | |
| Model 1 | Exam 5 CASI | -0.19, p = 0.003 | -0.06; p = 0.43 |
| Model 2 | Exam 6 CASI | -0.16, p = 0.037 | -0.18, p = 0.061 |
| Model 3 | Exam 7 CASI | -0.02, p = 0.858 | -0.29, p = 0.023 |
Standardized parameters. Restricted to subjects with unimpaired CASI > 70 at baseline and adjusted for age at baseline, and education.
The NFT data are distributed across three factors: EarlyNFT, LateNFT and Non- NP-related NFT. EarlyNFT and LateNFT are not significantly correlated with each other, nor are they significantly correlated with each other's residual in NP counts. Thus, they appear to be orthogonal. Together (with covariates), these factors account for 100% of the variance in CA1 NFT counts, 47% of the variance in subiculum NFT counts, 84% of the variance in temporal lobe counts, 74% of the variance in parietal lobe counts, 77% of the variance in frontal lobe counts, and 33% of the variance in occipital lobe counts.
The latent variables EarlyNP and LateNP load significantly on “EarlyNFT” and “LateNFT” respectively, suggesting that early and late NP pathology share variance with regional NFT counts. The amount of variance shared by early NP and early NFT is relatively modest (R = 0.14) compared with the variance shared between late NP and late NFT (R = 0.76). This suggests that neocortical NP and NFT formation are much more tightly bound than are allocortical NP and NFT formation.
There is also wide variation in the strengths of the regional loadings within the Early and Late NFT factors. In contrast to our hypothesis, NFT in neocortical regions (frontal lobe r = 0.62; parietal lobe r = 0.59, and temporal lobe r = 0.53) are most strongly associated with “Early” NP counts in the same structures. NFT in hippocampal regions (CA1 |r|= 0.58; subiculum |r|= 0.67) are most strongly associated with “Late” NP counts in those two structures.
In general, these factor loadings reveal that regional NP-related NFT counts share most variance with NP counts in the same structures. This does not seem surprising until one realizes that the observable corticofugal temporal spread of β-amyloid is incompatible with the hypothesized corticotropic temporal spread of NFT hypothesized by Braak, and confirmed in cross-sectional autopsy data [2].
Moreover, not all the variance in NFT is NP-related. Our model posits a third compartment of variance in NFT counts: “Non-NP-related NFT”. Model fit is substantially and significantly reduced if this factor is omitted from the model (χ2:df = 276.20:39, p <0.001; RMSEA = 0.043; CFI= 0.925; BCC = 469.17). Non- NP-related NFT is most strongly associated with NFT in CA1 (r = 0.78), the frontal lobe (r = 0.60) and the parietal lobe (r = 0.57) (all p <0.001).
The relative variation in regional NFT counts that is attributable to each latent construct can be simultaneously compared. CA1 NFT counts are significantly associated with all three latent variables, but most strongly associated with Non- NP-related NFT. Subiculum NFT are most strongly associated with Non-NP- related NFT and have no significant association with Early-NP-related NFT. This suggests that the subiculum might be involved late in the AD process, despite its close physical and neuroanatomical proximity to CA1. The temporal lobe seems to be vulnerable to all three NFT forming processes. In contrast, the frontal and parietal lobes are relatively unaffected by late NP-related NFT formation.
Finally, we examined the associations between all three latent NFT factors and Braak stage, in a multivariate regression model. Age at death, last CASI and education were entered first and explained 11% of the variance in Braak stage. 51% was explained after the addition of EarlyNFT, LateNFT and non-NP-related NFT to the model. LateNFT (partial |r|= 0.576) and non-NP-related NFT (r = 0.332) made significant independent contributions to Braak stage (both p <0.001). EarlyNFT made no significant independent contribution.
Discussion
This analysis offers revealing insights into NFT formation. First, at least two NFT forming processes appear to be responsible for the observed pattern of interregional NFT counts. Only one of these is NP-related. Together, NP-related and NP independent NFT formation account for the majority of variance in neocortical NFT counts, and essentially all the variance in CA1 counts.
