

Abstract
Background
In many cells, bile acids (BAs) have a multitude of effects, some of which may be mediated by specific receptors such the TGR5 or FXR receptors. In pancreas systemic BAs, as well as intra-ductal BAs from bile reflux, can affect pancreatic secretion. Extracellular ATP and purinergic signalling are other important regulators of similar secretory mechanisms in pancreas. The aim of our study was to elucidate whether there is interplay between ATP and BA signalling.
Results
Here we show that CDCA (chenodeoxycholic acid) caused fast and concentration-dependent ATP release from acini (AR42J) and duct cells (Capan-1). Taurine and glycine conjugated forms of CDCA had smaller effects on ATP release in Capan-1 cells. In duct monolayers, CDCA stimulated ATP release mainly from the luminal membrane; the releasing mechanisms involved both vesicular and non-vesicular secretion pathways. Duct cells were not depleted of intracellular ATP with CDCA, but acinar cells lost some ATP, as detected by several methods including ATP sensor AT1.03YEMK. In duct cells, CDCA caused reversible increase in the intracellular Ca2+ concentration [Ca2 +]i, which could be significantly inhibited by antagonists of purinergic receptors. The TGR5 receptor, expressed on the luminal side of pancreatic ducts, was not involved in ATP release and Ca2+ signals, but could stimulate Na+/Ca2+ exchange in some conditions.
Conclusions
CDCA evokes significant ATP release that can stimulate purinergic receptors, which in turn increase [Ca2+]i. The TGR5 receptor is not involved in these processes but can play a protective role at high intracellular Ca2+ conditions. We propose that purinergic signalling could be taken into consideration in other cells/organs, and thereby potentially explain some of the multifaceted effects of BAs.
Electronic supplementary material
The online version of this article (doi:10.1186/s12964-015-0107-9) contains supplementary material, which is available to authorized users.
Keywords: ATP release, P2 receptors, Bile acids, CDCA, TGR5, FXR, Ca2+, Pancreas, ATP sensor, AT1.03YEMK, FLIM-FRET
Lay abstract
In recent years there has been a growing interest for the role of bile acids as signalling molecules in many cells/organs. Several types of bile acids receptors have been identified, but some modulatory functions of bile acids remain unexplained. Here, we show that bile acids, in particular chenodeoxycholic acid, causes significant release of ATP from exocrine pancreatic cells. Extracellular ATP can then via purinergic receptors regulate or co-regulate epithelial functions, such as pancreatic duct secretion, which is important for normal digestive processes. Our study brings novel insights into regulation of pancreas functions. Moreover, we propose that purinergic signalling should be taken into consideration in other cells/organ types, as it could potentially explain some of the multifaceted effects of bile acids.
Background
Bile acids (BAs) are natural amphiphilic metabolites originating from cholesterol degradation. The main human primary bile acids are chenodeoxycholic acid (CDCA) and cholic acid (CA), which can be conjugated with glycine and taurine to form bile salts. The role of bile acids as signalling molecules and as targets for drug development gained interest during the last years, and several bile acid receptors were discovered, including the nuclear farnesoid receptor (FXR) and the membrane receptor TGR5 (GPBAR1) [1–4]. TGR5 is expressed in many cell types and regulates a variety of functions. For example, TGR5 modulates liver function, glucose metabolism and insulin sensitivity, and immune responses [4–7]. In several epithelia such as colon, respiratory epithelia and bile ducts, BAs regulate ion transport, at least partly via TGR5 or FXR receptors [8–10]. For example, in bile ducts, TGR5 stimulates biliary HCO3− and fluid secretion [11–14].
In pancreas, BAs have multiple effects. In the endocrine pancreas, tauroursodeoxycholate has a protective role on pancreatic islets as it decreases apoptosis and stimulates insulin secretion after stress conditions [15]. It is reported that BAs can stimulate both FXR and TGR5 receptors in mouse β-cells and induce fast insulin secretion [16, 17]. Furthermore, there is TGR5-dependent stimulation of glucagon-like-peptide-1 (GLP-1) release from enterocytes [18], and then systemic GLP-1 increases insulin secretion from β-cells [19]. In exocrine pancreas, BAs can exert effects on several levels, as they can reach pancreas not only systemically, but also via reflux of bile into pancreatic ducts. Pancreatic acini express BA transporters [20] and some BAs at low concentrations can activate Ca2+-independent cation currents [21]. At high concentrations BAs can evoke cytotoxicaly high intracellular Ca2+ concentrations, [Ca2+]i, in pancreatic acini. This is due to inhibition of the sacro/endoplasmatic reticulum Ca2+-ATPases (SERCA), release of Ca2+ from ER and acidic stores and granules, increased Ca2+ influx, and acini show cellular acidosis, enzyme activation and mitochondrial malfunction, which can eventually lead to development of (biliary) acute pancreatitis [20, 22–24]. In one study, TGR5/GPBAR1 receptor knockout mice had less severe pancreatitis after infusion of BAs [25]. For pancreatic ducts, it has been proposed that BAs can stimulate duct secretion and they can tolerate higher BA concentrations [26–28]. Several studies show that BAs stimulate Cl− and K+ channels; where the later have been identified as large conductance Ca2+-activated K+ channels (BK, KCa1.1), but identity of Cl− channels is not clear [26, 29, 30].
Another important regulatory system in pancreatic ducts is the purinergic signalling. Extracellular ATP can, via a number of P2 receptors that stimulate Ca2+ signalling, regulate Cl− and K+ channels and acid/base transporters and thereby modulate HCO3− and fluid secretion [31, 32]. Until now it has not been investigated whether pancreatic ducts release ATP, however, it is well established that ATP is released from pancreatic acini, which store ATP in zymogen granules, where it is accumulated by the Vesicular NUcleotide Transporter (VNUT, SLC17A9) [33]. Acinar ATP is released by exocytosis into the duct lumen in response to cholinergic or hormonal stimulation [34, 35]. In various other cells, ATP release can occur also via ion channels/transporters, such as maxi-anion channels, connexins, pannexins with/without P2X7 receptors [36, 37].
Considering that BAs and purinergic signalling appear to have similar paracrine effects on ion and fluid transport in exocrine pancreas, we hypothesized whether there was any interaction between these two intra-ductal regulatory systems. Therefore, we designed a study to test whether BAs, i.e., chenodeoxycholic acid (CDCA) and its glycine and taurine conjugated forms (GCDCA and TCDCA) can affect ATP release, and whether CDCA also influence intracellular ATP levels. A further aim was to elucidate whether BA signalling involves TGR5 or FXR receptors in the ATP release process and/or P2 receptors and downstream Ca2+ signalling. For this purpose we used duct and acini models (Capan-1 and AR42J cells), live cell luminescence assay for extracellular ATP, intracellular ATP sensors, AT1.03YEMK, and [Ca2+]i. imaging. Our study shows that CDCA indeed caused a very fast release of ATP from exocrine pancreatic cells via both non-exocytotic and vesicular pathways. Furthermore, a significant part of the CDCA effect on Ca2+ signalling is mediated via purinergic signalling. We also demonstrate the presence of the TGR5 and FXR receptors in human pancreatic ducts and show that TGR5 receptor can prevent intracellular Ca2+ overloads possibly by stimulation of the Na+/Ca2+ exchanger.
Results
CDCA but not GCDCA and TCDCA stimulates high ATP release from pancreatic cells
ATP release on the whole organ level is difficult to detect due to action of membrane-bound and soluble nucleotidases [35, 38]. Therefore, ATP release and autocrine/paracrine signalling is usually studied on isolated cells or cell lines. In the first series of experiments we investigated the effect of several bile acids: GCDCA (glycochenodeoxycholic acid); TCDCA (taurochenodeoxycholic acid); and CDCA (chenodeoxycholic acid) on ATP release from Capan-1 cells, which are a model of pancreatic ducts. Time resolved luminescence recordings revealed that 0.3 mM of GCDCA and TCDCA had minor effects on ATP release from these cells (Fig. 1 a-b). At higher concentrations (1 mM) GCDCA evoked significant but small increase in extracellular ATP, ATPe, by 4.2 ± 0.8 nM (n = 4) above the basal level. TCDCA (1 mM) had no effect on ATP release. In contrast to GCDCA and TCDCA, CDCA (0.3 mM) caused a fast and substantial ATP release in Capan-1 cells (Fig. 1c). Therefore, we investigated the effect of CDCA concentrations on ATP release from Capan-1 (Fig. 1d). Furthermore, we investigated the effect of CDCA on AR42J cells, which are a model for pancreatic acinar cells (Fig. 1 e, f). Fig. 1 c-f shows that the duct and acinar cells released ATP in a concentration-dependent manner in a narrow range from 0.1 to 1 mM CDCA, and EC50 values are 0.43 mM and 0.44 mM for Capan-1 and AR42J cells, respectively. The peak/maximum ATP release observed after stimulation with 1 mM CDCA was 848 ± 16 nM (n = 6) for Capan-1 and 614 ± 79 nM (n = 5) for AR42J cells. These data show that CDCA can increase extracellular ATP concentrations by a factor of 100–1000 above the baseline. In all the following experiments, Capan-1 cells were stimulated with 0.3 mM CDCA and AR42J cells with 0.5 mM CDCA.
Fig. 1.
