

Summary
In response to numerous pathologic stimuli, the myocardium undergoes a hypertrophic response characterized by increased myocardial cell size and activation of fetal cardiac genes. We show that cardiac hypertrophy is induced by the calcium-dependent phosphatase calcineurin, which dephosphorylates the transcription factor NF-AT3, enabling it to translocate to the nucleus. NF-AT3 interacts with the cardiac zinc finger transcription factor GATA4, resulting in synergistic activation of cardiac transcription. Transgenic mice that express activated forms of calcineurin or NF-AT3 in the heart develop cardiac hypertrophy and heart failure that mimic human heart disease. Pharmacologic inhibition of calcineurin activity blocks hypertrophy in vivo and in vitro. These results define a novel hypertrophic signaling pathway and suggest pharmacologic approaches to prevent cardiac hypertrophy and heart failure.
Introduction
Cardiac hypertrophy is an adaptive response of the heart to virtually all forms of cardiac disease, including those arising from hypertension, mechanical load, myocardial infarction, cardiac arrhythmias, endocrine disorders, and genetic mutations in cardiac contractile protein genes. While the hypertrophic response is initially a compensatory mechanism that augments cardiac output, sustained hypertrophy can lead to dilated cardiomyopathy, heart failure, and sudden death. In the United States, approximately half a million individuals are diagnosed with heart failure each year, with a mortality rate approaching 50%.
Despite the diverse stimuli that lead to cardiac hypertrophy, there is a prototypical final molecular response of cardiomyocytes to hypertrophic signals that involves an increase in cell size and protein synthesis, enhanced sarcomeric organization, up-regulation of fetal cardiac genes, and induction of immediate-early genes, such as c-fos and c-myc (reviewed in Chien et al., 1993; Sadoshima and Izumo, 1997). The causes and effects of cardiac hypertrophy have been extensively documented, but the underlying molecular mechanisms that couple hypertrophic signals initiated at the cell membrane to the reprogramming of cardiomyocyte gene expression remain poorly understood. Elucidation of these mechanisms is a central issue in cardiovascular biology and will be critical for designing new strategies for prevention or treatment of cardiac hypertrophy and heart failure.
Numerous studies have implicated intracellular Ca2+ as a signal for cardiac hypertrophy. In response to myocyte stretch or increased loads on working heart preparations, intracellular Ca2+ concentrations increase (Marban et al., 1987; Bustamante et al., 1991; Hongo et al., 1995), consistent with a role of Ca2+ in coordinating physiologic responses with enhanced cardiac output. A variety of humoral factors, including angiotensin II (AngII), phenylephrine (PE), and endothelin-1 (ET-1), which induce the hypertrophic response in cardiomyocytes (Karliner et al., 1990; Sadoshima and Izumo, 1993; Sadoshima et al., 1993; Leite et al., 1994), also share the ability to elevate intracellular Ca2+ concentrations.
Hypertrophic stimuli result in reprogramming of gene expression in the adult myocardium, such that genes encoding fetal protein isoforms like β-myosin heavy chain (MHC) and α-skeletal actin are up-regulated, whereas the corresponding adult isoforms, α-MHC and α-cardiac actin, are down-regulated. The natriuretic peptides, atrial natriuretic factor (ANF), and b-type natriuretic peptide (BNP), which decrease blood pressure by vasodilation and natriuresis, are also rapidly up-regulated in the heart in response to hypertrophic signals (reviewed in Komuro and Yazaki, 1993). The mechanisms involved in coordinately regulating these cardiac genes during hypertrophy are unknown, although binding sites for several transcription factors, including serum response factor (SRF), TEF-1, AP-1, and Sp1, are important for activation of fetal cardiac genes in response to hypertrophy (Sadoshima and Izumo, 1993; Sadoshima et al., 1993; Kariya et al., 1994; Karns et al., 1995; Kovacic-Milivojevic et al., 1996). Most recently, the cardiac-restricted zinc finger transcription factor GATA4 has also been shown to be required for transcriptional activation of the genes for Ang II type 1a receptor and β-MHC during hypertrophy (Herzig et al., 1997; Hasegawa et al., 1997; reviewed in Molkentin and Olson, 1997).
A number of intracellular signaling pathways have been implicated in transduction of hypertrophic stimuli. For example, occupancy of the cell surface receptors for AngII, PE, and ET-1 leads to activation of phospholipase C, resulting in the production of diacylglycerol and inositol triphosphate, which in turn results in mobilization of intracellular Ca2+ and activation of protein kinase C (PKC) (Sadoshima and Izumo, 1993; Yamazaki et al., 1996; Zou et al., 1996). There is also evidence that the Ras and mitogen-activated protein (MAP) kinase pathways are transducers of hypertrophic signals (Thorburn et al., 1993; reviewed in Force et al., 1996). The extent to which these signaling pathways are coordinated during cardiac hypertrophy is unknown. However, all of these pathways are associated with an increase in intracellular Ca2+, consistent with a central regulatory role of Ca2+ in coordinating the activities of multiple hypertrophic signaling pathways.
