

Abstract
Antisense oligonucleotides (AONs) used to reframe dystrophin mRNA transcripts for Duchenne muscular dystrophy (DMD) patients are tested in clinical trials. Here, AONs are administered subcutaneously and intravenously, while the less invasive oral route would be preferred. Oral delivery of encapsulated AONs supplemented with a permeation enhancer, sodium caprate, has been successfully used to target tumor necrosis factor (TNF)-α expression in liver. To test the feasibility of orally delivered AONs for DMD, we applied 2′-O-methyl phosphorothioate AONs (with or without sodium caprate supplementation) directly to the intestine of mdx mice and compared pharmacokinetics and -dynamics with intravenous, intraperitoneal, and subcutaneous delivery. Intestinally infused AONs were taken up, but resulted in lower plasma levels compared to other delivery routes, although bioavailability could be largely improved by supplementation of sodium caprate. After intestinal infusion, AON levels in all tissues were lower than for other administration routes, as were the ratios of target versus nontarget organ levels, except for diaphragm and heart where comparable levels and ratios were observed. For each administration route, low levels of exon skipping in triceps was observed 3 hours post-AON administration. These data suggest that oral administration of naked 2′-O-methyl phosphorothioate AONs may be feasible, but only when high AON concentrations are used in combination with sodium caprate.
Keywords: antisense oligonucleotides, bioavailability, Duchenne muscular dystrophy, dystrophin, oral administration
Introduction
Antisense oligonucleotides (AONs) are short chemically modified RNA molecules that can be used to modulate splicing. Splice modulation is currently in development as a potential treatment for multiple rare diseases, including Duchenne muscular dystrophy (DMD).1,2,3,4 DMD patients lack dystrophin due to frame-shifting or nonsense mutations in the DMD gene, which leads to premature truncation of protein synthesis during mRNA translation. Dystrophin connects actin filaments with the extracellular matrix in muscle and stabilizes muscle fibers during contractions. In the absence of dystrophin, muscle fibers are vulnerable to damage, resulting in progressive muscle wasting. This leads to wheelchair dependency and eventually death due to respiratory and heart failure.5 Mutations in the DMD gene that maintain the reading frame allow the synthesis of internally deleted but partly functional dystrophin proteins. These are present in Becker muscular dystrophy, which is generally characterized by a later disease onset and a slower disease progression.6,7 AONs can be used in DMD patients to hide signals that are required for exon recognition from the splicing machinery, leading to skipping of the targeted exon and restoration of the reading frame for deletion mutations or bypassing of small mutations in in-frame exons. Hereby, translation can continue, enabling synthesis of Becker muscular dystrophy-like dystrophin proteins.
Currently, two AON chemistries (2′-O-methyl phosphorothioate AONs (2OMePS) and phosphorodiamidate morpholino oligomers) are tested in clinical trials targeting DMD exons 51 and 53 (2OMePS and phosphorodiamidate morpholino oligomers) and exons 44 and 45 (2OMePS only). In these trials, AONs are administered subcutaneously (SC) or intravenously (IV). Notably, SC injections lead to injection side reactions for 2OMePS and repeated IV injections are invasive. Oral administration would obviously be more convenient and less burdensome to DMD patients. Naked phosphorodiamidate morpholino oligomers have high oral bioavailability,8 but naked AONs do not survive the hostile environment (low pH) of the stomach.9 Additionally, naked 2OMePS AONs are normally not taken up by the intestine (since they are high molecular weight, charged, hydrophilic compounds) making oral delivery challenging.9 To overcome these challenges, naked AONs should be protected (e.g., encapsulated) to allow safe passage through the gastrointestinal tract, while membrane permeation enhancers like sodium caprate can be used to improve uptake by the intestine.10
Previous studies have explored the potential of oral administration of 20-O-methoxy ethyl partially modified phosphorothioate oligonucleotides (ISIS104838) targeting the human tumor necrosis factor-α (TNF-α) mRNA in beagle dogs and healthy volunteers.10,11 In dogs receiving three enteric-coated tablets each containing 80.31 mg ISIS104838 and 330 mg sodium caprate or IV injections of 0.5 mg/kg ISIS104838 once daily for a week, oral plasma bioavailability was 1.4% relative to IV injection. The tissue distribution profile and tissue AON levels were similar for both routes, while tissue bioavailability ranged from 2 to 4.3% relative to IV injection. In humans receiving five capsids of 100 or 120 mg ISIS104848 and 330 mg sodium caprate, plasma bioavailability was on average 8.7 and 12.5% respectively across four formulations tested compared to SC dosing of 25 mg ISIS104848. Notably, AON doses for the oral route were respectively 48 and 20–28 times higher than that of the IV route tested in dogs and SC route tested in humans. In both studies, AONs survived the gastrointestinal tract and were still effective in target organs/cells associated with TNF-α synthesis (e.g., myeloid and macrophage cells of bone marrow, blood cells, and several tissues including liver).