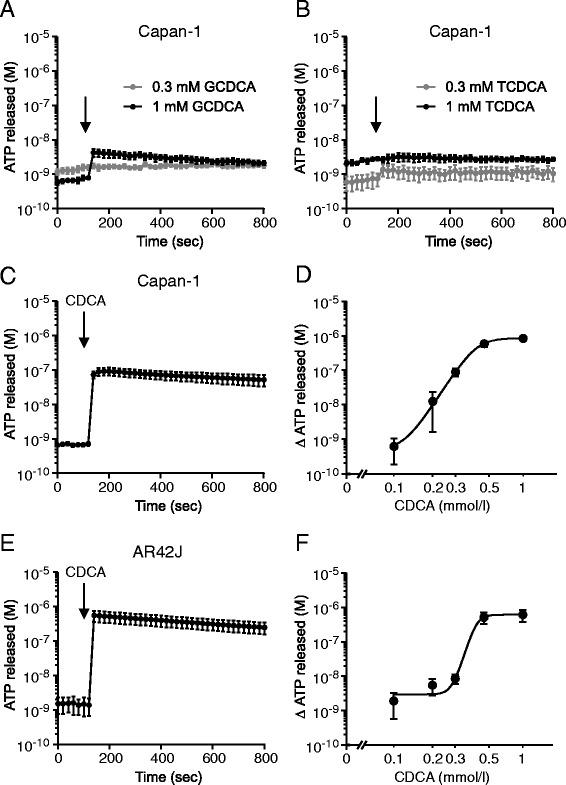
CDCA induced ATP release from exocrine pancreatic cells. The time-course of ATP release from Capan-1 cells in response to 0.3 and 1 mM of (a) GCDCA and (b) TCDCA (n = 4, 4). The time-course of ATP release from (c) Capan-1 and (e) AR42J after stimulation with 0.3 and 0.5 mM CDCA, respectively (n = 6, 5). Dose-dependent release of ATP from (d) Capan-1 and (f) AR42J cells in response to CDCA (n = 5, 6). The EC50 values of ATP release from AR42J and Capan-1 are 0.44 mM (pEC50 = 3.36 ± 0.03) and 0.43 mM (pEC50 3.36 ± 0.04) (n = 5, 6) respectively in the range of 0.1 – 1 mM CDCA. Y-axis shows ATP concentrations, which were corrected for 106 cells per 1 ml (see Methods). Data shown as mean values ± SEM. Arrows indicate addition of stimulants
Since Capan-1 cells are a model of human pancreatic duct epithelium, it was relevant to investigate whether ATP release is polarized, i.e., whether it occurs preferentially across the luminal or the basolateral membrane. Capan-1 cells were cultured as polarized monolayers, and CDCA was administered luminally or basolaterally. After the luminal stimulation with 0.3 mM CDCA (Fig. 2a), there was significantly higher ATP release from the luminal side (5.7 ± 1.3 nM, n = 8) compared to the basolateral side (1.3 ± 0.7 nM, n = 5). Interestingly, a similar high luminal ATP release was observed when monolayers were stimulated with basolateral CDCA (Fig. 2a). ATP released across the luminal side was 10.5 ± 2.9 nM (n = 3) compared to the basolateral side 0.6 ± 0.03 nM (n = 5). Notably, these ATP values made in offline analysis of samples were lower than in online measurements, most likely due to different sampling volumes and ATP hydrolysis by ecto-nucleotidases [39, 40].
Fig. 2.
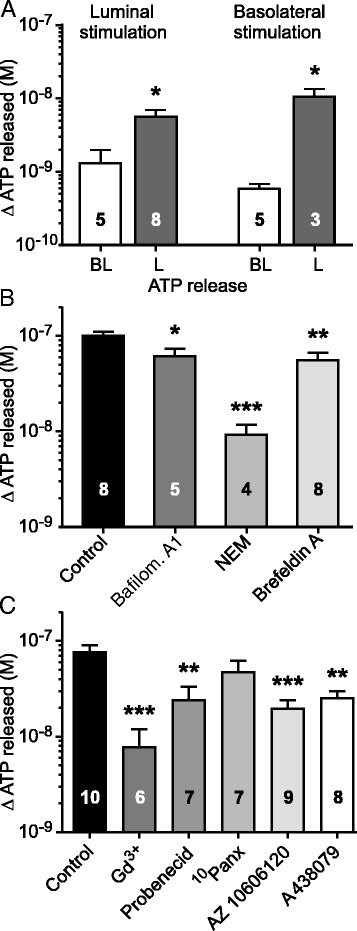
Sidedness of ATP release and the effect of vesicular and non-vesicular inhibitors on ATP release pathways. a Mean values of ATP released across basolateral (BL) and luminal (L) sides from Capan-1 cells after 1 min of apical or serosal stimulation with 0.3 mM CDCA. b Inhibitors of vesicular transport decreased ATP release from Capan-1 cells in response to 0.3 mM CDCA. Cells were incubated with vacuolar-type H+-ATPase inhibitor (bafilomycin A1, 1 μM; n = 5), vesicle fusion inhibitor N-Ethylmaleimide (NEM, 250 μM, n = 4) and brefeldin A (5 μg/ml; n = 8). c Influence of the non-vesicular transport inhibitors: gadolinium chloride (Gd3+, 50 μM; n = 6), probenecid (500 μM; n = 7), 10Panx - mimetic pannexin peptide (100 μM; n = 7), as well as P2X7 receptor inhibitors AZ10606120 (10 μM; n = 9) and A438079 (10 μM; n = 7) on CDCA-induced ATP release from Capan-1 cells is shown. Results are given as mean net values ± SEM. *= P < 0.05, **= P < 0.01, ***= P < 0.001
CDCA induces ATP release via multiple pathways
In order to identify the pathways involved in CDCA-induced ATP release, Capan-1 cells were incubated with vesicular and non-vesicular transport inhibitors. Fig. 2b shows that CDCA caused high ATP release of 100 ± 10 nM (n = 8), and after incubation with bafilomycin A1, an inhibitor for vacuolar-type H+-ATPase, ATP release was significantly reduced to 61 ± 12 nM (n = 5). Inhibition of CDCA-evoked vesicular ATP release was also observed in the presence of N-Ethylmaleimide, an inhibitor of vesicle fusion, which markedly supressed ATP release to 9 ± 3 nM (n = 4). In addition, reducing vesicular transport from ER to Golgi by brefeldin A also decreased the ATP release to 55 ± 11 nM (n = 8). In another series of experiments, we tested the effect of non-vesicular transport inhibitors (Fig. 2c). Data presented in Fig. 2c, show that Gd3+, which inhibits pannexin and connexin hemichannels in addition to maxi anion channels, markedly inhibited CDCA-induced ATP release to 8 ± 4 nM (n = 6) compared to control of 76 ± 15 nM (n = 10). When cells were treated with probenecid, there was a significant decrease in ATP release to 24 ± 9 nM (n = 7). The pannexin-1 inhibitor, 10Panx, tended to decrease ATP release to 47 ± 15 nM (n = 7), but no statistical significance was reached. Two P2X7 receptor antagonists significantly inhibited the ATP release to 19 ± 5 nM (AZ10606120, n = 9) and 25 ± 5 nM (A438079, n = 8). Taken together, present data indicate that both vesicular and non-vesicular mechanisms are involved in CDCA-induced ATP release from pancreatic epithelial cells.
Effect of CDCA on intracellular ATP
Since we observed that CDCA stimulated substantial ATP release from exocrine pancreatic cells, in the following experiments we investigated whether this was accompanied by a decrease of intracellular ATP concentrations, ATPi. We used the Magnesium Green (MgGreen), an indirect ATP sensor, as it increases in fluorescence when ATP concentrations decrease and the released Mg2+ can bind to the fluorophore. After CDCA stimulation, there was a fast and transient increase in the MgGreen F/F0 fluorescence ratio to 1.53 ± 0.21 in AR42J (n = 3), and then the ratio decreased but remained elevated by about 0.2 units above the basal value (Fig. 3a). Capan-1 cells also responded by a transient increase in F/F0 ratio to 1.34 ± 0.08 (n = 6), but after 300–400 s the signal recovered to pre-CDCA values (Fig. 3b). The transient changes in MgGreen fluorescence could have several explanations (see Discussion). Since we observed a tendency to partial depletion of ATPi in AR42J cells after prolonged stimulation with CDCA (Fig. 3a), we used a more direct method employing one of the ATeam sensors developed by Imamura and colleagues [41].
Fig. 3.
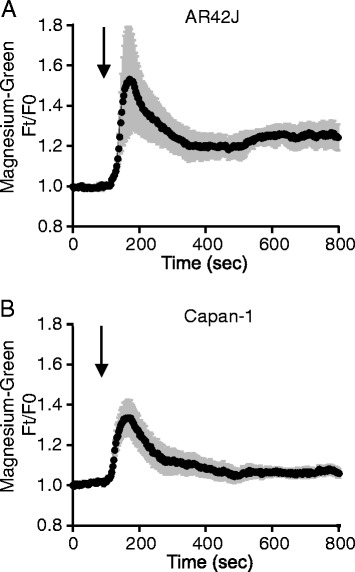
CDCA effect on Magnesium Green fluorescence in AR42J and Capan-1. Effect of 0.5 and 0.3 mM CDCA on intracellular ATP (ATPi) changes in AR42J (a) (n = 3) and Capan-1 (b) (n = 4) cells. Cells were loaded with Magnesium Green indicator (5 μM) for 20 min. Changes of ATPi are given as ratios of fluorescence at time t in relation to time 0 (Ft/F0), where the starting baseline is set to the 1. Results are shown as mean values ± SEM of 10 cells or group of acini per each individual experiment. Arrows indicate time of adding CDCA
AR42J cells were transfected with AT1.03YEMK and simultaneous images of YFP and CFP were used to build YFP/CFP ratio. Fig. 4a shows that there was a small decrease in the ratio with CDCA incubation, which indicates decreased FRET. In addition, in separate fluorescence lifetime imaging (FLIM) experiments we determined CFP fluorescence lifetime of AT1.03YEMK using the time-correlated single-photon counting technique. The data were used to generate a lifetime map by fitting each point to a double exponential decay. χ2 values were lying around 0.9-1.1. The sensor had a uniform lifetime distribution within the cell cytoplasm (Fig. 4b). It is well established that CFP has bi-exponential decay and the reported lifetimes lie around 1.1–1.3 ns and 2.8–2.9 ns [42]. For untreated AR42J cells we found two lifetimes of 0.876 ± 0.013 ns and 2.732 ± 0.026 ns (n = 25). These lifetimes were shorter than the reported lifetime for CFP due to fact that the interacting donor lifetime is shortened by ATP binding and causing FRET in the AT1.03YEMK sensor. After addition of CDCA the lifetimes increased to 0.950 ± 0.018 and 2.815 ± 0.033 in the same experiments, indicating that there is less FRET, likely corresponding to less ATP within the cell.
Fig. 4.
