

Abstract
Nicotinamide adenine dinucleotide (NAD+) plays a central role in cellular metabolism both as coenzyme for electron-transfer enzymes as well as a substrate for a wide range of metabolic pathways. In the current study NAD+ was detected on rat brain in vivo at 11.7 T by 3D localized 1H MRS of the NAD+ nicotinamide protons in the 8.7 – 9.5 ppm spectral region. Avoiding water perturbation was critical to the detection of NAD+ as strong, possibly indirect cross-relaxation between NAD+ and water would lead to a several-fold reduction of the NAD+ intensity in the presence of water suppression. Water perturbation was minimized through the use of LASER localization in combination with frequency-selective excitation. The NAD+ concentration in the rat cerebral cortex was determined at 296 ± 28 μmol/L, which is in good agreement with recently published 31P NMR-based results as well as results from brain extracts in vitro (355 ± 34 μmol/L). The T1 relaxation time constants of the NAD+ nicotinamide protons as measured by inversion recovery were 280 ± 65 ms and 1136 ± 122 ms in the absence and presence of water inversion, respectively. This confirms the strong interaction between NAD+ nicotinamide and water protons as observed during water suppression. The T2 relaxation time constants of the NAD+ nicotinamide protons were determined at 60 ± 13 ms after confounding effects of scalar coupling evolution were taken into account. The simplicity of the MR sequence together with the robustness of NAD+ signal detection and quantification makes the presented method a convenient choice for studies on NAD+ metabolism and function. As the method does not critically rely on magnetic field homogeneity and spectral resolution it should find immediate applications in rodents and humans even at lower magnetic fields.
Keywords: NAD+, 1H MRS, brain, T1, T2, water
Graphical abstract
The NAD+ nicotinamide H2, H4 and H6 resonances were detected and quantified in rat brain in vivo at 11.7 T. Minimizing water perturbation by frequency-selective excitation (B) was critical for NAD+ detection as cross-relaxation between NAD+ and water would lead to NAD+ signal destruction in the presence of water suppression (A). Detailed NAD+ T1 and T2 measurements further characterized the in vivo NAD+ signal.
Introduction
Nicotinamide adenine dinucleotide (NAD+) and its reduced form, NADH, have central roles in cellular metabolism and energy production as electron-accepting and electron-donating coenzymes. Reduction-oxidation (redox) reactions catalyzed by various NAD(H)-dependent dehydrogenases are vital for biochemical processes such as glycolysis and mitochondrial metabolism. In addition to its well-known role as a coenzyme for electron-transfer enzymes, NAD+ is also a substrate for ADPribose transferases, poly(ADP-ribose) polymerases, cADP-ribose synthases and sirtuins (1–3). Active biosynthesis via salvage or de novo pathways is thus required in order to maintain NAD+ levels. Taken together, the critical involvement of NAD+ in key cellular processes related to gene expression and repair, calcium mobilization, metabolism and aging (4, 5), cancer and cell death (6, 7) and the timing of metabolism via the circadian rhythm (8, 9) highlights the need for quantitative techniques to assess NAD+ levels non-invasively in vivo.
Until recently, the options for in vivo detection of NAD+ and NADH have been limited. The use of chemical, enzyme-linked fluorescence or HPLC assays to detect NAD+ and NADH directly or indirectly are available, but they require the use of tissue extracts and are therefore incompatible with (longitudinal) in vivo studies (10, 11). A less invasive approach utilizes the autofluorescence signal of NADH coupled with confocal microscopy (12). However, among other complications this method has limited tissue penetration and is unable to detect NAD+.
Lu et al (13) have recently detected NAD+ and NADH simultaneously through the use of in vivo 31P NMR spectroscopy. Even though the 31P NMR signal of the total NAD pool (i.e. NAD+ and NADH) has been observed since the early days of in vivo 31P NMR, the separation of the heavily overlapping NAD+ and NADH 31P NMR signals was deemed impossible. However, the availability of (ultra) high field magnets together with the use of spectral fitting routines makes it possible to disentangle the NAD+ and NADH contributions to the observed 31P NMR signals. With this novel 31P NMR method the concentrations of NAD+ and NADH, and thus the redox state, have been established in normal rat (13), cat (14) and human (15) brain as well as during ischemia in the rat brain (14).
Here we present a novel method for the detection of the NAD+ nicotinamide protons in the rat brain in vivo with 1H NMR spectroscopy. The method relies on the fact that the non-exchangeable NAD+ nicotinamide proton signals can be observed directly if perturbation of the water protons is minimized by frequency-selective excitation. The non-overlapping NAD+ nicotinamide proton signals are readily converted to concentrations and are compared to previously reported values. The T1 and T2 relaxation times of the NAD+ nicotinamide protons were measured on rat brain in situ. To demonstrate the importance of minimizing perturbation of the water protons, both frequency-selective and non-selective T1 measurements were performed.
Methods
Animal preparation
For in vivo studies three male Sprague-Dawley rats (215 ± 14 g, mean ± SD) were prepared in accordance to the guidelines established by the Yale Animal Care and Use Committee. The animals were tracheotomized and ventilated with a mixture of 70 % nitrous oxide and 28.5 % oxygen under 1.5 % isoflurane anesthesia. A femoral artery was cannulated for monitoring of blood gases (pO2, pCO2), pH and blood pressure. Physiological variables were maintained within normal limits by small adjustments in ventilation (pCO2 = 33–45 mm Hg; pO2 > 120 mm Hg; pH = 7.20–7.38; blood pressure = 90–110 mm Hg). After all surgery was completed, anesthesia was maintained by 0.3 – 0.7 % isoflurane in combination with 70 % nitrous oxide. During NMR experiments the animal core temperature was measured with a rectal thermosensor and was maintained at 37 ± 1 °C by means of a heated water pad.
