

Abstract
In the title compound, C17H10F3NS, the dihedral angle between the fused benzothiophene ring system (r.m.s. deviation = 0.042 Å) and the benzene ring is 29.78 (11)°. The crystal structure features C—H⋯F and very weak C—H⋯N hydrogen bonds, which generate (001) sheets.
Keywords: crystal structure, benzo[b]thiophene, hydrogen bonding
Related literature
For background to benzothiophene derivatives, see: Bettinetti et al. (2002 ▸); Roberts & Hartley (2004 ▸).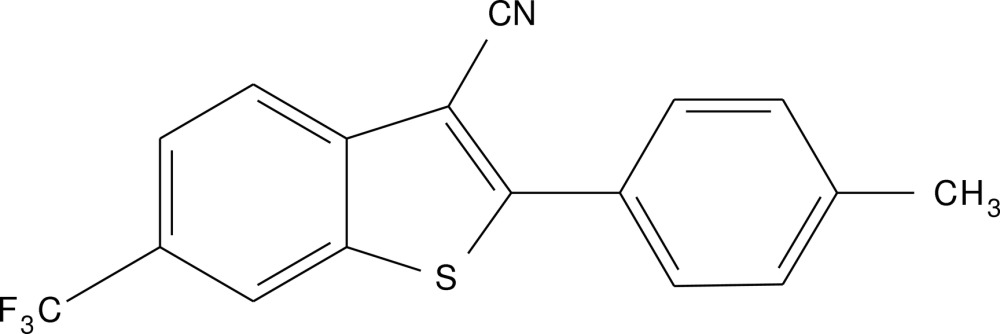
Experimental
Crystal data
C17H10F3NS
M r = 317.33
Monoclinic,

a = 13.7576 (5) Å
b = 14.5343 (6) Å
c = 7.1353 (3) Å
β = 92.817 (3)°
V = 1425.03 (10) Å3
Z = 4
Cu Kα radiation
μ = 2.29 mm−1
T = 293 K
0.30 × 0.27 × 0.25 mm
Data collection
Bruker X8 Proteum diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2013 ▸) T min = 0.546, T max = 0.598
7045 measured reflections
2316 independent reflections
1860 reflections with I > 2σ(I)
R int = 0.063
Refinement
R[F 2 > 2σ(F 2)] = 0.053
wR(F 2) = 0.141
S = 1.06
2316 reflections
200 parameters
H-atom parameters constrained
Δρmax = 0.43 e Å−3
Δρmin = −0.40 e Å−3
Data collection: APEX2 (Bruker, 2013 ▸); cell refinement: SAINT (Bruker, 2013 ▸); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▸); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▸); molecular graphics: PLATON (Spek, 2009 ▸); software used to prepare material for publication: PLATON.
Supplementary Material
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S2056989015008671/hb7416sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989015008671/hb7416Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989015008671/hb7416Isup3.cml
. DOI: 10.1107/S2056989015008671/hb7416fig1.tif
A view of the title compound with displacement ellipsoids drawn at the 50% probability level.
a . DOI: 10.1107/S2056989015008671/hb7416fig2.tif
A view along the a axis of the crystal packing of the title compound. Hydrogen bonds are shown as dashed lines (see Table 2 for details).
CCDC reference: 1063141
Additional supporting information: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (, ).
| DHA | DH | HA | D A | DHA |
|---|---|---|---|---|
| C3H3N11i | 0.93 | 2.62 | 3.411(4) | 143 |
| C22H22CF15ii | 0.96 | 2.45 | 3.375(4) | 162 |
Symmetry codes: (i)  ; (ii)
; (ii)  .
.
Acknowledgments
The authors are thankful to Institution of Excellence, University of Mysore, Mysore, for providing the single-crystal X-ray diffraction facility.
supplementary crystallographic information
S1. Comment
Benzo[b]thiophene derivatives are important heterocyclic compounds because of their various applications in medicinal chemistry. They represent an important heterocyclic core and are shown to display a range of promising pharmacological properties such as antipsychotic, antidepressive, antithrombolytic, dopamine receptor antagonist and 5-lipoxygenase inhibitor. Number of 2-arylbenzo[b]thiophene derivatives have indeed, these sulfur heterocycles are essential components of clinically important drugs such as Clopidogrel (Bettinetti et al., 2002), Raloxifene (Roberts & Hartley, 2004) and Zileuton.