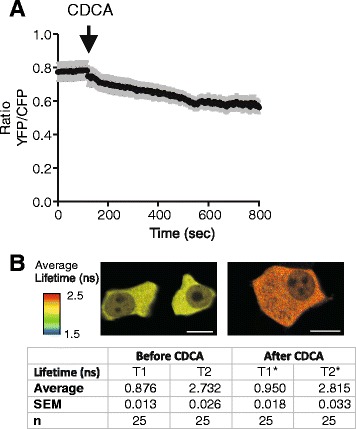
Effect of CDCA on ATP sensor (AT1.03YEMK) in AR42J cells. a Effect of 0.5 mM CDCA on FRET ratio of the ATP sensor in AR42J cells (n = 25). b Lifetime images of cells before and after treatment with CDCA. Images, where each pixel was analysed, are average of the two lifetime constants corrected for intensity scale. The insert table summarizes FLIM-FRET life constants T1 and T2 in 25 independent experiments. * P = 0.0001 for T1 and P = 0.007 for T1 and T2 comparison
The above ATPi measurement methods are dynamic, but difficult to calibrate in acinar cells. Therefore, we also used luciferin/luciferase assay to determine ATPi. Cell membranes were permeabilized with digitonin after CDCA treatment and intracellular ATPi was quantified (Fig. 5a). ATPi concentrations were measured at different time points (1 and 12 min), correlating with the peak and plateau for MgGreen (Fig. 3). Additionally, the long-term effect was also determined by incubating the cells with CDCA for 24 h. After stimulation with 0.3 mM CDCA, Capan-1 released 68 ± 26 nM ATP to extracellular medium, which corresponds to a calculated decrease of 0.08 ± 0.03 mM ATPi in a cell (n = 8). Fig. 5b shows that after incubation of duct cells for 1 min and 12 min, the total content of remaining ATPi was not significantly changed and remained at 2.47 ± 0.32 mM calculated per cell; (n = 8) and 2.22 ± 0.5 mM (n = 7) compared to their respect controls of 2.72 ± 0.38 mM and 2.46 ± 0.45 mM (n = 7). Furthermore, the long-term exposure of Capan-1 to 0.3 mM CDCA did not cause significant changes in ATPi concentrations, i.e., 2.08 ± 0.24 mM compared to control 1.85 ± 0.18 mM (n = 10). For AR42J cells, stimulation with 0.5 mM CDCA (Fig. 5c) caused ATP release of 358 ± 52 nM to extracellular medium, which corresponds to a calculated decrease of 0.68 ± 0.1 mM in the cell (n = 5). Similar to the Capan-1 cells, we did not observe significant changes in remaining ATPi concentrations after 1 min (1.36 ± 0.1 mM; n = 5). However, after 12 min there was a tendency, though not significant, of lower intracellular ATP levels (0.68 ± 0.06 mM; n = 5) with CDCA compared to their respective controls (1.12 ± 0.01 mM and 0.97 ± 0.13 mM; n = 5). Furthermore, after 24 h incubation of AR42J with 0.5 mM CDCA, there was a significant decrease of ATPi to 0.66 ± 0.06 mM compared to the control 1.3 ± 0.08 mM (n = 10).
Fig. 5.
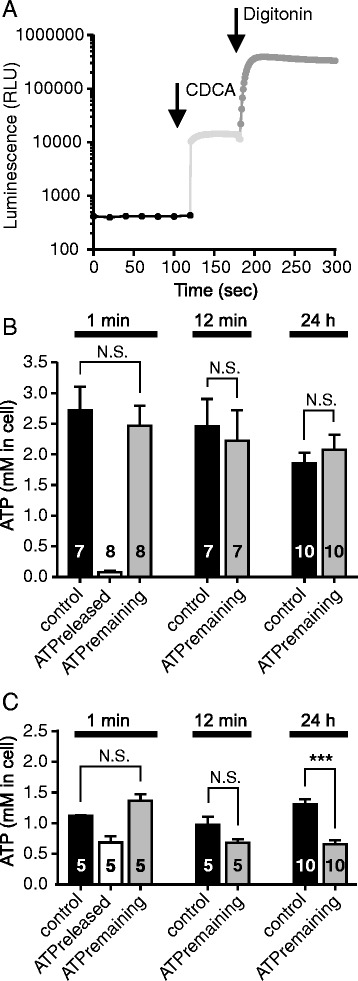
The acute and chronic effects of CDCA on intracellular ATP concentration in AR42J and Capan-1 cells. a Original trace for luminometric measurements of ATP based on Capan-1 measurement. Baseline values were recorded every 20 s for 2 min. Cells were then stimulated with 0.3 mM (Capan-1) or 0.5 mM (AR42J) with CDCA or with vehicle. Stimulated ATP release was recorded every 1 sec for 1 min directly after addition of CDCA, or after 12 min or 24 h of incubation. Finally, cells were permeabilized with digitonin (50 μM), added automatically using pump to release remaining ATP. Panel b and c show the values of released and remaining ATP in Capan-1 and AR42J cells, respectively. These values were calculated per cell after 1, 12 min and 24 h (n = 8, 7, 10 and n = 5, 5, 10) of incubation with CDCA. Data are shown as mean values ± SEM; ***= P < 0.001, N.S. - not significant
CDCA-induced intracellular Ca2+ responses are inhibited by P2 receptor inhibitors
Bile acids are reported to increase [Ca2+]i in pancreatic cells, but it is not clear which receptors are involved (see Introduction). Since we observed that CDCA induced ATP release, we hypothesized that the intracellular Ca2+ responses could be due to effect of released ATP on P2 receptors, which are well established regulators of stimulated Ca2+ influx and/or Ca2+ release in pancreatic ducts. Therefore, [Ca2+]i was monitored in Capan-1 cells in a chamber perfused with physiological solution containing CDCA followed by ATP (Fig. 6a,c). CDCA evoked a slow Δ[Ca2+]i increase (180 ± 24 nM, n = 5). Similar slow and small responses to other BAs were also reported for pancreatic acinar cells and cholangiocytes [25, 43]. In contrast, infusion of ATP caused fast and markedly higher Δ[Ca2+]i increase (772 ± 173 nM, n = 5). Further experiments were conducted without perfusion, in order to minimize mechanical stimulation and amount of inhibitors used. In standing bath CDCA evoked similar Δ[Ca2+]i response as in the perfused conditions, but the signal was delayed (Fig. 6b, d). Stimulation with ATP in the presence of CDCA, caused a lower but fast Δ[Ca2+]i response (302 ± 47 nM, n = 4), perhaps because P2 receptors were already desensitized by CDCA-induced ATP release as ATP was not washed away during perfusion. Therefore, for the following studies with P2R inhibitors, we conducted separate experiments for the ATP and CDCA stimuli.
Fig. 6.
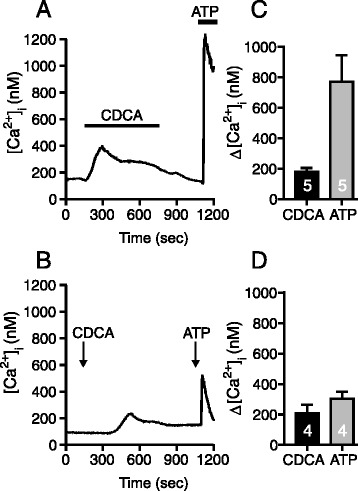
Effect of CDCA and ATP on [Ca2+]i responses in perfused and non-perfused conditions in Capan-1 cells. Representative recordings of intracellular Ca2+ concentrations [Ca2+]i in Capan-1 cells in perfused and non - perfused (standing bath) conditions. a A chamber with Capan-1 cells was perfused (1 ml/min) with physiological buffer containing 0.3 mM CDCA or 100 μM ATP. b Addition of CDCA to a standing bath showed delayed response to CDCA and diminished [Ca2+]i response induced by ATP (100 μM). c and d show the summary of data as mean values ± SEM, (n = 5, 4)
Figure 7a-d shows that the P2R inhibitors markedly reduced the ATP-stimulated Δ[Ca2+]i response from 937 ± 88 nM (n = 11) to 232 ± 47 nM (n = 9). The antagonists also caused significant inhibition of CDCA stimulated Δ[Ca2+]i (from 217 ± 54 to 90 ± 9 nM; n = 5). These data indicate that the Δ[Ca2+]i response evoked by the bile acid could be a result of P2 receptor stimulation by CDCA-induced ATP release. Thapsigargin, the inhibitor of SERCA [44], was added at the end of the experiments to inhibit re-uptake of Ca2+ to intracellular stores. Thapsigargin induced a small Δ[Ca2+]i increase after CDCA by 70 ± 17 nM (n = 5) but a large increase after ATP, i.e., 971 ± 143 nM, (n = 6). P2R inhibitors did not have effect on intracellular Ca2+ response induced by thapsigargin after ATP (Fig. 7c). Interestingly, when thapsigargin was applied after CDCA in the presence of the P2R inhibitors (Fig. 7d), Δ[Ca2+]i was significantly higher (270 ± 35 nM, n = 5).
Fig. 7.
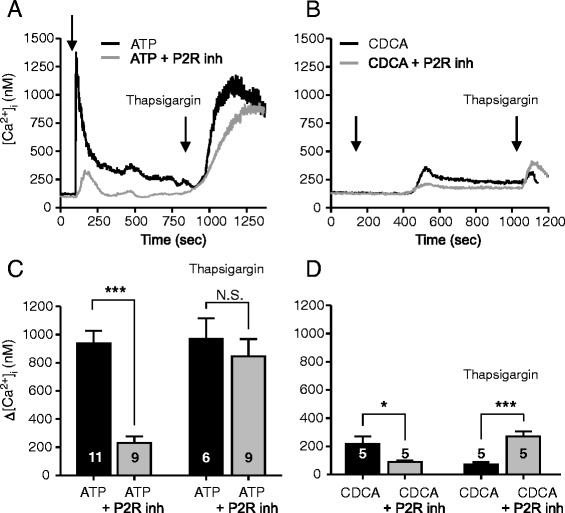
Effect of P2 receptor inhibitors on intracellular Ca2+ responses induced by CDCA in Capan-1 cells. a and b Representative recordings of [Ca2+]i transients in Capan-1 cells with or without P2 receptors inhibitors (standing bath). Cell were incubated with a mix of P2R inhibitors: PPADS (250 μM), suramin (250 μM), and 10 μM of AZ 10606120 and 10 μM of A438079 for 25 min. a, c The presence of P2R antagonists markedly inhibited [Ca2+]i response induced by ATP (100 μM), but had no effect on Thapsigargin (1 μM) induced Ca2+ response. b, d Incubation cells with P2R antagonists inhibited [Ca2+]i transient induced by 0.3 mM CDCA but not by Thapsigargin (1 μM). c, d Change in [Ca2+]i above baseline are given as mean values ± SEM of 7–15 cells per each independent experiment (n). Arrows indicate the time of adding the stimuli. *= P < 0.05, ***= P < 0.001, N.S = not significant
Expression of the TGR5 and FXR receptors in exocrine pancreatic cells
It is not known whether TGR5 and FXR receptors are expressed in the human pancreatic ducts and could account for the effects seen in our study. Also from studies on animal pancreatic tissue, it is not certain whether TGR5 is expressed in ducts [25, 29]. We therefore investigated TGR5 expression in AR42J and Capan-1 cells using RT-PCR and Western Blot. Fig. 8a, b shows that TGR5 is expressed in both cell lines. This observation was confirmed by immunostaining of the acinar cells and on a polarized monolayer of Capan-1 (Fig. 8c, d). In duct epithelium it appears that the receptor is localized mainly on the luminal membrane. Since TGR5 is expressed in pancreatic cells, we wanted to determine whether the receptor has any effect on ATP release and for this purpose we used GPBAR-A, a specific TGR5 receptor agonist that upon binding stimulates cAMP synthesis [45]. Using the luminescence method, we did not detect ATP release (Fig. 8e) from Capan-1 cells after stimulation with 3 or 30 μM GPBAR-A.
Fig. 8.