For the in situ studies four male Sprague-Dawley rats (208 ± 15 g, mean ± SD) were euthanized by microwave irradiation using the commercially available 10 kW Muromachi Microwave Fixation System (Stoelting Co, Wood Dale, IL) in accordance to the guidelines established by the Yale Animal Care and Use Committee. Under light isoflurane anesthesia the animals were place in dedicated animal holders to ensure proper positioning within the device. As previously described (16), the optimal fixation power and duration represents a compromise between stopping metabolism and the generation of microscopic air bubbles that severely compromise the magnetic field homogeneity. The current group of animals was irradiated at 4.5 kW in 1.1 s. Following microwave fixation all extracranial tissues were removed after which the skull and brain were immersed in a 17 mm diameter, 55 mm long acrylic tube. In order to minimize magnetic susceptibility-related artifacts the tube was filled with Fluorinert FC-43 (3M, St. Paul, MN), an inert, clear fluid with a magnetic susceptibility close to water (17).
A separate group of animals (n = 4, 212 ± 13 g, mean ± SD) were euthanized by microwave irradiation, after which specific brain areas were dissected. Dissected tissue sections from various brain locations (50 – 100 mg) were homogenized with 0.1 M HCl/methanol (2:1 vol/wt) followed by extraction with ethanol. The supernatant was clarified by centrifugation after which potential heavy metal contaminants were removed on Chelex-100 columns. No efforts were made to determine the extraction efficiency. Samples were lyophilized for long-term storage at −80°C.
Sample preparation
High-resolution 1H NMR studies were performed on solutions of 2 mM NAD+ or 2 mM NADH (Sigma Aldrich, St. Louis, MO, USA), 0.1 mM 2,2-dimethyl-2-silapentane-5-sulfonate-D6 (DSS-D6) and 50 mM phosphate buffer (pH = 7.2) dissolved in 95/5% H2O/D2O.
Lyophilized brain extracts were resuspended in 700 uL of phosphate-buffered (50 mM, pH 7.2) H2O/D2O (15/85%) solution containing 0.5 mM DSS-D6.
1H NMR spectroscopy
All in vivo and in situ experiments were performed on an 11.74 T Magnex magnet (Magnex Scientific Ltd, Oxford, UK) interfaced to an Agilent Direct Drive Spectrometer (Agilent Technologies, Santa Clara, CA) equipped with 9.0 cm diameter Magnex gradients capable of switching 395 mT/m in 180 us. For in vivo experiments RF transmission and reception was performed with a 14 mm diameter surface coil tuned to the proton NMR frequency (499.8 MHz). For all in situ experiments RF transmission and reception was achieved with a five-turn, 25 mm diameter solenoidal coil of 45 mm length.
In vitro experiments were performed on a Bruker Avance III HD spectrometer (Bruker Instruments, Billerica, MA) operating at 500.13 MHz for 1H and equipped with a 5-mm triple resonance probe incorporating triple-axis gradient coils. The magnetic field homogeneity on each sample was optimized with an automated 3D field mapping algorithm capable of adjusting up to fifth order spherical harmonics. Following non-selective excitation with a 12 us hard pulse, free induction decays (FIDs) were acquired as 65,536 complex points over 6.55 s (TR = 30 s, NA = 128). 1H NMR spectra were obtained through zero-filling to 256K points, fast Fourier transformation (no apodization) and zero-order phase correction, giving a spectral width of 10 kHz. Water suppression was achieved with low-power presaturation over 23.40 s. 1H NMR spectra were acquired at 298 K and 310 K. 31P NMR spectra were acquired as 16,384 points over a 10 kHz spectral width (TR = 5 s, NA = 2048, 30° excitation). 2D NMR spectra were acquired to aid in the spectral assignment of NAD+. Specifically, 2D gradient-enhanced double-quantum-filtered (DQF) correlation spectroscopy (COSY) spectra ((18) 4,096 × 512 data matrix over 5 × 5 kHz, TR = 5 s, NA = 8) were acquired to establish 1H-1H connectivities. In addition, 2D gradient-enhanced 1H-31P heteronuclear multiple quantum coherence (HMQC) spectra (4,096 × 256 data matrix over 5 × 1 kHz, TR = 5 s, NA = 8) were acquired to establish 1H-31P connectivities.
All in vivo and in situ experiments were performed with a LASER sequence (19) consisting of six 0.75 ms adiabatic full passage pulses (R = bandwidth x pulse length = 20, sech/tanh modulation) surrounded by 0.5 ms 40 G/cm magnetic field crushers, giving a minimum echo-time TE of 14 ms. Non-selective and selective signal excitations were performed with 0.375 ms and 3.0 ms minimum-phase Shinnar-Le Roux optimized pulses ((20) R = 6.8), respectively. During non-selective excitation the transmitter offset was centered on the water resonance, whereas selective excitation was centered on 9.1 ppm. VAPOR water suppression (21) was employed during studies with non-selective excitation, whereas the minimal water perturbation during selective excitation did not require any additional water suppression. The magnetic field homogeneity was optimized up to second order with a multi-slice B0 mapping method as previously described (22).
