

Abstract
The triphenylethylene antiestrogen, tamoxifen, can be an effective inhibitor of sphingolipid metabolism. This off-target activity makes tamoxifen an interesting ancillary for boosting the apoptosis-inducing properties of ceramide, a sphingolipid with valuable tumor censoring activity. Here we show for the first time that tamoxifen and metabolite, N –desmethyltamoxifen (DMT) block ceramide glycosylation and inhibit ceramide hydrolysis (by acid ceramidase, AC) in human acute myelogenous leukemia (AML) cell lines and in AML cells derived from patients. Tamoxifen (1-10 μM) inhibition of AC in AML cells was accompanied by decreases in AC protein expression. Tamoxifen also depressed expression and activity of sphingosine kinase 1 (SphK1), the enzyme catalyzing production of mitogenic sphingosine 1-phosphate (S1-P). Results from mass spectroscopy showed that tamoxifen and DMT, i ) increased the levels of endogenous C16:0- and C24:1 ceramide molecular species, ii) nearly totally halted production of respective glucosylceramide (GC) molecular species, iii ) drastically reduced levels of sphingosine ( to 9% of control), and iv ) reduced levels of S1-P by 85%, in vincristine-resistant HL-60/VCR cells. Co-administration of tamoxifen with either N-(4-hydroxyphenyl)retinamide (4-HPR), a ceramide-generating retinoid, or a cell-deliverable form of ceramide, C6-ceramide, resulted in marked decreases in HL-60/VCR cell viability that far exceeded single agent potency. Combination treatments resulted in synergistic apoptotic cell death as gauged by increased Annexin V binding and DNA fragmentation and activation of caspase-3. These results show the versatility of adjuvant triphenylethylene with ceramide-centric therapies for magnifying therapeutic potential in AML. Such drug regimens could serve as effective strategies, even in the multidrug resistant setting.
Keywords: leukemia, sphingolipid metabolism, tamoxifen, triphenylethylenes, ceramide
1. Introduction
Sphingolipid metabolism is an area of cancer biology that has risen to prominence over the past decade and a half [1,2,3,4] .This is because ceramide, the aliphatic backbone of sphingolipids, acts as a powerful tumor censor, whereas its glycosylated product, GC, formed by the action of glucosylceramide synthase (GCS), is anti-apoptotic and a biomarker of multidrug resistance [5]. Ceramidase, in particular acid ceramidase (AC), another sentinel enzyme regulator of cancer cell growth, has been identified as candidate gene for development of new cancer diagnostics and touted as a therapeutic target in metastatic cancer [6,7] . Much like GCS, AC dampens the tumor suppressor properties of ceramide via ceramide hydrolysis, leading to the formation of sphingosine 1-phosphate (S 1-P), a problematic mitogen in cancer[8]. Thus, sphingolipid metabolism is a dynamic process with complex orchestration, impact, and clinical applications, presenting druggable targets for exploitation.
GCS, AC, and sphingosine kinase −1 (SphK1), which catalyzes formation of S 1-P, are all important regulators of the tumor suppressor actions of ceramide [2,9] . Numerous studies have demonstrated that inhibition of these enzymes drives apoptotic responses and/or subdues mitogenicity [7,10]. Although many inhibitors of these enzymes have been studied, rarely does one find a single agent that demonstrates multi-hit capacity. The present study shows, in human acute myelogenous leukemia (AML) cell lines and in AML cells derived from patients, that tamoxifen inhibits ceramide glycosylation, AC activity, and Sphk1 activity, and downregulates AC and SphK-1 expression, and sensitizes AML cells to ceramide-centric therapeutics. These unique actions place tamoxifen and associated chemical relatives, N-desmethyltamoxifen (DMT) and raloxifene, in an interesting place for study, specifically as regards “off-target” activities of tirphenylethylenes in cancer therapy.
Our earlier studies with tamoxifen showed that this gold standard therapy for treatment of estrogen receptor-positive breast cancer, acted as a potent inhibitor of ceramide glycosylation [11], an action in common with other P-glycoprotein (P-gp) antagonists such as verapamil and cyclosporin A [12]. Whereas tamoxifen is not a specific GCS inhibitor, addition to cells in culture results in reduced glucosylceramide (GC) levels [12] and in some instances, increases in intracellular ceramide levels [13]. Another commonality of tamoxifen with sphingolipid metabolism is inhibition of AC, a property associated with elicitation of lysosomal membrane permeability and subsequent AC proteolysis by cathepsin B [14].
The focus of the present study is AML, the most common type of leukemia in adults. AML is aggressive, and only about 25% of patients that experience remission with cytotoxic chemotherapy remain disease- free. Drug resistance is a major problem in treatment of AML [15], and new therapeutic approaches are needed. The use of P-gp antagonists to overcome drug resistance has had limited clinical success in treatment of leukemia [16].
An allied function of drug transporters such as P-gp is glycolipid trafficking [17], an area that has not been explored from a therapeutic standpoint. Building upon what has been learned from past work on the effects of tamoxifen on ceramide glycosylation [11,12], it appears appropriate that triphenylethylene anti-estrogens could have therapeutic application in AML, application divorced from their use as modulators of multidrug resistance and divorced from anti-estrogenic activities. Herein we report for the first time that tamoxifen targets three important junctures in sphingolipid metabolism in AML, ceramide glycosylation, ceramide hydrolysis, and sphingosine phosphorylation. This work is an introduction to future research on the use of cell-deliverable ceramides and/or ceramide-generating agents in combination with triphenylethylenes in treatment of cancer and supportive of combination therapy with standard of care approaches to treating cancer.
2. Materials and Methods
2.1 Cell lines, patient samples, and reagents
Human AML cell lines HL-60 and KG-1 were obtained from the American Type Culture Collection (ATCC), Manassas, VA. The cells were expanded and cryo-preserved in liquid nitrogen in the investigator's laboratory. Cell lines were authenticated by documentation provided by the ATCC, which include antigen expression, DNA profile, and cytogenic analysis. HL-60/VCR cells were provided by A.R. Safa (Indiana University School of Medicine, Indianapolis, IN); cells were grown in medium containing 1.0 μg vincristine sulfate/mL culture medium. Cell lines were maintained for approximately 30 passages in RPMI-1640 GlutaMAX medium (Life Technologies, Carlsbad, CA), supplemented with 10% FBS (Atlanta Biologicals, Atlanta, GA) and 100 units/mL of penicillin and 100 μg/mL streptomycin. For experiments with HL-60/VCR cells, vincristine was removed from the medium. Cells were cultured in a humidified atmosphere, 95% air, 5% CO2 at 37 °C.
AML patient samples with 20% or greater blast count were obtained with informed consents signed for sample collection according to a protocol approved by the Institutional Review Board of the Milton S. Hershey Medical Center. Cells were cryo-preserved before use, and after thawing, cells were seeded for experiments based upon viable cell counts.