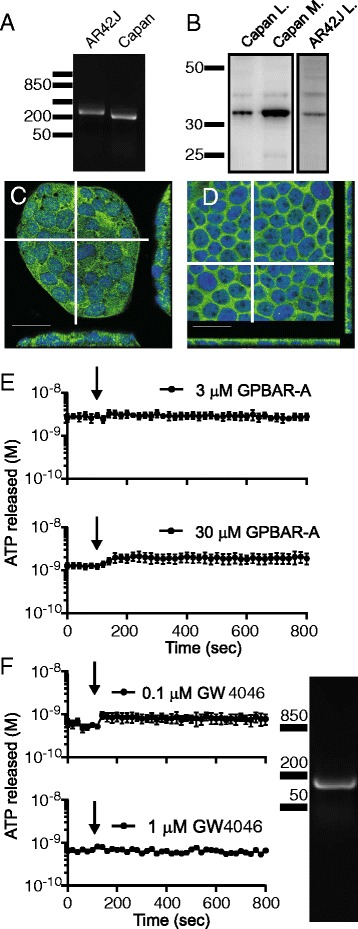
TGR5 and FXR receptors expression in exocrine pancreatic cells. a RT-PCR and b Western Blot analysis of the TGR5 receptor expression in AR42J and Capan-1 cells, in lysates (L) and membrane microdomain enriched fraction (M) shows a clear band at 33 kDa. c - d Immunocytochemistry of TGR5 in AR2J and Capan-1 cells. Scale bars are 25 μm. White lines indicate where the z scan was taken. e. The effect of the TGR5 receptor agonist GPBAR-A at 3 and 30 μM (n = 3, 4) on ATP release from Capan-1 cells. f Expression of FXR in Capan-1 cells showed by RT-PCR. Activation of FXR with the specific agonist GW4046 at 0.1 and 1 μM (n = 3, 2) did not have effect on ATP release from duct cells. Arrows indicate when the agonists were added
In another series of experiments we investigated whether Capan-1 cells express the nuclear type of bile acid receptor, FXR, and whether its stimulation might induce ATP release. We indeed found the transcript for FXR in Capan-1 cells using RT-PCR (Fig. 8f). However, activation of the receptor with the specific agonist GW4064 (0.1 and 1 μM) did not cause significant ATP release (Fig. 8f). Based on these observations, we concluded that stimulation of BAs receptors TGR5 and FXR with specific pharmacological agonists does not induce ATP release from duct cells.
Above we observed that CDCA appeared to protect Capan-1 cells from thapsigargin effects (Fig. 7d, f). This could occur if, for example, CDCA stimulated SERCA (or protected the pumps from thapsigargin) or if CDCA stimulated alternative Ca2+ efflux pathways. Our hypothesis was that the TGR5 receptor could be involved in these effects and we tested this in the following experiments. First, intracellular Ca2+ stores were emptied by thapsigargin in low Ca2+ medium (Fig. 9a). Thereafter, extracellular Ca2+ was reintroduced, and since SERCA pumps were inhibited, [Ca2+]i increased to very high levels in control cells. Notably, perfusion of the cells with CDCA caused a fast and marked reduction in [Ca2+]i from 1253 ± 117 to 244 ± 28 nM (Fig. 9 a, b, n = 3). A similar response was observed with perfusion of GPBAR-A (Fig. 9a-b), which lowered the [Ca2+]i from 1249 ± 69 nM to 647 ± 71 nM (n = 5).
Fig. 9.
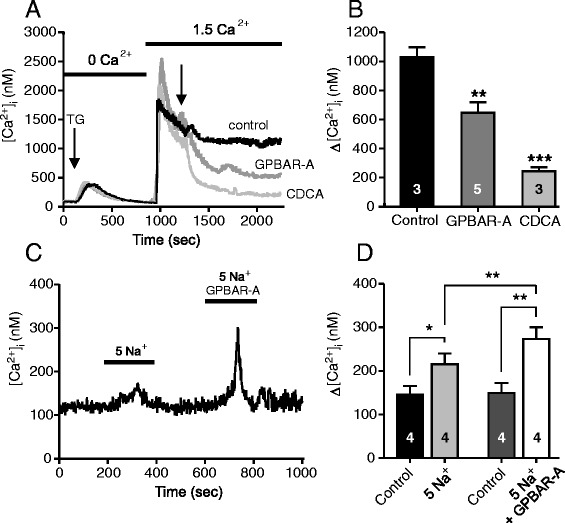
Effect of the TGR5 ligands on [Ca2+]i transients in duct epithelia. Capan-1 cells were incubated with nominal 0 mM Ca2+ buffer (a, b) and thapsigargin (1 μM) to deplete intracellular Ca2+ stores. Thereafter, cells were gently perfused with physiological buffer to refill intracellular Ca2+ stores and after the fluorescence was relatively stable, solutions were changed to GPBAR-A (30 μM), CDCA (0.3 mM), or control. c, d The contribution of the sodium-calcium exchanger (NCX) was tested. Cells were perfused with 5 mM Na+ buffer which increased [Ca2+]i and this response was potentiated in the presence of GPBAR-A (30 μM). b, d Summary of data given as mean values ± SEM of 7–15 cells per each independent experiment (n). *= P < 0.05, ***= P < 0.001, N.S = not significant
In next experiments we addressed the question whether the lowering of [Ca2+]i caused by CDCA could involve Ca2+ efflux via the Na+/Ca2+ exchanger (NCX), which is expressed in duct epithelia [46, 47]. Since CDCA evokes Ca2+ transients itself, we used, GPBAR-A, which showed no effect on Ca2+ responses when given alone (see Additional file 1: Figure S1). Capan-1 cells were perfused with a buffer containing 5 mM Na+, which favours Ca2+ influx via NCX as observed in Fig. 9c. This effect of low Na+ on [Ca2+]i was increased with GPBAR-A (30 μM), which is consistent with TGR5 stimulation of NCX.
Discussion
The present study shows that some of the BA (i.e., CDCA) effects are due to stimulation of purinergic signalling. Firstly, acini and ducts (AR42J and Capan-1 cells) release substantial amounts of ATP in response to CDCA, and this process can be inhibited by several inhibitors of ATP release. Secondly, CDCA effects on intracellular Ca2+ could be significantly inhibited by P2 receptor antagonists, indicating cross-activation of the two signalling pathways. Furthermore, the TGR5 receptor affected intracellular Ca2+ indirectly by activating NCX. Below, we discuss these results and propose that interplay between BAs and purinergic signalling may be important in physiological regulation of pancreatic function.
In order to evaluate physiological or pathological effects of BAs, it is important to consider what BAs concentrations pancreas may encounter. In healthy human, plasma BAs concentrations are <5 μM in resting state and about 10–15 μM post-prandial. However, in case of liver or pancreatic diseases, BAs concentrations can increase to around 300 μM [48, 49]. In normal bile, the concentration of bile acids is higher than 100 mM, and CDCA contributes to about 50 % [50]. Therefore, pancreas could be exposed to low BA concentrations as well as to higher concentrations resulting from bile reflux into pancreatic duct tree following outflow obstruction, e.g., gallstones. In the current studies, we used concentrations that have been considered stimulatory on pancreatic ducts [27, 28].
The most important finding is that primary bile acids can cause ATP release and therefore stimulate purinergic signalling in exocrine pancreatic cells. The unconjugated CDCA had the most pronounced effects compared to far less effective glycine- and taurine-conjugated forms of the acid (Fig. 1). Thus CDCA caused fast and large ATP release from both acinar and duct cells (Fig. 1). In comparison, cholinergic or hormonal stimulation or cell swelling of pancreatic cells induced significantly lower ATP release [34, 35]. We investigated the mechanisms of ATP release and find that both exocytotic pathways and ion channels/receptors seemed to be involved in this process (Fig. 2b, c). Our findings with bafilomycin, brefeldin and NEM strongly support participation of a vesicular component in ATP release, e.g., VNUT (Fig. 2b), which is also expressed in duct cells (unpublished data). In addition, the inhibitor data (Fig. 2C) indicate that connexin hemichannels and/or pannexin with P2X7R could also be involved in ATP release. One of the most potent blockers was Gd3+ that can inhibit pannexin/connexins, Ca2+ influx and hence exocytosis, and it is known inhibitor of maxi-anion channels which may be a part of osmosensitive ATP release with hypotonic stress [51, 52]. We cannot exclude that CDCA either can activate these maxi-anion channels directly or it induce cell volume changes, though cell volume changes due to hypotonic shock (unpublished data) have much smaller impact on ATP release than CDCA. The P2X7 receptors may also have positive effects on CDCA-induced ATP release (Fig. 2c), and modulating effects of the receptor on exocytosis and/or pannexin-1 have been described in other cells [53, 54]. CFTR has also been proposed as a channel/regulator for ATP release [36] and in bile ducts the secondary ursodeoxycholic acid stimulates CFTR-dependent ATP secretion [43]. In Capan-1 cells, CFTR does not seem to contribute to ATP release (unpublished data). Taken together, we propose that several ATP releasing mechanism contribute to CDCA-evoked ATP release, and result in extracellular ATP increase by 100–1000 fold, which is the largest increase compared to that observed with other stimuli (unpublished data).
One important point to consider was how the BA-induced ATP release is triggered. Since GPBAR-A and GW4046 had no effects on ATP release, we presume that the TGR5 and FXR receptors are not involved. An alternative mechanism could be BA induced membrane depolarisation [21], which may be mediated by activation of bile acid-sensitive ion channel (BASIC), which belongs to the DEG/ENaC family, recently identified in bile ducts [55]. In addition, CDCA may incorporate into membranes, increase membrane fluidity [56] and thereby affect one or more of ATP release mechanisms as proposed above. Nevertheless, we show that the ATP release is clearly directed towards the lumen of pancreatic ducts (Fig. 2a), and therefore it is likely that there is a trigger mechanism and luminal exocytosis/transport mechanisms. Also in other epithelia, e.g., kidney tubuli and airway epithelia, ATP is released preferentially to the apical/luminal side in response to a number of stimuli [57–60].
Regarding effect of BA in pancreas, we propose that BA stimulates release of ATP towards lumen and then ATP binds to P2 receptors and therefore stimulates Ca2+ signalling pathways, and these can potentially increase Cl− and K+ conductances, which are necessary to initiate and acid/base transport and thus ductal fluid secretion. Indeed it is well documented that several P2 receptors regulate ion channels such as TMEM16A/ANO1, CFTR and KCa3.1 and KCa1.1 [32, 61]. The more traditional view is that BAs acting directly on BA receptors can affect epithelial transport [10, 26, 62]. For example, in airway epithelial cells, taurodeoxycholic acid (TDCA) stimulates CFTR and Ca2+-activated Cl− currents and these effects seems to be mediated by the basolateral TGR5 receptor [10]. From published studies on pancreatic duct epithelia, it was not clear whether BA receptors were expressed. Nevertheless, it is reported that in dog pancreatic duct epithelial cells TDCA also increased Cl− and K+ fluxes [26], and in guinea pig pancreatic ducts BAs stimulate KCa1.1 channels, but accompanying Cl− channels have not been detected [27, 30].