In B and T cells, the Ca2+, calmodulin-dependent phosphatase calcineurin has been shown to link intracellular signaling pathways that result in elevation of intracellular Ca2+ with activation of the immune response. Calcineurin regulates immune response genes through dephosphorylation of a family of transcription factors known as NF-ATs (nuclear factors of activated T cells) (reviewed in Rao et al., 1997). Once dephosphorylated by calcineurin, NF-AT transcription factors translocate to the nucleus and directly activate immune response genes (Flanagan et al., 1991; Loh et al., 1996a, 1996b). The immunosuppressant drugs cyclosporin A (CsA) and FK 506 suppress the immune response by inhibiting calcineurin’s ability to activate NF-AT transcription factors (Shaw et al., 1995; Loh et al., 1996b).
Here we show that the myocardium utilizes a calcineurin-dependent pathway to activate the transcription factor NF-AT3 during hypertrophy, suggesting a conservation in signaling between immune cells and cardiomyocytes as a mechanism for responding to environmental stress. Our results show that activated calcineurin is both necessary and sufficient to induce cardiac hypertrophy in vitro and in vivo. Activation of calcineurin results in dephosphorylation of NF-AT3 and the subsequent induction of fetal cardiac genes via a combinatorial mechanism involving its direct interaction with GATA4. Transgenic mice that express activated forms of calcineurin or NF-AT3 in the heart develop cardiac hypertrophy that progresses to dilated cardiomyopathy, with interstitial fibrosis, congestive heart failure, and sudden death. CsA and FK 506 block the morphologic and molecular responses of primary cardiomyocytes to Ang II and PE, indicating that calcineurin is a component of the signaling pathways whereby these factors induce hypertrophy. Moreover, CsA administration can prevent cardiac hypertrophy and associated pathology in calcineurin transgenic mice. These results define a novel signaling pathway that couples hypertrophic signals at the cell membrane to changes in cardiac gene expression and suggest opportunities for pharmacologic intervention into cardiac hypertrophy and heart failure.
Results
Interaction between NF-AT3 and GATA4
Our initial interest was to identify proteins, using the yeast two-hybrid system, that might act as cofactors for GATA4 in the heart. The GATA4 bait consisted of amino acids 130–409 fused in-frame to the yeast GAL4 protein (Figure 1A). This region of GATA4 encompasses the two zinc fingers and most of the carboxyl terminus but lacks the amino-terminal transcription activation domain, and therefore does not activate transcription on its own in yeast. Screening of a 10.5-day mouse embryo cDNA library resulted in the identification of numerous GATA4-interacting factors, one of which was NF-AT3. The other GATA4-interacting factors identified in this screen will be described elsewhere.
Figure 1. Interactions between GATA4 and NF-AT3 in the Two-Hybrid System.

(A) Schematic diagrams of GATA4 and NF-AT3 proteins. The portion of GATA4 used as bait in the two-hybrid system encompassed amino acids 130–409 and is shown beneath the full-length protein. The portion of NF-AT3 recovered in the yeast two-hybrid screen spanned amino acids 522–902. The Rel homology domain (RHD) extends from amino acids 404–694 and the conserved phosphorylation domain from 145–275.
(B) Amino acids 522–902 of NF-AT3 were fused in-frame to the GAL4 DNA-binding domain (DBD) and used as bait in a two-hybrid assay in transfected 10T1/2 cells.
NF-AT3 is a member of a multigene family containing four members, NF-ATc, NF-ATp, NF-AT3, and NF-AT4 (McCaffrey et al., 1993; Northrop et al., 1994; Ho et al., 1995; Hoey et al., 1995; Masuda et al., 1995; Park et al., 1996). These factors bind the consensus DNA sequence GGAAAAT as monomers or dimers through a Rel homology domain (RHD) (Rooney et al., 1994; Hoey et al., 1995). Three of the NF-AT genes are restricted in their expression to T cells and skeletal muscle, whereas NF-AT3 is expressed in a variety of tissues including the heart (Hoey et al., 1995).
Given the ability of NF-AT factors to mediate changes in gene expression in response to Ca2+ signaling in T cells, we were especially interested in the finding that GATA4, a known effector of cardiac gene expression, and NF-AT3 were able to interact. This interaction suggested a potential mechanism for coupling Ca2+ signaling to cardiac transcription, as is known to occur during cardiac hypertrophy.
NF-AT3 is a 902-amino acid protein with a regulatory domain at its amino terminus that mediates nuclear translocation and the Rel homology domain near its carboxyl terminus that mediates DNA binding (Figure 1A). The region of NF-AT3 recovered from the yeast two-hybrid screen extended from amino acid 522, which is near the middle of the Rel homology domain, to the carboxyl terminus.
The specificity of interaction between GATA4 and NF-AT3 was tested by retransforming yeast with the rescued NF-AT3-GAL4 activation domain plasmid and various GAL4 DNA-binding domain bait plasmids. In this assay, NF-AT3 was also found to interact with residues 133–265 of GATA5, which encompass only the zinc finger DNA-binding domain. However, NF-AT3 did not interact with the basic helix-loop-helix protein E12 or with the GAL4 DNA-binding domain alone (data not shown).