S2. Experimental
To a solution of 2-(2-chlorophenyl)acetonitrile (1.0 mmol), methyl benzodithioate (1.0 mmol) in DMF (2 ml), K3PO4 (2.0 mmol), pivalic acid (1.5 mmol), cuprous iodide (0.2 mmol) were added. The mixture was stirred at 80°C and progress was monitored by TLC. When the dithioesters could no longer be detected, the reaction mixture was extracted with EtOAc (3 × 10 ml). The organic layer was dried over anhydrous Na2SO4. The solvent was then removed under reduced pressure and the residue was purified by silica gel chromatography. White solid single crystals were obtained from slow evaporation of its solvent.
S3. Refinement
All H atoms were positioned geometrically and allowed to ride on their parent atom, with C–H = 0.93–0.97 Å, and with Uiso(H) = 1.2–1.5Ueq(C).
Figures
Fig. 1.

A view of the title compound with displacement ellipsoids drawn at the 50% probability level.
Fig. 2.

A view along the a axis of the crystal packing of the title compound. Hydrogen bonds are shown as dashed lines (see Table 2 for details).
Crystal data
| C17H10F3NS | F(000) = 648 |
| Mr = 317.33 | Dx = 1.479 Mg m−3 |
| Monoclinic, P21/c | Cu Kα radiation, λ = 1.54178 Å |
| Hall symbol: -P 2ybc | Cell parameters from 2316 reflections |
| a = 13.7576 (5) Å | θ = 6.4–64.7° |
| b = 14.5343 (6) Å | µ = 2.29 mm−1 |
| c = 7.1353 (3) Å | T = 293 K |
| β = 92.817 (3)° | Block, colourless |
| V = 1425.03 (10) Å3 | 0.30 × 0.27 × 0.25 mm |
| Z = 4 |
Data collection
| Bruker X8 Proteum diffractometer | 2316 independent reflections |
| Radiation source: Rotating Anode | 1860 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.063 |
| Detector resolution: 18.4 pixels mm-1 | θmax = 64.7°, θmin = 6.4° |
| φ and ω scans | h = −15→15 |
| Absorption correction: multi-scan (SADABS; Bruker, 2013) | k = −15→16 |
| Tmin = 0.546, Tmax = 0.598 | l = −8→6 |
| 7045 measured reflections |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.053 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.141 | H-atom parameters constrained |
| S = 1.06 | w = 1/[σ2(Fo2) + (0.0726P)2 + 0.2459P] where P = (Fo2 + 2Fc2)/3 |
| 2316 reflections | (Δ/σ)max < 0.001 |
| 200 parameters | Δρmax = 0.43 e Å−3 |
| 0 restraints | Δρmin = −0.40 e Å−3 |
Special details
| Geometry. Bond distances, angles etc. have been calculated using the rounded fractional coordinates. All su's are estimated from the variances of the (full) variance-covariance matrix. The cell e.s.d.'s are taken into account in the estimation of distances, angles and torsion angles |
| Refinement. Refinement on F2 for ALL reflections except those flagged by the user for potential systematic errors. Weighted R-factors wR and all goodnesses of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The observed criterion of F2 > σ(F2) is used only for calculating -R-factor-obs etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R-factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| S1 | 0.51842 (5) | 0.17833 (5) | 0.23250 (10) | 0.0214 (2) | |