All T1 and T2 measurements were performed in situ on an 8 × 4 × 8 mm3 = 256 uL localized volume encompassing parts of the cerebral cortex, corpus callosum and most of the hippocampus. T1 measurements were made with an inversion recovery method preceding the LASER localization sequence. A repetition time of 5120 ms was used, while the inversion time TI was varied as 1, 10, 20, 40, 80, 160, 320, 640, 1280, 2560 and 5120 ms. While all T1 measurements employed selective excitation, the inversion pulse was either selective (4.5 ms adiabatic full passage, R = 10, sech/tanh modulation, centered on 9.1 ppm) or non-selective (1.0 ms pulse centered on the water resonance). T2 measurements were made by varying the LASER echo-time TE according to 14, 20, 30, 40, 50, 60, 80, 100, 120 and 140 ms. All T2 measurements employed selective excitation with a repetition time TR = 2560 ms. The number of averages for each measurement point (TI or TE) was 640. All in situ measurements were performed at circa 310 K by blowing warm air through the bore of the magnet.
In vivo measurements were performed on a 6 × 1.5 × 5 mm3 = 45 uL volume encompassing cerebral cortex using TR/TE = 2000/14 ms. Using the fully excited water signal from a separate scan, the integrated NAD+ signals obtained in vivo were converted to absolute concentrations under the assumption of a 82% water content for cerebral gray matter (22). The NAD+ signals obtained from rat cerebral cortex extracts in vitro were converted to absolute concentrations under the assumption of a 10 mM total creatine concentration. The NAD+ signals obtained in situ were not converted to absolute concentrations as the large volume encompassed a large range of cerebral structures.
Data processing
All FIDs were processed in off-line NMR processing software written in Matlab 8.0 (The Mathworks, Natick, MA, USA). Processing included time-domain B0 correction using a water reference scan (23), zero-filling, exponential line broadening (0, 1 and 2 Hz for in vitro, in vivo and in situ 1H NMR data, respectively), Fourier transformation, phase correction and chemical shift referencing. The spectral areas of the NAD+ H2, H4 and H6 resonances were obtained by baseline correction and integration. Even though the water resonance was typically less than 1% of the full water intensity, removal of the residual water signal was deemed necessary for improved baseline correction and display. The residual water signal was removed by a Hankel SVD algorithm (24).
The T1 relaxation times were obtained with a least-squares three parameter fit (M0, Minv, T1) of the inversion times TI versus the observed signal intensity Mobs according to Mobs = M0 – Minv.exp(-TI/T1). The T2 relaxation times were obtained with a least-squares two parameter fit (M0, T2) of the echo-times TE versus the observed signal intensity Mobs according to Mobs = M0.F(J).exp(-TE/T2). F(J) is a signal modulation function that depends on the scalar couplings J for the particular resonance under investigation.
The spectral assignment of the high-resolution NAD+ NMR spectra was aided by density matrix simulations written in Matlab. Initial estimates for chemical shifts and scalar couplings were obtained by manual inspection of the 1H NMR spectrum. The final chemical shifts and scalar couplings were obtained by a least-squares optimization of the experimental and simulated 1H NMR spectra.
Results
Fig 1A shows the structure of NAD+. Only the carbon and phosphorus nuclei are labeled. Figs 1B–D show various parts of the high resolution 1H NMR spectrum of NAD+ at 298 K, while Fig. 1E shows the 31P NMR spectrum. The complete spectral assignment of the NAD+ 1H and 31P NMR spectra is summarized in Table 1. The peak height intensities for the ribose H1 and H1′ are different due to small line width variations.
Figure 1.

(A) Chemical structure of NAD+. (B–D) Various regions of the 1H NMR spectrum of NAD+ acquired at 500 MHz. All chemical shifts are referenced against the methyl protons in DSS-D6 at 0.00 ppm. The spectral regions show resonances from (B) the nicotinamide and adenine protons, (C) the ribose H1 and H1′ protons and (D) the ribose H2–H5 and H3′–H5′ protons. The ribose H2′ proton resonating at circa 4.75 ppm is eliminated by the water suppression. Note the 2x vertical scale expansion of (C, D) relative to (B). (E) 31P NMR spectrum of NAD+. Besides the 31P-31P scalar coupling the spectrum reveals extensive 1H-31P scalar couplings.
Table 1.
Proton chemical shifts and scalar coupling constants for NAD+
| Moiety | proton | chemical shift (ppm) | scalar coupling (Hz) 1 | ||||
|---|---|---|---|---|---|---|---|
| 298K | 310K | ||||||
| Adenine | H2 | 8.162 | 8.184 | ||||
| H8 | 8.420 | 8.415 | |||||
| Nicotinamide | H2 | 9.330 | 9.334 | 4JH2H4 | 1.3 | 3JH4H5 | 8.1 |
| H4 | 8.831 | 8.849 | 5JH2H5 | 0.7 | 4JH4H6 | 1.8 | |
| H5 | 8.191 | 8.210 | 4JH2H6 | 0.5 | 3JH5H6 | 6.3 | |
| H6 | 9.142 | 9.158 | 2JH2N | 1.8 | 2JH6N | 1.1 | |
| Ribose (adenine) | H1′ | 6.030 | 6.040 | 3JH1′H2′ | 5.9 | 4JH4′P2 | 2.2 |
| H2′ | 4.762 | 4.749 | 3JH2′H3′ | 5.2 | 2JH5′AH5′B | −11.7 | |
| H3′ | 4.502 | 4.498 | 3JH3′H4′ | 3.6 | 3JH5′AP2 | 4.8 | |
| H4′ | 4.372 | 4.364 | 3JH4′H5′A | 2.7 | 3JH5′BP2 | 5.3 | |
| H5′(A) | 4.249 | 4.242 | 3JH4′H5′B | 3.4 | |||
| H5′(B) | 4.199 | 4.196 | |||||
| Ribose (nicotinamide) | H1 | 6.080 | 6.091 | 3JH1H2 | 5.5 | 4JH4P1 | 2.9 |
| H2 | 4.484 | 4.494 | 3JH2H3 | 5.1 | 2JH5AH5B | −11.9 | |
| H3 | 4.427 | 4.432 | 3JH3H4 | 2.9 | 3JH5AP1 | 4.3 | |
| H4 | 4.539 | 4.543 | 3JH4H5A | 2.4 | 3JH5BP1 | 5.5 | |
| H5(A) | 4.360 | 4.353 | 3JH4H5B | 2.5 | |||
| H5(B) | 4.228 | 4.228 | |||||
Scalar coupling constants did not display a significant temperature dependence in the range 298 – 310K.