Previous studies show that CDCA (0.5 mM) caused ATP depletion in pancreatic acinar cells and colon epithelial cells, presumed to be due to inhibited metabolism [63, 64]. Since CDCA induced such a large ATP release in our exocrine cells (Fig. 1), it was relevant to examine whether CDCA could also affect intracellular ATP. We have used several techniques to study this, including intracellular ATP sensors. The transient increase in MgGreen fluorescence could indicate transient decrease in ATPi. However, since CDCA and ATP caused transient increase in [Ca2+]i, and the fluorophore can also bind Ca2+, the MgGreen signals can have several components. In addition, in AR42J cells there was a slow increase in MgGreen fluorescence occurring long after Ca2+ peak. There was a similar slow effect of CDCA on AT1.03YEMK ratio, as well as a change in the lifetime constants of the sensor. These data together indicate that there was a decrease in ATPi. Using luciferase assay, we find that in the first minute after CDCA stimulation acinar cells release significant amount of cellular ATP (Fig. 5c), though ATPi seems to decrease first after 12 min, as also indicated by the AT1.03YEMK measurements (Fig. 4). Nevertheless, it is only after long-term incubation of AR42J cells with 0.5 mM CDCA that significant depletion in ATPi was detected by luminescence assay (Fig. 5c). This depletion could be due to decreased mitochondrial ATP production and/or secondary effects caused by digestive enzymes released by CDCA-induced exocytosis [20, 63, 65] and by exocytosis/release of ATP, as we show in the present study. In contrast to acini, pancreatic ducts seem very robust. In duct cells, CDCA caused release of less than 3 % of total intracellular ATP, and ATP was presumably replenished (Fig. 5b), which also agrees with total recovery of MgGreen fluorescence with continued CDCA stimulation. Interestingly, in pancreatic islets, BAs (e.g., tauroursodeoxycholate) are not detrimental but enhance ATPi concentration [15].
It is extensively documented that BAs (0.1 – 1 mM) cause an increase in [Ca2+]i in many cells, including pancreatic cells and mechanisms include increased release from ER, inhibition of SERCA and increased Ca2+ entry [20, 21, 27, 66]. Regarding TGR5, it is well established that the receptor interacts with Gs protein and leads to stimulation of adenylate cyclase and cAMP signalling [5, 12, 45, 67]. In addition, several reports show that BAs (high μM concentrations) show small and slow increases in intracellular Ca2+ [10, 18, 21, 23, 25, 45]. Underlying mechanisms are unclear, as seen in a number of studies, though one study has suggested the involvement of TGR5 [10, 18, 21, 23, 25, 45]. In pancreatic acinar cells genetic deletion of TRG5 still leaves BA-induced Ca2+ transients in many cells and BA-stimulated amylase release is relatively undisturbed [25], indicating that there is not very tight coupling between TGR5 and Ca2+ signalling. Since GPBAR-A had no effect on [Ca2+]i in our duct cells expressing the receptor, it seems that TGR5 receptor is not involved in initiating simple Ca2+ transients in given experimental conditions. We propose that CDCA-evoked ATP release leads to P2 receptor activation and thereby increases in [Ca2+]i. Indeed, CDCA-stimulated Ca2+ increase was inhibited by a cocktail of P2R antagonists (Fig. 7d). This observation indicates that a significant part of Ca2+-dependent BA effects could be due to ATP release and subsequent stimulation of P2 receptors expressed in duct cells. These processes and potential effects on ion transport are acute (seconds to minutes). Our study did not address the question whether BAs via activation of FXR and TGR5 could regulate purinergic receptor expression on a longer time-scale.
The differences in the thapsigargin induced Ca2+ response after stimulation with ATP or CDCA lead us to speculate whether there is a possible protective role of CDCA during high [Ca2+]i stress conditions in pancreas. It has been shown that CDCA can protect cells when ER is depleted of Ca2+ [68]. We observed that CDCA markedly decreased the thapsigargin-induced high [Ca2+]i and that the GPBAR-A was similarly effective (Fig. 9a). Others have shown that bile acids could prevent thapsigargin-evoked ER stress in liver, adipocytes and β-cells [15, 69]. Our observations are in the line with these and we suggest that the “protective effect” of CDCA is mediated via TGR5 activation. Pancreatic ducts express NCX, which is stimulated by cAMP and Ca2+ [47]. We propose that some of the CDCA-induced decrease in high [Ca2+]i conditions could be due to NCX activation. Indeed stimulation of TGR5 with GPBAR-A increased NCX activity (Fig. 9c). It should be noted that since NCX is electrogenic (exchanging 1 Ca2+ : 3 Na+), it can transport Ca2+ in or out of the cell, depending in electrochemical potential, and this could vary depending on the cell type and its stimulation.
Conclusions
In conclusion, as summarized in Fig. 10, the most important finding in our study is that unconjugated BA evokes significant ATP release from pancreatic exocrine cells, this ATP in turn can stimulate P2R, which thereby increases [Ca2+]i. We show the expression of the TGR5 receptor in a human duct cell line, where it could play a protective role at high intracellular Ca2+ conditions. Taken together, for pancreas we envisage that purinergic signalling is a significant part of the cellular response to BA and could support the physiological function such as secretion. Finally, we propose that purinergic signalling, i.e., ATP release and cell/organ P2 receptor involvement, should be taken into consideration in other cell/organ types, as it could potentially explain the multifaceted and ubiquitous effects of BAs.
Fig. 10.
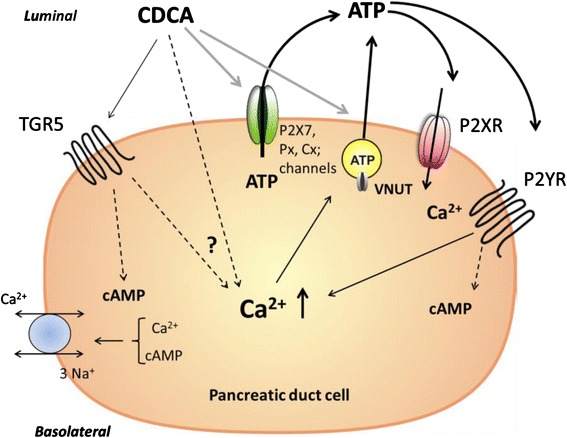
Proposed model of CDCA-induced cellular responses in pancreatic duct cells. CDCA acting on the plasma cell membrane induces ATP release via non-vesicular releasing mechanisms (pannexin, connexin, P2X7R, other ion channels) and vesicular exocytosis of ATP. Released ATP may in turn activate P2X receptors/cation channels and P2Y receptors, which allow Ca2+ influx directly (P2XR) or via G-protein coupled signalling mediate Ca2+ release from intracellular stores and Ca2+ influx (P2YR). Furthermore, CDCA may activate TGR5 receptor, which leads to stimulation of adenylyl cyclase and cAMP production, possibly increase in Ca2+ in some cells by yet undefined mechanisms. The sodium-calcium exchanger (NCX) can be stimulated by TGR5 resulting of transport of Ca2+ out of the cell, or into the cell, depending on prevailing electrochemical gradients
Methods
Chemicals
All chemicals were purchased from Sigma-Aldrich unless otherwise specified. In present study following chemicals were used: chenodeoxycholic acid (CDCA sodium salt, 0.1 – 1 mM), glycochenodeoxycholic acid (GCDCA sodium salt, 0.3 and 1 mM), taurochenodeoxycholic acids (TCDCA sodium salt, 0.3 and 1 mM), ATP (100 μM), digitonin (50 μM), thapsigargin (1 μM), 4-[[3,5-Bis(trifluoromethyl)phenyl]methyl]-6-(2-fluorophenyl)-4,5-dihydro-pyrido[3,2-f]-1,4-oxazepin-3(2H)-one (GPBAR-A, 30 μM, Tocris) dexamethasone (50 nM). Cells were pre-treated/incubated with inhibitors and fluorescent indicators as follow: bafilomycin A1 (1 μM), N-Ethylmaleimide (NEM, 250 μM), brefeldin A (5 μg/ml; Molecular Probes- Life Technology), pannexin inhibitor 10Panx (100 μM, Tocris), gadolinium chloride (Gd3+, 50 μM), probenecid (500 μM), P2X7 inhibitors: AZ10606120 (10 μM, Tocris) and A438079 (10 μM, Tocris), pyridoxal phosphate-6-azo(benzene-2,4-disulfonic acid) tetrasodium salt hydrate (PPADS, 250 μM), suramin 250 μM, Magnesium Green indicator (MgGreen; 5 μM, Invitrogen), Fura-2 AM calcium indicator (5 μM, Teflabs), pluronic F127 (Molecular Probes- Life Technology), ATP kit SL 144–041 (BioThema), cell counting kit-8 (CCK-8, DOJINDO).
Cell cultures
Pancreatic rat acinar (AR42J, CRL-1492) and human duct (Capan-1, HTB-79) cell lines were obtained from ATTC (Manassas, VA) and cultured according to recommended procedures. For imaging experiments WillCo-dishes (WillCo Wells BV, Amsterdam, the Netherlands) were used and for luminescence, 96-well NUNC white plates. For luminescence recordings of ATP release, 50,000 Capan-1 cells were cultured to 80 % confluence and for imaging 35.000 cells were cultured for 2 days. For all experiments for AR42J, 10,000 cells (or 40,000 cells for FRET-FLIM analysis) were plated and grown for 48 h, followed by 48 h with 50 nM dexamethasone to induce an acinar phenotype, which increases formation of zymogen granules [70]. All experiments were conducted at 37 °C.
Measurement of ATP concentrations
Different protocols were used to measure ATP: in extracellular fluid; in single cells; permeabilized cells and polarized cells. Extracellular ATP (ATPe) was monitored in using luciferase + luciferin luminescence reaction in extracellular medium. Capan-1 and AR42J cells were washed and allowed to rest in 65 μl of physiological buffer that contained (in mM): 140 NaCl, 1 MgCl2 · 6H2O, 1.5 CaCl2, 0.4 KH2PO4, 1.6 K2HPO4 · 3H2O, 10 HEPES and 20 glucose (Capan-1) or 10 glucose (AR42J); pH = 7.4. After 30 min, 25 μl of 2 × concentrated luciferase/luciferin mix was added into the wells. Measurements were made using plate mode (20-s sampling rate with 1-s integration) in FLUOstar Optima (BMG, Labtech). CDCA (10 μl) was gently added manually. To quantify CDCA-induced intracellular ATP (ATPi), similar protocol was repeated but after 1 min of recordings, cells were permeabilized with 25 μl of digitonin (50 μM). For each experimental protocol, standard curves were made with ATP concentrations 10−9 to 10−5 M. The bile acids were included in all standards. Also the effect of the inhibitors on luciferase/luciferine was tested independently on standard curves (Additional file 1: Figure S2 a-b). Cell number was determined by cell counting kit and ATP concentrations were corrected for 106 cells per 1 ml. To calculate the single cell ATPi concentration, volumes of Capan-1 and AR42J were estimated assuming a spherical shape with diameter of 12 μm and 10 μm, respectively.
