

Abstract
Rapid recruitment of neutrophils to sites of infection and their ability to phagocytose and kill microbes is an important aspect of the innate immune response. Challenges associated with imaging of these cells include their short lifespan and small size and the fact that unstimulated cells are nonadherent. In addition, although cytoplasmic granules are plentiful, the abundance of many other organelles is diminished. Here we reprise methods for analysis of resting and activated cells using immunofluorescence and confocal microscopy, including kinetic analysis of phagosome maturation and degranulation, and detection of intraphagosomal superoxide accumulation. We describe approaches for rapid cell fixation and permeabilization that maximize antigen detection and discuss other variables that also affect data interpretation and image quality (such as cell spreading, degranulation, and phagocytosis). Finally, we show that these methods are also applicable to studies of neutrophil interactions with the extracellular matrix.
Keywords: Neutrophil, Immunofluorescence, Confocal microscopy, Phagosome, Granule, Talin, Myeloperoxidase, Fibrinogen, Bacteria, Zymosan
1 Introduction
Distinctive characteristics of neutrophils (polymorphonuclear leukocytes, PMNs) are their small size, lobed nucleus, and abundant cytoplasmic granules. Fundamental initial insight into the composition of gelatinase and specific and azurophilic granules and the subcellular localization of NADPH oxidase subunits in resting, primed, and activated cells was obtained using subcellular fractionation [1, 2]. However, the application of fluorescence microscopy approaches to studies of neutrophils has lagged behind other cell types. In part for this reason, much less is known about fundamental processes such as phagosome maturation in PMNs as compared with macrophages [3].
Summarized here are methods we developed originally for analysis of macrophages [4, 5] and then optimized for immunofluorescence and confocal analysis of human neutrophils [6–12]. Our approaches address general issues such as methods of cell fixation and permeabilization that are also relevant to studies of other cell types. In addition, we address factors particular to neutrophils such as cell size, adhesion, spreading, degranulation, and phagocytosis in the context of image analysis using immunofluorescence and confocal microscopy.
2 Materials
2.1 Cell Culture
Neutrophils: isolate from the peripheral blood of healthy donors using dextran sedimentation and density gradient separation on Ficoll-Hypaque [13]. Cell purity should be >95 % PMNs.
Fresh human serum: to coat coverslips and also as a source of active complement factors that can be used for particle opsonization. Transfer a sample of non-heparinized venous blood (10 ml) to a sterile glass tube and incubate for 1 h at room temperature followed by 1 h on ice. Detach the clot from the tube wall using a sterile pipette tip and centrifuge at 1,500 × g for 15 min at 4 °C. Transfer the serum layer to a sterile polypropylene tube and keep on ice to preserve bioactivity. Fresh serum should be used within 2 h or stored in single-use aliquots at −80 °C for future use. Either autologous serum or serum pooled from several donors is suitable for this purpose.
Heat-inactivated fetal bovine serum (HI-FBS): incubate serum at 56 °C for 30 min with occasional swirling. Filter (0.45 μm) to remove any particulates and store at −20 °C.
Endotoxin-free, HEPES-buffered RPMI-1640 medium containing L -glutamine. Store at 4 °C. Supplement with HI-FBS or human serum to 10 % final concentration.
10 mM HEPES solution, pH 7.2.
10 mM glucose solution.
Endotoxin-free Hank’s buffered salt solution containing calcium and magnesium (HBSS), supplemented with 10 mM HEPES and 10 mM glucose. Sterile filter (0.2 μm) and store at room temperature.
35 mm tissue culture dishes.
Sterile 15 and 50 ml conical polypropylene tubes.
Rectangular, flat-bottom aluminum pans (approximately 7 × 11 in.).
Refrigerated tissue culture centrifuge with swinging bucket rotor and microplate carriers such as the Allegra 6KR (Beckman Coulter).
2.2 Particulate and Soluble Stimuli
Zymosan (yeast cell wall particles, Sigma-Aldrich, St. Louis, MO): rehydrate 200 mg zymosan in 20 ml sterile PBS in a 50 ml sterile conical polypropylene tube. Vortex briefly and then place in a water bath sonicator for 5 min. Transfer zymosan to a boiling water bath for 10 min, collect particles by centrifugation (400 × g for 10 min), decant PBS, and repeat the sonication and boiling steps twice using fresh changes of PBS. Resuspend the final zymosan pellet in 10 ml tissue culture medium (without serum) or sterile HBSS, and store in 250 μl (5 mg) aliquots at −20 °C. Prepare a working stock solution by diluting one aliquot of zymosan with three volumes of buffer or tissue culture medium (5 mg/ml final concentration). Before each use, briefly sonicate the thawed working stock to disperse any aggregates (see Note 1).
Complement-opsonized zymosan (COZ) particles are obtained by incubating an aliquot of the zymosan working stock with fresh human serum (50 % final concentration) at 37 °C for 30 min. Pellet COZ using a microfuge (2 min, 16,000 × g) and then wash twice with PBS or serum-free tissue culture medium. Keep COZ on ice and use within 2 h. IgG-opsonized zymosan (IgG-Z) particles are prepared using Molecular Probes/ Invitrogen/Life Technologies (Grand Island, NY) zymosan opsonizing reagent. Store IgG-Z in small aliquots at −20 °C. Dynabeads precoated with IgG are available from life technologies. Bacteria can also be opsonized with complement factors, antibodies, or immune serum (see Note 1).