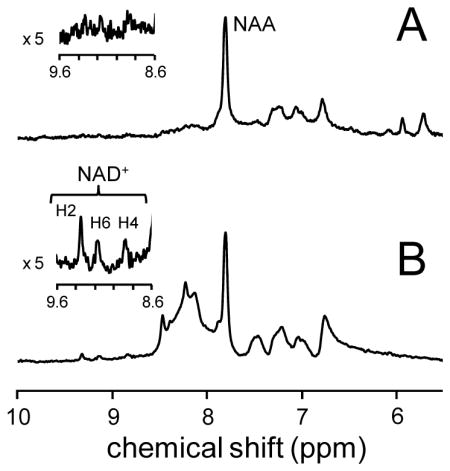
Fig. 2 shows in vivo 1H NMR spectra acquired with (A) non-selective excitation and VAPOR water suppression and (B) selective excitation without water suppression. The upfield region (0 – 4.2 ppm) in (A) is rich in chemical information content and shows the typically observed resonances from N-acetyl aspartate (NAA), glutamate (Glu), glutamine (Gln), total creatine (tCr = sum of creatine (Cr) and phosphocreatine (PCr)), taurine (Tau) and myo-inositol (mI). The information content in the downfield region (5.5 – 10 ppm) is relatively sparse with the most dominant signal arising from the NAA amide group at circa 7.9 ppm. The 1H NMR spectrum acquired with selective excitation centered on 9.1 ppm, thereby not perturbing the water, shows greatly enhanced signals in the downfield region. Most importantly, three well-separated signals can be observed at 8.8, 9.1 and 9.3 ppm in close correspondence to the three signals coming from NAD+ shown in Fig. 1B. Note that the upfield region in (B) is completely empty as the selective excitation pulse only excites signals between circa 6.8 and 11.4 ppm. The in vivo and in vitro NAD+ concentrations were determined as 296 ± 28 (n = 3) and 355 ± 34 μmol/L (n = 4, mean ± standard deviation), respectively.
Figure 2.

Localized 1H NMR spectra obtained from rat cerebral cortex in vivo (45 μL, TR/TE = 2000/14 ms, 768 averages, LASER localization) employing (A) VAPOR and (B) no water suppression. Water excitation in (B) was avoided by using a 3.0 ms frequency-selective excitation pulse (4.6 ppm bandwidth) centered at 9.1 ppm, whereas non-selective excitation in (A) was achieved with a 0.375 ms pulse (36.8 ppm bandwidth) centered on the water resonance.
Fig. 3 shows in situ 1H NMR spectra obtained during the T1 measurement employing an (A) selective and (B) non-selective inversion pulse. It can be seen that while the three NAD+ resonances cross the zero-intensity point around 160 ms when measured with a selective inversion pulse, the NAD+ T1 relaxation times are significantly prolonged in the presence of a non-selective inversion pulse (zero-intensity crossing around 640 ms). Figs. 3B–D graphically summarize the T1 relaxation recovery curves in the presence of selective and non-selective inversion pulses for (C) NAD+ H2, (D) NAD+ H4 and (E) NAD+ H6. Table 2 summarizes the quantitative T1 relaxation time constants. For all three resonances the T1 relaxation time constant in the presence of water inversion is circa four times longer than in the absence of a water perturbation. A similar lengthening of the observed T1 relaxation recovery can be seen for most other downfield resonances. The water T1 relaxation time constants obtained with inversion pulses that were selective and non-selective for the water protons were 1970 ± 55 and 2019 ± 71 ms, respectively.
Figure 3.

Localized 1H NMR spectra obtained from rat brain in situ (256 μL, TR/TE = 5120/14 ms, 640 averages) during (A) selective and (B) non-selective T1 measurements. (C–E) Integrated signal intensities as a function of inversion time for NAD+ nicotinamide protons (C) H2, (D) H4 and (E) H6 as measured by selective (black dots) and non-selective (gray dots) inversion. The black and gray lines represents the best three-parameter, single-exponential fit to the selective and non-selective data, respectively. Whereas all eleven relaxation recovery delays TI were used in the determination of T1, the longest delay of 5120 ms was omitted from (C–E) for an improved graphical display.
Table 2.