C6-ceramide (N-hexanoyl-D-erythro-sphingosine) was obtained from Avanti Polar Lipids, Alabaster, AL, and dissolved in 100% ethanol (10 mM stock) and stored at −20 °C. N-(4-hydroxyphenyl)retinamide (4-HPR), a product of Calbiochem (San Diego, CA), was dissolved in DMSO (10 mM stock) in amber glass vials and stored at −20 °C. Tamoxifen-HCl and DMT were purchased from Sigma Chemical Company, St. Louis, MO; all were dissolved 100% ethanol (10 mM stock) and stored at −20 °C. LY117018, a raloxifene analog, obtained from Eli Lilly, Indianapolis, IN, was dissolved and stored, as were the other triphenylethylenes. LCL204 ((1R,2R) 2-(N-tetradecylamino)-1-(4-NO2)-phenyl- 1,3-dihydroxy-propane HCl) was generously provided by Dhimant Desai and Shantu Amin, Penn State College of Medicine, Organic Synthesis Core Facility, Hershey , PA. Radiolabeled [9,10-3H] palmitic acid, 40-60 Ci/mmol, was purchased from American Radiolabeled Chemicals, St. Louis, MO, and stored at −20 °C. Solvents, HPLC-grade, for thin-layer chromatography (TLC) were purchased from ThermoFisher Scientific (Waltham, MA), and Silica Gel G plates were purchased from Analtech, Newark, DE. Ecolume liquid scintillation fluid was a product of MP Biomedicals, Solon, OH.
2.2 Acid ceramidase activity assays
AC activity was measured in intact cells by fluorogenic assay as described [18] , with slight modification. Briefly, cells (20,000/well) were seeded into 96-well plates, in 50 μl 10% FBS-containing culture medium. After 24 hr, tamoxifen or DMT was added to the cells (in medium containing 1% FBS) to a final volume of 100 μL (controls contained ethanol vehicle), and cells were placed in a tissue culture incubator at 37 °C , 5% CO2, for 24 hr. Cell viability assays were conducted in parallel (see methods below). Fluorogenic substrate (ethanol vehicle) was then added to the wells to a final concentration of 16 μM (125 μL final well volume), and the cells were incubated for 3 hr. At this point, the plates with cells were placed at −20 °C, and the chemistry was conducted the following day. To complete the chemistry, 50 μL methanol and 100 μL NaIO4 (2.5 mg/mL) in 0.1 M glycine buffer, pH 10.6, was added, and the plates were incubated in the dark for 2 hr at 37 °C. Fluorescence was measured in the UV range (365 nm excitation/410-460 nm emission) using a GloMax multi-detection system (Promega, Madison, WI).
2.3 Sphk1 activity assays
Sphk1 activity was quantified by using a commercial Sphingosine Kinase Activity Assay Kit (Echelon Biosciences, Salt Lake City, UT) as the manufacturer instructed. In brief, 1 × 106 cells were incubated with tamoxifen for the indicated times, at 37 °C in culture medium containing 5% FBS, collected by centrifugation, and washed in ice-cold PBS. Cells were then lysed by freeze-thaw cycles in the reaction buffer provided, and lysates were then incubated in reaction buffer containing 100 μM sphingosine and 10 μM ATP for 1 hr at 37 °C. Luminescence-attached ATP detector was then added to stop the reaction. Luminescence was determined by using a Perkin Elmer Victor-3 1420 microplate reader.
2.4 Cell viability assays
Viability was assessed using the CellTiter 96 One Solution Cell Proliferation Assay Kit (MTS) (Promega, Milwaukee, WI). Viability was calculated as the mean (n=3 or n=6) absorbance (minus vehicle control) at 490 nm, using a Perkin Elmer Victor-3 1420 microplate reader.
2.5 Ceramide analysis by thin-layer chromatography
The effect of 4-HPR on cellular ceramide levels was determined using [3H]palmitic acid labeling. Briefly, HL-60/VCR cells (2 × 106) were seeded into 6-well plates in 2.0 mL medium containing 5% FBS with 2.0 μCi [3H]palmitic acid/mL. Cells were treated without (ethanol vehicle) or with 4-HPR for 24 hr. Cells were then harvested by centrifugation, washed 2-times with ice-cold PBS, and total lipids were extracted using chloroform, methanol, and water [5]. 3H-Ceramides were resolved from total lipid extracts by TLC in a solvent system containing chloroform/acetic acid (90:10, v/v). Commercial brain ceramides (Avanti Polar Lipids, Inc.) and monooleoylglycerol (Nu Chck Prep, Alysian ,MN) were co-chromatographed, the latter because monoacylglycerols run just below ceramide in this solvent system. Tritiated ceramide was quantitated by liquid scintillation counting as described [19] .
2.6 Determination of apoptosis
Apoptosis was detected using the ApoDETECT Annexin V-FITC Kit (Life Technologies, Carlsbad, CA), following the manufacturers protocol. Briefly, cells were seeded in 6-well plates at 1 × 106 cells/well in medium containing 5% FBS. Cells were treated with indicated agents for 18 hr, collected by centrifugation and washed with PBS and stained with Annexin V and propidium iodide (PI) using 1× Annexin binding buffer (provided in the kit). Following, the cells were washed with 1× binding buffer and fixed in 2% paraformaldehyde overnight. Cells were then washed in binding buffer and the percent of Annexin V-positive cells determined by flow cytometry. Data was assessed using FCS Express 4, from De Novo Software (Glendale, CA). Apoptosis was also determined by flow cytometric analysis of DNA fragmentation of PI-stained nuclei following published protocol [20] . Cells were seeded as above and treated with the indicated agents for 24 hr. Cells were collected by centrifugation, washed with ice-cold PBS containing 0.2% bovine serum albumin, Fraction V (Sigma Chemical Company), and DNA was stained for 2 hr in the dark with 0.5ml hypotonic PI buffer (0.1% sodium citrate, 50 μg/mL DNase-free RNase, 0.1% Triton X-100). DNA content was analyzed using a FACScan, and sub G0 (apoptosis marker) was quantitated using FCS Express 4 software.
2.7 Caspase-3 activity assay
Caspase-3 proteolytic activity was measured (after 12 hr treatment of intact cells) in cell lysates using the Caspase-3 DEVD-R110 Fluorometric HTS Assay Kit (catalog #30009, Biotium Inc. Hayward, CA) according to the manufacturer's instructions.
2.8 Mass spectroscopy
HL-60/VCR cells (8 × 106) were seeded in 5 mL RPMI-1640 medium containing 5% FBS in 10-cm culture dishes. After 2 hr in a 37 °C tissue culture incubator, the cells were treated with either 5 or 10 μM tamoxifen or 10 μM DMT by adding 5 mL of the appropriate drug-containing media (final dish volume, 10 mL). Cultures were incubated for 18 hr, cells were collected by centrifugation, washed 3-times with ice cold PBS, and cell pellets were immediately frozen at −80 C. The samples were overnight freighted, on dry ice, for analysis (Penn State/Hershey College of Medicine). Lipids were extracted with ethyl acetate/isopropanol/water (60:30:10, v/v), without phase partitioning and the solvents were evaporated (azeotroph) under a steam of nitrogen. Internal standards containing C17-sphingosine 1-phosphate, C17-sphingosine, C17-sphinganine, C17-sphinganine 1-phosphate, C12-ceramide, and C12-glucosylceramide (Avanti Polar Lipids) were used. Lipid samples were chromatographically separated on an Agilent 1100 HPLC system and analyzed on a 4000 QTRAP (AB Sciex) using the electrospray ionization-tandem mass spectrometry (ESI-MS/MS) method as previously described[21]. Quantification of each sphingolipid subspecies was calculated against the internal standards and normalized to protein content.