Phorbol myristate acetate (PMA) is dissolved in endotoxin-free, tissue culture grade DMSO (Sigma-Aldrich) at a final concentration of 5 mM. Store at −80 °C in small aliquots. Because PMA is rapidly inactivated in aqueous solution, thaw stocks at room temperature and use immediately after dilution into buffer or medium.
f-Met-Leu-Phe (fMLF) and TNFα are dissolved in tissue culture grade, endotoxin-free DMSO at a final concentration of 4 mM and 10 μM, respectively, and stored in aliquots at −80 °C.
2.3 Microscopy Supplies
Round glass coverslips (12 mm diameter).
Unlabeled fibrinogen (Sigma-Aldrich): 0.1 mg/ml stock solution in sterile HBSS, store at −20 °C.
Oregon Green-labeled fibrinogen (Molecular Probes/Invitrogen/Life Technologies): 0.1 mg/ml stock solution in sterile HBSS, store at −20 °C.
Straight needle-point stainless steel forceps.
Glass Petri dishes (60 mm), store at −20 °C.
Disposable microbeakers (10 ml capacity).
Pre-cleaned microscopy slides with frosted marking area.
Lids from 24-well tissue culture cluster plates. These do not need to be sterile and can be lids saved after use in other experiments.
Plastic flat-bottom plastic boxes with lids (Tupperware type), large enough to accommodate one–three lids from 24-well tissue culture dishes.
Cardboard slide folders.
2.4 Buffers and Other Reagents
Nitric acid.
95 % ethanol.
70 % ethanol: dilute 95 % ethanol using tissue culture grade sterile deionized water.
10 % neutral buffered formalin solution (see Note 2).
Acetone: store at room temperature and chilled rapidly to −20 °C prior to use (see Note 3).
Phosphate-buffered saline (PBS): 137 mM NaCl, 2.68 mM KCl, 8.1 mM Na2 HPO4, and 1.47 mM KH2PO4 (pH 7.4).
Blocking buffer: PBS supplemented with 5 mg/ml bovine serum albumin (BSA), 0.5 mg/ml NaN3, and 10 % horse serum (see Note 4). Sterile (0.2 μm) filter and store at 4 °C.
Washing buffer (PBS-azide-BSA, “PAB”): blocking buffer without horse serum, store at 4 °C.
Antibodies directed against specific marker proteins of neutrophils obtained from commercial sources or prepared in your own laboratory (see Note 5).
DyLight 488 and rhodamine-conjugated F(ab′)2 secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove, PA): rehydrate with sterile deionized water according to the manufacturer’s directions and store at 4 °C. Secondary antibodies conjugated to other fluors can also be used (see Note 6).
Gelvatol mounting medium (see Note 7): mix 2.4 g polyvinyl alcohol with 6 g glycerol, 6 ml deionized water, and 12 ml 200 mM Tris–HCl (pH 8.0) in a beaker on a stir plate for several hours until dissolved. Heat the solution to 50 °C for 10 min and then clarify by centrifugation at 5,000 × g for 15 min. Decant the supernatant into a new 50 ml polypropylene tube and add 625 mg 1,4-diazobicyclo-[2.2.2]-octane (DABCO). Invert the tube to dissolve DABCO. Store in 1 ml aliquots in Eppendorf tubes in a frost-free freezer at −20 or −80 °C. DABCO reduces photobleaching of fluorescence.
3 Methods
3.1 Preparation of Acid-Washed Coverslips
Acid washing is essential since it removes manufacturing residue, oils, and other contaminants such as LPS that may inadvertently activate PMNs, impair cell adhesion, and/or cause high nonspecific background fluorescence after antibody staining.
Transfer one package of 12 mm round glass coverslips into a small glass bottle.
Inside a fume hood, pour nitric acid over the coverslips making sure they are all submerged.
Cap the bottle and swirl gently to ensure all coverslips are in contact with the acid.
Incubate in the fume hood for at least 48 h.
Carefully decant the acid and then rinse the coverslips with 15–20 changes of sterile deionized tissue culture grade water (pour water over the coverslips, cap bottle, rotate gently to rinse coverslips, decant water, and repeat).
Rinse the coverslips with two changes of 95 % ethanol to remove residual water.
Store coverslips in 70 % ethanol at room temperature. Use after ≥16 h in ethanol.
3.2 Preparation of Coated Coverslips
Bloodstream neutrophils are nonadherent, whereas PMNs at a site of infection have attached to, and migrated along, the extracellular matrix. Freshly isolated peripheral blood PMNs will associate with glass coverslips precoated with serum proteins or purified fibrinogen (see Note 8). Adherent PMN are preferred since these cells are more similar to neutrophils at sites of infection and because it is easier to synchronize phagocytosis of adherent cells than cells in suspension.
3.2.1 Flaming of Coverslips and Dispersal in 35 mm Dishes
-
1
Set up 35 mm tissue culture dishes in a rectangular metal pan. Each dish will hold three coverslips which provide triplicate samples for each experimental condition or time point.
-
2
Use forceps to remove a few coverslips from their ethanol storage bottle and place on a pile of Kimwipes.
-
3
Fill one small microbeaker with 70 % ethanol and light a Bunsen burner.
-
4
Working with one coverslip at a time, grasp with forceps, dunk into ethanol, and drain off excess ethanol by touching edge of the coverslip to the pile of Kimwipes.
-
5
Pass the coverslip through the flame and then place it into a 35 mm dish. Because the 35 mm dishes are opened only briefly, this procedure can be performed on the bench top. Failure to remove excess ethanol will cause coverslips to shatter when passed through the flame.
3.2.2 Coating Coverslips with Serum or Fibrinogen
-
6
Transfer 35 mm dishes containing coverslips into a tissue culture hood.