Based on these results, we here explored the potential of oral 2OMePS AON delivery in the mdx mouse (the most commonly used mouse model for DMD). Mdx mice carry a nonsense mutation in exon 23 of the Dmd gene abolishing the production of functional dystrophin.12 AONs targeting the in-frame exon 23 lead to the skipping of this exon, thus bypassing the mutation while maintaining the open reading frame and enabling the synthesis of internally deleted but functional Becker muscular dystrophy-like dystrophin proteins.13,14,15 Because the encapsulation needed for stomach passage results in large pellets, oral delivery in mice is challenging, as their small pylorus (estimated size 250 µm) precludes administration.16 However, since proof of concept for encapsulated AONs has been shown in dogs, pigs and humans,10,11,17 we focused on pharmacokinetic/dynamic (PK/PD) properties of 2OMePS after intestinal uptake in mdx mice instead.
This study is the first to describe the PK/PD properties of intraintestinal (INT) administration of naked 2OMePS. We directly compared this to equal doses delivered with IV, intraperitoneal (IP) and SC injections and show that oral delivery is feasible when supplemented with sodium caprate, but that local high AON concentrations are needed in the intestine.
Results
2OMePS plasma pharmacokinetics for different administration routes
To test the feasibility of oral AON administration, mdx mice were either IV, IP, SC, or INT administered with 250 or 1,000 mg/kg 2OMePS AONs targeting exon 23 of the Dmd gene. One mouse receiving 250 mg/kg IP was not considered in the analyses as plasma AON levels were <0.4 µg/ml (compared to >130 µg/ml for similarly treated mice) suggesting misplacement of the injection. For the INT route, one mouse received 353 mg/kg instead of the intended 250 mg/kg as the pump was not stopped on time. One mouse of the 1,000 mg/kg group died 40 minutes after the infusion started, likely because of the anesthetics, therefore the infused dose was 762 mg/kg. These mice were not included in further analyzes. Results were calculated for the animals receiving the complete dose (250 mg/kg (n = 3 for IV and SC, n = 2 for IP and INT) or 1,000 mg/kg (n = 3 for all groups).
In all injected mice, plasma AON levels peaked slightly earlier for the low dose compared to the high dose (Table 1). Maximum AON concentrations (Cmax) in plasma of mice INT infused with 250 mg/kg were ~26, 5.4, and 5 times lower compared to IV, IP, and SC injected mice respectively, while Cmax was 242, 13, and 7.5 times lower for mice infused with 1,000 mg/kg (Figure 1). Besides Cmax, the area under the curve (AUC) at t = 180 minutes also increased in a dose-dependent manner for all delivery routes, being highest for IV injected and lowest for INT infused mice. This increase was more than dose proportional for the IV and IP groups (Cmax 21 and 5.8 times, AUC 23 and 7.2 times respectively), whereas this was equal or less than dose proportional for the SC and INT groups (Cmax 3.5 and 2.4 times, AUC 4 and 2.5 times respectively). Greater than dose proportional increase was especially noted when Cmax levels were above 500 ng/ml, suggesting saturable nonlinear kinetics above this plasma concentration in mice. Since plasma samples were collected in a relatively short time frame (up to 3 hours), extrapolations could not be made.