T1 and T2 relaxation of NAD+ H2, H4 and H6 1
| NAD+ H2 | NAD+ H4 | NAD+ H6 | ||
|---|---|---|---|---|
| T1, selective 2 | mean | 207 | 299 | 333 |
| std | 64 | 50 | 50 | |
| summed 4 | 203 | 295 | 332 | |
| T1, non-selective 3 | mean | 1004 | 1157 | 1246 |
| std | 80 | 99 | 173 | |
| summed 4 | 998 | 1151 | 1178 | |
| T2 | mean | 54 | 51 | 75 |
| std | 5 | 2 | 9 | |
| summed 4 | 58 | 54 | 70 |
Values are in ms with the mean and standard deviation (std) calculated over n = 4.
T1 relaxation measured with a selective inversion recovery method.
T1 relaxation measured with a non-selective inversion recovery method.
Represents a single fit of the summed data from all animals (n = 4)
Fig. 4A shows in situ 1H NMR spectra obtained during a T2 measurement. The signals from NAD+ H4 and H6 appear to decrease significantly faster than the signal from NAD+ H2. However, this is largely due to the more extensive scalar coupling in NAD+ H4 and H6. To a first approximation, NAD+ H2 behaves as a singlet, whereas NAD+ H4 and H6 behave as doublets. An inverted signal for NAD+ H4 can be seen at TE = 80 ms. A similar signal modulation can also be observed for the NAA amide peak which is scalar coupled to the aspartate-H2 proton. Figs. 4B–D graphically summarize the T2 relaxation decay curves for the three NAD+ protons. For (C) NAD+ H4 and (D) NAD+ H6 a clear signal modulation with negative signal intensity can be seen. As the scalar coupling constants are quantitatively known (Table 1), the T2 relaxation time constants can be unambiguously determined and are summarized in Table 2. The water T2 relaxation time constant was determined at 32 ± 5 ms.
Figure 4.

(A) Localized 1H NMR spectra obtained from rat brain in situ (256 μL, TR/TE = 2000/14 ms, 640 averages) during a T2 measurement. (B–D) Integrated signal intensities (black dots) as a function of echo time for NAD+ nicotinamide protons (B) H2, (C) H4 and (D) H6. The solid line represents the best two-parameter, single-exponential fit to the data. For the H4 and H6 protons the T2 decay curve is modulated by the scalar couplings listed in Table 1.
Discussion
Here it has been shown that 1H NMR spectroscopy allows the unambiguous detection of NAD+ in rat brain in vivo with simple, but robust methods. The three NAD+ nicotinamide protons H2, H4 and H6 offer a straightforward route to detect NAD+ without spectral overlap from other compounds. It was found that a single frequency-selective excitation pulse centered on 9.1 ppm provided clear NAD+ signals with minimal excitation of the water resonance. Preliminary efforts in generating multi-frequency pulses that excite both downfield and upfield spectral regions simultaneously also significantly perturbed the water resonance, thereby leading to unacceptable water suppression. However, providing broadband excitation without affecting the water resonance is certainly possible and can be achieved through the use of binomial or multi-frequency RF pulses (22).
One of the reasons that NAD+ has not been detected previously by in vivo 1H NMR spectroscopy is that the NAD+ signal intensity depends on the state of the water magnetization. In the majority of in vivo NMR studies the water signal is suppressed prior to excitation using methods like CHESS (25) or VAPOR (21). As was demonstrated in Fig. 2, suppression of the water signal by VAPOR leads to a strong depression of the NAD+ signals. As the NAD+ concentration is already low (circa 300 uM), any suppression of the NAD+ signals will result in a decreased ability to detect NAD+.
The interaction between NAD+ and water protons could also be observed during the T1 measurements. The T1 relaxation time constants for the NAD+ protons measured in the absence of water perturbation were about four times shorter than measured in the presence of water perturbation (Table 2 and Fig. 3). A qualitative explanation is that a large reservoir of magnetically unperturbed water can quickly restore the perturbed metabolic magnetization through chemical exchange and/or cross-relaxation. When the water magnetization has also been perturbed, e.g. by excitation or inversion, the replenishment of the metabolic magnetization is much slower and will be affected by the water T1 relaxation time. The interaction between water and labile, exchangeable protons and the effect on NMR signal intensities and apparent T1 relaxation times is well-known from biomolecular NMR studies on proteins and other macromolecules (26, 27). By using frequency-selective RF pulses or water ‘flip-back’ pulses significant sensitivity gains for exchangeable protons could be realized. For in vivo NMR the interaction between water and exchangeable protons has also been observed for water suppression in NMR spectroscopy (28) and chemical exchange saturation transfer (CEST) MRI (29). Of course, the NAD+ nicotinamide protons are non-exchangeable protons and as such the interaction with water is more complicated than simple chemical exchange and likely involves cross-relaxation and interactions between NAD+ protons and immobilized macromolecular proton pools. Magnetization transfer effects between mobile and immobile pools of creatine (30) and lactate (31) involving water as an intermediate have been observed in vivo. Similar to the NAD+ results obtained here, significant shortening of T1 relaxation times have recently been observed for non-labile protons of creatine, choline and N-acetyl aspartate during frequency-selective excitation and inversion (32).
Further studies will be needed to establish the exact mechanisms and relative contributions of chemical exchange and cross-relaxation underlying the observed T1 relaxation effects. However, in order to maximize the detection sensitivity of NAD+ nicotinamide proton signals it is crucial to minimize perturbation of the water protons. In this case the NAD+ T1 relaxation times are circa 300 ms, thereby allowing a fast repetition rate of 1000 ms without introducing any significant signal saturation.