To validate further the interaction between GATA4 and NF-AT3, the rescued NF-AT3 cDNA fragment was fused to the GAL4 DNA-binding domain and tested for its ability to interact with full-length GATA4 in transfected mammalian cells. As a reporter plasmid, we used pG5E1bCAT, which contains five tandem GAL4 DNA-binding sites upstream of the minimal E1b promoter linked to CAT. This reporter was not significantly activated by either GAL4-NF-AT3 or GATA4 alone but was strongly activated by the two factors together in 10T1/2 fibroblasts (Figure 1B), as well as in primary neonatal rat cardiomyocytes (data not shown). Full-length NF-AT3 also interacted with the GAL4-GATA4 bait in the mammalian transfection assay (data not shown).
Mapping the Protein Determinants of GATA4-NF-AT3 Interaction
To define further the interaction between GATA4 and NF-AT3, we tested whether interactions between the corresponding 35S-methionine-labeled in vitro translation products could be detected. Cotranslation of full-length GATA4 with the NF-AT3 deletion mutant containing residues 522–902 fused to a Flag epitope at the C terminus, followed by immunoprecipitation with anti-Flag antibody and SDS-PAGE, showed that the two proteins coimmunoprecipitated (Figure 2A, lane 5). The anti-Flag antibody did not immunoprecipitate GATA4 in the absence of NF-AT3-Flag (lane 4).
Figure 2. Coimmunoprecipitation of GATA4 and NF-AT3.

(A) NF-AT3 with a Flag epitope tag and GATA4, as indicated, were translated in a rabbit reticulocyte lysate in the presence of 35S-methionine. Anti-Flag antibody was then used for coimmunoprecipitation assays. Proteins were resolved by SDS-PAGE. Lanes 1 and 2 show the in vitro translation products. Lanes 3–5 show the coimmunoprecipitations. The anti-Flag antibody selectively immunoprecipitates NF-AT3 (lane 3) but does not recognize GATA4 (lane 4). However, when NF-AT3 is mixed with GATA4, GATA4 is coimmunoprecipitated (asterisk in lane 5).
(B) The domain of GATA4 that interacts with NF-AT3 was mapped by translating NF-AT3-Flag with a series of GATA4 deletion mutants (shown in [C]), followed by coimmunoprecipitation. Even-numbered lanes show the GATA4 or GATA6 (lane 14) deletion mutants loaded directly on the gel. The other lanes contain the indicated GATA4 or GATA6 (lane 15) deletion mutants plus NF-AT3 and were immunoprecipitated with anti-Flag antibody. All GATA4 deletion mutants except 80–441/d265–294, which lacks the second zinc finger, were coimmunoprecipitated. A GATA6 deletion mutant containing the two zinc fingers was also coimmunoprecipitated with NF-AT3.
(C) Summary of coimmunoprecipitation results. F1 and F2 denote the two zinc fingers and NLS designates the nuclear localization signal.
(D) The C-terminal region of NF-AT3, encompassing the Rel homology domain (RHD), was translated separately or together with GATA4 deletion mutant 80–328, as indicated. Lanes 1 and 2 show the individual in vitro translation products loaded directly on the gel. Lanes 3–5 show the results of immunoprecipitation with anti-NFAT antibody that recognizes the NF-AT3 RHD. This region is sufficient for interaction with GATA4 (indicated by an asterisk in lane 5).
To map the determinants of this interaction more precisely, we tested a series of GATA4 deletion mutants for the ability to be coimmunoprecipitated with NF-AT3-Flag. Residues 181–328 of GATA4, which encompass the two zinc fingers and NLS, interacted with NF-AT3 as efficiently as full-length GATA4 (Figure 2B, lane 11). Residues 239–441 of GATA4, which extend from the second zinc finger to the C terminus, also interacted with NF-AT3 (lane 7), whereas an internal deletion mutant lacking the second zinc finger (80–441/d265–294) did not (lane 13). These experiments demonstrated that the second zinc finger of GATA4 was essential for interaction with NF-AT3, whereas the N terminus, the first zinc finger, and the C terminus were unimportant for this interaction (Figure 2C). Also of note, the zinc finger region (amino acids 130–350) of GATA6 was immunoprecipitated with NF-AT3 (lane 15).
We also tested a deletion mutant of NF-AT3 that encompassed only the Rel homology domain, residues 404–694. This region was sufficient to interact with GATA4 (Figure 2D, lane 5). Together, these results indicated that the Rel homology region of NF-AT3 contained determinants that mediate interaction with the second zinc finger of GATA4.
Synergistic Activation of the BNP Gene by GATA4 and NF-AT3
To begin to investigate whether the GATA4-NF-AT3 interaction had a functional role in cardiac gene expression, we tested the ANF, BNP, and cardiac troponin I promoters, which are up-regulated during hypertrophy, for their responsiveness to these factors in transfected neonatal rat cardiomyocytes. The BNP promoter showed a dramatic response and was therefore analyzed further. For these experiments, we also used a cDNA expression plasmid encoding a constitutively active form of the calcineurin catalytic A subunit lacking the C-terminal autoinhibitory domain (O’Keefe et al., 1992). This calcineurin mutant functions as a Ca2+-independent phosphatase but retains sensitivity to CsA and FK506. As shown in Figure 3A, the BNP promoter was activated greater than 100-fold in the presence of GATA4, NF-AT3, and calcineurin. GATA4 alone was also able to activate this promoter, as reported previously (Grepin et al., 1994), but the extent of activation was less than one-tenth that when NF-AT3 and calcineurin were also present. Since GATA4 and NF-AT3 are expressed in neonatal rat cardiomyocytes, it seems they are limiting in this type of transfection assay, making it necessary to express the exogenous proteins to see the maximal response of the BNP promoter.