Table 1. PK analysis of plasma AON levels.
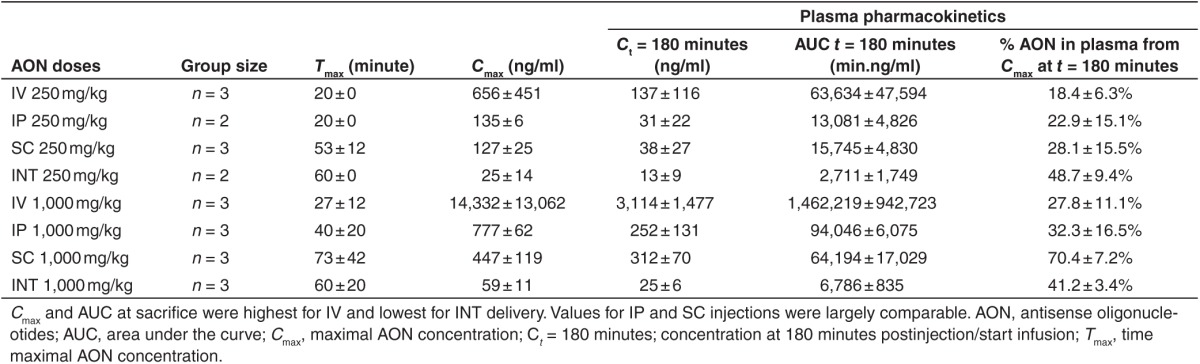
Figure 1.
Antisense oligonucleotides (AON) levels in plasma. Plasma was collected at seven time points post-AON injection or start of the infusion. Plasma AON levels of mdx mice treated with both 250 mg/kg (a) and 1,000 mg/kg AON (b) were highest for IV and lowest for INT administration. Dotted lines in a and b resemble single mice infused with 353 and 762 mg/kg respectively. The mouse receiving 762 mg/kg died 40 minutes after the start of the infusion due to anesthetics. Data are represented in a logarithmic scale as mean ± standard error of the mean. INT, intraintestinal; IV, intravenous; IP, intraperitoneal; SC, subcutaneous.
2OMePS tissue distribution for different administration routes
AON levels in several target organs (gastrocnemius, triceps, diaphragm, heart, and intestine) and nontarget organs (spleen, kidney, and liver) were determined for both doses of each administration route at t = 180 minutes (Figure 2). Delivery of 1,000 mg/kg AON resulted in tissue AON levels which were on average 4.0 ± 1.1 (target organs) and 2.9 ± 1.4 (nontarget organs) times higher than in mice treated with 250 mg/kg (Table 2). Regardless of AON dose and delivery route, the majority of AON was rapidly taken up by kidney. Inherent to the delivery route, AON levels were high in diaphragm and intestine for IP injected and INT infused mice, respectively. Highest AON levels in skeletal muscles and kidney were observed after SC injection. Levels in skeletal muscle were low upon IV injection considering the high plasma Cmax, suggesting saturable uptake at these dose levels. Compared to the injected mice, INT infused mice had lower AON levels in gastrocnemius, triceps, kidney, and liver independent of the dose, while more comparable levels were detected for diaphragm, heart, and spleen despite the lower plasma Cmax and AUC.
Figure 2.

Antisense oligonucleotides (AON) levels in target and nontarget organs. AON levels in muscle and nontarget organs of mice treated with 250 or 1,000 mg/kg. The high dose resulted in higher AON levels in target and nontarget organs. Highest AON levels in diaphragm and intestine were observed after IP injection and INT infusion respectively. AON levels were lower in skeletal muscles and nontarget organs after INT infusion, corresponding to plasma AON levels, while levels were comparable in diaphragm and heart. Gastro, gastrocnemius; INT, intraintestinal; IP, intraperitoneal; IV, intravenous; SC, subcutaneous.
Table 2. Dose-dependent increment of tissue AON levels.