Second, NP-related NFT are co-localized with NP in the same regions. Since the temporal development of NP can be estimated by longitudinal in vivo β-amyloid imaging, this finding has implications for the temporal evolution of NP-related tangleopathy. Like β-amyloid, NP-related NFT may develop pre-clinically in the neocortex and propagate corticofugally towards the allocortex. This speculation is supported by CA1 and notably the subiculum's relatively strong associations with late, as opposed to early NP-related NFT, the relatively strong associations of EarlyNFT to neocortical structures, and the lack of any significant association between late NP-related NFT formation and NFT counts in the parietal lobe. The subiculum is a major center for hippocampal corticotropic output [24]. Its specific involvement by late as opposed to early NFT formation supports the concept of corticofugal retrograde transynaptic propagation of neocortical NFT.
We have previously shown in HAAS data that NP are arranged corticofugally in space [2] and that neocortical NFT develop contemporaneously with neocortical NP over the course of CASI surveillance [11]. This supports our conclusion that NP-related NFT formation is corticofugal, and suggests that the process has clinical consequences.
Several previous studies support the possibility that only NP-related NFT formation has clinical consequences. Neocortical NFT are more clinically salient [25]. NFT counts in the entorhinal cortex and hippocampus are associated with the cognitive performance of normal elders, whereas NFT formation in neocortical structures is associated with clinical AD [26-28].
We have related neocortical NFT and NP to longitudinal change in cognition [11]. We found that the cognitively-salient fractions of NP and NFT develop concurrently, a decade or less before death, and not before. Since those lesions were measured in the neocortex, cognitively salient NFT formation also probably represents NP-related NFT formation, which we have found here to propagate corticofugally. Therefore, our findings appear to contradict the progressive hierarchical propagation of NFT described by Braak, and incorporated into current dementia staging [1, 29]. On the other hand, since mean NFT counts nevertheless appear to be corticotropically oriented on average, NP-related NFT formation may not be the dominant tangleopathy in this cohort.
The obvious candidate for corticotropic NFT formation would be non-NP-related NFT formation. The temporospatial development of that process cannot be determined from this analysis but, if it were found to be the case, Braak's system would describe an unlikely AD-related tangleopathy. First, non-NP-related NFT formation is by definition not NP-related, and second, allocortical NFT are less clinically salient. However, if non-NP-related NFT formation is in fact the dominant corticotropically distributed process, that might explain why a significant fraction of autopsies present “outside the box” of expected corticotropic NP- related NFT propagation [30]. Moreover, since both processes contribute independently to the observed NFT counts in any region, it would be impossible to distinguish them by visual inspection, especially in a single case or small case-series.
Furthermore, since Braak stage at autopsy is significantly associated with two latent constructs, it too risks to confound NP-related and NP-independent tangleopathies, especially if they co-occur in the same brains. That possibility cannot be determined by this analysis. Both processes might co-occur in the same brains, or either could manifest uniquely in subgroups of the HAAS participants.
It has been suggested that neocortical NFT in the absence of NP may represent a distinct pathological entity [31]. Non-NP-related NFT's strong associations with CA1, the frontal and parietal lobes suggests that this process may be a candidate to explain one of the atypical frontal lobe “AD” syndromes [32]. However, NFT counts in the absence of NP also appear to be less strongly associated with dementia risk [33] and might be due to normal aging or other factors. Finally, a very late onset NFT-“only” dementia has been described, that is largely limited to the allocortex [34]. Thus, our non-NP-related NFT factor may encompass several different conditions.
In contrast to neocortical NFT, allocortical NFT in the absence of NP is a common autopsy finding. This is often interpreted as a pre-clinical manifestation of AD [35]. However, non-demented cases with allocortical NFT are more likely to be due to the non-NP-related NFT forming process because NP-related NFT formation appears to originate in the neocortex and because neocortical NFT are clinically salient, and therefore potentially dementing. Neocortical NFT formation in the absence of neocortical NP is rare, but has also been reported [31, 33].
Since isolated neocortical NFT formation is rare, while isolated allocortical NFT formation is not, either non-NP-related NFT formation begins the allocortex and is trapped there, or it frequently progresses to the neocortex and is overrun by corticofugal NP-related NFT formation.
The first possibility is supported by the subiculum's relatively weak association with non-NP related NFT. If hippocampal NFT were propagating coticotropically, the subiculum would be strongly affected. In HAAS, subiculum counts are relatively low compared with CA1, although those structures are separated by a single synapse. Regardless, subiculum NFT are much “closer” to neocortical neuropathology [2]. This suggests a “barrier” to corticotropic NFT propagation between CA1 and the subiculum (and /or corticofugal entry of the hippocampus via the subiculum in a relatively small subset of cases).