2.9 Immunoblotting
Cells, KG-1 or HL-60/VCR were seeded at 5 × 106 cells/well in a 6-well plate in RPMI-1640 medium containing 5% FBS and treated with tamoxifen (ethanol vehicle) or LCL204 (DMSO vehicle) after 1 hr, for 2, 4 or 24 hr. Controls were exposed to vehicle only. For analysis by Western blot, cells were lysed using RIPA buffer containing phosphatase inhibitor cocktail and protease inhibitor P8340 according to the manufacturers protocol (Sigma Chemical Company). Proteins were resolved on 10% SDS-PAGE gels and transferred onto PVDF membranes (Millipore, Billerica, MA). Primary antibodies used were as follows: AC (BD Biosciences, San Jose, CA), SphK1 (#3297, Cell Signaling, Danvers, MA), GAPDH (#2118, Cell Signaling), and beta-actin (#3700, Cell Signaling). For secondary antibodies, HRP-conjugated goat anti-mouse or goat anti-rabbit IgG (Cell Signaling) were used, and Pierce Enhanced Chemiluminescence (Thermo Scientific) was applied to blots according to manufacturer's protocol.
2.10 Data analysis
Results are expressed as means ± S.E. and were analyzed by ANOVA. Differences among the treatment groups were assessed by Tukey post hoc test. Differences were considered significant at P≤0.05. A combination index (CI), based on the Chou-Talalay method [22], was calculated as described, using the following formula: CI = sum of specific apoptosis of single agent treatment/specific apoptosis upon combined treatment. When CI was < 1, =1, or >1, the effects were defined as synergistic, additive, and infra-additive, respectively. The percentage of specific apoptosis was determined by using the following formula: specific apoptosis = (drug-induced apoptosis - spontaneous apoptosis)/ (100 – spontaneous apoptosis) × 100%.
3. Results
3.1 Suppression of AC enzymatic activity in AML cells by triphenylethylenes
Ceramidase, in particular AC, is a sentinel enzyme regulator of cancer cell growth. Much like GCS, AC dampens the tumor suppressor properties of ceramide through hydrolysis. The first experiments were designed to determine the effect of tamoxifen on AC activity in AML, using human AML cell lines and samples derived from patients. Although we have previously demonstrated that tamoxifen inhibits AC in cancer cells [14], hematological malignancies have not been investigated. The data in Fig. 1A-D show that tamoxifen and the major in vivo metabolite, DMT, inhibit AC activity in the promyeloblast/acute promyelocytic leukemia cell line, HL-60, in its Vinca alkaloid-resistant counterpart, HL-60/VCR, and in KG-1 cells, a macrophage/acute myelogenous leukemia. As pictured, the dose-response for inhibition of AC by tamoxifen and DMT in KG-1 cells was similar (Fig. 1C,D). Viability was not majorly influenced after 24 hr exposure to tamoxifen, as shown by the upper curves, with the exception of KG-1 cells, which demonstrated a decrease to 70% viability upon DMT exposure. Therefore, inhibition of AC by tamoxifen had little cytotoxic impact at 24 hr. However, extended-time experiments showed that tamoxifen and DMT were cytotoxic after a 72 hr exposure (10 μM), where upon cell viability was reduced to 23 and 8% of control, respectively (data not shown). The selective estrogen receptor modulator LY117018, a raloxifene analogue, also inhibited AC activity in KG-1 cells (Fig. 1C, inset). The data in Fig. 1E demonstrate the influence of tamoxifen and DMT on AC activity in AML samples derived from patients. For comparison, we have included the AC inhibitor, LCL204 [23]. As shown, after a 4 hr exposure, both tamoxifen and DMT inhibited AC activity in all samples, albeit to differing degrees. LCL204 showed similar inhibitory activity.
Fig. 1.

Effect of triphenylethylenes and AC inhibitor LCL204 on AC activity in cultured human AML cell lines and in AML cells derived from patients. (A-D) Cell lines. Cells (20,000/well) were seeded into 96-well plates in medium containing 10% heat-inactivated FBS one day prior to treatment. Tamoxifen, DMT, and raloxifene analog LY117018 were administered (in culture medium containing 1% FBS) for 24 hr at the concentrations indicated, and AC activity was then measured in intact cells using fluorescent substrate as detailed in Materials and Methods. Cell viability was measured at 24 hr in parallel experiments. n=6 for each experimental point; values are the mean ± S.E. Repeated experiments yielded like results. (E) Cells derived from patients. Cryo-preserved cells were thawed and viabilities assessed by trypan blue staining. Cells were seeded (30,000 live cells/well), and after 3 hr equilibration (tissue culture incubator), cells were treated as indicated for 4 hr. AC activity was then measured. n=5 wells per treatment; values are ± S.E. Micromolar concentrations in parentheses. CTR, control; DMT, N-desmethyltamoxifen; Tam, tamoxifen; pt, patient.
3.2 Downregulation of AC and SphK1 by tamoxifen
Prior studies with the lysosomotropic AC inhibitor, LCL204, demonstrated that enzyme inhibition ensued via downregulation of AC expression in the prostate cancer cell line, DU145 [23]. To determine whether inhibition of AC by tamoxifen in leukemia cells followed a similar track, we exposed KG-1 cells to tamoxifen and measured AC expression by Western blotting. Fig. 2A shows that tamoxifen downregulated AC expression in a dose-dependent manner in KG-1 cells. Notably, there was an approximate 70% reduction in AC expression in response to 2.5 μM tamoxifen at 24 hr and near disappearance at 10 μM. Moreover, tamoxifen exposure also resulted in downregulated expression of SphK1 (Fig. 2A); although not as robust as the dose-response effects on AC, 10 μM tamoxifen exacted near-total elimination of SphK1 expression at 24 hr. Diminution of Sphk1 expression was accompanied by time- and dose-dependent reductions in enzyme activity in KG-1 cells (Fig. 2B). As illustrated, a short time exposure (4 hr) to 10 μM tamoxifen resulted in a >50% decrease in enzyme activity as measured in cell lysates, and similar with the Western blot of Sphk1 expression (Fig. 2A), a 24 hr exposure to 5 and 10 μM tamoxifen resulted in reciprocating 40 and 95% decreases in enzyme activity, respectively (Fig. 2B). Subsequent experiments were conducted to determine whether tamoxifen and LCL204 similarly influenced AC expression. For these experiments we chose short-time exposure and used multidrug resistant HL-60/VCR cells. Figure 2C shows that both LCL204 and tamoxifen, at a concentration of 10 μM, elicited early (2 hr) downregulation of AC expression (with tamoxifen appearing slightly more potent than LCL204).
Fig. 2.

Impact of tamoxifen and LCL204 on AC and Sphk1 expression and Sphk1 activity in AML cell lines. (A) Tamoxifen dose-response in KG-1 cells. KG-1 cells were seeded in 6-well plates, 5 × 106/well, in media containing 5% FBS. After 60 min, cells were treated with ethanol vehicle or tamoxifen for 24 hr. AC and SphK1 were detected by Western blot as detailed in Materials and Methods. (B) Cell-free Sphk1 activity. KG-1 cells were exposed to crescendo tamoxifen doses for the times shown, after which Sphk1 activity was measured in cell lysates. Error < 5% of the mean. (C) LCL204 and tamoxifen time course. HL-60/VCR cells were seeded (in vincristine-free media) as above, and treated for the times shown with either LCL204 or tamoxifen (both at 10 μM). AC expression was determined by Western blot. AC, acid ceramidase; SphK1, sphingosine kinase-1; CTR, control.