For both AON doses, the ratios between AON levels in target versus nontarget organs were determined (Figure 3). In mice INT infused with 250 mg/kg AONs, ratios of skeletal muscle versus kidney and liver were lower compared to IV, IP, or SC injected mice, however ratios for diaphragm and heart were comparable or higher. Similarly, INT infusion with 1,000 mg/kg resulted in lower or similar ratios of skeletal muscle versus kidney or liver and higher ratios for diaphragm and heart.
Figure 3.

Ratio of antisense oligonucleotides (AON) in muscle versus nontarget organs. The ratio of AON in skeletal muscles (average of gastrocnemius and triceps), diaphragm and heart versus nontarget organs kidney (a) and liver (b) was determined for each delivery route. Ratios were higher in mice receiving the highest AON dose. INT, intraintestinal; IP, intraperitoneal; IV, intravenous; SC, subcutaneous; Sk. muscles, skeletal muscles.
To determine whether AONs were taken up more efficiently by the intestine when delivered combined with sodium caprate, two mdx mice were infused with a 1,000 mg/kg AON solution that was not supplemented with sodium caprate. Without sodium caprate supplementation, AONs were still detectable both in plasma and skeletal muscle, however, levels were much lower (310 and 8.1 times for plasma at t = 60 minutes and tibialis anterior at t = 180 minutes respectively (Table 3)). This indicates that for efficient uptake of 2OMePS AONs by the intestine, an uptake enhancer like sodium caprate is required.
Table 3. Improved intestinal uptake with sodium caprate administration.
2OMePS pharmacodynamics for different administration routes
Exon 23 skipping levels were determined after nested polymerase chain reaction (PCR) for the gastrocnemius, triceps, diaphragm and intestine (see Figure 4 for examples of triceps and diaphragm). Although AONs were only administered 3 hours before mice were sacrificed, exon skipping was observed in the majority of muscle samples obtained from mice receiving either 250 or 1,000 mg/kg delivered via IV, IP, and SC injection. Corresponding to the lower AON levels detected after INT infusion and the shorter circulation time of the total doses (2 versus 3 hours), exon skipping was only detected in the triceps of one mouse infused with 250 mg/kg AON but not in mice infused with 1,000 mg/kg AON.
Figure 4.
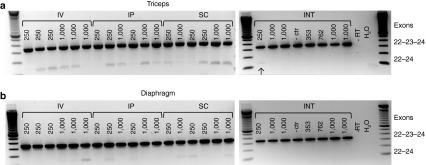
Exon 23 skipping detectable in muscles 3 hours after injection or infusion. Low levels of exon 23 skipping as seen by the presence of a smaller band were already observed 3 hours after antisense oligonucleotides administration, regardless of the administration route or doses in the triceps (a) and diaphragm (b). Bands represent each of the individual mice in the study. INT, intraintestinal; IP, intraperitoneal; IV, intravenous; SC, subcutaneous.
Discussion
AONs are widely studied as a tool to modulate splicing, e.g., to potentially treat neuromuscular disorders like DMD. AONs targeting the dystrophin transcript are delivered IV or SC in clinical trials, but unfortunately due to skin reactions at the injection site and mandatory hospital visits for SC and IV delivery, respectively, these delivery routes are not optimal.1,18 Obviously, oral administration of encapsulated AONs, which are protected from the hostile gastrointestinal tract, would be preferred. Notably, proof of concept for oral delivery of AONs has been obtained for different AON chemistries targeting other diseases,10,11,19 while for DMD, oral delivery of 2OMePS AONs bound to nanoparticles has been previously tested in mdx mice.20 Our study is the first to INT infuse mdx mice with naked 2OMePS AONs (with or without sodium caprate supplementation) and directly compare PK and PD profiles with IV, IP, and SC injections in detail.