Figure 3. Regulation of the BNP Promoter by NF-AT3 in Primary Cardiomyocytes.

(A) Primary rat cardiomyocytes were transiently transfected with a CAT reporter gene linked to the BNP 5′-flanking region and expression vectors encoding NF-AT3, activated calcineurin, or GATA4, as indicated. Forty-eight hours later, cells were harvested and CAT activity was determined. In the lane labeled −927 site mutant, a BNP-CAT reporter gene, in which the NF-AT3 site at −927 was mutated, was used.
(B) 32P-labeled oligonucleotide probes corresponding to the potential NF-AT binding sites from the IL-2 promoter and from −927, −327, and −27 bp 5′ of the BNP gene were used in gel mobility shift assays with unprogrammed reticulocyte lysate (lanes 1–4) or in vitro translated NF-AT3 (lanes 5–8). Only the IL-2 and BNP-927 probes yielded a specific DNA–protein complex.
(C) A 32P-labeled oligonucleotide probe corresponding to the −927 BNP site was used in gel mobility shift assays with protein extracts from neonatal rat cardiomyocytes. Multiple specific complexes were generated (lane 1), which were competed by unlabeled cognate site (lane 2), but not by an unrelated E-box oligonucleotide (lane 3). Addition of NF-AT3 antibody eliminated the major DNA–protein complex and created a minor ternary complex (asterisk in lane 5), whereas nonimmune rabbit serum had no effect (lane 4). Free probe was electrophoresed off the bottom of the gel in both experiments.
Given the dramatic responsiveness of the BNP promoter to NF-AT3, we examined the 1800 bp promoter region used in the above transfection assays for potential NF-AT consensus binding sites (GGAAAAT). Three sequences related to this site were identified at −927 (TGGAAAACAA), −327 (TGGAAAAGGC), and −27 (AGG ATAAAAG). The −27 site also binds GATA4 and is required for BNP expression (Grepin et al., 1994). Using oligonucleotide probes corresponding to these sequences, we used gel mobility shift assays to test for binding to in vitro translated NF-AT3 protein generated in a rabbit reticulocyte lysate. The putative site at −927 bound NF-AT3 as avidly as the consensus NF-AT site from the IL-2 promoter, whereas no binding was detected to the −327 or −27 sites (Figure 3B).
To confirm that NF-AT3 from cardiomyocytes could also bind the −927 site from the BNP promoter, we used cardiac protein extracts in a gel mobility shift assay with the −927 site as a probe. Cardiac extract gave rise to multiple complexes that could be eliminated in the presence of an excess of the same unlabeled oligonucleotide or by a sequence corresponding to the NF-AT site in the IL-2 promoter (data not shown), but not by nonspecific sequences (Figure 3C). The cardiomyocyte complex could also be largely eliminated using an NF-AT3-specific antibody (lane 5).
To determine whether the −927 site was required for transcriptional activation by NF-AT3, we mutated this site and found that the mutant promoter was insensitive to NF-AT3 (Figure 3A). These results demonstrate that the BNP promoter is a direct transcriptional target for synergistic activation by GATA4 and NF-AT3 in cardiomyocytes.
CsA and FK506 Inhibit the Hypertrophic Effects of AngII and PE
Exposure of primary cardiomyocytes to AngII and PE results in an increase in intracellular Ca2+ and a hypertrophic response. To determine whether the hypertrophic response of cardiomyocytes to these agonists was mediated by calcineurin, we exposed neonatal rat cardiomyocytes to AngII (10 nM) or PE (10 μM) in the presence and absence of CsA or FK 506. Cardiomyocytes demonstrated a dramatic increase in size and sarcomeric assembly after 72 hr of exposure to AngII or PE (Figures 4B and 4E). In the presence of CsA (Figures 4C and 4F) or FK 506 (data not shown), the response to AngII was completely abolished and the response to PE was dramatically reduced.
Figure 4. Inhibition of AngII- and PE-Dependent Hypertrophy of Primary Cardiocytes by CsA and FK506.

(A–F) Primary rat cardiocytes in serum-free medium were stimulated with AngII (10 nM) or PE (10 μM) for 72 hr. Cells were then fixed and stained with anti-α-actinin antibody to reveal sarcomeres and Hoechst stain to reveal nuclei. CsA (500 ng/ml) was added to one set of cultures at the time of AngII addition.
(G) Total RNA was isolated from primary cardiocyte cultures treated with AngII in the presence or absence of CsA as in (A) and analyzed for expression of GAPDH and ANF transcripts by dot blot.
(H) Primary rat cardiomyocytes were transiently transfected with an NF-AT-dependent luciferase reporter gene. Cells were then treated with AngII or PE in the presence or absence of CsA, as described above. Forty-eight hours later, cells were harvested and luciferase activity was determined.
To determine whether changes in cardiomyocyte gene expression in response to AngII were also controlled by a calcineurin-dependent signaling pathway, we examined by dot blot assay the expression of ANF mRNA in cardiomyocytes treated with AngII in the presence and absence of CsA. Exposure to AngII resulted in a 15-fold increase in ANF mRNA, which was completely blocked by CsA (Figure 4G). GAPDH mRNA was measured as a control. Together, these morphologic and molecular data demonstrate that the AngII and PE hypertrophic signaling pathways are CsA-/FK 506-sensitive and therefore involve calcineurin activation.