To investigate the polarity of ATP release, i.e., to the luminal or basolateral sides of polarized epithelium, 200,000–400,000 Capan-1 cells/cm2 were seeded on collagen-coated Snapwells (no. 3407; Corning). When resistance of the monolayer was ~ 500–600 Ω*cm2, Snapwells were put into a holder, and monolayers were incubated with 500 μl of physiological solution on each side (luminal and basolateral). 150 μl of fluid was collected for a baseline value, and cells were then luminally or basolaterally stimulated with 0.3 mM CDCA. 150 μl samples collected from apical and basolateral sides and heated for 1 min in 98 °C and kept on ice before analysed for ATP.
Magnesium-Green fluorescence measurement
ATPi was estimated indirectly using Magnesium Green (MgGreen) fluorescence. Cells were incubated with 5 μM MgGreen in presence of 0.02 % pluronic F127 for 20 min and gently washed 15 min before experiments. MgGreen was excited with an argon laser (488 nm) and emission was monitored at 498–581 nm using a confocal laser scanning microscope (TCS SP 5X, Leica Microsystems, Heidelberg). Images were captured every 3 sec using a 20X N.A. 0.7 immersion (water) objective. Images analysis was performed on 10 single cells or group of AR42J per dish. Changes of ATPi responses are presented as ratios at time t in relation to time 0 (Ft/F0) and baseline is set to 1.
ATP sensor FRET-FLIM microscopy
ATPi was estimated more specifically using FRET sensor based on ε subunit of bacterial F0F1-synthase, i.e., AT1.03YEMK [41]. AR42J cells were transfected using FuGENE transfection kit (Promega) according to manufacturer’s protocol. Media was changed the day after transfection to media containing dexametasome. Experiments were conducted on AR42J cells bathed in physiological saline containing 10 mM glucose and the temperature was 37 °C and images were collected with a 63 x water objective (HCX PLAPO, 1.2 NA) in a TCS SP5X Confocal microscope). Two types of recordings were made: FRET ratio analysis and FRET-FLIM analysis. In time resolved protocols, 405 nm laser was used for excitation of the CFP, the fluorescence of CFP and excited YFP was collected simultaneously at 460–500 nm for CFP and 530–570 nm for YFP. Images were taken at 3 s intervals. The individual measurements were corrected for background and YFP/CFP ratio was continuously recorded. In separate experiments, FRET was estimated using fluorescence lifetime imaging (FLIM). The SP5X microscope, equipped with a time-correlated single-photon counting (TCSPC) module (PicoHarp 300, PicoQuant, Berlin, Germany), allows FLIM analysis using a Mai-Tai Ti-Sapphire laser (850 nm) providing 80 ps pulses. The CFP lifetime was recorded with the emission filter: 450–500 nm and detected by FLIM PMT. The laser power was reduced so the counting was kept under 800 kcounts to avoid build up effects. Photons were collected continuously for 2 min for each image. The decay curve was analysed with the SymProTime software (PicoQuant) via a tail-fitting procedure. The counts around the peak were typically around 103 photons and the quality of the fit was judged on the basis of the chi-squared statistic, χ2, and reduced randomness of residuals. Three to five different positions in each dish with cells were analysed before and after addition of CDCA.
Calcium signals
Capan-1 cells were incubated with Fura-2 AM in the presence of 0.02 % pluronic F127 and probenecid (0.25 mM) in physiological buffer for 30 min. For perfusion experiments the perfusion rate was 1 ml/min. For standing bath (non-perfused) conditions cells were incubated for 25 min with or without P2R antagonists. In some experiments, Capan-1 cells were stimulated with Thapsigargin in nominal Ca2+-free buffer containing (in mM): 115 NaCl, 25 Na-gluconate, 1 MgCl2 · 6H2O, 5 HEPES, 1.6 K2HPO4 · 3H2O, 0.4 KH2PO4, 20 glucose, 5 EGTA. Subsequently, cells were gently perfused with buffer containing Ca2+ and after 6–8 min cells were perfused with physiological buffer including CDCA or GPBAR-A. In order to examine the role of the sodium-calcium exchanger (NCX) on [Ca2+]i changes, Capan-1 cells were perfused with 5 mM Na+ buffer, where the rest of NaCl was substituted with 140 N-Methyl-D-Glucamine (NMDG) titrated with HCl. Fura-2 recording was performed on a system described earlier [71]. Fura-2 ratios were calibrated in situ to calcium concentrations based on formula described by Grynkiewicz [72] with Kd for Fura-2: 224 nM.
Reverse transcription PCR
RNA was isolated using RNeasy Mini Kit (Qiangen 74104) following the manufacturer’s instructions. RT-PCR was analysed with QIAGEN OneStep RT-PCR Kit (210212) with amplification parameters as follows: one cycle at 50 °C for 30 min and one cycle at 95 °C for 15 min followed by 37 cycles at 95 °C for 30 s, 58 °C for 30 s, 72 °C for 40 s, and one final cycle at 72 °C for 10 min. The following primers were designed using Primer BLAST and used for TGR5 amplification: human TGR5 forward 3′ TCCTGCCTCCTCGTCTACTT 5′ human TGR5 reverse 3′ GGTAGGGGGCTGGGAAGATA 5′(247 bp), human FXR forward 3′AGAGATGGGAATGTTGGCTGAA 5′ human FXR reverse 3′ GTGAGTTCAGTTTTCTCCCTG 5′(186 bp), rat TGR5 forward 3′ GCTACTGGAGTGGTAGGCAG 5′ rat TGR5 reverse 3′ TCAGTCTTGGCCTATGAGCG 5′(225 bp). All primers were synthesised by TAG Copenhagen A/S (Denmark).
Western blot
Protein lysates were prepared by adding lysis buffer (50 mM TrisBase, 0.25 M NaCl, 5 mM EDTA, 1 % Triton X-100, and 4 mM NaF) containing protease inhibitor. Cell lysates were centrifuged at 15,000 g for 15 min at 4 °C. To obtain the membrane microdomain enriched samples the lysate was centrifuged at 200,000 g for 1 h (Beckman Ultracentrifuge Ti 70.1 Rotor) [61]. Western blot samples were denatured by heating to 37 °C in 50 mM dithiothreitol for 30 min and run on precast gels from Invitrogen. The membranes were blocked overnight at 4 °C in 0.5 % milk powder and 1 % BSA. Primary antibody for TGR5 (1:400 rabbit, Abcam ab72608) were added in blocking buffer for 1.5 h. The goat anti-rabbit secondary antibody conjugated to horse-radish peroxidase (1:2.500) was added in blocking buffer, for 1 h. EZ-ECL chemiluminescence detection kit for HRP (BI, Biological Industries) was added and blots were viewed on Fusion FX Vilber Lourmat.
Immunocytochemistry
AR42J cells were grown on glass coverslips (similar as for dishes, see above) and Capan-1 cells were seeded on collagen coated Snapwells. The cells were gently washed with physiological PBS and fixed in 4 % paraformaldehyde in PBS for 15 min, treated with 0.1 M TRIS-glycine (pH 7.4) for 15 min, and then rinsed in PBS and permeabilized for 10 min in PBS with 0.5 % TritonX-100. Cells were blocked with 10 % BSA in PBS for 45 min and then incubated with TGR5 (1:400; Abcam) for 1.5 h. Slides were washed for 10 min and then incubated 1 h with 1:400 goat anti-rabbit secondary antibody conjugated to Alexa 488 (Life Technology). For nuclear staining, DAPI was used (1:400) and mounted with DAKO fluorescent mounting medium. Slides were viewed using a 40X N.A 1.3 objective with TCS SP 5X.
Statistics
Data are shown as the mean values ± S.E.M. To test the statistical significance between two conditions, unpaired two-tail Student’s t test was applied. For multiple conditions, one-way ANOVA with Bonferroni’s Multiple Comparison Test was used. P < 0.05 was considered statistically significant. For FLIM-FRET analysis and statistics see above.
Acknowledgements
ATP sensor was kindly provided by Professor Hiromi Imamura, Japan Science and Technology Agency. Imaging experiments were done in the Center for Advanced Bioimaging (CAB), University of Copenhagen, Denmark. The technical assistance of Pernille Roshof is greatly acknowledged.
Abbreviations
- ATPi
Intracellular Adenosine 5′ – triphosphate
- ATPe
Extracellular Adenosine 5′ – triphosphate
- BA(s)
Bile acid(s)
- CDCA
Chenodeoxycholic acid
- GCDCA
Glycochenodeoxycholic acid
- CFTR
Cystic fibrosis transmembrane conductance regulator
- [Ca2+]i
Intracellular Ca2+ concentration
- FLIM
Fluorescence lifetime imaging
- FRET
Fluorescence resonance energy transfer
- FXR
Farnesoid X receptor
- GPBAR-A
4-[[3,5-Bis(trifluoromethyl)phenyl]methyl]-6-(2-fluorophenyl)-4,5-dihydro-pyrido[3,2-f]-1,4-oxazepin-3(2H)-one
- MgGreen
Magnesium Green indicator
- NCX
Sodium/calcium exchanger
- NEM
N-Ethylmaleimide
- P2R
P2 purinergic receptor
- PPADS
Pyridoxal phosphate-6-azo(benzene-2,4-disulfonic acid) tetrasodium salt hydrate
- SERCA
Sacro/endoplasmatic reticulum Ca2+ ATPases
- TCDCA
Taurochenodeoxycholic acid
- TGR5
G protein-coupled bile acid receptor
- VNUT
Vesicular NUcleotide Transporter
Additional file
Effect of GPBAR-A on intracellular Ca2+ response from Capan-1 cells. Figure S2. Effect of (A) CDCA and (B) ATP release inhibitors on standard curves.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Author contribution
The study was planned by IN, JMK, KAH and NMC. JMK performed all experiments and analysis on Capan-1 cells. KAH performed experiments on AR42J cells. NMC and IN performed experiments on ATP sensor. IN concept of the study. The manuscript was written by JMK and IN. All authors were involved in critically revising and approved the final version of the manuscript.
Contributor Information
Justyna M. Kowal, Email: justyna.kowal@bio.ku.dk
Kristian A. Haanes, Email: kristian.agmund.haanes@regionh.dk
Nynne M. Christensen, Email: nmchristensen@bio.ku.dk
Ivana Novak, Phone: + 45 353-30275, Email: inovak@bio.ku.dk.