The latter possibility is supported by strong associations between non-NP-related NFT and neocortical NFT counts. Moreover, corticotropic non-NP-related NFT formation would have to be usually overrun by corticofugal NP-related NFT formation, because corticotropic NFT formation appears to dominate the autopsy record and otherwise there would be many cases with isolated NFT lesions at high Braak stages. This suggests that NP-related and non-related NFT formation may be parts of a coordinated process, co-localized in the same brains.
Our analysis has several significant limitations. HAAS may not represent an unbiased distribution of regional NP and NFT burdens. The data would be unbiased only if they were sampled from a typically aged-distributed population. However, the autopsy panel favors older decedents and demented decedents [10]. Moreover, HAAS as a whole is limited to Japanese-American males. Despite these limitations, the HAAS is a population-based cohort and much closer to this ideal than many comparable large convenience samples.
Brain harvesting, cutting, and staining are vulnerable to myriad technical errors. However, not only did HAAS exert systematic quality controls, latent variables are inherently resistant to non-systematic measurement error of any kind [36]. Another potential limitation arises from the fact that β-amyloid imaging is sensitive to both neuritic and diffuse plaques (DP). We did not model DP in this analysis, although such data is available for future analysis. Finally, we also ignored Lewy bodies, which are commonly found with NFT and might interact with some of these constructs.
In summary, the NP-related fraction of NFT appears to be co-localized spatially with NP. That finding has implications for the temporal spread of NFT. Since NP evolve corticofugally in time, this suggests that NP-related NFT do so as well. This challenges the presumed corticotropic evolution of AD lesions, which is the foundation of Braak staging. Non-NP-related NFT formation may also occur, and could indeed be corticotropic. If so, then it must be the dominant tangle forming process. Never the less, it seems unlikely to be clinically salient and very likely to be “overwritten” by clinically salient corticofugal NP-related NFT formation. This suggests that the two processes are related. Clinically silent corticotropic NFT formation may therefore trigger clinically salient NP-related cofticofugal NFT formation. Further analyses are required to better understand the temporal spread of NFT. If these findings stand, they may have broad implications for AD staging, and our conceptualization of that disease's natural history.
Acknowledgments
HAAS data collection has been supported by U01 AG019349 from the National Institute on Aging, contract N01-AG-4-2149, and National Heart Lung and Blood Institute R21 Grant NS048123-01. HAAS data were provided by Lon R. White (LRW), the HAAS Principal Investigator, but HAAS staff were not further involved in its analysis or interpretation. DRR was also funded by the Julia and Van Buren Parr professorship in Aging and Geriatric Psychiatry. Portions of this work were presented at the 2011 AAICAD. Paris, France. The authors accept full responsibility for all analyses, results and interpretations.
This work has been supported by NINDS R21 Grant NS048123-01, contract N01- AG-4-2149, and grant U01 AG019349 from the National Institute on Aging. Portions of this work were presented at the 2011 AAICAD. Paris, France.
Footnotes
Conflicts of interest: None.
References