If NF-AT activation mediates Ca2+-dependent hypertrophic signaling, then AngII and PE would be expected to induce NF-AT activity. To test this, we transfected primary rat cardiomyocytes with an NF-AT-dependent luciferase reporter containing three copies of the NF-AT consensus sequence linked to the thymidine kinase minimal promoter. In the presence of AngII (10 nM) or PE (10 μM), reporter gene expression was up-regulated (Figure 4H). This up-regulation was completely abolished in the presence of CsA or FK 506, supporting the conclusion that AngII and PE activate NF-AT through a calcineurin-dependent signal transduction pathway.
Induction of Cardiac Hypertrophy In Vivo by Activated Calcineurin
To determine whether the calcineurin signal transduction pathway could also operate in the myocardium in vivo, we generated transgenic mice that expressed the constitutively active form of the calcineurin catalytic subunit in the heart, using the α-MHC promoter to drive expression. Previous studies have shown that this cardiac-specific promoter is active in the ventricular chambers primarily after birth (Jones et al., 1994). A total of ten independent founder transgenic mice were generated, which contained between 2 and 68 copies of the α-MHC-calcineurin transgene (Table 1).
Table 1.
Summary of α-MHC-Calcineurin Transgenic Lines
| Transgenic Line | Transgene Copy # | Cause of Death | Age at Death | Heart Weight Tg./Control | Cardiac Phenotype |
|---|---|---|---|---|---|
| 46 | 8 | Sacrificed | 18 days | 2.2 | Hypertrophic |
| 22 | 22 | Sudden | 10 weeks | 2.3 | Dilated |
| 110 | 3 | Sudden | 4 weeks | 2.6 | Hypertrophic |
| 106 | 2 | Sudden | 9 weeks | ND | ND |
| 108 | 3 | Still alive | (14 weeks) | — | — |
| 41 | 68 | Still alive | (24 weeks) | — | — |
| 37 | 15 | Still alive | (23 weeks) | — | — |
| 37-1 | 15 | Sacrificed | 5 weeks | 2.3 | Hypertrophic |
| 37-2 | 15 | Sudden | 4 weeks | ND | Hypertrophic |
| 37-3 | 15 | Sudden | 3 weeks | 2.5 | Hypertrophic |
| 37-4 | 15 | Still alive | (8 weeks) | — | — |
| 37-5 | 15 | Still alive | (8 weeks) | — | — |
| 37-6 | 15 | Sudden | 12 weeks | 2.9 | Dilated |
| 39 | 3 | Sudden | 11 weeks | 2.7 | Hypertrophic |
| 39-1 | 3 | Sudden | 3 weeks | ND | Hypertrophic |
| 39-2 | 3 | Sudden | 4 weeks | ND | ND |
| 39-3 | 3 | Still alive | (10 weeks) | — | — |
Heart weights were determined and are expressed as the relative weight of the transgenic heart compared to nontransgenic litter mates. Ages of mice that are still alive are shown in parentheses as of 2/18/98. 37 and 39 were founder transgenics and, mice designated as 37-and 39- were their offspring. ND, not determined.
Every calcineurin transgenic mouse analyzed showed a dramatic increase in heart size relative to nontransgenic littermates. The heart-to-body weight ratio averaged 2- to 3-fold greater in the calcineurin transgenics compared to control littermates, even as early as 18 days postnatally (Figure 5, Table 1). Histological analysis showed concentric hypertrophy wherein the cross sectional areas of the ventricular walls and interventricular septum were dramatically increased (Figure 5C). The left ventricle was most affected, but the right ventricle and the atrial chambers were also enlarged. In contrast to the well-organized, striated musculature of the normal ventricular wall, cardiomyocytes from the calcineurin transgenic hearts were disorganized and obviously hypertrophic (Figures 5D and 5E). The hypertrophic cardiomyocytes often had dramatic karyomegaly. Measurement of cross sectional areas of myocytes within the left ventricular wall showed a greater than 2-fold increase in calcineurin transgenics compared to controls (data not shown).
Figure 5. Hypertrophy of α-MHC-Calcineurin Transgenic Hearts.

(A) Control and α-MHC-calcineurin transgenic littermates were sacrificed at 18 days of age and hearts were removed and photographed. (B and C) Hearts shown in (A) were sectioned longitudinally. The left ventricular wall and septum are extensively hypertrophied in the calcineurin transgenic (C). (D and E) High magnification views of left ventricular walls of hearts shown in (B) and (C), respectively. (F and G) Transverse sections through hearts of control and calcineurin transgenics at 9 weeks of age stained with H and E. The calcineurin transgenic died suddenly and shows hypertrophy and dilatation. (H) Sections from the calcineurin transgenic heart shown in (G) were stained with Masson trichrome to reveal collagen. Note the extensive interstitial collagen deposits surrounding degenerated cardiomyocytes. Ca, coronary artery; la, left atrium; lv, left ventricle; ra, right atrium; rv, right ventricle. Bars in (B), (C), (F), (G), and (H) = 1 μm. Bars in (D) and (E) = 50 μm.