-
7
Open all dishes and, using a sterile probe (such as a P200 Pipetman tip), position the coverslips in each dish such that they are not touching the side of the dish or touching one another (Fig. 1).
-
8
Pipette 50 μl of 0.1 mg/ml fibrinogen or 50 μl fresh serum directly onto each coverslip. The coating agent will spread evenly over the acid-washed glass, and surface tension holds the domes of liquid in place.
-
9
Close all dishes, transfer back into the metal pan, and place inside a 37 °C tissue culture incubator for 30–60 min.
-
10
Rinse coverslips by flooding each dish with about 3 ml of sterile HBSS or PBS. Note that failure to remove excess fibrinogen may cause coverslips to become glued to the tissue culture dish if they air dry.
Fig. 1.
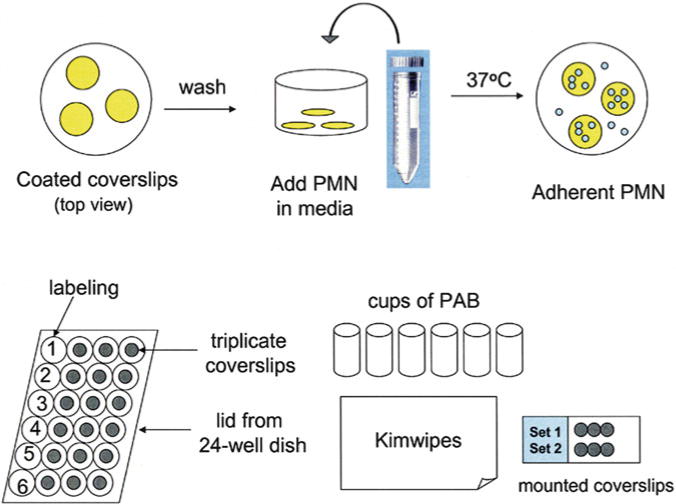
Cell plating and staining schematic. The process of coverslip coating and neutrophil plating is shown (top row) as well as the process of cell staining and washing and coverslip mounting (bottom row). Reprinted from [12] with permission from Humana press
3.3 Plating Neutrophils on Coated Coverslips
Dilute PMNs to 1 × 10 6 cells per ml in tissue culture medium in a conical tube.
Invert the tube gently to mix cells, do not vortex.
Place 1 ml of this suspension in each 35 mm dish containing coated coverslips. Be sure that the coverslips are not overlapping one another.
Push any floating coverslips to the bottom of the dish using a sterile pipette tip.
Place dishes in a 37 °C incubator for 1 h. Neutrophils will adhere to the coated coverslips in preference to the uncoated tissue culture dish (Fig. 1). For microscopy, a moderate cell density is optimal.
3.4 Stimulation of Adherent PMNs
Because PMNs are only loosely adherent, it is important to add stimuli to dishes carefully so as not to detach the cells. For this same reason, cells are not fluid changed before addition of stimuli. Rather, particles or soluble stimuli are added to adherent neutrophils in a volume of 1 ml to achieve a total volume of 2 ml per 35 mm dish. Most stimuli will enhance PMN adhesion and spreading via their ability to trigger upregulation of surface β2 integrins (see Notes 8 and 9).
3.4.1 Soluble Stimuli
Human PMNs are optimally activated by exposure to 200 nM PMA or 1–10 μM fMLF and can be primed or activated by TNFα depending on the concentration used.
Dilute each agonist in tissue culture medium to achieve to twice the desired final concentration.
Vortex to mix.
Add 1 ml of the desired stimulus to each dish.
Transfer to 37 °C incubator for the desired amount of time (generally 0.5–5 min for fMLF and 5–30 min for PMA).
3.4.2 Synchronized Phagocytosis of Opsonized Zymosan
Centrifugation of opsonized zymosan particles or bacteria onto adherent neutrophils at low temperature allows particle binding but not phagocytosis, and rapid transfer of samples to 37 °C supports synchronized ingestion.
Dilute COZ in cold (4 °C) tissue culture medium (use 1–2 μl of 5 mg/ml COZ per dish to achieve ~2–3 phagosomes per cell).
Add 1 ml of cold, diluted COZ to each dish of PMN (already containing 1 ml warm medium). Mixing warm and cold medium will reduce the overall temperature below the threshold for phagocytosis (16 °C).
Rapidly and carefully transfer the dishes onto plate carriers in a refrigerated tissue culture centrifuge cooled to 12–15 °C.
Centrifuge for 2 min at 2,000 rpm (~600 × g) with maximum braking. For smaller particles such as bacteria, increase the centrifugation time to 3.5–4 min.
Carefully transfer dishes to a 37 °C incubator to allow phagocytosis or fix cells immediately (t = 0 min). If samples remained cold during centrifugation, the particles or bacteria in the 0 min samples should be cell associated but not internalized, and forming phagosomes are typically apparent within 1 min of transfer to 37 °C [5, 6], though this is subject to manipulation by pathogenic microbes [14, 15]. For Fcγ receptor-mediated phagocytosis, samples can be cooled to 4 °C instead of 15 °C, but in our hands the lower temperature does not support tight binding to CD11b/CD18.
3.5 Fixation
3.6 Permeabilization
Fill chilled glass Petri dishes with −20 °C acetone.
Using forceps, transfer coverslips (one at a time) into chilled acetone.
After 5 min, rehydrate coverslips by transferring into new tissue culture dishes filled with room temperature PBS. Longer incubations in acetone (at least up to 30 min) will not harm cells.