References
- 1.Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, Fukusumi S, Habata Y, Itoh T, Shintani Y, Hinuma S, Fujisawa Y, Fujino M. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003;278:9435–40. doi: 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- 2.Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, Lustig KD, Mangelsdorf DJ, Shan B. Identification of a nuclear receptor for bile acids. Science. 1999;284:1362–5. doi: 10.1126/science.284.5418.1362. [DOI] [PubMed] [Google Scholar]
- 3.Maruyama T, Miyamoto Y, Nakamura T, Tamai Y, Okada H, Sugiyama E, Nakamura T, Itadani H, Tanaka K. Identification of membrane-type receptor for bile acids (M-BAR) Biochem Biophys Res Commun. 2002;298:714–9. doi: 10.1016/S0006-291X(02)02550-0. [DOI] [PubMed] [Google Scholar]
- 4.Schaap FG, Trauner M, Jansen PL. Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol. 2014;11:55–67. doi: 10.1038/nrgastro.2013.151. [DOI] [PubMed] [Google Scholar]
- 5.Maruyama T, Tanaka K, Suzuki J, Miyoshi H, Harada N, Nakamura T, Miyamoto Y, Kanatani A, Tamai Y. Targeted disruption of G protein-coupled bile acid receptor 1 (Gpbar1/M-Bar) in mice. J Endocrinol. 2006;191:197–205. doi: 10.1677/joe.1.06546. [DOI] [PubMed] [Google Scholar]
- 6.Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, Messaddeq N, Harney JW, Ezaki O, Kodama T, Schoonjans K, Bianco AC, Auwerx J. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439:484–9. doi: 10.1038/nature04330. [DOI] [PubMed] [Google Scholar]
- 7.Pols TW, Noriega LG, Nomura M, Auwerx J, Schoonjans K. The bile acid membrane receptor TGR5 as an emerging target in metabolism and inflammation. J Hepatol. 2011;54:1263–72. doi: 10.1016/j.jhep.2010.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keely SJ, Scharl MM, Bertelsen LS, Hagey LR, Barrett KE, Hofmann AF. Bile acid-induced secretion in polarized monolayers of T84 colonic epithelial cells: Structure-activity relationships. Am J Physiol Gastrointest Liver Physiol. 2007;292:G290–7. doi: 10.1152/ajpgi.00076.2006. [DOI] [PubMed] [Google Scholar]
- 9.Mroz MS, Keating N, Ward JB, Sarker R, Amu S, Aviello G, Donowitz M, Fallon PG, Keely SJ. Farnesoid X receptor agonists attenuate colonic epithelial secretory function and prevent experimental diarrhoea in vivo. Gut. 2014;63:808–17. doi: 10.1136/gutjnl-2013-305088. [DOI] [PubMed] [Google Scholar]
- 10.Hendrick SM, Mroz MS, Greene CM, Keely SJ, Harvey BJ. Bile acids stimulate chloride secretion through CFTR and calcium-activated Cl− channels in Calu-3 airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2014;307:L407–18. doi: 10.1152/ajplung.00352.2013. [DOI] [PubMed] [Google Scholar]
- 11.Keitel V, Gorg B, Bidmon HJ, Zemtsova I, Spomer L, Zilles K, Haussinger D. The bile acid receptor TGR5 (Gpbar-1) acts as a neurosteroid receptor in brain. Glia. 2010;58:1794–805. doi: 10.1002/glia.21049. [DOI] [PubMed] [Google Scholar]
- 12.Masyuk AI, Huang BQ, Radtke BN, Gajdos GB, Splinter PL, Masyuk TV, Gradilone SA, LaRusso NF. Ciliary subcellular localization of TGR5 determines the cholangiocyte functional response to bile acid signaling. Am J Physiol Gastrointest Liver Physiol. 2013;304:G1013–24. doi: 10.1152/ajpgi.00383.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beuers U, Hohenester S, de Buy Wenniger LJ, Kremer AE, Jansen PL, Elferink RP. The biliary HCO3− umbrella: a unifying hypothesis on pathogenetic and therapeutic aspects of fibrosing cholangiopathies. Hepatology. 2010;52:1489–96. doi: 10.1002/hep.23810. [DOI] [PubMed] [Google Scholar]
- 14.Keitel V, Haussinger D. TGR5 in cholangiocytes. Curr Opin Gastroenterol. 2013;29:299–304. doi: 10.1097/MOG.0b013e32835f3f14. [DOI] [PubMed] [Google Scholar]
- 15.Lee YY, Hong SH, Lee YJ, Chung SS, Jung HS, Park SG, Park KS. Tauroursodeoxycholate (TUDCA), chemical chaperone, enhances function of islets by reducing ER stress. Biochem Biophys Res Commun. 2010;397:735–9. doi: 10.1016/j.bbrc.2010.06.022. [DOI] [PubMed] [Google Scholar]
- 16.Dufer M, Horth K, Wagner R, Schittenhelm B, Prowald S, Wagner TF, Oberwinkler J, Lukowski R, Gonzalez FJ, Krippeit-Drews P, Drews G. Bile acids acutely stimulate insulin secretion of mouse beta-cells via farnesoid X receptor activation and KATP channel inhibition. Diabetes. 2012;61:1479–89. doi: 10.2337/db11-0815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kumar DP, Rajagopal S, Mahavadi S, Mirshahi F, Grider JR, Murthy KS, Sanyal AJ. Activation of transmembrane bile acid receptor TGR5 stimulates insulin secretion in pancreatic beta cells. Biochem Biophys Res Commun. 2012;427:600–5. doi: 10.1016/j.bbrc.2012.09.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, Macchiarulo A, Yamamoto H, Mataki C, Pruzanski M, Pellicciari R, Auwerx J, Schoonjans K. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009;10:167–77. doi: 10.1016/j.cmet.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meloni AR, DeYoung MB, Lowe C, Parkes DG. GLP-1 receptor activated insulin secretion from pancreatic beta-cells: mechanism and glucose dependence. Diabetes Obes Metab. 2013;15:15–27. doi: 10.1111/j.1463-1326.2012.01663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim JY, Kim KH, Lee JA, Namkung W, Sun AQ, Ananthanarayanan M, Suchy FJ, Shin DM, Muallem S, Lee MG. Transporter-mediated bile acid uptake causes Ca2 + −dependent cell death in rat pancreatic acinar cells. Gastroenterology. 2002;122:1941–53. doi: 10.1053/gast.2002.33617. [DOI] [PubMed] [Google Scholar]
- 21.Voronina SG, Gryshchenko OV, Gerasimenko OV, Green AK, Petersen OH, Tepikin AV. Bile acids induce a cationic current, depolarizing pancreatic acinar cells and increasing the intracellular Na+ concentration. J Biol Chem. 2005;280:1764–70. doi: 10.1074/jbc.M410230200. [DOI] [PubMed] [Google Scholar]
- 22.Petersen OH, Sutton R. Ca2+ signalling and pancreatitis: effects of alcohol, bile and coffee. Trends Pharmacol Sci. 2006;27:113–20. doi: 10.1016/j.tips.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 23.Gerasimenko JV, Flowerdew SE, Voronina SG, Sukhomlin TK, Tepikin AV, Petersen OH, Gerasimenko OV. Bile acids induce Ca2+ release from both the endoplasmic reticulum and acidic intracellular calcium stores through activation of inositol trisphosphate receptors and ryanodine receptors. J Biol Chem. 2006;281:40154–63. doi: 10.1074/jbc.M606402200. [DOI] [PubMed] [Google Scholar]
- 24.Wan MH, Huang W, Latawiec D, Jiang K, Booth DM, Elliott V, Mukherjee R, Xia Q. Review of experimental animal models of biliary acute pancreatitis and recent advances in basic research. HPB (Oxford) 2012;14:73–81. doi: 10.1111/j.1477-2574.2011.00408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perides G, Laukkarinen JM, Vassileva G, Steer ML. Biliary acute pancreatitis in mice is mediated by the G-protein-coupled cell surface bile acid receptor Gpbar1. Gastroenterology. 2010;138:715–25. doi: 10.1053/j.gastro.2009.10.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Okolo C, Wong T, Moody MW, Nguyen TD. Effects of bile acids on dog pancreatic duct epithelial cell secretion and monolayer resistance. Am J Physiol Gastrointest Liver Physiol. 2002;283:G1042–50. doi: 10.1152/ajpgi.00436.2001. [DOI] [PubMed] [Google Scholar]
- 27.Venglovecz V, Rakonczay Z, Jr, Ozsvari B, Takacs T, Lonovics J, Varro A, Gray MA, Argent BE, Hegyi P. Effects of bile acids on pancreatic ductal bicarbonate secretion in guinea pig. Gut. 2008;57:1102–12. doi: 10.1136/gut.2007.134361. [DOI] [PubMed] [Google Scholar]
- 28.Hegyi P, Petersen OH. The exocrine pancreas: the acinar-ductal tango in physiology and pathophysiology. Rev Physiol Biochem Pharmacol. 2013;165:1–30. doi: 10.1007/112_2013_14. [DOI] [PubMed] [Google Scholar]
- 29.Venglovecz V, Hegyi P, Rakonczay Z, Jr, Tiszlavicz L, Nardi A, Grunnet M, Gray MA. Pathophysiological relevance of apical large-conductance Ca2+-activated potassium channels in pancreatic duct epithelial cells. Gut. 2011;60:361–9. doi: 10.1136/gut.2010.214213. [DOI] [PubMed] [Google Scholar]
- 30.Ignath I, Hegyi P, Venglovecz V, Szekely CA, Carr G, Hasegawa M, Inoue M, Takacs T, Argent BE, Gray MA, Rakonczay Z., Jr CFTR expression but not Cl− transport is involved in the stimulatory effect of bile acids on apical Cl−. Pancreas. 2009;38:921–9. doi: 10.1097/MPA.0b013e3181b65d34. [DOI] [PubMed] [Google Scholar]
- 31.Burnstock G, Novak I. Purinergic signalling in the pancreas in health and disease. J Endocrinol. 2012;213:123–41. doi: 10.1530/JOE-11-0434. [DOI] [PubMed] [Google Scholar]
- 32.Novak I, Haanes KA, Wang J. Acid–base transport in pancreas-new challenges. Front Physiol. 2013;4:380. doi: 10.3389/fphys.2013.00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Haanes KA, Novak I. ATP storage and uptake by isolated pancreatic zymogen granules. Biochem J. 2010;429:303–11. doi: 10.1042/BJ20091337. [DOI] [PubMed] [Google Scholar]
- 34.Haanes KA, Kowal JM, Arpino G, Lange SC, Moriyama Y, Pedersen PA, Novak I. Role of vesicular nucleotide transporter VNUT (SLC17A9) in release of ATP from AR42J cells and mouse pancreatic acinar cells. Purinergic Signal. 2014, [DOI] [PMC free article] [PubMed]
- 35.Sorensen CE, Novak I. Visualization of ATP release in pancreatic acini in response to cholinergic stimulus. Use of fluorescent probes and confocal microscopy. J Biol Chem. 2001;276:32925–32. doi: 10.1074/jbc.M103313200. [DOI] [PubMed] [Google Scholar]