The NAD+ T2 relaxation measurements revealed relatively long T2 relaxation time constants for all three NAD+ nicotinamide protons. The signal decay for the H4 and H6 protons appeared visually much faster than for the H2 proton (Fig. 4), but this was caused by the more extensive scalar coupling evolution for the H4 and H6 protons. When the scalar coupling evolution was taken into account according to the scalar couplings summarized in Table 1, the intrinsic T2 relaxation time constants were similar for all NAD+ nicotinamide protons.
Due to the long measurement durations required, the T1 and T2 measurements have been performed on rat brain in situ. The rat brains were prepared by focused beam microwave irradiation (FBMI), a method that very rapidly (< 1 s) heats up the brain tissue to circa 80 °C thereby instantly halting cerebral metabolism (33). Previous studies have shown identical 1H NMR spectral signatures, T2 weighted MRIs and diffusion tensor images between in vivo and in situ rat brains (16). As such we are confident that the measured T1 and T2 relaxation times for NAD+ are an accurate reflection of the in vivo situation. This is further supported by the measured T1 and T2 relaxation times for water which correspond well to previously reported values at 11.7 T (34).
The in vivo concentration of NAD+ (296 ± 28 umol/L) as determined by 1H NMR spectroscopy is in good correspondence to previously reported values ((13) and references therein). The acquisition of the in vivo 1H NMR spectrum shown in Fig. 1 was achieved from a 45 μL volume over circa 54 minutes with TR = 2000 ms. Given the short T1 relaxation times for NAD+ established in this study, the same spectrum could have been acquired in only 27 minutes with TR = 1000 ms. When these results are extrapolated, it appears that 1H NMR spectroscopy has sufficient sensitivity to acquire high-quality NAD+ spectra from 50–100 uL volumes in the span of circa 10 minutes. And while a direct comparison with other techniques is inherently difficult, the sensitivity, ease-of-use and non-invasive nature of in vivo 1H NMR make it a highly competitive technique for NAD+ detection. NAD+ was readily detected in rat brain extracts with a similar, albeit slightly higher concentration (355 ± 34 μmol/L) than determined in vivo. Even though NADH has a unique spectral signature it was not detected in rat brain extracts in vitro. It is well known (35) that NAD+ and NADH are rapidly degraded in alkaline and acidic environments, respectively. High-resolution 1H NMR studies on pure NADH at pH 2 have shown that NADH is completely degraded within 10 min. During the degradation NADH is not oxidized to NAD+, but rather to a cyclic form of NADH (36). As the current brain extraction protocol has a short period of low pH (pH < 2), it is likely that NADH was degraded during extraction. However, the brain extraction protocol can be modified to prevent highly acidic or alkaline conditions, thereby opening the possibility of an in vitro assay of NAD+ and NADH using high-resolution 1H NMR.
A recent report by Lu et al (13) used in vivo 31P NMR spectroscopy to detect the 31P NMR signals of NAD+ and NADH in the rat cerebral cortex. Even though the 31P NMR spectra of NAD+ and NADH exhibit strong spectral overlap, the two compounds could be separately detected on the basis of differences in chemical shift and scalar coupling patterns. The separation involves the spectral deconvolution of a strongly coupled doublet of doublets for NAD+ from a singlet resonance for NADH by means of spectral fitting. While the currently presented method cannot detect the NADH pool, the 1H NMR method has a number of advantages over the 31P NMR method. Firstly, while every MR system in the world is capable of 1H NMR detection, the detection of 31P NMR signals is still largely limited to research-dedicated MR systems. Secondly, the 1H NMR method is potentially more sensitive, does not require (ultra) high magnetic fields and is relatively insensitive to experimental parameters like the magnetic field homogeneity. However, the choice of detection method will be ultimately be dictated by experimental availability and the need to detect NADH in addition to NAD+.
It should be noted that Lu et al (13) modeled the NAD+ 31P NMR spectrum as a simple two-spin-system involving only phosphorus nuclei. However, visual inspection of Fig. 1E and the scalar connectivities in Table 1 reveal that the 31P NMR spectrum should be modeled as an eight-spin-system, involving two phosphorus nuclei and six protons in two ribose rings. However, giving the typical 31P line widths observed by Lu et al (13), the additional 1H-31P scalar couplings may have limited practical importance. The application of proton decoupling would lead to a large (circa fourfold) increase in sensitivity and resolution under high-resolution conditions as shown in Fig. 1E. With the broader lines found in vivo, the value of proton decoupling for NAD+ and NADH detection may be limited.
1H NMR spectroscopy would, in principle, also allow the in vivo detection of NADH. Unfortunately, the three nicotinamide protons (H2, H4 and H6) that allow the unambiguous detection of NAD+ are shifted to lower chemical shifts for NADH (H2 = 6.9 ppm, H4 = 2.7 ppm and H6 = 6.0 ppm). The NADH-H2 proton signal thus overlaps with broad signals from glutamine and other amide signals, whereas the NADH-H6 proton will overlap with signals from ribose H1 protons in ATP and other compounds. The NADH-H4 protons form a strongly coupled multiplet around 2.7 ppm. Given the relatively low spectral overlap at that chemical shift (primarily aspartate) and under the assumption that the NAD+ and NADH T2 relaxation times are similar, the NADH-H4 protons may become detectable through the use of spectral editing methods. This is the subject of ongoing research.
As the 1H NMR detection of NAD+ is not heavily reliant on the magnetic field strength, magnetic field homogeneity or water suppression, it should be possible to detect the nicotinamide H2, H4 and H6 signals in the human brain at lower magnetic fields. Preliminary results obtained in the human brain at 7 T (data not shown) indicate that NAD+ can indeed be detected with a short TE sequence (TE = 15 ms) employing frequency-selective excitation. These results will be presented in a separate publication.