3.3 Evaluation of tamoxifen and DMT effects on sphingolipid metabolism in HL-60/VCR cells
Tamoxifen has been shown to be an effective inhibitor of ceramide glycosylation [11,12], activity that is an asset for enhancing ceramide-driven cell death cascades. As little is known regarding the impact of tamoxifen and tamoxifen metabolites on GC metabolism in leukemia, we next sought to address this issue using drug resistant HL-60/VCR cells. Firstly, tamoxifen dose-dependently increased over control values, the levels of C16:0- and C24:1-ceramide molecular species (Fig. 3A). Specifically, at 5 and 10 μM tamoxifen, levels of C16:0- ceramide increased 148 and 234% over control, respectively, and levels of C24:1-ceramide increased 160 and 265% over control, respectively. Of note, DMT was as effective as tamoxifen in increasing levels of C16:0-ceramide (223% over control) but did not as prominently increase the amount of C24:1-ceramide (160% over control), compared with 10 μM tamoxifen. There was a striking effect of tamoxifen and DMT on cerebroside (GC) production, as shown in Fig. 3B, namely, C16:0- and C24:1-GC synthesis was all but halted by tamoxifen and DMT. For example, levels of C16:0- and C24:1-GC fell to 7 and 4% of control, respectively, in cells exposed to 10 μM tamoxifen. Sphingomyelin, which is a major cellular lipid component, increased with treatment (Fig. 3C). For instance, levels of C16:0-SM, the predominant molecular species, increased to approximately 140% of control, upon exposure to both 5 and 10 μM tamoxifen; this was on par with response to DMT (134% of control). Likewise, levels of C24:1-SM increased to approximately 168% of control with exposures. Tamoxifen dose-dependently reduced levels of sphingosine to 43 and 9% of control, respectively, whereas levels of S1-P were only diminished with exposure to 10 μM tamoxifen (Fig. 3D). DMT was also effective in reducing the levels of sphingosine and S1-P.
Fig. 3.

The effect of tamoxifen and DMT on sphingolipid composition in HL-60/VCR cells. Cells (8 × 106) were seeded in 10-cm dishes in 10 ml vincristine-free medium containing 5% FBS. After a 2 hr equilibration period in a tissue culture incubator, cells were treated as indicated, for 18 hr. After harvesting and washing with ice-cold PBS, lipids were analyzed by mass spectroscopy as detailed in Materials and Methods. Micromolar concentrations given in parentheses. CTR, control; Tam, tamoxifen; DMT, N-desmethyltamoxien; LCB, long-chain base; S1-P, sphingosine 1-phosphate; Sph, sphingosine.
3.4 Efficacy of triphenylethylenes in combination regimens in AML
One advantage of having a drug that interferes with both ceramide hydrolysis and glycosylation is the potential as adjuvant to ceramide-based therapies. In order to explore this asset, we studied the effects of pairing tamoxifen with a ceramide-generating drug, 4-HPR, and with C6-ceramide, a cell-deliverable analog of natural ceramide, on cytotoxicity in HL-60/VCR cells. The data in Fig. 4A show that when HL-60/VCR cells were exposed to 4-HPR (5 μM), [3H](dihydro)ceramide levels (radiolabeling with palmitic and analysis by TLC does not distinguish dihydroceramide from ceramide) increased approximately 15-fold over control. In spite of this large elevation, viability studies showed that HL-60/VCR cells were nearly refractory to 4-HPR over a concentration range of 1.25-5 μM (Fig. 4B). However, the inclusion of tamoxifen, which as a single agent displayed minimal cytotoxic effects (80% of control), promoted 4-HPR cytotoxicity in a dose-dependent fashion (Fig. 4B, crosshatch). Moreover, DMT, when paired with 4-HPR, was as effective as tamoxifen (Fig. 4B, check). Providing ceramide in the form of C6-ceramide, in place of utilizing a ceramide generating agent such as 4-HPR, again showed little activity as single agent (Fig. 4C); however, the inclusion of tamoxifen resulted in dose-dependent enhancement of C6-ceramide cytotoxicity (Fig. 4C, stipple). Because single agents showed only slight cytotoxicity, we were interested in determining the type of cell death resulting from combination treatment. For this work, we chose to study 4-HPR, which is currently under clinical investigation [24]. As revealed by flow cytometry data (Fig. 5A), co-administration of 4-HPR and tamoxifen elicited increased Annexin V binding (right shift in fluorescence), indicative of apoptosis. The percent of specific apoptosis takes into account spontaneous apoptosis, a factor in untreated cells. With concentrations of 2.5 μM 4-HPR plus 10 μM tamoxifen and 5 μM 4-HPR plus 10 μM tamoxifen, specific apoptosis levels (based on Annexin V binding, Fig. 5A) were 42 and 67% over untreated control values, respectively (Fig. 5B). Apoptosis was also determined by DNA fragmentation, and as shown, single agents produced little damage compared to combination treatments (Fig. 5C). Based on the DNA fragmentation data, combination index (CI) values, a measure of drug synergy, were calculated for the 4-HPR-tamoxifen regimens, and as shown in Fig. 5D, CI values were <1.0, indicative of a synergistic response in this model of multidrug resistant HL-60/VCR cells. The apoptotic mechanism was documented by assessing the effect of the 4-HPR-tamoxifen combination on executioner caspase activity, and as shown in Fig. 5E, strong activation of caspase-3 ensued in a dose-dependent manner. For example, a 12-hr exposure to low-dose 4-HRR or high-dose 4-HPR, in the presence of tamoxifen resulted in 4- and 12-fold increases in caspase-3 activation, respectively.
Fig. 4.

The effect of tamoxifen and DMT on HL-60/VCR cell response to ceramide-centric therapies. (A) Effect of 4-HPR on cellular ceramide levels. Cells (2 × 106) were seeded in 6-well plates in 2.0 ml 5% FBS medium containing 2.0 μCi [3H]palmitic acid/ml, in the absence and presence of 4-HPR (5.0 μM) for 24 hr. Cells were then harvested by centrifugation, pellets washed in ice-cold PBS, and total lipids were extracted, and radiolabeled ceramide content was assessed by TLC as detailed in Materials and Methods. (B) Effect of tamoxifen and DMT on 4-HPR cytotoxicity. Cells, seeded in 96-well plates (20,000/well/0.2 ml 5% FBS-containing media), were exposed to the agents shown (micromolar concentrations in parentheses) for 24 hr. Cell viabilities were determined by MTS. n=6 for each experimental group; values shown are ± S.D. Repeated experiments yielded similar results. (C) Effect of tamoxifen on C6-ceramide cytotoxicity. Experiments were conducted exactly as in “b” above. Micromolar concentrations in parentheses. CTR, control; Tam, tamoxifen; DMT, N-desmethyltamoxifen; C6-cer, C6-ceramide
Fig. 5.

Combination tamoxifen-4-HPR elicits Annexin binding, DNA fragmentation, and activation of caspase-3 in HL-60/VCR cells. (A) Effect of drug combination on Annexin binding. Cells (1 × 106), seeded in 6-well plates were pretreated with tamoxifen for 2 hr, after which 4-HPR was added for 18 hr; drug concentrations given in parentheses. Annexin binding was quantitated by flow cytometry as detailed in Materials and Methods. (B). Specific apoptosis, as calculated from data in panel “A”. (C) Effect of drug combinations on DNA fragmentation. Cells (1 × 106), seeded in 6-well plates were pretreated with tamoxifen for 2 hr after which 4-HPR was added for 24 hr. Cells were then processed for PI staining, a measure of DNA fragmentation, as described in Materials and Methods. *denotes groups with significant difference, P ≤ 0.05. (D) Combination index for tamoxifen-4-HPR regimen. Data calculated from DNA fragmentation data of panel “C”. (E) Caspase-3 activation. Cells were exposed to the drug combinations shown for 12 hr, after which caspase-3 activation was measured in cell lysates. Micromolar concentrations given in parentheses. Tam, tamoxifen.