- 36.Lazarowski ER. Vesicular and conductive mechanisms of nucleotide release. Purinergic Signal. 2012;8:359–73. doi: 10.1007/s11302-012-9304-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lohman AW, Isakson BE. Differentiating connexin hemichannels and pannexin channels in cellular ATP release. FEBS Lett. 2014;588:1379–88. doi: 10.1016/j.febslet.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yegutkin GG, Samburski SS, Jalkanen S, Novak I. ATP-consuming and ATP-generating enzymes secreted by pancreas. J Biol Chem. 2006;281:29441–7. doi: 10.1074/jbc.M602480200. [DOI] [PubMed] [Google Scholar]
- 39.Okada SF, Nicholas RA, Kreda SM, Lazarowski ER, Boucher RC. Physiological regulation of ATP release at the apical surface of human airway epithelia. J Biol Chem. 2006;281:22992–3002. doi: 10.1074/jbc.M603019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yegutkin GG, Mikhailov A, Samburski SS, Jalkanen S. The detection of micromolar pericellular ATP pool on lymphocyte surface by using lymphoid ecto-adenylate kinase as intrinsic ATP sensor. Mol Biol Cell. 2006;17:3378–85. doi: 10.1091/mbc.E05-10-0993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Imamura H, Nhat KP, Togawa H, Saito K, Iino R, Kato-Yamada Y, Nagai T, Noji H. Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc Natl Acad Sci U S A. 2009;106:15651–6. doi: 10.1073/pnas.0904764106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Becker W, Bergmann A, Hink MA, Konig K, Benndorf K, Biskup C. Fluorescence lifetime imaging by time-correlated single-photon counting. Microsc Res Tech. 2004;63:58–66. doi: 10.1002/jemt.10421. [DOI] [PubMed] [Google Scholar]
- 43.Fiorotto R, Spirli C, Fabris L, Cadamuro M, Okolicsanyi L, Strazzabosco M. Ursodeoxycholic acid stimulates cholangiocyte fluid secretion in mice via CFTR-dependent ATP secretion. Gastroenterology. 2007;133:1603–13. doi: 10.1053/j.gastro.2007.08.071. [DOI] [PubMed] [Google Scholar]
- 44.Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proc Natl Acad Sci U S A. 1990;87:2466–70. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parker HE, Wallis K, le Roux CW, Wong KY, Reimann F, Gribble FM. Molecular mechanisms underlying bile acid-stimulated glucagon-like peptide-1 secretion. Br J Pharmacol. 2012;165:414–23. doi: 10.1111/j.1476-5381.2011.01561.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hansen MR, Krabbe S, Ankorina-Stark I, Novak I. Purinergic receptors stimulate Na+/Ca2+ exchange in pancreatic duct cells: possible role of proteins handling and transporting Ca2+ Cell Physiol Biochem. 2009;23:387–96. doi: 10.1159/000218185. [DOI] [PubMed] [Google Scholar]
- 47.Ankorina-Stark I, Amstrup J, Novak I. Regulation of the Na+/Ca2+ exchanger in rat pancreatic ducts. J Membr Biol. 2002;186:43–53. doi: 10.1007/s00232-001-0134-x. [DOI] [PubMed] [Google Scholar]
- 48.Neale G, Lewis B, Weaver V, Panveliwalla D. Serum bile acids in liver disease. Gut. 1971;12:145–52. doi: 10.1136/gut.12.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brufau G, Bahr MJ, Staels B, Claudel T, Ockenga J, Boker KH, Murphy EJ, Prado K, Stellaard F, Manns MP, Kuipers F, Tietge UJ. Plasma bile acids are not associated with energy metabolism in humans. Nutr Metab (Lond) 2010;7:73. doi: 10.1186/1743-7075-7-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fisher MM, Yousef IM. Sex differences in the bile acid composition of human bile: studies in patients with and without gallstones. Can Med Assoc J. 1973;109:190–3. [PMC free article] [PubMed] [Google Scholar]
- 51.Liu HT, Toychiev AH, Takahashi N, Sabirov RZ, Okada Y. Maxi-anion channel as a candidate pathway for osmosensitive ATP release from mouse astrocytes in primary culture. Cell Res. 2008;18:558–65. doi: 10.1038/cr.2008.49. [DOI] [PubMed] [Google Scholar]
- 52.Sabirov RZ, Dutta AK, Okada Y. Volume-dependent ATP-conductive large-conductance anion channel as a pathway for swelling-induced ATP release. J Gen Physiol. 2001;118:251–66. doi: 10.1085/jgp.118.3.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gutierrez-Martin Y, Bustillo D, Gomez-Villafuertes R, Sanchez-Nogueiro J, Torregrosa-Hetland C, Binz T, Gutierrez LM, Miras-Portugal MT, Artalejo AR. P2X7 receptors trigger ATP exocytosis and modify secretory vesicle dynamics in neuroblastoma cells. J Biol Chem. 2011;286:11370–81. doi: 10.1074/jbc.M110.139410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pelegrin P, Surprenant A. The P2X7 receptor-pannexin connection to dye uptake and IL-1beta release. Purinergic Signal. 2009;5:129–37. doi: 10.1007/s11302-009-9141-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wiemuth D, Sahin H, Falkenburger BH, Lefevre CM, Wasmuth HE, Grunder S. BASIC–a bile acid-sensitive ion channel highly expressed in bile ducts. FASEB J. 2012;26:4122–30. doi: 10.1096/fj.12-207043. [DOI] [PubMed] [Google Scholar]
- 56.Schroder O, Rathner W, Caspary WF, Stein J. Bile acid-induced increase of rat colonic apical membrane fluidity and proton permeability. Z Gastroenterol. 1996;34:365–70. [PubMed] [Google Scholar]
- 57.Seminario-Vidal L, Kreda S, Jones L, O’Neal W, Trejo J, Boucher RC, Lazarowski ER. Thrombin promotes release of ATP from lung epithelial cells through coordinated activation of rho- and Ca2+-dependent signaling pathways. J Biol Chem. 2009;284:20638–48. doi: 10.1074/jbc.M109.004762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wilson PD, Hovater JS, Casey CC, Fortenberry JA, Schwiebert EM. ATP release mechanisms in primary cultures of epithelia derived from the cysts of polycystic kidneys. J Am Soc Nephrol. 1999;10:218–29. doi: 10.1681/ASN.V102218. [DOI] [PubMed] [Google Scholar]
- 59.Bjaelde RG, Arnadottir SS, Overgaard MT, Leipziger J, Praetorius HA. Renal epithelial cells can release ATP by vesicular fusion. Front Physiol. 2013;4:238. doi: 10.3389/fphys.2013.00238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Praetorius HA, Leipziger J. Released nucleotides amplify the cilium-dependent, flow-induced [Ca2+]i response in MDCK cells. Acta Physiol (Oxf) 2009;197:241–51. doi: 10.1111/j.1748-1716.2009.02002.x. [DOI] [PubMed] [Google Scholar]
- 61.Wang J, Haanes KA, Novak I. Purinergic regulation of CFTR and Ca2+-activated Cl− channels and K+ channels in human pancreatic duct epithelium. Am J Physiol Cell Physiol. 2013;304:C673–84. doi: 10.1152/ajpcell.00196.2012. [DOI] [PubMed] [Google Scholar]
- 62.Kelly OB, Mroz MS, Ward JB, Colliva C, Scharl M, Pellicciari R, Gilmer JF, Fallon PG, Hofmann AF, Roda A, Murray FE, Keely SJ. Ursodeoxycholic acid attenuates colonic epithelial secretory function. J Physiol. 2013;591:2307–18. doi: 10.1113/jphysiol.2013.252544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Voronina SG, Barrow SL, Simpson AW, Gerasimenko OV, da Silva X, Rutter GA, Petersen OH, Tepikin AV. Dynamic changes in cytosolic and mitochondrial ATP levels in pancreatic acinar cells. Gastroenterology. 2010;138:1976–87. doi: 10.1053/j.gastro.2010.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ignacio BJ, Olmo N, Perez-Ramos P, Santiago-Gomez A, Lecona E, Turnay J, Antonia LM. Deoxycholic and chenodeoxycholic bile acids induce apoptosis via oxidative stress in human colon adenocarcinoma cells. Apoptosis. 2011;16:1054–67. doi: 10.1007/s10495-011-0633-x. [DOI] [PubMed] [Google Scholar]
- 65.Duan RD, Erlanson-Albertsson C. Effect of bile salt on amylase release from rat pancreatic acini. Scand J Gastroenterol. 1985;20:1239–45. doi: 10.3109/00365528509089283. [DOI] [PubMed] [Google Scholar]
- 66.Nguyen A, Bouscarel B. Bile acids and signal transduction: role in glucose homeostasis. Cell Signal. 2008;20:2180–97. doi: 10.1016/j.cellsig.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 67.Keitel V, Reinehr R, Gatsios P, Rupprecht C, Gorg B, Selbach O, Haussinger D, Kubitz R. The G-protein coupled bile salt receptor TGR5 is expressed in liver sinusoidal endothelial cells. Hepatology. 2007;45:695–704. doi: 10.1002/hep.21458. [DOI] [PubMed] [Google Scholar]
- 68.Xie Q, Khaoustov VI, Chung CC, Sohn J, Krishnan B, Lewis DE, Yoffe B. Effect of tauroursodeoxycholic acid on endoplasmic reticulum stress-induced caspase-12 activation. Hepatology. 2002;36:592–601. doi: 10.1053/jhep.2002.35441. [DOI] [PubMed] [Google Scholar]
- 69.Cummings BP, Bettaieb A, Graham JL, Kim J, Ma F, Shibata N, Stanhope KL, Giulivi C, Hansen F, Jelsing J, Vrang N, Kowala M, Chouinard ML, Haj FG, Havel PJ. Bile-acid-mediated decrease in endoplasmic reticulum stress: a potential contributor to the metabolic benefits of ileal interposition surgery in UCD-T2DM rats. Dis Model Mech. 2013;6:443–56. doi: 10.1242/dmm.010421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Logsdon CD, Moessner J, Williams JA, Goldfine ID. Glucocorticoids increase amylase mRNA levels, secretory organelles, and secretion in pancreatic acinar AR42J cells. J Cell Biol. 1985;100:1200–8. doi: 10.1083/jcb.100.4.1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hansen MR, Krabbe S, Novak I. Purinergic receptors and calcium signalling in human pancreatic duct cell lines. Cell Physiol Biochem. 2008;22:157–68. doi: 10.1159/000149793. [DOI] [PubMed] [Google Scholar]
- 72.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–50. [PubMed] [Google Scholar]