NADP+ has an identical structure to NAD+ with the exception of a phosphate group that replaces the H2′ proton in the ribose moiety. It is therefore not surprising that the downfield region of the 1H NMR spectrum of NADP+ is essentially identical to that of NAD+ as shown in Fig. 1. As such the resonances of NAD+ and NADP+ cannot be distinguished by 1H NMR and the observed signal represents their sum. However, it is well-known that in normal brain NADP+ levels are only 10% of NAD+ levels (37), such that the observed 1H NMR signal is heavily weighted towards NAD+. This is also supported by the results of Lu et al (13) who were unable to detect the 2′-phosphate signal from NADP+ by 31P NMR spectroscopy. Nicotinamide, one of the breakdown products of NAD+, also has resonances in the downfield region. However, none of the resonances of pure nicotinamide are overlapping with the NAD+ nicotinamide protons detected in this study.
Acknowledgments
The authors like to thank Xiaoxian Ma for his assistance with animal preparation. Part of this research was funded by NIH grants R01-MH095104 and P30-NS052519.
Abbreviations
- CEST
Chemical exchange saturation transfer
- CHESS
Chemical shift selective
- DSS
2, 2-dimethyl-2-silapentane-5-sulfonate
- Gln
Glutamine
- Glu
Glutamate
- HPLC
High-performance liquid chromatography
- LASER
Localization by adiabatic spin-echo refocusing
- mI
Myo-inositol
- NAA
N-acetyl aspartate
- NAD+
Nicotinamide adenine dinucleotide, oxidized form
- NADH
Nicotinamide adenine dinucleotide, reduced form
- NADP+
Nicotinamide adenine dinucleotide phosphate. oxidized form
- SVD
Singular value decomposition
- Tau
Taurine
- tCr
total creatine (sum of creatine (Cr) and phosphocreatine (PCr))
- VAPOR
Variable pulse power and optimized relaxation delays
References
- 1.Houtkooper RH, Canto C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev. 2010;31:194–223. doi: 10.1210/er.2009-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stein LR, Imai S. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol Metab. 2012;23:420–428. doi: 10.1016/j.tem.2012.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bai P, Canto C. The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell Metab. 2012;16:290–295. doi: 10.1016/j.cmet.2012.06.016. [DOI] [PubMed] [Google Scholar]
- 4.Imai S. The NAD World: a new systemic regulatory network for metabolism and aging--Sirt1, systemic NAD biosynthesis, and their importance. Cell Biochem Biophys. 2009;53:65–74. doi: 10.1007/s12013-008-9041-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Imai S, Yoshino J. The importance of NAMPT/NAD/SIRT1 in the systemic regulation of metabolism and ageing. Diabetes Obes Metab. 2013;15:26–33. doi: 10.1111/dom.12171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu TF, McCall CE. Deacetylation by SIRT1 reprograms inflammation and cancer. Genes Cancer. 2013;4:135–147. doi: 10.1177/1947601913476948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chiarugi A, Dolle C, Felici R, Ziegler M. The NAD metabolome--a key determinant of cancer cell biology. Nat Rev Cancer. 2012;12:741–752. doi: 10.1038/nrc3340. [DOI] [PubMed] [Google Scholar]
- 8.Nakahata Y, Shara S, Astarita G, Kaluzova M, Sassone-Corsi P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science. 2009;324:654–657. doi: 10.1126/science.1170803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Peek CB, Affinati AH, Ramsey KM, Kuo HY, Yu W, Sena LA, Ilkayeva O, Marcheva B, Kobayashi Y, Omura C, Levine DC, Bacsik DJ, Gius D, Newgard CB, Goetzman E, Chandel NS, Denu JM, Mrksich M, Bass J. Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science. 2013;342:1243417. doi: 10.1126/science.1243417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Williamson DH, Lund P, Krebs HA. The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem J. 1967;103:514–527. doi: 10.1042/bj1030514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yoshino J, Imai S. Accurate measurement of nicotinamide adenine dinucleotide (NAD+) with high-performance liquid chromatography. Methods Mol Biol. 2013;1077:203–215. doi: 10.1007/978-1-62703-637-5_14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chance B, Cohen P, Jobsis F, Schoener B. Intracellular oxidation-reduction states in vivo. Science. 1962;137:499–508. doi: 10.1126/science.137.3529.499. [DOI] [PubMed] [Google Scholar]
- 13.Lu M, Zhu XH, Zhang Y, Chen W. Intracellular redox state revealed by in vivo 31P MRS measurement of NAD and NADH contents in brains. Magn Reson Med. 2013 doi: 10.1002/mrm.24859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu X-H, Lu M, Zhang Y, Chen W. In vivo 31P MRS imaging of intracellular NAD contents and NAD+/NADH redox states in normal and ischemic brains. Proc Int Soc Magn Reson Med. 2013;21:861. [Google Scholar]