Alternatively, cells can be permeabilized using a 1:1 mixture of −20 °C acetone–methanol which is compatible with tissue culture plastic (see Notes 2 and 3).
3.7 Blocking
Aspirate PBS from each dish and cover the fixed and permeabilized cells with 2–4 ml blocking buffer.
Incubate at room temperature for 1 h or overnight at 4 °C.
3.8 Antibody Staining for Fluorescence Microscopy
For each antibody the optimal concentration for fluorescence microscopy must be determined empirically. As a general rule, higher concentrations of antibodies are needed for microscopy than for immunoblotting. It is also essential to determine whether the antibodies of choice exhibit nonspecific binding to opsonized zymosan, bacteria, or other particulate stimuli you may be using. In most cases, nonspecific staining can be eliminated by antibody affinity purification.
3.8.1 Primary Antibodies
Dilute primary antibodies in blocking buffer to the desired final concentration (see Note 11). Each coverslip requires 30 μl of antibody. For double or triple staining, primary antibodies can be mixed together. Prepare only what is needed for each experiment since diluted antibodies are unstable and cannot be reused on subsequent days.
Centrifuge diluted antibodies (10 min, 4 °C, 10,000 × g) to pellet any aggregates.
Label a 24-well dish lid and set up a pile of Kimwipes and six microbeakers of PAB (Fig. 1).
Using forceps, remove one coverslip from blocking buffer.
Touch the edge of coverslip to the pile of Kimwipes to drain off liquid, rinse (swish) in one microbeaker of PAB, and drain again.
Dry the back (non-cell side) of the coverslip using a fresh Kimwipes and place it on the 24-well dish lid. Immediately cover with 30 μl antibody (do not let coverslips dry out).
Repeat this process for other coverslips.
Place coverslips into a plastic box lined with moist paper towels and incubate for 1 h.
3.8.2 Washing and Secondary Antibody Staining
Prepare diluted secondary antibodies in blocking buffer. Appropriate dilutions will not stain cells in the absence of primary antibody. For example, we use the DyLight 488- or rhodamine- conjugated Jackson ImmunoResearch F(ab′)2 antibodies at 1:600 and 1:200 dilution, respectively. Once again, 30 μl of antibody is used per coverslip. Prepare the amount needed for each experiment. Secondary antibodies generally do not need to be centrifuged prior to use.
Lift the first coverslip off of the 24-well dish lid using forceps (grasp as close to the edge as possible) and drain primary antibody into Kimwipes.
Wash the coverslip sequentially in six microbeakers of PAB with thorough draining between washes.
After the final rinse, carefully blot dry the back of the coverslip and return it to the 24-well dish lid.
Cover cells with 30 μl secondary antibody. Repeat for other coverslips.
Incubate for 1 h in a covered box lined with moist paper towels.
3.8.3 Non-antibody Stains
Actin filaments can be detected by staining cells with fluorophore-conjugated phalloidins. Several nucleic acid stains can be used to label cell nuclei as well as microbes. Phalloidin conjugates can be added along with primary or secondary antibodies. For most DNA stains, a 5–10-min incubation is sufficient. Wash samples thoroughly before proceeding (see Note 6).
3.8.4 Mounting Coverslips Onto Slides
Label microscopy slides in the frosted marking area. Up to six coverslips will fit onto one slide (Fig. 1). It is important to place the coverslips as close as possible to the frosted marking area of the slide, as the other edge supports the slide by resting on the microscope stage and as a result any coverslips in this area cannot viewed.
Thaw one tube of mounting medium.
Working with coverslips for one slide at a time, place six small drops (~15 μl) of gelvatol mounting medium onto the glass slide (see Note 7).
Wash each coverslip in six beakers of PAB, as described above, followed by a final rinse in deionized water. The water rinse prevents a dried salt crust from forming on mounted coverslips.
Blot excess water off the back of the coverslip and then invert it (cell side down) onto a bead of mounting medium (start from one edge of the coverslip and lower it slowly to avoid trapping air bubbles). Repeat for the other coverslips on this slide.
Gently press down on the top of each coverslip to ensure that their edges are not overlapping (this will also push out small air bubbles). If necessary, excess mounting medium can be removed using an aspirator.
Place slides in a cardboard slide folder; this keeps the slides flat and protected from light.
Let the mounting medium set overnight at 4 °C. Leftover mounting medium can be refrozen for future use. Very high humidity will prevent the mounting medium from hardening, so do not store samples in a cold room or refrigerator near dripping or condensed water.
3.9 Microscopy, Image Acquisition, and Analysis
Neutrophil shape, phagocytosis, and recruitment of granule markers and other proteins to large phagosomes, such as those containing opsonized zymosan particles, can be evaluated using conventional fluorescence microscopy [6]. The phase contrast objectives that are present on most fluorescence microscopes are our preferred method for analysis of cell morphology, as greater detail is revealed relative to the Nomarski (differential interference contrast, DIC) objectives that are typically found on confocal microscopes.
We prefer phase contrast objectives for analysis of intraphagosomal superoxide accumulation revealed by nitroblue tetrazolium staining, as we find that color images obtained in this manner can reveal details and subtleties that are more difficult to discern in similar grey-scale NBT-DIC merged images that we obtain by confocal microscopy [7, 9–11].
Analysis of granule distribution [12] and the composition of small bacterial phagosomes [7–10] is more readily analyzed by, and may require, the enhanced imaging capacity that is available with confocal microscopy. Advantages of confocal microscopy include the ability to zoom in on single cells and the ability to examine cells as sequential (or merged stacks) of thin optical sections. Using this approach, specific and azurophilic granules can be detected in the vicinity of COZ phagosomes prior to phagosome-granule fusion (Fig. 2) [12].