In humans, cardiac hypertrophy frequently progresses to ventricular dilatation, heart failure, and sudden death. Similarly, in calcineurin transgenic mice, we observed dilatation of the ventricular chambers with increasing age (Figures 5F and 5G). Calcineurin transgenic mice were also highly susceptible to sudden death. This occurred spontaneously, as well as during handling or anesthesia. The mice that died from sudden death showed right and left ventricular dilatation indicative of heart failure. Histology of the lungs also revealed extensive perivascular edema and intraalveolar macrophages containing red blood cells (data not shown), findings consistent with heart failure. One of the hallmarks of heart failure is fibrosis of the ventricular wall. The hearts of calcineurin transgenics contained extensive, primarily interstitial, deposits of collagen, as revealed by trichrome staining (Figure 5H). In foci with marked fibrosis, myofiber degeneration was evident.
Activation of the Molecular Response to Hypertrophy In Vivo by Calcineurin
We used a quantitative dot blot assay to examine RNA from hearts of calcineurin transgenic and nontransgenic littermates to determine whether activated calcineurin induced changes in cardiac gene expression characteristic of hypertrophy and heart failure. Consistent with reactivation of the fetal program of gene expression, β-MHC, α-skeletal actin, and BNP transcripts were dramatically up-regulated in transgenic hearts, whereas α-MHC was down-regulated (Figure 6). Transcripts for sarcoplasmic reticulum Ca2+-ATPase (SERCA) and phospholamban (PLB) have been shown previously to be down-regulated during heart failure, as the failing myocardium exhibits defective Ca2+ handling (Schwinger et al., 1995); both transcripts were decreased in calcineurin transgenics. There was no significant change in GAPDH expression.
Figure 6. Changes in Cardiac Gene Expression in α-MHC-Calcineurin Transgenic Mice.

Total RNA was isolated from hearts of control and α-MHC-calcineurin transgenic mice at 6 weeks of age. The indicated transcripts were detected by dot blot analysis, and their levels in transgenic hearts relative to controls are shown.
Induction of Cardiac Hypertrophy In Vivo by Activated NF-AT3
While activation of NF-AT3 proteins is a well-characterized mechanism of action of calcineurin in T cells, and NF-AT was able to synergize with GATA4 and calcineurin to activate the BNP promoter in cultured cardiomyocytes, it was formally possible that the hypertrophic response to calcineurin in vivo could involve a NF-AT-independent mechanism. To determine whether activated NF-AT3 could substitute for all upstream elements in the hypertrophic signaling cascade, we created a constitutively active NF-AT3 mutant by deleting the N-terminal regulatory domain. This mutant, referred to as NF-AT3Δ317, lacked the first 317 amino acids of the protein but retained the Rel homology and transactivation domains (Figure 7A).
Figure 7. Hypertrophy of α-MHC-NF-AT3 Transgenic Hearts.

(A) Structure of NF-AT3 and NF-AT3Δ317 mutant. RHD, Rel homology domain; Reg, regulatory domain. Amino acid positions are indicated.
(B and C) Primary cardiomyocytes were transfected with full-length NF-AT3 and NF-AT3Δ317 mutant cDNAs, respectively, and cells were stained with anti-NF-AT antibody. NF-AT3 is localized to the cytoplasm whereas NF-AT3Δ317 is localized to the nucleus.
(D and E) Hearts from nontransgenic and NF-AT3Δ317 littermates at 4 weeks of age were sectioned longitudinally and stained with H and E. La, left atrium; lv, left ventricle; ra, right atrium; rv, right ventricle. Bars in (D) and (E) = 2 μm.
When NF-AT3Δ317 was expressed in transfected cardiomyocytes, it became constitutively localized to the nucleus, in contrast to the wild-type protein, which required calcineurin signaling for nuclear localization (Figures 7B and 7C). The NF-AT3Δ317 mutant also activated the NF-AT-dependent reporter construct in transient transfection assays (data not shown). We therefore expressed this mutant in the hearts of transgenic mice, under control of the α-MHC promoter. Three independent founder transgenic mice were obtained, and all showed pronounced left and right ventricular concentric hypertrophy (Figures 7D and 7E). Like the calcineurin transgenics, the ventricular walls of the NF-AT3Δ317 transgenics showed extensive fibrosis, with myofiber disarray and cardiomyocyte enlargement. In contrast, expression of wild-type NF-AT3 under control of the α-MHC promoter did not lead to hypertrophy (data not shown). Thus, activated NF-AT3 alone is sufficient to substitute for Ca2+ signals in the heart and evoke a hypertrophic response in vivo.
Prevention of Cardiac Hypertrophy with CsA
To begin to determine whether inhibition of calcineurin activity in vivo might be an effective means of preventing cardiac hypertrophy, we tested whether subcutaneous injection of CsA could prevent cardiac dysfunction in calcineurin transgenic mice. For these experiments, we used eight transgenic littermates from a litter of transgenic mouse #37 (see Table 1). Four transgene-positive offspring were injected twice daily with CsA (25 mg/kg body weight) and four were injected with vehicle alone. Four nontransgenic littermates were also treated with CsA to control for potential toxic effects or cardiac abnormalities induced by CsA. CsA treatment was initiated at 9 days of age and animals were sacrificed 16 days later. As shown in Figure 8, the hearts of vehicle-treated animals were highly hypertrophic and dilated by day 25, whereas those from CsA-treated littermates were not significantly different in size from nontransgenic controls. The mean heart-to-body weight ratios for calcineurin transgenics were nearly 3-fold larger than those of CsA-treated transgenics and nontransgenics. CsA treatment also prevented fibrosis of the hearts of calcineurin transgenics (data not shown).