- 15.Zhu X-H, Lu M, Lee B-Y, Ugurbil K, Chen W. In vivo NMR of intracellular NAD contents and redox state in healthy human brain. Proc Int Soc Magn Reson Med. 2013;21:859. [Google Scholar]
- 16.de Graaf RA, Chowdhury GM, Brown PB, Rothman DL, Behar KL. In situ 3D magnetic resonance metabolic imaging of microwave-irradiated rodent brain: a new tool for metabolomics research. J Neurochem. 2009;109:494–501. doi: 10.1111/j.1471-4159.2009.05967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olson DL, Peck TL, Webb AG, Magin RL, Sweedler JV. High-resolution microcoil 1H NMR for mass-limited nanoliter-volume samples. Science. 1995;270:1967–1970. [Google Scholar]
- 18.Shaw AA, Salaun C, Dauphin J-F, Ancian B. Artifact-free PFG-enhanced double-quantum-filtered COSY experiments. J Magn Reson A. 1996;120:110–115. [Google Scholar]
- 19.Garwood M, DelaBarre L. The return of the frequency sweep: designing adiabatic pulses for contemporary NMR. J Magn Reson. 2001;153:155–177. doi: 10.1006/jmre.2001.2340. [DOI] [PubMed] [Google Scholar]
- 20.Pauly J, Le Roux P, Nishimura D, Macovski A. Parameter relations for the Shinnar-Le Roux selective excitation pulse design algorithm. IEEE Trans Med Imaging. 1991;10:53–65. doi: 10.1109/42.75611. [DOI] [PubMed] [Google Scholar]
- 21.Tkac I, Starcuk Z, Choi IY, Gruetter R. In vivo 1H NMR spectroscopy of rat brain at 1 ms echo time. Magn Reson Med. 1999;41:649–656. doi: 10.1002/(sici)1522-2594(199904)41:4<649::aid-mrm2>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 22.de Graaf RA. In Vivo NMR Spectroscopy. Principles and Techniques. 2. John Wiley; Chichester: 2007. [Google Scholar]
- 23.Klose U. In vivo proton spectroscopy in presence of eddy currents. Magn Reson Med. 1990;14:26–30. doi: 10.1002/mrm.1910140104. [DOI] [PubMed] [Google Scholar]
- 24.de Beer R, van Ormondt D. Analysis of NMR data using time domain fitting procedures. In: Diehl P, Fluck E, Gunther H, Kosfeld R, Seelig J, editors. NMR Basic Principles and Progress. Vol. 26. Springer-Verlag; Berlin: 1992. pp. 201–248. [Google Scholar]
- 25.Haase A, Frahm J, Hanicke W, Matthaei D. 1H NMR chemical shift selective (CHESS) imaging. Phys Med Biol. 1985;30:341–344. doi: 10.1088/0031-9155/30/4/008. [DOI] [PubMed] [Google Scholar]
- 26.Schanda P, Brutscher B. Very fast two-dimensional NMR spectroscopy for real-time investigation of dynamic events in proteins on the time scale of seconds. J Am Chem Soc. 2005;127:8014–8015. doi: 10.1021/ja051306e. [DOI] [PubMed] [Google Scholar]
- 27.Schanda P. Fast-pulsing longitudinal relaxation optimized techniques: Enriching the toolbox of fast biomolecular NMR spectroscopy. Prog NMR Spectroscopy. 2009;55:238–265. [Google Scholar]
- 28.MacMillan EL, Chong DG, Dreher W, Henning A, Boesch C, Kreis R. Magnetization exchange with water and T1 relaxation of the downfield resonances in human brain spectra at 3. 0 T. Magn Reson Med. 2011;65:1239–1246. doi: 10.1002/mrm.22813. [DOI] [PubMed] [Google Scholar]
- 29.van Zijl PC, Yadav NN. Chemical exchange saturation transfer (CEST): what is in a name and what isn’t? Magn Reson Med. 2011;65:927–948. doi: 10.1002/mrm.22761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Graaf RA, van Kranenburg A, Nicolay K. Off-resonance metabolite magnetization transfer measurements on rat brain in situ. Magn Reson Med. 1999;41:1136–1144. doi: 10.1002/(sici)1522-2594(199906)41:6<1136::aid-mrm9>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 31.Luo Y, Rydzewski J, de Graaf RA, Gruetter R, Garwood M, Schleich T. In vivo observation of lactate methyl proton magnetization transfer in rat C6 glioma. Magn Reson Med. 1999;41:676–685. doi: 10.1002/(sici)1522-2594(199904)41:4<676::aid-mrm5>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- 32.Shemesh N, Dumez JN, Frydman L. Longitudinal relaxation enhancement in 1H NMR spectroscopy of tissue metabolites via spectrally selective excitation. Chemistry. 2013;19:13002–13008. doi: 10.1002/chem.201300955. [DOI] [PubMed] [Google Scholar]
- 33.Stavinoha WB, Pepelko B, Smith PW. Microwave irradiation to inactivate cholinesterase in the rat brain prior to analysis for acetylcholine. Pharmacologist. 1970;12:257. [Google Scholar]
- 34.de Graaf RA, Brown PB, McIntyre S, Nixon TW, Behar KL, Rothman DL. High magnetic field water and metabolite proton T1 and T2 relaxation in rat brain in vivo. Magn Reson Med. 2006;56:386–394. doi: 10.1002/mrm.20946. [DOI] [PubMed] [Google Scholar]
- 35.Lowry OH, Passonneau JV, Rock MK. The stability of pyridine nucleotides. J Biol Chem. 1961;236:2756–2759. [PubMed] [Google Scholar]
- 36.Oppenheimer NJ, Kaplan NO. Structure of the primary acid rearrangement product of reduced nicotinamide adenine dinucleotide (NADH) Biochemistry. 1974;13:4675–4685. doi: 10.1021/bi00720a001. [DOI] [PubMed] [Google Scholar]
- 37.Lowry OH, Passonneau JV, Schulz DW, Rock MK. The measurement of pyridine nucleotides by enzymatic cycling. J Biol Chem. 1961;236:2746–2755. [PubMed] [Google Scholar]