The ability to capture several images using identical confocal and laser settings allows distinct fixation and permeabilization conditions [12], or effects of different stimuli (Fig. 3) to be compared directly, and the software packages associated with confocal microscopes allow extensive image analyses such as measurement of pixel intensity at different points within a single image or on a single phagosome [7, 16].
When testing new antibodies or particulate stimuli, all samples should be carefully evaluated for specificity of intracellular staining (if the antigen distribution is known) and absence of nonspecific staining of zymosan or bacteria. We have used the methods described here to study neutrophils from persons with chronic granulomatous disease and to analyze PMNs that have ingested opsonized zymosan, Neisseria meningitidis, Helicobacter pylori, Staphylococcus aureus, and Francisella tularensis [6–10, 12]. As reported previously [12], we find that that acetone or methanol–acetone permeabilization significantly enhances the sensitivity of detection of many granule proteins and NADPH oxidase subunits in PMNs (see Note 3) and, in general, cytoplasmic granules are dispersed more evenly under these conditions as compared with detergent-permeabilized neutrophils. This approach also enhances the sensitivity of detection of lamp-1, a prominent marker of late endosomes and multivesicular bodies, in macrophages as well as neutrophils (data not shown). Neutrophil spreading and degranulation can also affect antigen detection [12] (see Note 11).
Fig. 2.
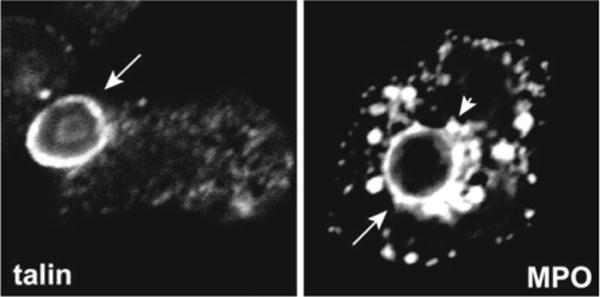
Images of nascent and mature phagosomes. Confocal images of PMNs on serum-coated coverslips show the accumulation of the cytoskeletal protein talin on 1 min COZ phagosomes and the azurophilic granule protein MPO on 15 min COZ phagosomes. Arrows indicate phagosomes; arrowhead indicates periphagosomal azurophilic granules
Fig. 3.
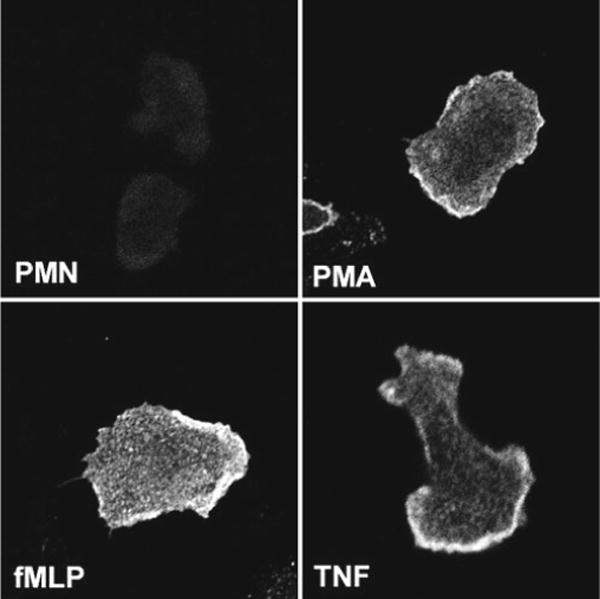
Detecting upregulation of proteins at the cell surface. Neutrophils plated on fibrinogen-coated coverslips were left untreated or stimulated with 200 nM PMA, 1 μM fMLF, or 0.02 μM TNFα as indicated. Fixed, intact cells were stained with mAb 7D5 to show upregulation of flavocytochrome b558 at the cell surface. All four images were acquired using identical confocal settings
Fig. 4.
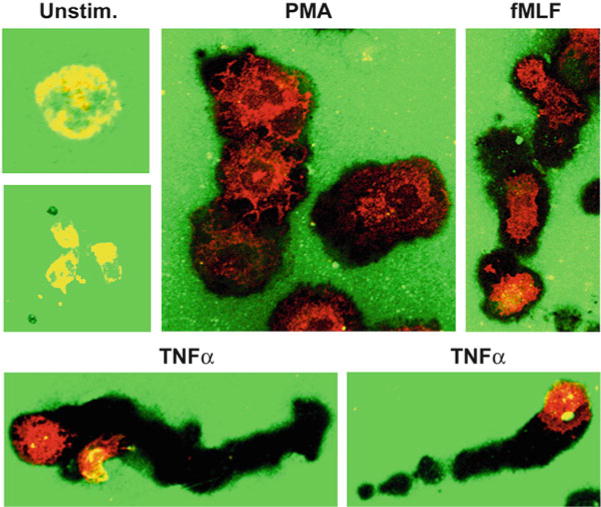
Neutrophil modulation of the extracellular matrix. PMNs were plated on coverslips coated with Oregon Green-labeled fibrinogen and then left untreated or stimulated with PMA, TNFα, or fMLF as indicated. PMNs were counterstained with mAb to gp91phox after fixation and permeabilization. Fluorescence microscopy images show the ability of activated cells to bleach and/or degrade fibrinogen. Differential effects of these stimuli on cell migration are also apparent
Fig. 5.