Figure 6 illustrates the multifunctional impact of tamoxifen on ceramide-centric therapies wherein drug combinations work in a synergistic fashion to magnify downstream, endpoint apoptosis. By dually blocking AC activity and the conversion of ceramides to GC, not only is the ceramide signal perpetuated, we propose that the mitogenic impact of S1-P can be lessened by this approach.
Fig.6.
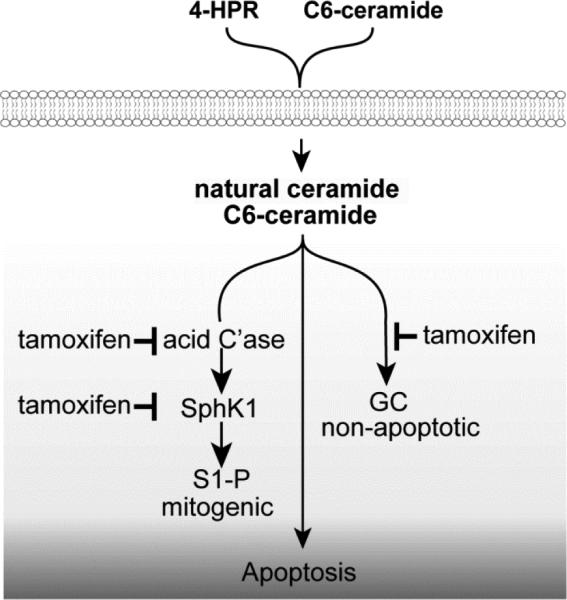
Magnification of ceramide-centric therapeutics by tamoxifen. We propose that tamoxifen's impact on sphingolipid metabolism (inhibition of ceramide glycosylation and hydrolysis ) propels ceramide-driven apoptosis. Additionally, downregulation of SphK1 could blunt the mitogenic capacity of S1-P. C'ase, acid ceramidase; SphK1, sphingosine kinase-1; S1-P, sphingosine 1-phosphate; GC, glucosylceramide.
4. Discussion
Ceramide-orchestrated cancer cell death can be countered by a number of sphingolipid metabolic events [2]. With this, it becomes clear that interfering with glycosylation and hydrolysis, important anabolic and catabolic junctures, could provide an entrée for strategies to improve ceramide-centric therapies. In the present work we show that tamoxifen and metabolite, DMT, enhance the action of ceramide-centric approaches in models of human AML.
Apropos to this discussion are tactical and metabolic issues encountered with implementation of ceramide-driven therapies in cancer. Firstly, ceramide as a biologic is difficult to deliver due to strong hydrophobicity. The solubility barrier can be overcome by use of short-chain, cell-permeable ceramide analogs like C6-ceramide[25], also used successfully in nanoliposomal formulation in in vivo studies [26]. In lieu of delivery of natural ceramide, ceramide-generating agents can be employed. For example, 4-HPR [27] as was used herein, PSC 833 [28,29], and etoposide [30] are but a few of many drugs that promote ceramide production; these drugs act by targeting ceramide synthetic machinery. However, as with C6-ceramide, natural ceramides, produced by ceramide-generating drugs might require partnering agents to limit constructive and destructive ceramide metabolism (glycosylation, hydrolysis), the strategy employed here.
The GCS inhibitors, D-threo-1-phenyl-2-palmitoylamino-3-morpholino-1-propanol (PPMP) and D-threo-1-phenyl-2-decanoylamino-3-morpholino-1-proapnol (PDMP) [31], have been used widely to block ceramide glycosylation. Reports demonstrate that these inhibitors can be of utility in enhancing chemotherapy and ceramide cytotoxicity [12,32,33,34,35]. Relevant to these findings are newer works positing that the chemosensitizing activity of this class of GCS inhibitor is not directly related to inhibition of GCS [13,36,37] , but rather to interaction with P-glycoprotein that is localized on the Golgi surface, a site of glycolipid trafficking into the Golgi lumen [17]. Thus, halting GC transit, post-synthesis, appears to be a mechanism underlying sensitization to ceramide and to ceramide-generating chemotherapies by the PPMP-class of inhibitors in intact cancer cells. A case in point is the study by Norris-Cervetto et al [36], who showed that the N-alkylated iminosugar inhibitors of GCS failed to reverse drug resistance when compared with PDMP. Several classical P-gp antagonist have also been shown to inhibit ceramide metabolism [12], indicating that use of these compounds should be revisited in the context of ceramide sensitizers, an action divorced from blocking the efflux of chemotherapy drugs from cancer cells. These findings support our works [13,38] and the work of Chai et al [37] that allied P-glycoprotein with PPMP.
AC inhibitors are also achieving notoriety in translational cancer research. Highlighting this designation are studies in prostate cancer [39,40], melanoma [41], and leukemia [42]. Similar to inhibitors of ceramide glycosylation, AC inhibitors exhibit beneficial effects in cancer models. For example, DM102 magnifies cytotoxic responses to 4-HPR and to PSC 833 in prostate and pancreatic cancer cells, respectively [29,43], and the lysomotrophic AC inhibitor, LCL204, alone induces apoptosis in prostate cancer cells [23]. In the present work we show that LCL204 and tamoxifen similarly downregulate AC expression in AML cells, although as a single agent, tamoxifen was not cytotoxic until prolonged exposure (72 hr).
Pairing tamoxifen with ceramide-centric agents, 4-HPR and C6-ceramide, revealed activity against HL-60/VCR cells that was low with single agents (see Fig. 4). In keeping with both the increases in Annexin V binding and DNA fragmentation was the increase in caspase-3 activation (see Fig. 5). Interestingly, in CCRF-CEM acute lymphoblastoid leukemia cells, Morales et al [44] showed that single agent 4-HPR was sufficient to trigger cytotoxicity through ceramide-induced mitochondrial oxidative stress. These authors astutely suggested the potential of modulators of ceramide metabolism to enhance the effects of 4-HPR-based therapies, much like our study demonstrates. Other works mirror this approach [45,46,47,48,49] . With high-dose regimens, serum levels of tamoxifen in excess of 4.0 μM are clinically achievable [50] ; however, use of nanoliposomal tamoxifen will enhance pharmacokinetics and tumor targeting [20,49].
It is noteworthy that tamoxifen and DMT alone were not cytotoxic (at 24 hr), even though both doubled steady-state levels of endogenous ceramides (see Fig. 3A). These levels of ceramide and/or the molecular species generated may be insufficient to elicit anti-proliferative responses, a type of threshold effect, and we propose that this effect can be cell type-specific. However, it is also possible that “tamoxifen ceramides” have hindered access to apoptosis effector molecules. Additionally, a recent study by Holliday et al [51] employing 4-HPR, demonstrated that T-cell ALL cytotoxicity was attributable to increases in discrete dihydroceramide species and not total dihydroceramide mass. This type of level-dependent cytotoxicity was promoted specifically by C22:0- and C24:0- dihydroceramides.
The increases in SM, accompanying tamoxifen and DMT exposure, suggest that the majority of ceramide generated was converted to the respective SM molecular species (see Fig. 3C); in this instance SM synthase would be exerting a protective effect. It is important to mention that aside from the increases in ceramide that arise by blockade of GC synthesis by tamoxifen, ceramide levels can also increase by recycling of sphingosine, albeit limited, and via stimulation of de novo ceramide synthesis by tamoxifen [52,53] . In addition, because tamoxifen is a potent inhibitor or cholesterol esterification [54] it is possible that resultant free palmitate is channeled into the de novo ceramide pathway.