As previously reported, plasma AON levels were highest after IV injection. However, because of the high doses used, the serum protein AON-binding capacity was reached, leading to the majority of AON to be rapidly filtered out via the kidneys, and relatively less to be taken up by organs.21 Plasma AON levels upon IP and SC injections were lower due to more gradual uptake from intraperitoneal cavity or subcutaneous depot, respectively, allowing a relatively higher amount of AONs to bind serum proteins and less clearance of unbound AONs and better bioavailability at these high dose levels in mice. We observed that plasma AON levels were even lower upon INT infusion. Here, AONs need to pass the intestinal wall, a slow and incomplete process of absorption resulting in lower tissue uptake (mainly by well perfused organs as diaphragm and heart) and the relatively longer circulation time compared to injections may support this process. For INT infusion, the bioavailability was greatly improved when the AON solution was supplemented with sodium caprate as shown before, e.g., in pigs.10,17,22 It is important to note that upon INT infusion, the entire AON dose was delivered over a period of 60 minutes, while this was within a minute for the other routes. Therefore a direct comparison between INT infusion and other routes of administration is not possible, since this difference has influenced PK and the slow infusion allows the entire dose to circulate in the body for a longer timeframe. It should also be noted that the AON doses used in this study are very high doses and do not directly correspond to the clinical situation in which much lower doses are used. From previous studies in which lower AON doses were injected (up to 200 mg/kg), it is known that plasma AON levels drop to ~5% of their Cmax levels within 5 hours postinjection.13,21 The higher doses (250 and 1,000 mg/kg) used in this study probably have overloaded the system with a Cmax of >500 ng/ml in IV and IP injected mice, resulting in nonlinear PK. Consequently, AON levels dropped faster in IV and IP injected mice compared to the SC and INT administered mice within the 3 hours postinjection studied, where Cmax did not exceed 500 ng/ml leading to a more advantageous linear PK.
We show that AONs were efficiently taken up by target and nontarget organs for all administration routes. Although Cmax was lower for the INT infused mice, AON levels in well perfused organs like diaphragm and heart were comparable to the other routes, and levels in liver and kidney were more favorable. We observed high AON concentrations in kidney for SC and IP injected mice, while as shown previously, five IV, IP, or SC injections with 250 mg/kg within a week lead to higher levels in kidney of IV injected mice.21 This might be caused by differences in AON uptake from the delivery routes and because five weekly doses cannot be directly compared to one single dose.
To our knowledge, we are the first to study PK and PD 3 hours post-AON injection. Interestingly, within this short time span AON administration already resulted in low exon 23 skipping levels as shown by nested PCR. Skip levels were very low and were observed across various muscles, doses, and administration routes.
The chronic use of permeation enhancers is essential to improve bioavailability of AONs. However chronic exposure to sodium caprate could directly or indirectly damage the intestine, i.e., there is an increased risk that toxins will also be taken up by the intestine.22 To gain a better understanding of the safety, applicability and the long-term bioavailability and effects of oral administration and to be able to conclude whether oral delivery is favored over the other delivery routes or could be used in conjunction, larger studies would be needed using extended administration periods. Unfortunately, applying a 1,000 mg/kg AON solution supplemented with sodium caprate in Alzet pumps is impossible as high viscosity of this solution combined with small diameter tubes of the pump system precludes studying this in mice.
Even though studies on the long-term effects should be undertaken and higher dosing and the use of sodium caprate is needed for comparable AON levels as seen with other injection routes, intraintestinal AON administration seems to be feasible and results in favorable muscle/nontarget organ ratios.
Materials and Methods
Animal care. Mice were bred at the animal facility of the Leiden University Medical Center. They were housed in individually ventilated cages with 12-hour light–dark cycles and had ad libitum access to standard chow and water. Studies were performed in 12-week-old male and female mdx (C57BL/10ScSn-mdx/J) mice, which were randomly assigned over the treatment groups. All experiments followed the guidelines of and were approved by the Animal Ethics Committee (DEC) of the Leiden University Medical Center.
AON administration and sample collection. Mdx mice received a single dose of 250 or 1,000 mg/kg M23D(+2–18) (a 2′-O-methyl phosphorothioate AON described previously)15 administered either IV, IP, SC, or INT. For each dosing and administration route, three mice were used, except for the INT 1,000 mg/kg group which consisted of four mice.