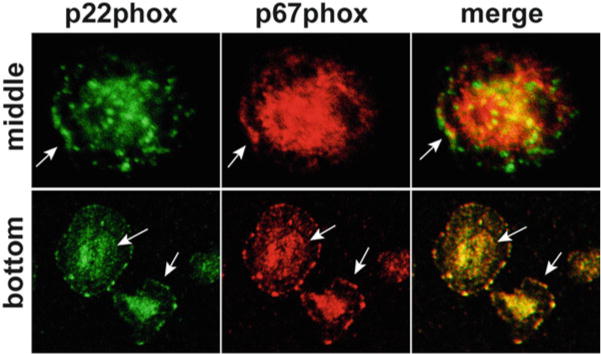
Localization of NADPH oxidase subunits. Neutrophils were plated on fibrinogen-coated coverslips and then stimulated with TNFα for 30 min. Fixed and permeabilized cells were double stained to detect p22phox (green) and p67phox (red). Confocal sections taken through the center of cells or at the substrate-adherent surface indicate colocalization of NADPH oxidase subunits at the leading edge and throughout the plasma membrane (arrows). In contrast, p67phox was not detected on p22phox -positive specific granules (arrowheads)
Acknowledgments
This work was supported by funds from the National Institutes of Health (R01AI073835, P01AI044642) and a VA Merit Review Grant.
Footnotes
Other particulate stimuli can be substituted for zymosan including live or killed microorganisms, latex beads, or opsonized sheep red blood cells. Zymosan, latex beads, and red blood cells are visible using phase contrast, bright field, or DIC optics [6]. On the other hand, the small size of many bacteria makes them more difficult to detect in the absence of a direct probe (such as green-fluorescent protein or a specific antibody) [7, 9, 10, 17]. Opsonization of encapsulated bacteria can be inefficient, and particle to serum ratios may need to be adjusted to ensure that complement deposition or antibody biding is sufficient to confer phagocytosis [17]. When using IgG or immune serum, it is also important to ensure that particles are not aggregated [10]. Prelabeled fluorescent beads, Escherichia coli, or other BioParticles should be used with caution as we find that the intense fluorescence of these particles overwhelms all other signals and appears as nonspecific fluorescence (bleed through) across a wide spectrum of wavelengths or channels using either conventional or confocal immunofluorescence microscopy.
Neutral buffered solutions of 10 % formalin are equipotent to 4 % paraformaldehyde. We prefer formalin because it does not need to be prepared fresh each day. However, the small amount of methanol used to stabilize formalin makes it unsuitable as a fixative when intact cell membranes are required, for example, when fixing cells that have pinocytosed fluorescent dextrans or Lucifer yellow [18] or when strict detection of a surface-exposed (but not intracellular) antigen or epitopes is desired [7] (Fig. 3). In these instances, cells should be fixed using fresh 4 % paraformaldehyde. Direct exposure of live cells to cold methanol (which can fix and permeabilize neutrophils in one step) is compatible with some (but not all) antigens.
Detergents such as Triton X-100 are commonly used to permeabilize fixed cells for fluorescence microscopy. However, this approach is prone to artifacts, particularly when used to detect soluble proteins [19]. We find that permeabilization of formalin-fixed cells with −20 °C acetone (or acetone–methanol mixtures) provides superior preservation of cell morphology and enhances the sensitivity of detection of many antigens including p22phox and CD66b [12]. These data are noteworthy since the epitope on p22phox that is recognized by monoclonal antibody 44.1 is cytosolic [20, 21]. As such, the limited detection conferred by 0.1 % Triton X-100 cannot be explained by differential permeabilization of granule and plasma membranes by this detergent. Similarly, we have shown previously that acetone permeabilization enhances detection of lamp-1 on Helicobacter pylori phagosomes in macrophages [16]. On the other hand, detergent permeabilization is preferred if cells contain pinocytosed dextrans or Lucifer yellow (see Note 2).
In our hands, the type of serum used in blocking buffer is not critical. We find that Sigma-Aldrich horse serum works well and is cost effective (as compared with FBS, goat serum, or donkey serum (see also Note 6)).
We routinely use the following antibodies for immunofluorescence microscopy of human neutrophils [6–10]. To detect NADPH oxidase components, antibodies specific for p22phox (mAb 44.1) and gp91phox (mAb 54.1) [20, 21] are now available from Santa Cruz Biotechnology (Santa Cruz, CA); mAb 7D5 detects an epitope of gp91phox present in mature flavocytochrome b558 [22] and can be used on intact PMNs to detect protein upregulation at the cell surface [7]; a rabbit mAb specific for human p40phox (Epitomics, Burlingame, CA); an antibody that specifically detects active Rac (NewEast Biosciences, Malvern, PA); and rabbit antisera from William Nauseef (University of Iowa) specific for p47phox and p67phox [6]. Mouse mAbs specific for human CD63, lamp-1, and CD11b are obtained from the University of Iowa Developmental Studies Hybridoma Bank. Mouse mAb to human lactoferrin and myeloperoxidase are purchased from Meridian Life Sciences (Saco, ME) or other vendors.