| F13 | 0.10569 (13) | −0.01265 (15) | 0.0632 (4) | 0.0518 (8) | |
| F14 | 0.14077 (13) | 0.11887 (16) | −0.0469 (3) | 0.0450 (7) | |
| F15 | 0.11752 (13) | 0.10360 (17) | 0.2439 (3) | 0.0484 (8) | |
| N11 | 0.64047 (19) | −0.14957 (19) | 0.3600 (4) | 0.0297 (9) | |
| C2 | 0.4251 (2) | 0.0996 (2) | 0.1944 (4) | 0.0202 (9) | |
| C3 | 0.3272 (2) | 0.1178 (2) | 0.1485 (4) | 0.0212 (9) | |
| C4 | 0.2634 (2) | 0.0444 (2) | 0.1373 (4) | 0.0210 (9) | |
| C5 | 0.2957 (2) | −0.0459 (2) | 0.1679 (4) | 0.0217 (9) | |
| C6 | 0.3920 (2) | −0.0643 (2) | 0.2103 (4) | 0.0203 (8) | |
| C7 | 0.4583 (2) | 0.0087 (2) | 0.2249 (4) | 0.0178 (8) | |
| C8 | 0.5601 (2) | 0.0069 (2) | 0.2816 (4) | 0.0174 (8) | |
| C9 | 0.6018 (2) | 0.0928 (2) | 0.2949 (4) | 0.0183 (8) | |
| C10 | 0.6079 (2) | −0.0785 (2) | 0.3267 (4) | 0.0204 (9) | |
| C12 | 0.1574 (2) | 0.0636 (2) | 0.0993 (4) | 0.0253 (9) | |
| C16 | 0.7014 (2) | 0.1180 (2) | 0.3613 (4) | 0.0182 (8) | |
| C17 | 0.7798 (2) | 0.0588 (2) | 0.3430 (4) | 0.0198 (9) | |
| C18 | 0.8717 (2) | 0.0826 (2) | 0.4144 (4) | 0.0236 (9) | |
| C19 | 0.8890 (2) | 0.1655 (2) | 0.5074 (4) | 0.0235 (9) | |
| C20 | 0.8107 (2) | 0.2255 (2) | 0.5220 (4) | 0.0217 (9) | |
| C21 | 0.7190 (2) | 0.2025 (2) | 0.4506 (4) | 0.0194 (8) | |
| C22 | 0.9878 (2) | 0.1887 (2) | 0.5939 (5) | 0.0306 (10) | |
| H3 | 0.30560 | 0.17760 | 0.12600 | 0.0250* | |
| H5 | 0.25120 | −0.09410 | 0.15940 | 0.0260* | |
| H6 | 0.41310 | −0.12450 | 0.22910 | 0.0240* | |
| H17 | 0.77040 | 0.00260 | 0.28230 | 0.0240* | |
| H18 | 0.92320 | 0.04210 | 0.39990 | 0.0280* | |
| H20 | 0.82050 | 0.28200 | 0.58110 | 0.0260* | |
| H21 | 0.66800 | 0.24390 | 0.46190 | 0.0230* | |
| H22A | 0.99640 | 0.15870 | 0.71340 | 0.0460* | |
| H22B | 0.99300 | 0.25410 | 0.61070 | 0.0460* | |
| H22C | 1.03700 | 0.16810 | 0.51280 | 0.0460* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| S1 | 0.0194 (4) | 0.0164 (4) | 0.0281 (4) | −0.0001 (3) | −0.0009 (3) | 0.0010 (3) |
| F13 | 0.0215 (9) | 0.0385 (13) | 0.0941 (17) | −0.0028 (9) | −0.0107 (10) | −0.0047 (12) |
| F14 | 0.0293 (10) | 0.0607 (15) | 0.0442 (12) | 0.0123 (10) | −0.0052 (9) | 0.0178 (11) |
| F15 | 0.0285 (10) | 0.0782 (17) | 0.0390 (11) | 0.0161 (10) | 0.0059 (9) | −0.0160 (11) |
| N11 | 0.0321 (15) | 0.0196 (16) | 0.0369 (16) | 0.0022 (12) | −0.0027 (12) | 0.0019 (13) |
| C2 | 0.0237 (15) | 0.0198 (16) | 0.0171 (14) | −0.0014 (12) | 0.0024 (11) | −0.0003 (13) |
| C3 | 0.0214 (14) | 0.0211 (17) | 0.0210 (14) | 0.0051 (12) | 0.0011 (12) | −0.0005 (13) |
| C4 | 0.0223 (15) | 0.0258 (17) | 0.0149 (13) | −0.0007 (13) | 0.0005 (11) | −0.0005 (13) |
| C5 | 0.0244 (15) | 0.0211 (17) | 0.0196 (14) | −0.0038 (12) | −0.0001 (12) | −0.0010 (13) |
| C6 | 0.0268 (15) | 0.0145 (15) | 0.0198 (14) | −0.0004 (12) | 0.0026 (12) | 0.0003 (12) |