- 1.Braak H, Braak E. Neuropathological staging of Alzheimer related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 2.Royall DR, Palmer RF, White LR, Petrovitch H, Ross GW, Masaki K. Modeling regional vulnerability to Alzheimer pathology. Neurobiol Aging. 2012;33:1556–1563. doi: 10.1016/j.neurobiolaging.2011.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li Y, Rinne JO, Mosconi L, Pirraglia E, Rusinek H, DeSanti S, Kemppainen N, Någren K, Kim BC, Tsui W, de Leon MJ. Regional analysis of FDG and PIB-PET images in normal aging, mild cognitive impairment, and Alzheimer's disease. Eur J Nucl Med Mol Imaging. 2008;35:2169–2181. doi: 10.1007/s00259-008-0833-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jack CR, Jr, Lowe VJ, Weigand SD, Wiste HJ, Senjem ML, Knopman DS, Shiung MM, Gunter JL, Boeve BF, Kemp BJ, Weine M, Petersen RC. Serial PiB and MRI in normal, mild cognitive impairment and Alzheimer's disease: Implications for sequence of pathological events in Alzheimer's disease. Brain. 2009;132:1355–1365. doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Villain N, Chételat G, Grassiot B, Bourgeat P, Jones G, Ellis KA, Ames D, Martins RN, Eustache F, Salvado O, Masters CL, Rowe CC, Villemagne VL. Regional dynamics of amyloid-β deposition in healthy elderly, mild cognitive impairment and Alzheimer's disease: a voxelwise PiB-PET longitudinal study. Brain. 2012;135:2126–2139. doi: 10.1093/brain/aws125. [DOI] [PubMed] [Google Scholar]
- 6.Villemagne VL, Rowe CC. Amyloid imaging. Int Psychogeriatr. 2011;23(Suppl 2):S41–49. doi: 10.1017/S1041610211000895. [DOI] [PubMed] [Google Scholar]
- 7.Davis DG, Schmitt FA, Wekstein DR, Markesbery WR. Alzheimer neuropathic alterations in aged cognitively normal subjects. J Neuropath Exp Neurol. 1999;58:376–388. doi: 10.1097/00005072-199904000-00008. [DOI] [PubMed] [Google Scholar]
- 8.Fukuktani Y, Cairns NJ, Shiozawa M, Sasaki K, Sudo S, Isaki K, Lantos PL. Neuronal loss and neurofibrillary degeneration in late-onset sporadic Alzheimer's disease. Psychiatr Clin Neurosci. 2000;54:523–529. doi: 10.1046/j.1440-1819.2000.00747.x. [DOI] [PubMed] [Google Scholar]
- 9.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 10.White L. Brain lesions at autopsy in older Japanese-American men as related to cognitive impairment and dementia in the final years of life: a summary report from the Honolulu-Asia aging study. J Alz Dis. 2009;18:713–725. doi: 10.3233/JAD-2009-1178. [DOI] [PubMed] [Google Scholar]
- 11.Royall DR, Palmer RF. Estimating the temporal evolution of Alzheimer's disease pathology with autopsy data. J Alz Dis. 2012;32:23–32. doi: 10.3233/JAD-2012-120430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.White L, Small BJ, Petrovitch H, Ross GW, Masaki K, Abbott RD, Hardman J, Davis D, Nelson J, Markesbery W. Recent clinical-pathologic research on the causes of dementia in late life: update from the Honolulu-Asia Aging Study. J Geriatr Psychiatry Neurol. 2005;18:224–227. doi: 10.1177/0891988705281872. [DOI] [PubMed] [Google Scholar]
- 13.White L, Petrovitch H, Hardman J, Nelson J, Davis DG, Ross GW, Masaki K, Launer L, Markesbery WR. Cerebrovascular pathology and dementia in autopsied Honolulu-Asia Aging Study participants. Ann NY Acad Sci. 2002;977:9–23. doi: 10.1111/j.1749-6632.2002.tb04794.x. [DOI] [PubMed] [Google Scholar]
- 14.Petrovitch H, White L, Ross GW, Steinhorn SC, Li CY, Masaki KH, Davis DG, Nelson J, Hardman J, Curb JD, Blanchette PL, Launer LJ, Yano K, Markesbery WR. Accuracy of clinical criteria for AD in the Honolulu-Asia Aging Study, a population-based study. Neurology. 2001;57:226–234. doi: 10.1212/wnl.57.2.226. [DOI] [PubMed] [Google Scholar]
- 15.Hasegawa K, Honma A, Ima Y. An epidemiological study of age-related dementia in the community. Int J Geriatr Psychiatr. 1986;1:45–55. [Google Scholar]
- 16.Teng EL, Hasegawa K, Homma A, Imai Y, Larson E, Graves A, Sugimoto K, Yamaguchi T, Sasaki H, Chiu D. The Cognitive Abilities Screening Instrument (CASI): a practical test for cross-cultural epidemiological studies of dementia. Int Psychogeriatr. 1994;6:45–58. doi: 10.1017/s1041610294001602. [DOI] [PubMed] [Google Scholar]
- 17.Arbuckle JL. Analysis of Moment Structures-AMOS (Version 7.0) [Computer Program] Chicago: SPSS; 2006. [Google Scholar]
- 18.Schafer JL, Graham JW. Missing data: Our view of the state of the art. Psychol Methods. 2002;7:147–177. [PubMed] [Google Scholar]
- 19.Graham JW. Missing Data Analysis: Making it work in the real world. Ann Rev Psychol. 2009;6:549–576. doi: 10.1146/annurev.psych.58.110405.085530. [DOI] [PubMed] [Google Scholar]