For dual and triple staining, optimal results are obtained using secondary antibodies that are cross-absorbed against other species (such as those available from Jackson ImmunoResearch Labs), and F(ab′)2 antibodies are preferred to whole immunoglobulins. If whole IgG are used, it may be necessary to use blocking buffer containing serum matched to the species of secondary antibody in order to prevent nonspecific binding to Fcγ receptors. On the other hand, the choice of fluorescent conjugate is largely a matter of personal preference. For double staining “green–red” combinations are standard. In this regard, DyLight 488 (Jackson ImmunoResearch) or Alexa 488 (Molecular Probes/Invitrogen/Life Technologies) is brighter, more photostable, and less pH sensitive than FITC. Rhodamine and similar DyLight or Alexa conjugates emit strong red– orange fluorescence that often appears brighter than the darker red of Alexa 594 and Alexa 610 conjugates. For triple labeling it is typical to include a far red dye such as Cy5, Cy7, or Alexa 635, which is then false-colored blue in merged images.
The ultraviolet-excitable dye DAPI is often used to stain cell nuclei [17], and fluor-conjugated phalloidins are used to detect actin filaments [6]. However, antibodies to cytoskeletal proteins such as talin (Fig. 2) or coronin [6] may give superior results in neutrophils for detection of forming phagosomes, as actin filaments in this cell type are less robust than in macrophages.
We prefer the polyvinyl alcohol base of gelvatol mounting medium because it hardens rapidly and completely. A common alternative to gelvatol is buffered glycerol (supplemented with an anti-fading agent). Because glycerol mounting solutions do not harden, coverslips must be attached to microscope slides using nail polish (which is more time consuming to apply and more apt to leak).
We described previously the phenotype of PMNs attached loosely to serum- or fibrinogen-coated coverslips [6–10, 12]. Here, we extended these results to show that the ability of PMNs to digest or bleach and migrate along surfaces coated with extracellular matrix proteins can also be analyzed (Figs. 4 and 5). Because these interactions are physiological, they are also preferred to the nonspecific adhesion obtained using polylysine-coated surfaces. Moreover, the general “stickiness” of polylysine can impair phagocytosis of particulate stimuli and increase background due to trapping of debris. If polylysine is used, 0.1 mg/ml poly- D -lysine is less toxic to most cell types than poly-L-lysine.
For several reasons we also prefer coated coverslips to chamber slides (Nunc). First, coverslips are more cost effective, in part because only six of the eight wells on each chamber slide can be used (because the wells on the far right side of the slide rest on the microscope stage). Second, neutrophils distribute evenly over coated coverslips yet bind preferentially near the edges of each chamber due to the meniscus of liquid in each well. Third, the binding interactions that mediate cell attachment to the proprietary slide coating are unclear and can trigger cell activation. Therefore, chamber slides should be coated with serum proteins or fibrinogen before use. Finally, 70 μl of antibody is needed for each well as compared with 30 μl for each 12 mm coverslip (despite similar surface area).
To stimulate neutrophils in suspension, dilute cells to 1 × 106 PMN/ml in desired medium or buffer and disperse into sterile polypropylene tubes. Add particulate or soluble stimuli and incubate at 37 °C (with tumbling). Terminate incubations by rapidly diluting each sample into 20 volumes of ice cold PBS and keep on ice. Attach cells to acid-washed coverslips using a cytocentrifuge (such as a Shandon CytoSpin). Assemble slides and filter papers as directed by the manufacturer and place a dry (uncoated) coverslip between the microscope slide and the absorbent paper with the coverslip positioned to receive the sample. Centrifuge 50,000–100,000 PMN onto each coverslip. Transfer coverslips to a tissue culture dish, cover with fixative, and continue as described for adherent cells. Do not fix and stain cells prior to cytocentrifugation. It is also important not to submerge the coverslips in dishes prefilled with fixative as they can cause cells to detach. Precise synchronization of phagocytosis is not achieved using this method.
Both the time and temperature of cell fixation are important. Room temperature fixation preserves cell morphology, whereas chilled fixatives can cause cells to contract or detach. Cells incubated in fixative for prolonged periods of time will appear dark using phase contrast optics. Inadequately fixed cells will have poor organelle morphology and, because antigens are not appropriately fixed in place, antibody staining may appear less distinct or poorly localized as compared with properly fixed cells. It is also important to note that aldehyde fixation is reversible. For this reason paraformaldehyde or formalin-fixed cells that have been permeabilized with detergents must be analyzed within a day or so. In contrast, aldehyde fixed samples that have been permeabilized using methanol and/or acetone are more stable and can be stored at 4 °C for 2–3 weeks.
Appropriate antibody concentrations vary with changes in cell morphology and the extent of degranulation. Concentrations of antibody appropriate for activated, spread PMN that have undergone some degranulation can be too high when applied to round, unstimulated cells. Similarly, the amount of antibody needed to detect high concentrations of granule-associated proteins may not be sufficient to detect the same marker once it is distributed over a large phagosome or throughout the plasma membrane [12]. In this regard it is important to evaluate each antibody separately and also to realize that the appropriate dilution of primary antibody may vary depending on the fluor conjugated to the secondary antibody.