For INT administration, mice were fasted overnight to prevent food from obstructing the intestine. The next morning, mice were anesthetized (acepromazin (6.25 mg/kg; Sanofi Sant Nutrition Animale, Libourne Cedex, France), midazolam (6.25 mg/kg; Roche, Mijdrecht, the Netherlands), and fentanyl (0.31 mg/kg; Janssen-Cilag, Tilburg, the Netherlands) and placed on a heat mat. Anesthesia and body temperature were maintained throughout the procedure. The abdominal cavity was partly opened to access the duodenum in which a small incision was made ~0.5 cm distal to the stomach. A cannula (diameter 13 mm) was inserted into the intestine, 2 cm distal from the stomach. Stitches were made around the cannula forcing it to maintain its position and at the stomach part of the duodenum to prevent stomach fluids from leaking out. The cannula was connected to a peristaltic pump (11 Plus Syringe Pump, Harvard Apparatus, Kent, UK), which pumped AONs in the intestine at a rate of 5 µl/minute for 1 hour. This infusion rate was well tolerated by the intestine. To facilitate passage of AONs through the intestinal villi, the AON solution was dissolved in sodium caprate (1,000 mg/kg solution: 66 mg/ml AON in 206 mmol/l sodium caprate, 250 mg/kg solution: 40 mg/ml AON in 206 mmol/l sodium caprate). The abdominal cavity was closed with a continuous stitch and the wound was kept wet with saline during the experiment. AONs were infused for 1 hour to reach the maximum infusion dose of 250 or 1,000 mg/kg. One mdx mouse was taken along as a control and was infused with 206 mmol/l sodium caprate only. Two mdx mice were infused with 1,000 mg/kg AON without supplementation of sodium caprate.
For all mice, blood from the tail vein was collected in Minicollect tubes (0.8 Lithium Heparin Sep, Greiner bio-one, Alphen aan de Rijn, the Netherlands) at 20, 40, 60, 80, 100, 120, and 180 minutes after the INT infusion was started or after the IV, IP, or SC injection. Samples were centrifuged for 5 minutes at 1,700g at 4 °C, plasma was transferred to an Eppendorf tube and stored at −80 °C. After collection of the last blood sample, mice were sacrificed by cervical dislocation. Several tissues (gastrocnemius, triceps, tibialis anterior, diaphragm, heart, spleen, kidney, liver, and intestine) were isolated, snap frozen in liquid nitrogen, and stored at −80 °C.
AON hybridization-ligation assay. AON concentration in plasma and tissue lysates were determined using a method that is based on a previously published hybridization-ligation assay.23 To determine AON concentration in plasma, all samples were diluted 100–10,000 times (depending on the administration route, AON dose and time point of sample collection) in plasma of nontreated mdx mice that was 100 times diluted in phosphate-buffered saline.
To determine AON concentration in organs, small pieces (~60 mg) were weighed and corresponding amounts of buffer containing proteinase K (2 mg/ml Invitrogen, Bleiswijk, the Netherlands), 0.1 mol/l Tris-HCl (pH 8.5), 0.2 mol/l NaCl, 0.2% sodium dodecyl sulfate and 5 mmol/l ethylenediaminetetraacetic acid were added for tissue lysate preparation. Samples were ground in MagNa Lyser green beads tubes in the MagNa Lyser (Roche Diagnostics, Almere, the Netherlands) and kept at 55 °C overnight under gentle agitation. Samples and calibration curves were diluted 60 times in pooled lysates from corresponding tissues isolated from nontreated mdx mice. All samples were measured in duplicate. For mice treated with 250 mg/kg, muscles and spleen lysates were diluted 600 and 1,200 times, liver and kidney lysates 5,000 and 10,000 times, and the intestine lysates 100,000 and 1,000,000 times, respectively. For mice treated with 1,000 mg/kg, muscles and spleen lysates were diluted 1,000 and 5,000 times, liver and kidney lysates 10,000 and 50,000 times and the intestine lysates 1,000,000 and 10,000,000 times, respectively.