Herein we also show for the first time that tamoxifen exposure downregulates the expression of SphK1, a reaction that in and of itself can limit the mitogenic impact of S1-P, although a redundant step in light of the AC-downregulating action of tamoxifen. It is noteworthy that downregulation of Sphk1 expression was accompanied by reductions in enzymatic activity (see Fig. 2A,B). Complementary and with clinical implications is the work of Bonhoure et al [55] demonstrating that inhibition of Sphk1 elicited production of apoptotic markers and downregulated expression of anti-apoptotic XIAP in AML cells.
In summary, the present report demonstrates that the triphenylethylene antiestrogen, tamoxifen and metabolite, DMT, are potent regulators of ceramide metabolism in human AML cells, limiting ceramide glycosylation, hydrolysis, and sphingosine phosphorylation. Moreover, the triphenylethylenes are effective adjuvants to ceramide-based therapeutics in our AML models. As C6-ceramide-based therapy is on the horizon [49,56], adjuvant tamoxifen or better, DMT, which has an extended serum half-life [57], could provide enhanced clinical efficacy. AML is the most common type of leukemia in adults and a leukemia for which there have been few treatment advances in 40 years. The disease is aggressive, and response to conventional chemotherapy is poor. Targeting sphingolipid metabolism by the employ of triphenylethylenes could provide a beneficial therapeutic option.
Highlights.
A tamoxifen regimen to enhance ceramide cytotoxicity in AML is demonstrated.
When used with ceramide-centric therapies, tamoxifen amplifies end-point apoptosis.
Tamoxifen blocks ceramide glycosylation and hydrolysis.
Curbing ceramide metabolism provides a unique indication for tamoxifen.
Acknowledgements
This work was supported by the National Institutes of Health (NCI) grant number 5 P01 CA171983-02. We wish to thank Drs. Gemma Fabrias, Antonio Delgado, and Josefina Casas, Department of Biomedical Chemistry, Institute for Advanced Chemistry of Catalunya, Barcelona, Spain, for providing the AC fluorogenic substrate, CB14C12.
Abbreviations
- DMT
N–desmethyltamoxifen
- AC
acid ceramidase
- AML
acute myelogenous leukemia
- SphK1
sphingosine kinase 1
- S1-P
sphingosine 1-phosphate
- GC
glucosylceramide
- 4-HPR
fenretinide
- GCS
glucosylceramide synthase
- P-gp
P-glycoprotein
- ATCC
American Type Culture Collection
- C6-ceramide
N-hexanoyl-D-erythro-sphingosine
- DMSO
Dimethyl sulfoxide
- LCL204
(1R,2R) 2-(N-tetradecylamino)-1-(4-NO2)-phenyl- 1,3-dihydroxy-propane HCl
- TLC
thin-layer chromatography
- ESI-MS/MS
electrospray ionization tandem mass spectrometry
- PBS
phosphate buffered saline
- FBS
fetal bovine serum
- PI
propidium iodide
- HPLC
high-performance liquid chromatography
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- PVDF
polyvinyl difluoride
- GADPH
glyceraldehyde 3-phosphate dehydrogenase
- ANOVA
analysis of variance
- CI
combination index
- PPMP
D-threo-1-phenyl-2-palmitoylamino-3-morpholino-1-propanol
- PDMP
D-threo-1-phenyl-2-decanoylamino-3-morpholino-1-proapnol
- SM
sphingomyelin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
No conflict of interest
References
- 1.Canals D, Hannun YA. Novel chemotherapeutic drugs in sphingolipid cancer research. Handb Exp Pharmacol. 2013:211–238. doi: 10.1007/978-3-7091-1368-4_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morad SA, Cabot MC. Ceramide-orchestrated signalling in cancer cells. Nat Rev Cancer. 2013;13:51–65. doi: 10.1038/nrc3398. [DOI] [PubMed] [Google Scholar]
- 3.Savic R, Schuchman EH. Use of acid sphingomyelinase for cancer therapy. Adv Cancer Res. 2013;117:91–115. doi: 10.1016/B978-0-12-394274-6.00004-2. [DOI] [PubMed] [Google Scholar]
- 4.Dimanche-Boitrel M-T, Rebillard A. Sphingolipids and response to chemotherapy. Springer; 2013. [DOI] [PubMed] [Google Scholar]
- 5.Lavie Y, Cao H, Bursten SL, Giuliano AE, Cabot MC. Accumulation of glucosylceramides in multidrug-resistant cancer cells. J Biol Chem. 1996;271:19530–19536. doi: 10.1074/jbc.271.32.19530. [DOI] [PubMed] [Google Scholar]
- 6.Musumarra G, Barresi V, Condorelli DF, Scire S. A bioinformatic approach to the identification of candidate genes for the development of new cancer diagnostics. Biol Chem. 2003;384:321–327. doi: 10.1515/BC.2003.037. [DOI] [PubMed] [Google Scholar]
- 7.Zeidan YH, Jenkins RW, Korman JB, Liu X, Obeid LM, Norris JS, Hannun YA. Molecular targeting of acid ceramidase: implications to cancer therapy. Curr Drug Targets. 2008;9:653–661. doi: 10.2174/138945008785132358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Orr Gandy KA, Obeid LM. Targeting the sphingosine kinase/sphingosine 1-phosphate pathway in disease: review of sphingosine kinase inhibitors. Biochim Biophys Acta. 2013;1831:157–166. doi: 10.1016/j.bbalip.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Senchenkov A, Litvak DA, Cabot MC. Targeting ceramide metabolism--a strategy for overcoming drug resistance. J Natl Cancer Inst. 2001;93:347–357. doi: 10.1093/jnci/93.5.347. [DOI] [PubMed] [Google Scholar]
- 10.Truman JP, Garcia-Barros M, Obeid LM, Hannun YA. Evolving concepts in cancer therapy through targeting sphingolipid metabolism. Biochim Biophys Acta. 2014;1841:1174–1188. doi: 10.1016/j.bbalip.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cabot MC, Giuliano AE, Volner A, Han TY. Tamoxifen retards glycosphingolipid metabolism in human cancer cells. FEBS Lett. 1996;394:129–131. doi: 10.1016/0014-5793(96)00942-8. [DOI] [PubMed] [Google Scholar]
- 12.Lavie Y, Cao H, Volner A, Lucci A, Han TY, Geffen V, Giuliano AE, Cabot MC. Agents that reverse multidrug resistance, tamoxifen, verapamil, and cyclosporin A, block glycosphingolipid metabolism by inhibiting ceramide glycosylation in human cancer cells. J Biol Chem. 1997;272:1682–1687. doi: 10.1074/jbc.272.3.1682. [DOI] [PubMed] [Google Scholar]
- 13.Chapman JV, Gouaze-Andersson V, Cabot MC. Expression of P-glycoprotein in HeLa cells confers resistance to ceramide cytotoxicity. Int J Oncol. 2010;37:1591–1597. doi: 10.3892/ijo_00000813. [DOI] [PubMed] [Google Scholar]