References
- 1.Faurschou M, Borregaard N. Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. 2003;5:1317–1327. doi: 10.1016/j.micinf.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 2.Nauseef WM. Assembly of the phagocyte NADPH oxidase. Histochem Cell Biol. 2004;122:277–291. doi: 10.1007/s00418-004-0679-8. [DOI] [PubMed] [Google Scholar]
- 3.Nordenfelt P, Tapper H. Phagosome dynamics during phagocytosis by neutrophils. J Leukoc Biol. 2011;90:271–284. doi: 10.1189/jlb.0810457. [DOI] [PubMed] [Google Scholar]
- 4.Allen L-AH, Aderem A. A role for MARCKS, the α isozyme of protein kinase C and myosin I in zymosan phagocytosis by macrophages. J Exp Med. 1995;182:829–840. doi: 10.1084/jem.182.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Allen L-AH, Aderem A. Molecular definition of distinct cytoskeletal structures involved in complement- and Fc receptor- mediated phagocytosis in macrophages. J Exp Med. 1996;184:627–637. doi: 10.1084/jem.184.2.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allen L-AH, DeLeo FR, Gallois A, Toyoshima S, Suzuki K, Nauseef WM. Transient association of the nicotinamide adenine dinucleotide phosphate oxidase subunits p47phox and p67phox with phagosomes in neutrophils from patients with X-linked chronic granulomatous disease. Blood. 1999;93:3521–3530. [PubMed] [Google Scholar]
- 7.Allen L-AH, Beecher BR, Lynch JT, Rohner OV, Wittine LM. Helicobacter pylori disrupts NADPH oxidase targeting in human neutrophils to induce extracellular superoxide release. J Immunol. 2005;174:3658–3667. doi: 10.4049/jimmunol.174.6.3658. [DOI] [PubMed] [Google Scholar]
- 8.DeLeo FR, Allen L-AH, Apicella M, Nauseef WM. NADPH oxidase activation and assembly during phagocytosis. J Immunol. 1999;163:6732–6740. [PubMed] [Google Scholar]
- 9.McCaffrey RL, Allen L-AH. Pivotal advance: Francisella tularensis evades killing by human neutrophils via inhibition of the respiratory burst and phagosome escape. J Leukoc Biol. 2006;80:1224–1230. doi: 10.1189/jlb.0406287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCaffrey RL, Schwartz JT, Lindemann SR, Moreland JG, Buchan BW, Jones BD, Allen L-AH. Multiple mechanisms of NADPH oxidase inhibition by type A and type B Francisella tularensis. J Leukoc Biol. 2010;88:791–805. doi: 10.1189/jlb.1209811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schulert GS, McCaffrey RL, Buchan BW, Lindemann SR, Hollenback C, Jones BD, Allen L-AH. Francisella tularensis genes required for inhibition of the neutrophil respiratory burst and intramacrophage growth identified by random transposon mutagenesis of strain LVS. Infect Immun. 2009;77:1324–1336. doi: 10.1128/IAI.01318-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allen L-AH. Immunofluorescence and confocal microscopy of neutrophils. Methods Mol Biol. 2007;412:273–287. doi: 10.1007/978-1-59745-467-4_18. [DOI] [PubMed] [Google Scholar]
- 13.Nauseef WW. Isolation of human neutrophils from venous blood. Methods Mol Biol. 2007;412:15–20. doi: 10.1007/978-1-59745-467-4_2. [DOI] [PubMed] [Google Scholar]
- 14.Allen L-AH, Schlesinger LS, Kang B. Virulent strains of Helicobacter pylori demonstrate delayed phagocytosis and stimulate homotypic phagosome fusion in macrophages. J Exp Med. 2000;191:115–127. doi: 10.1084/jem.191.1.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allen L-AH. Rate and extent of Helicobacter pylori phagocytosis. Methods Mol Biol. 2008;431:147–157. doi: 10.1007/978-1-60327-032-8_12. [DOI] [PubMed] [Google Scholar]
- 16.Schwartz JT, Allen L-AH. Role of urease in megasome formation and Helicobacter pylori survival in macrophages. J Leukoc Biol. 2006;79:1214–1225. doi: 10.1189/jlb.0106030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schwartz JT, Barker JH, Long ME, Kaufman J, McCracken J, Allen L-AH. Natural IgM mediates complement-dependent uptake of Francisella tularensis by human neutrophils via complement receptors 1 and 3 in nonimmune serum. J Immunol. 2012;189:3064–3077. doi: 10.4049/jimmunol.1200816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Botelho RJ, Tapper H, Furuya W, Mojdami D, Grinstein S. FcγR-mediated phagocytosis stimulates localized pinocytosis in human neutrophils. J Immunol. 2002;169:4423–4429. doi: 10.4049/jimmunol.169.8.4423. [DOI] [PubMed] [Google Scholar]
- 19.Melan MA, Sluder G. Redistribution and differential extraction of soluble proteins in permeabilized cultured cells: implications for immunofluorescence microscopy. J Cell Sci. 1992;101:731–743. doi: 10.1242/jcs.101.4.731. [DOI] [PubMed] [Google Scholar]
- 20.Burritt JB, Quinn MT, Jutila MA, Bond CW, Jesaitis AJ. Topological mapping of neutrophil cytochrome b epitopes with phage-display libraries. J Biol Chem. 1995;270:16974–16980. doi: 10.1074/jbc.270.28.16974. [DOI] [PubMed] [Google Scholar]
- 21.Burritt JB, Busse SC, Gizachew D, Siemsen DW, Quinn MT, Bond CW, Dratz EA, Jesaitis AJ. Antibody imprint of a membrane protein surface. Phagocyte flavocytochrome b. J Biol Chem. 1998;273:24847–24852. doi: 10.1074/jbc.273.38.24847. [DOI] [PubMed] [Google Scholar]
- 22.Yamauchi A, Yu LX, Potgens AJG, Kuribayashi F, Nunoi H, Kanegasaki S, Roos D, Malech HL, Dinauer MC, Nakamura M. Location of the epitope for 7D5, a monoclonal antibody raised against human flavocyto-chrome b558, to the extracellular peptide portion of primate gp91(phox) Microbiol Immunol. 2001;45:249–257. doi: 10.1111/j.1348-0421.2001.tb02614.x. [DOI] [PubMed] [Google Scholar]