Figure 8. Prevention of Calcineurin-Dependent Hypertrophy by CsA.
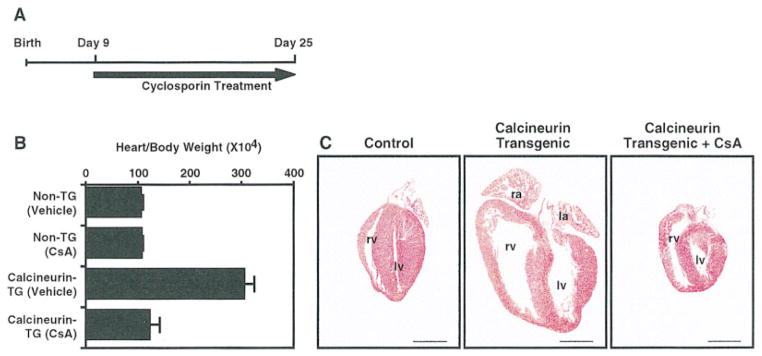
(A) The regimen for CsA treatment is shown.
(B) α-MHC-calcineurin transgenic (TG) and nontransgenic mice were treated with or without CsA (25 mg/kg body weight), as indicated. Heart-to-body weight ratios are expressed ± standard deviations. Transgenic littermates obtained from male calcineurin transgenic #37 (see Table 1) were injected subcutaneously twice daily with CsA or vehicle alone beginning at 9 days of age. At 25 days of age, animals were sacrificed and hearts were removed and sectioned longitudinally.
(C) H and E sections of hearts from nontransgenic (control) and transgenic mice treated with vehicle (center panel) or CsA (right panel). La, left atrium; lv, left ventricle; ra, right atrium; rv, right ventricle. The right atrium was removed from the control, and both atria were removed from the transgenic treated with CsA. Bar = 2 μm.
At a cellular level, the hypertrophic response of cardiomyocytes in the calcineurin transgenics was largely inhibited by CsA, although there were isolated areas of myofiber disarray and scattered cells with prominent hyperchromatic nuclei. Whether these represent cells that were already hypertrophic at the time CsA administration was initiated or whether there are a few cells that escaped the effects of CsA will require further investigation. Nevertheless, CsA treatment prevented gross cardiac hypertrophy and associated pathology in response to activated calcineurin in vivo.
Discussion
Our results define an intracellular pathway for induction of cardiac hypertrophy that links Ca2+ signaling in the cytoplasm with changes in cardiac gene expression. Activation of this hypertrophic pathway either in the cytoplasm or in the nucleus, with activated forms of calcineurin or NF-AT3, respectively, results in molecular and pathophysiologic changes in the hearts of transgenic mice that mimic those associated with cardiac hypertrophy and heart failure in humans.
Calcineurin, a Ca2+-Sensitive Regulator of Hypertrophy
It is well established that elevation in intracellular Ca2+ is associated with the initiation of mechanical or agonist-induced cardiac hypertrophy (Marban et al., 1987; Bustamante et al., 1991; Le Guennec et al., 1991; Saeki et al., 1993; Peurreault et al., 1994; Hongo et al., 1995). Remarkably, however, the possibility that calcineurin might participate in the transduction of hypertrophic signals in cardiomyocytes has not been addressed.
Calcineurin is a ubiquitously expressed serine/threonine phosphatase that exists as a heterodimer, comprised of a 59 kDa calmodulin-binding catalytic A subunit and a 19 kDa Ca2+-binding regulatory B subunit (Stemmer and Klee, 1994; Su et al., 1995). Calcineurin is uniquely suited to mediate the prolonged hypertrophic response of a cardiomyocyte to Ca2+ signaling because the enzyme is activated by a sustained Ca2+ plateau and is insensitive to transient Ca2+ fluxes such as occur in response to cardiomyocyte contraction (Dolmetsch et al., 1997).
Activation of calcineurin is mediated by binding of Ca2+ and calmodulin to the regulatory and catalytic subunits, respectively. Previous studies showed that over-expression of calmodulin in the heart also results in hypertrophy, but the mechanism involved was not determined (Gruver et al., 1993). Our results suggest that calmodulin acts through the calcineurin pathway to induce the hypertrophic response.
In T cells, many of the changes in gene expression in response to calcineurin activation are mediated by members of the NF-AT family of transcription factors, which translocate to the nucleus following dephosphorylation by calcineurin. Three independent observations support the conclusion that NF-AT is also an important mediator of cardiac hypertrophy in response to calcineurin activation. (1) NF-AT activity was induced by treatment of cardiomyocytes with AngII and PE. This induction was blocked by CsA and FK 506, indicating that it is calcineurin-dependent. (2) NF-AT3 synergized with GATA4 to activate the BNP promoter in cardiomyocytes. (3) Expression of activated NF-AT3 in the heart was sufficient to bypass all upstream elements in the hypertrophic signaling pathway and evoke a hypertrophic response.