- 20.Bollen KA, Long JS. Testing Structural Equation Models. Sage Publications; Thousand Oaks, California: 1993. [Google Scholar]
- 21.Browne M, Cudeck R. Alternative ways of assessing model fit. In: Bollen KA, Long JS, editors. Testing Structural Equation Models. Sage Publications; Thousand Oaks, California: 1993. pp. 136–162. [Google Scholar]
- 22.Bentler PM. Comparative fit indexes in structural models. Psychol Bull. 1990;107:238–246. doi: 10.1037/0033-2909.107.2.238. [DOI] [PubMed] [Google Scholar]
- 23.Browne MW, Cudeck R. Single sample cross-validation indices for covariance structures. Multivariate Behav Res. 1989;24:445–455. doi: 10.1207/s15327906mbr2404_4. [DOI] [PubMed] [Google Scholar]
- 24.O'Mara SM, Commins S, Anderson M, Gigg J. The subiculum: A review of its form, physiology and function. Prog Neurobiol. 2001;64:129–155. doi: 10.1016/s0301-0082(00)00054-x. [DOI] [PubMed] [Google Scholar]
- 25.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not amyloid plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 26.Riley KP, Snowdon DA, Markesbery WR. Alzheimer's neurofibrillary pathology and the spectrum of cognitive function: findings from the Nun Study. Ann Neurol. 2002;51:567–577. doi: 10.1002/ana.10161. [DOI] [PubMed] [Google Scholar]
- 27.Giannakopoulos P, Herrmann FR, Bussiere T, Bouras C, Kovari E, Perl DP, Morrison JH, Gold G, Hof PR. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology. 2003;60:1495–1500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- 28.Guillozet AL, Weintraub S, Mash D, Mesulam M. Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Arch Neurol. 2003;60:729–736. doi: 10.1001/archneur.60.5.729. [DOI] [PubMed] [Google Scholar]
- 29.Bierer LM, Hof PR, Purohit DP, Carlin L, Schmeidler J, Davis KL, Perl DP. Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer's disease. Arch Neurol. 1995;52:81–88. doi: 10.1001/archneur.1995.00540250089017. [DOI] [PubMed] [Google Scholar]
- 30.Nelson PT, Kukull WA, Frosch MP. Thinking outside the box: Alzheimer's-type pathology that does not map directly onto current consensus recommendations. J Neuropath Exp Neurol. 2010;69:449–454. doi: 10.1097/NEN.0b013e3181d8db07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nelson PT, Abner EL, Schmitt FA, Kryscio RJ, Jicha GA, Santacruz K, Smith CD, Patel E, Markesbery WR. Brains with medial temporal lobe neurofibrillary tangles but no neuritic amyloid plaques are a diagnostic dilemma but may have pathogenetic aspects distinct from Alzheimer disease. J Neuropathol Exp Neurol. 2009;68:774–784. doi: 10.1097/NEN.0b013e3181aacbe9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Murray ME, Graff-Radford NR, Ross OA, Petersen RC, Duara R, Dickson DW. Neuropathologically defined subtypes of Alzheimer's disease with distinct clinical characteristics: a retrospective study. Lancet Neurol. 2011;10:785–796. doi: 10.1016/S1474-4422(11)70156-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petrovitch H, Ross GW, Hea Q, Uyehara-Lock J, Markesbery WR, Davis DG, Nelson J, Masaki KH, Launer LJ, White LR. Characterization of Japanese- American men with a single neocortical AD lesion type. Neurobiol Aging. 2008;29:1448–1455. doi: 10.1016/j.neurobiolaging.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jellinger KA, Bancher C. Senile dementia with tangles (Tangle predominant form of senile dementia) Brain Pathol. 1998;8:367–376. doi: 10.1111/j.1750-3639.1998.tb00160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haroutunian V, Purohit DP, Perl DP, Marin D, Kahn K, Lantz M, Davis KL, Mohs RC. Neurofibrillary tangles in non-demented elderly subjects and mild Alzheimer disease. Arch Neurol. 1999;56:713–718. doi: 10.1001/archneur.56.6.713. [DOI] [PubMed] [Google Scholar]
- 36.Palta M, Lin CY. Latent variables, measurement error and methods for analyzing longitudinal binary and ordinal data. Stat Med. 1999;18:385–396. doi: 10.1002/(sici)1097-0258(19990228)18:4<385::aid-sim25>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]