| C7 | 0.0216 (14) | 0.0207 (16) | 0.0111 (13) | 0.0011 (12) | 0.0023 (11) | 0.0016 (12) |
| C8 | 0.0203 (14) | 0.0170 (15) | 0.0149 (13) | 0.0012 (12) | 0.0017 (11) | −0.0007 (12) |
| C9 | 0.0220 (14) | 0.0191 (16) | 0.0142 (13) | 0.0006 (12) | 0.0041 (11) | −0.0006 (12) |
| C10 | 0.0199 (14) | 0.0220 (18) | 0.0191 (15) | −0.0039 (13) | −0.0011 (12) | −0.0006 (13) |
| C12 | 0.0240 (15) | 0.0254 (18) | 0.0265 (16) | 0.0002 (13) | 0.0002 (13) | −0.0017 (14) |
| C16 | 0.0206 (14) | 0.0174 (16) | 0.0166 (13) | −0.0004 (12) | 0.0005 (11) | 0.0015 (12) |
| C17 | 0.0219 (15) | 0.0169 (16) | 0.0209 (14) | −0.0001 (12) | 0.0034 (12) | −0.0013 (13) |
| C18 | 0.0229 (15) | 0.0240 (17) | 0.0241 (15) | 0.0042 (13) | 0.0026 (12) | −0.0007 (13) |
| C19 | 0.0237 (15) | 0.0293 (18) | 0.0175 (14) | −0.0019 (13) | 0.0003 (12) | 0.0033 (13) |
| C20 | 0.0240 (15) | 0.0223 (17) | 0.0187 (14) | −0.0044 (13) | 0.0011 (12) | −0.0009 (13) |
| C21 | 0.0204 (14) | 0.0186 (16) | 0.0195 (14) | 0.0003 (12) | 0.0032 (11) | 0.0022 (13) |
| C22 | 0.0251 (16) | 0.037 (2) | 0.0295 (16) | 0.0014 (14) | −0.0015 (13) | −0.0015 (16) |
Geometric parameters (Å, º)
| S1—C2 | 1.731 (3) | C16—C17 | 1.391 (4) |
| S1—C9 | 1.735 (3) | C16—C21 | 1.399 (4) |
| F13—C12 | 1.335 (4) | C17—C18 | 1.384 (4) |
| F14—C12 | 1.328 (4) | C18—C19 | 1.390 (4) |
| F15—C12 | 1.326 (4) | C19—C20 | 1.394 (4) |
| N11—C10 | 1.146 (4) | C19—C22 | 1.503 (4) |
| C2—C3 | 1.396 (4) | C20—C21 | 1.378 (4) |
| C2—C7 | 1.411 (4) | C3—H3 | 0.9300 |
| C3—C4 | 1.381 (4) | C5—H5 | 0.9300 |
| C4—C5 | 1.399 (4) | C6—H6 | 0.9300 |
| C4—C12 | 1.496 (4) | C17—H17 | 0.9300 |
| C5—C6 | 1.371 (4) | C18—H18 | 0.9300 |
| C6—C7 | 1.400 (4) | C20—H20 | 0.9300 |
| C7—C8 | 1.439 (4) | C21—H21 | 0.9300 |
| C8—C9 | 1.375 (4) | C22—H22A | 0.9600 |
| C8—C10 | 1.434 (4) | C22—H22B | 0.9600 |
| C9—C16 | 1.474 (4) | C22—H22C | 0.9600 |
| C2—S1—C9 | 92.44 (14) | C17—C16—C21 | 117.9 (3) |
| S1—C2—C3 | 127.7 (2) | C16—C17—C18 | 120.6 (3) |
| S1—C2—C7 | 111.3 (2) | C17—C18—C19 | 121.6 (3) |
| C3—C2—C7 | 121.0 (3) | C18—C19—C20 | 117.6 (3) |
| C2—C3—C4 | 118.1 (3) | C18—C19—C22 | 121.4 (3) |
| C3—C4—C5 | 121.3 (3) | C20—C19—C22 | 121.0 (3) |
| C3—C4—C12 | 118.5 (3) | C19—C20—C21 | 121.2 (3) |
| C5—C4—C12 | 120.2 (3) | C16—C21—C20 | 121.1 (3) |
| C4—C5—C6 | 120.9 (3) | C2—C3—H3 | 121.00 |
| C5—C6—C7 | 119.2 (3) | C4—C3—H3 | 121.00 |
| C2—C7—C6 | 119.6 (3) | C4—C5—H5 | 120.00 |
| C2—C7—C8 | 111.3 (2) | C6—C5—H5 | 120.00 |
| C6—C7—C8 | 129.0 (3) | C5—C6—H6 | 120.00 |
| C7—C8—C9 | 113.6 (3) | C7—C6—H6 | 120.00 |
| C7—C8—C10 | 120.6 (3) | C16—C17—H17 | 120.00 |
| C9—C8—C10 | 125.8 (3) | C18—C17—H17 | 120.00 |
| S1—C9—C8 | 111.4 (2) | C17—C18—H18 | 119.00 |
| S1—C9—C16 | 119.8 (2) | C19—C18—H18 | 119.00 |
| C8—C9—C16 | 128.7 (3) | C19—C20—H20 | 119.00 |
| N11—C10—C8 | 175.6 (3) | C21—C20—H20 | 119.00 |
| F13—C12—F14 | 106.3 (2) | C16—C21—H21 | 120.00 |