- 14.Morad SA, Levin JC, Tan SF, Fox TE, Feith DJ, Cabot MC. Novel off-target effect of tamoxifen--inhibition of acid ceramidase activity in cancer cells. Biochim Biophys Acta. 2013;1831:1657–1664. doi: 10.1016/j.bbalip.2013.07.016. [DOI] [PubMed] [Google Scholar]
- 15.Gouaze-Andersson V, Cabot MC. Sphingolipid metabolism and drug resistance in hematological malignancies. Anticancer Agents Med Chem. 2011;11:891–903. doi: 10.2174/187152011797655069. [DOI] [PubMed] [Google Scholar]
- 16.Mahadevan D. Will MDR-1/P-gp modulators provide clinical benefit in hematologic malignancies? Leuk Res. 2006;30:1077–1078. doi: 10.1016/j.leukres.2006.03.014. [DOI] [PubMed] [Google Scholar]
- 17.Borst P, Zelcer N, van Helvoort A. ABC transporters in lipid transport. Biochim Biophys Acta. 2000;1486:128–144. doi: 10.1016/s1388-1981(00)00053-6. [DOI] [PubMed] [Google Scholar]
- 18.Bedia C, Casas J, Garcia V, Levade T, Fabrias G. Synthesis of a novel ceramide analogue and its use in a high-throughput fluorogenic assay for ceramidases. Chembiochem. 2007;8:642–648. doi: 10.1002/cbic.200600533. [DOI] [PubMed] [Google Scholar]
- 19.Chapman JV, Gouaze-Andersson V, Messner MC, Flowers M, Karimi R, Kester M, Barth BM, Liu X, Liu YY, Giuliano AE, Cabot MC. Metabolism of short-chain ceramide by human cancer cells--implications for therapeutic approaches. Biochem Pharmacol. 2010;80:308–315. doi: 10.1016/j.bcp.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morad SA, Levin JC, Shanmugavelandy SS, Kester M, Fabrias G, Bedia C, Cabot MC. Ceramide--antiestrogen nanoliposomal combinations--novel impact of hormonal therapy in hormone-insensitive breast cancer. Mol Cancer Ther. 2012;11:2352–2361. doi: 10.1158/1535-7163.MCT-12-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fox TE, Bewley MC, Unrath KA, Pedersen MM, Anderson RE, Jung DY, Jefferson LS, Kim JK, Bronson SK, Flanagan JM, Kester M. Circulating sphingolipid biomarkers in models of type 1 diabetes. J Lipid Res. 2011;52:509–517. doi: 10.1194/jlr.M010595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chou TC. Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res. 2010;70:440–446. doi: 10.1158/0008-5472.CAN-09-1947. [DOI] [PubMed] [Google Scholar]
- 23.Holman DH, Turner LS, El-Zawahry A, Elojeimy S, Liu X, Bielawski J, Szulc ZM, Norris K, Zeidan YH, Hannun YA, Bielawska A, Norris JS. Lysosomotropic acid ceramidase inhibitor induces apoptosis in prostate cancer cells. Cancer Chemother Pharmacol. 2008;61:231–242. doi: 10.1007/s00280-007-0465-0. [DOI] [PubMed] [Google Scholar]
- 24.Maurer BJ, Kang MH, Villablanca JG, Janeba J, Groshen S, Matthay KK, Sondel PM, Maris JM, Jackson HA, Goodarzian F, Shimada H, Czarnecki S, Hasenauer B, Reynolds CP, Marachelian A. Phase I trial of fenretinide delivered orally in a novel organized lipid complex in patients with relapsed/refractory neuroblastoma: a report from the New Approaches to Neuroblastoma Therapy (NANT) consortium. Pediatr Blood Cancer. 2013;60:1801–1808. doi: 10.1002/pbc.24643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abe A, Wu D, Shayman JA, Radin NS. Metabolic effects of short-chain ceramide and glucosylceramide on sphingolipids and protein kinase C. Eur J Biochem. 1992;210:765–773. doi: 10.1111/j.1432-1033.1992.tb17479.x. [DOI] [PubMed] [Google Scholar]
- 26.Zolnik BS, Stern ST, Kaiser JM, Heakal Y, Clogston JD, Kester M, McNeil SE. Rapid distribution of liposomal short-chain ceramide in vitro and in vivo. Drug Metab Dispos. 2008;36:1709–1715. doi: 10.1124/dmd.107.019679. [DOI] [PubMed] [Google Scholar]
- 27.Maurer BJ, Metelitsa LS, Seeger RC, Cabot MC, Reynolds CP. Increase of ceramide and induction of mixed apoptosis/necrosis by N-(4-hydroxyphenyl)- retinamide in neuroblastoma cell lines. J Natl Cancer Inst. 1999;91:1138–1146. doi: 10.1093/jnci/91.13.1138. [DOI] [PubMed] [Google Scholar]
- 28.Cabot MC, Han TY, Giuliano AE. The multidrug resistance modulator SDZ PSC 833 is a potent activator of cellular ceramide formation. FEBS Lett. 1998;431:185–188. doi: 10.1016/s0014-5793(98)00744-3. [DOI] [PubMed] [Google Scholar]
- 29.Morad SA, Messner MC, Levin JC, Abdelmageed N, Park H, Merrill AH, Jr., Cabot MC. Potential role of acid ceramidase in conversion of cytostatic to cytotoxic end-point in pancreatic cancer cells. Cancer Chemother Pharmacol. 2013;71:635–645. doi: 10.1007/s00280-012-2050-4. [DOI] [PubMed] [Google Scholar]
- 30.Perry DK, Carton J, Shah AK, Meredith F, Uhlinger DJ, Hannun YA. Serine palmitoyltransferase regulates de novo ceramide generation during etoposide-induced apoptosis. J Biol Chem. 2000;275:9078–9084. doi: 10.1074/jbc.275.12.9078. [DOI] [PubMed] [Google Scholar]
- 31.Lee L, Abe A, Shayman JA. Improved inhibitors of glucosylceramide synthase. J Biol Chem. 1999;274:14662–14669. doi: 10.1074/jbc.274.21.14662. [DOI] [PubMed] [Google Scholar]
- 32.Sietsma H, Veldman RJ, Kolk D, Ausema B, Nijhof W, Kamps W, Vellenga E, Kok JW. 1-phenyl-2-decanoylamino-3-morpholino-1-propanol chemosensitizes neuroblastoma cells for taxol and vincristine. Clin Cancer Res. 2000;6:942–948. [PubMed] [Google Scholar]
- 33.Olshefski RS, Ladisch S. Glucosylceramide synthase inhibition enhances vincristine-induced cytotoxicity. Int J Cancer. 2001;93:131–138. doi: 10.1002/ijc.1301. [DOI] [PubMed] [Google Scholar]
- 34.Furlong SJ, Ridgway ND, Hoskin DW. Modulation of ceramide metabolism in T-leukemia cell lines potentiates apoptosis induced by the cationic antimicrobial peptide bovine lactoferricin. Int J Oncol. 2008;32:537–544. [PubMed] [Google Scholar]
- 35.Watters RJ, Fox TE, Tan SF, Shanmugavelandy S, Choby JE, Broeg K, Liao J, Kester M, Cabot MC, Loughran TP, Liu X. Targeting glucosylceramide synthase synergizes with C6-ceramide nanoliposomes to induce apoptosis in natural killer cell leukemia. Leuk Lymphoma. 2013;54:1288–1296. doi: 10.3109/10428194.2012.752485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Norris-Cervetto E, Callaghan R, Platt FM, Dwek RA, Butters TD. Inhibition of glucosylceramide synthase does not reverse drug resistance in cancer cells. J Biol Chem. 2004;279:40412–40418. doi: 10.1074/jbc.M404466200. [DOI] [PubMed] [Google Scholar]