RNA isolation and RT-PCR. Tissues were minced in TriPure isolation reagent (Roche Diagnostics) in MagNa Lyser green beads with the MagNa Lyser (Roche Diagnostics) according to the manufacturer's instructions. Total RNA was isolated with chloroform and precipitated with isopropanol. cDNA synthesis was performed with 1,000 ng RNA using BioScript reverse transcriptase (Bioline, Essex, UK) and specific primers (sequences on request) in 20 μl at 55 °C for 30 minutes. Primary PCR was performed on 3 μl cDNA for 20 cycles of 94 °C (40 seconds), 60 °C (40 seconds), and 72 °C (80 seconds). A nested PCR was performed on 1.5 μl of PCR product in a 50 μl mixture by 32 cycles of 94 °C (40 seconds), 60 °C (60 seconds) and 72 °C (60 seconds). PCR products were visualized on 2% agarose gels.
Statistics. Plasma AON levels were analyzed with a noncompartmental PK plasma analysis using Phoenix WinNOnlin 6.2 (Pharsight, Sheffield, UK). Unless stated differently, all data are represented as mean ± standard deviation.
Acknowledgments
This study was financially supported by TREAT-NMD and ZonMw. We are grateful to Ella van Huizen for her technical support. Sd.K. reports being an employee and shareholder of Prosensa Therapeutics. A.A.R. reports being employed by Leiden University Medical Center, which has patent applications on exon skipping, some of which have been licensed to Prosensa Therapeutics. As coinventor on some of these patents, A.A.R. is entitled to a share of royalties.
References
- Goemans NM, Tulinius M, van den Akker JT, Burm BE, Ekhart PF, Heuvelmans N, et al. Systemic administration of PRO051 in Duchenne's muscular dystrophy. N Engl J Med. 2011;364:1513–1522. doi: 10.1056/NEJMoa1011367. [DOI] [PubMed] [Google Scholar]
- Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K, et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378:595–605. doi: 10.1016/S0140-6736(11)60756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havens MA, Duelli DM, Hastings ML. Targeting RNA splicing for disease therapy. Wiley Interdiscip Rev RNA. 2013;4:247–266. doi: 10.1002/wrna.1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntoni F, Wood MJ. Targeting RNA to treat neuromuscular disease. Nat Rev Drug Discov. 2011;10:621–637. doi: 10.1038/nrd3459. [DOI] [PubMed] [Google Scholar]
- Kohler M, Clarenbach CF, Bahler C, Brack T, Russi EW, Bloch KE. Disability and survival in Duchenne muscular dystrophy. J Neurol Neurosurg Psychiatry. 2009;80:320–325. doi: 10.1136/jnnp.2007.141721. [DOI] [PubMed] [Google Scholar]
- Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 1988;2:90–95. doi: 10.1016/0888-7543(88)90113-9. [DOI] [PubMed] [Google Scholar]
- Emery AE. The muscular dystrophies. Lancet. 2002;359:687–695. doi: 10.1016/S0140-6736(02)07815-7. [DOI] [PubMed] [Google Scholar]
- Arora V, Knapp DC, Reddy MT, Weller DD, Iversen PL. Bioavailability and efficacy of antisense morpholino oligomers targeted to c-myc and cytochrome P-450 3A2 following oral administration in rats. J Pharm Sci. 2002;91:1009–1018. doi: 10.1002/jps.10088. [DOI] [PubMed] [Google Scholar]
- Geary RS, Khatsenko O, Bunker K, Crooke R, Moore M, Burckin T, et al. Absolute bioavailability of 2'-O-(2-methoxyethyl)-modified antisense oligonucleotides following intraduodenal instillation in rats. J Pharmacol Exp Ther. 2001;296:898–904. [PubMed] [Google Scholar]
- Raoof AA, Chiu P, Ramtoola Z, Cumming IK, Teng C, Weinbach SP, et al. Oral bioavailability and multiple dose tolerability of an antisense oligonucleotide tablet formulated with sodium caprate. J Pharm Sci. 2004;93:1431–1439. doi: 10.1002/jps.20051. [DOI] [PubMed] [Google Scholar]