| F13—C12—F15 | 106.1 (2) | C20—C21—H21 | 119.00 |
| F13—C12—C4 | 112.7 (2) | C19—C22—H22A | 109.00 |
| F14—C12—F15 | 106.5 (2) | C19—C22—H22B | 109.00 |
| F14—C12—C4 | 112.6 (2) | C19—C22—H22C | 109.00 |
| F15—C12—C4 | 112.2 (2) | H22A—C22—H22B | 109.00 |
| C9—C16—C17 | 121.9 (3) | H22A—C22—H22C | 110.00 |
| C9—C16—C21 | 120.1 (3) | H22B—C22—H22C | 109.00 |
| C9—S1—C2—C3 | 175.6 (3) | C2—C7—C8—C9 | 0.5 (4) |
| C9—S1—C2—C7 | −1.4 (2) | C2—C7—C8—C10 | 177.7 (3) |
| C2—S1—C9—C8 | 1.7 (2) | C6—C7—C8—C9 | −175.0 (3) |
| C2—S1—C9—C16 | −175.3 (2) | C6—C7—C8—C10 | 2.2 (5) |
| S1—C2—C3—C4 | −175.5 (2) | C7—C8—C9—S1 | −1.6 (3) |
| C7—C2—C3—C4 | 1.2 (4) | C7—C8—C9—C16 | 175.2 (3) |
| S1—C2—C7—C6 | 176.7 (2) | C10—C8—C9—S1 | −178.6 (2) |
| S1—C2—C7—C8 | 0.8 (3) | C10—C8—C9—C16 | −1.8 (5) |
| C3—C2—C7—C6 | −0.5 (4) | S1—C9—C16—C17 | −154.1 (2) |
| C3—C2—C7—C8 | −176.4 (3) | S1—C9—C16—C21 | 27.8 (4) |
| C2—C3—C4—C5 | −1.0 (4) | C8—C9—C16—C17 | 29.4 (5) |
| C2—C3—C4—C12 | 176.5 (3) | C8—C9—C16—C21 | −148.7 (3) |
| C3—C4—C5—C6 | 0.1 (4) | C9—C16—C17—C18 | −177.0 (3) |
| C12—C4—C5—C6 | −177.4 (3) | C21—C16—C17—C18 | 1.2 (4) |
| C3—C4—C12—F13 | 170.3 (3) | C9—C16—C21—C20 | 176.8 (3) |
| C3—C4—C12—F14 | 50.1 (4) | C17—C16—C21—C20 | −1.5 (4) |
| C3—C4—C12—F15 | −70.0 (3) | C16—C17—C18—C19 | 0.4 (4) |
| C5—C4—C12—F13 | −12.2 (4) | C17—C18—C19—C20 | −1.8 (4) |
| C5—C4—C12—F14 | −132.4 (3) | C17—C18—C19—C22 | 176.6 (3) |
| C5—C4—C12—F15 | 107.5 (3) | C18—C19—C20—C21 | 1.5 (4) |
| C4—C5—C6—C7 | 0.7 (4) | C22—C19—C20—C21 | −176.9 (3) |
| C5—C6—C7—C2 | −0.5 (4) | C19—C20—C21—C16 | 0.1 (4) |
| C5—C6—C7—C8 | 174.6 (3) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C3—H3···N11i | 0.93 | 2.62 | 3.411 (4) | 143 |
| C22—H22C···F15ii | 0.96 | 2.45 | 3.375 (4) | 162 |
Symmetry codes: (i) −x+1, y+1/2, −z+1/2; (ii) x+1, y, z.
Footnotes
Supporting information for this paper is available from the IUCr electronic archives (Reference: HB7416).
References
- Bettinetti, L., Schlotter, K., Hübner, H. & Gmeiner, P. (2002). J. Med. Chem. 45, 4594–4597. [DOI] [PubMed]
- Bruker (2013). APEX2, SAINT and SADABS. Bruker AXS Inc., Madison, Wisconsin, USA.
- Roberts, C. F. & Hartley, R. C. (2004). J. Org. Chem. 69, 6145–6148. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S2056989015008671/hb7416sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S2056989015008671/hb7416Isup2.hkl
Supporting information file. DOI: 10.1107/S2056989015008671/hb7416Isup3.cml
. DOI: 10.1107/S2056989015008671/hb7416fig1.tif
A view of the title compound with displacement ellipsoids drawn at the 50% probability level.
a . DOI: 10.1107/S2056989015008671/hb7416fig2.tif
A view along the a axis of the crystal packing of the title compound. Hydrogen bonds are shown as dashed lines (see Table 2 for details).
CCDC reference: 1063141
Additional supporting information: crystallographic information; 3D view; checkCIF report