- 37.Chai L, McLaren RP, Byrne A, Chuang WL, Huang Y, Dufault MR, Pacheco J, Madhiwalla S, Zhang X, Zhang M, Teicher BA, Carter K, Cheng SH, Leonard JP, Xiang Y, Vasconcelles M, Goldberg MA, Copeland DP, Klinger KW, Lillie J, Madden SL, Jiang YA. The chemosensitizing activity of inhibitors of glucosylceramide synthase is mediated primarily through modulation of P-gp function. Int J Oncol. 2011;38:701–711. doi: 10.3892/ijo.2010.888. [DOI] [PubMed] [Google Scholar]
- 38.Chapman JV, Gouaze-Andersson V, Karimi R, Messner MC, Cabot MC. P-glycoprotein antagonists confer synergistic sensitivity to short-chain ceramide in human multidrug-resistant cancer cells. Exp Cell Res. 2011;317:1736–1745. doi: 10.1016/j.yexcr.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Camacho L, Meca-Cortes O, Abad JL, Garcia S, Rubio N, Diaz A, Celia-Terrassa T, Cingolani F, Bermudo R, Fernandez PL, Blanco J, Delgado A, Casas J, Fabrias G, Thomson TM. Acid ceramidase as a therapeutic target in metastatic prostate cancer. J Lipid Res. 2013;54:1207–1220. doi: 10.1194/jlr.M032375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beckham TH, Cheng JC, Lu P, Marrison ST, Norris JS, Liu X. Acid ceramidase promotes nuclear export of PTEN through sphingosine 1-phosphate mediated Akt signaling. PLoS One. 2013;8:e76593. doi: 10.1371/journal.pone.0076593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bedia C, Casas J, Andrieu-Abadie N, Fabrias G, Levade T. Acid ceramidase expression modulates the sensitivity of A375 melanoma cells to dacarbazine. J Biol Chem. 2011;286:28200–28209. doi: 10.1074/jbc.M110.216382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shah MV, Zhang R, Irby R, Kothapalli R, Liu X, Arrington T, Frank B, Lee NH, Loughran TP., Jr. Molecular profiling of LGL leukemia reveals role of sphingolipid signaling in survival of cytotoxic lymphocytes. Blood. 2008;112:770–781. doi: 10.1182/blood-2007-11-121871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gouaze-Andersson V, Flowers M, Karimi R, Fabrias G, Delgado A, Casas J, Cabot MC. Inhibition of acid ceramidase by a 2-substituted aminoethanol amide synergistically sensitizes prostate cancer cells to N-(4-hydroxyphenyl) retinamide. Prostate. 2011;71:1064–1073. doi: 10.1002/pros.21321. [DOI] [PubMed] [Google Scholar]
- 44.Morales MC, Perez-Yarza G, Rementeria NN, Boyano MD, Apraiz A, Gomez-Munoz A, Perez-Andres E, Asumendi A. 4-HPR-mediated leukemia cell cytotoxicity is triggered by ceramide-induced mitochondrial oxidative stress and is regulated downstream by Bcl-2. Free Radic Res. 2007;41:591–601. doi: 10.1080/10715760701218558. [DOI] [PubMed] [Google Scholar]
- 45.Maurer BJ, Melton L, Billups C, Cabot MC, Reynolds CP. Synergistic cytotoxicity in solid tumor cell lines between N-(4-hydroxyphenyl)retinamide and modulators of ceramide metabolism. J Natl Cancer Inst. 2000;92:1897–1909. doi: 10.1093/jnci/92.23.1897. [DOI] [PubMed] [Google Scholar]
- 46.Pallis M, Russell N. Strategies for overcoming p-glycoprotein-mediated drug resistance in acute myeloblastic leukaemia. Leukemia. 2004;18:1927–1930. doi: 10.1038/sj.leu.2403511. [DOI] [PubMed] [Google Scholar]
- 47.Batra S, Reynolds CP, Maurer BJ. Fenretinide cytotoxicity for Ewing's sarcoma and primitive neuroectodermal tumor cell lines is decreased by hypoxia and synergistically enhanced by ceramide modulators. Cancer Res. 2004;64:5415–5424. doi: 10.1158/0008-5472.CAN-04-0377. [DOI] [PubMed] [Google Scholar]
- 48.Devalapally H, Duan Z, Seiden MV, Amiji MM. Modulation of drug resistance in ovarian adenocarcinoma by enhancing intracellular ceramide using tamoxifen-loaded biodegradable polymeric nanoparticles. Clin Cancer Res. 2008;14:3193–3203. doi: 10.1158/1078-0432.CCR-07-4973. [DOI] [PubMed] [Google Scholar]
- 49.Jiang Y, DiVittore NA, Kaiser JM, Shanmugavelandy SS, Fritz JL, Heakal Y, Tagaram HR, Cheng H, Cabot MC, Staveley-O'Carroll KF, Tran MA, Fox TE, Barth BM, Kester M. Combinatorial therapies improve the therapeutic efficacy of nanoliposomal ceramide for pancreatic cancer. Cancer Biol Ther. 2011;12:574–585. doi: 10.4161/cbt.12.7.15971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O'Day SJ, Boasberg PD, Kristedja TS, Martin M, Wang HJ, Fournier P, Cabot M, DeGregorio MW, Gammon G. High-dose tamoxifen added to concurrent biochemotherapy with decrescendo interleukin-2 in patients with metastatic melanoma. Cancer. 2001;92:609–619. doi: 10.1002/1097-0142(20010801)92:3<609::aid-cncr1361>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- 51.Holliday MW, Jr., Cox SB, Kang MH, Maurer BJ. C22:0- and C24:0-dihydroceramides confer mixed cytotoxicity in T-cell acute lymphoblastic leukemia cell lines. PLoS One. 2013;8:e74768. doi: 10.1371/journal.pone.0074768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Scarlatti F, Bauvy C, Ventruti A, Sala G, Cluzeaud F, Vandewalle A, Ghidoni R, Codogno P. Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J Biol Chem. 2004;279:18384–18391. doi: 10.1074/jbc.M313561200. [DOI] [PubMed] [Google Scholar]
- 53.Pattingre S, Bauvy C, Carpentier S, Levade T, Levine B, Codogno P. Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J Biol Chem. 2009;284:2719–2728. doi: 10.1074/jbc.M805920200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.de Medina P, Payre BL, Bernad J, Bosser I, Pipy B, Silvente-Poirot S, Favre G, Faye JC, Poirot M. Tamoxifen is a potent inhibitor of cholesterol esterification and prevents the formation of foam cells. J Pharmacol Exp Ther. 2004;308:1165–1173. doi: 10.1124/jpet.103.060426. [DOI] [PubMed] [Google Scholar]
- 55.Bonhoure E, Pchejetski D, Aouali N, Morjani H, Levade T, Kohama T, Cuvillier O. Overcoming MDR-associated chemoresistance in HL-60 acute myeloid leukemia cells by targeting sphingosine kinase-1. Leukemia. 2006;20:95–102. doi: 10.1038/sj.leu.2404023. [DOI] [PubMed] [Google Scholar]
- 56.Barth BM, Cabot MC, Kester M. Ceramide-based therapeutics for the treatment of cancer. Anticancer Agents Med Chem. 2011;11:911–919. doi: 10.2174/187152011797655177. [DOI] [PubMed] [Google Scholar]
- 57.Berman E, McBride M, Tong W. Comparative activity of tamoxifen and N-desmethyltamoxifen in human multidrug resistant leukemia cell lines. Leukemia. 1994;8:1191–1196. [PubMed] [Google Scholar]