- Tillman LG, Geary RS, Hardee GE. Oral delivery of antisense oligonucleotides in man. J Pharm Sci. 2008;97:225–236. doi: 10.1002/jps.21084. [DOI] [PubMed] [Google Scholar]
- Sicinski P, Geng Y, Ryder-Cook AS, Barnard EA, Darlison MG, Barnard PJ. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244:1578–1580. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- Verhaart IE, Tanganyika-de Winter CL, Karnaoukh TG, Kolfschoten IG, de Kimpe SJ, van Deutekom JC, et al. Dose-dependent pharmacokinetic profiles of 2'-O-methyl phosphorothioate antisense oligonucleotidesin mdx mice. Nucleic Acid Ther. 2013;23:228–237. doi: 10.1089/nat.2012.0398. [DOI] [PubMed] [Google Scholar]
- Betts C, Saleh AF, Arzumanov AA, Hammond SM, Godfrey C, Coursindel T, et al. Pip6-PMO, A New Generation of Peptide-oligonucleotide Conjugates With Improved Cardiac Exon Skipping Activity for DMD Treatment. Mol Ther Nucleic Acids. 2012;1:e38. doi: 10.1038/mtna.2012.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann CJ, Honeyman K, McClorey G, Fletcher S, Wilton SD. Improved antisense oligonucleotide induced exon skipping in the mdx mouse model of muscular dystrophy. J Gene Med. 2002;4:644–654. doi: 10.1002/jgm.295. [DOI] [PubMed] [Google Scholar]
- Jang SF, Goins BA, Phillips WT, Santoyo C, Rice-Ficht A, McConville JT. Size discrimination in rat and mouse gastric emptying. Biopharm Drug Dispos. 2013;34:107–124. doi: 10.1002/bdd.1828. [DOI] [PubMed] [Google Scholar]
- Raoof AA, Ramtoola Z, McKenna B, Yu RZ, Hardee G, Geary RS. Effect of sodium caprate on the intestinal absorption of two modified antisense oligonucleotides in pigs. Eur J Pharm Sci. 2002;17:131–138. doi: 10.1016/s0928-0987(02)00162-8. [DOI] [PubMed] [Google Scholar]
- Kinali M, Arechavala-Gomeza V, Feng L, Cirak S, Hunt D, Adkin C, et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009;8:918–928. doi: 10.1016/S1474-4422(09)70211-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteleone G, Fantini MC, Onali S, Zorzi F, Sancesario G, Bernardini S, et al. Phase I clinical trial of Smad7 knockdown using antisense oligonucleotide in patients with active Crohn's disease. Mol Ther. 2012;20:870–876. doi: 10.1038/mt.2011.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falzarano MS, Passarelli C, Bassi E, Fabris M, Perrone D, Sabatelli P, et al. Biodistribution and molecular studies on orally administered nanoparticle-AON complexes encapsulated with alginate aiming at inducing dystrophin rescue in mdx mice. Biomed Res Int. 2013;2013:527418. doi: 10.1155/2013/527418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heemskerk HA, de Winter CL, de Kimpe SJ, van Kuik-Romeijn P, Heuvelmans N, Platenburg GJ, et al. In vivo comparison of 2'-O-methyl phosphorothioate and morpholino antisense oligonucleotides for Duchenne muscular dystrophy exon skipping. J Gene Med. 2009;11:257–266. doi: 10.1002/jgm.1288. [DOI] [PubMed] [Google Scholar]
- Maher S, Leonard TW, Jacobsen J, Brayden DJ. Safety and efficacy of sodium caprate in promoting oral drug absorption: from in vitro to the clinic. Adv Drug Deliv Rev. 2009;61:1427–1449. doi: 10.1016/j.addr.2009.09.006. [DOI] [PubMed] [Google Scholar]
- Yu RZ, Baker B, Chappell A, Geary RS, Cheung E, Levin AA. Development of an ultrasensitive noncompetitive hybridization-ligation enzyme-linked immunosorbent assay for the determination of phosphorothioate oligodeoxynucleotide in plasma. Anal Biochem. 2002;304:19–25. doi: 10.1006/abio.2002.5576. [DOI] [PubMed] [Google Scholar]