Combinatorial Interactions between GATA and NF-AT Proteins
Our results demonstrate that the C-terminal portion of the Rel homology domain of NF-AT3 interacts with the second zinc finger of GATA4, as well as with GATA5 and GATA6, which are also expressed in the heart. The crystal structure of the DNA binding region of NF-ATc has revealed that the C-terminal portion of the Rel homology domain projects away from the DNA-binding site and also mediates interaction with AP-1 in immune cells (Wolfe et al., 1997).
What is the significance of the NF-AT3-GATA4 interaction for activation of the hypertrophic response? Cooperative activation of the BNP promoter by NF-AT3 and GATA4 requires NF-AT binding to a target sequence in the BNP upstream region. Previous studies have demonstrated that GATA4 binding sites located near the proximal BNP promoter are also required for activation of the gene (Grepin et al., 1994). Thus, on this specific hypertrophic-responsive gene, and perhaps others, these factors act combinatorially to activate transcription. NF-AT proteins regulate certain T cell genes by binding a composite DNA sequence in conjunction with AP-1 (Wolfe et al., 1997). In the case of the BNP promoter, we have no evidence for this type of joint DNA binding between GATA4 and NF-AT3, since the binding sites for these factors are not immediately adjacent and sites for both factors are required for synergistic activation. Moreover, in DNA binding assays, we have found no evidence for binding of GATA4 and NF-AT3 together to either type of site (data not shown).
A variety of transcription factors have been implicated in cardiac hypertrophy, including TEF-1 (Kariya et al., 1994; Karns et al., 1995), SRF (Sadoshima and Izumo, 1993; Sadoshima et al., 1993), AP-1 (Kovacic-Milivojevic et al., 1996), and GATA4 (Hasegawa et al., 1997; Herzig et al., 1997). In light of the cooperation between NF-AT and AP-1 in the control of T cell gene expression, it is tempting to speculate that a similar mechanism could regulate certain cardiac genes in response to hypertrophy. Whether NF-AT interacts with or regulates the expression of other transcription factors that control cardiac genes warrants further investigation.
Six GATA transcription factors have been identified in vertebrate species, each of which contains a highly conserved DNA-binding domain consisting of two zinc fingers of the motif Cys-X2-Cys-X17-Cys-X2-Cys (reviewed in Evans, 1997). Based on sequence homology and expression patterns, the GATA proteins can be divided into two subfamilies. GATA1/2/3 are expressed in hematopoietic cells, while GATA4/5/6 are expressed primarily in the heart and vascular system, as well as in visceral endodermal derivatives. Given the importance of GATA1/2/3 in hematopoietic cells and the well-documented roles of NF-AT proteins in T cells, it will be of interest to determine whether these two families of transcription factors can interact in these cells.
A Transcriptional Pathway for Cardiac Hypertrophy
Previous studies have demonstrated important roles for Ras, MAP kinase, and PKC signaling pathways in the hypertrophic response. All of these signal transduction pathways are associated with an inotropic increase in intracellular Ca2+ concentration. In T cells, the calcineurin signaling pathway is activated independently of, but is integrated with, the Ras/MAP kinase and PKC pathways. Full induction of IL-2 transcription requires co-stimulation via the calcineurin and Ras pathways, which result in activation of NF-AT and AP-1, respectively, and their convergence on a common downstream target sequence (reviewed in Rao et al., 1997). This type of integrated signaling bears obvious similarities to the mechanisms for induction of cardiac hypertrophy. While our results demonstrate that the calcineurin-NFAT3 signaling pathway is sufficient to induce hypertrophy in vivo, it also seems likely that this pathway and the Ras/MAP kinase pathway may be interdependent in cardiomyocytes, as in immune cells.
CsA and FK 506 bind the immunophilins cyclophilin and FK 506–binding protein (FKBP12), respectively, forming complexes that bind the calcineurin catalytic subunit and inhibit its activity. Our results show that CsA and FK 506 block the ability of cultured cardiomyocytes to undergo hypertrophy in response to AngII and PE. Both of these hypertrophic agonists have been shown to act by elevating intracellular Ca2+, which results in activation of the PKC and MAP kinase signaling pathways (Sadoshima and Izumo, 1993; Sadoshima et al., 1993; Zou et al., 1996; Kudoh et al., 1997; Yamazaki et al., 1997). CsA does not interfere with early signaling events at the cell membrane, such as PI turnover, Ca2+ mobilization, or PKC activation (Emmel et al., 1989). Thus, its ability to abrogate the hypertrophic responses of AngII and PE suggests that calcineurin activation is an essential step in the AngII and PE signal transduction pathways.
Our results are consistent with a molecular pathway for cardiac hypertrophy as shown in Figure 9. According to this model, hypertrophic stimuli such as AngII and PE, which lead to an elevation of intracellular Ca2+, result in activation of calcineurin. NF-AT3 within the cytoplasm is dephosphorylated by calcineurin, enabling it to translocate to the nucleus where it can interact with GATA4. While calcineurin is essential for AngII- and PE-dependent hypertrophy in vitro, and activation of either calcineurin or NF-AT3 is sufficient to evoke the hypertrophic response in vivo, it is also conceivable that calcineurin could act through an NF-AT-independent mechanism to regulate hypertrophy. Indeed, calcineurin could potentially have other substrates in cardiomyocytes that play regulatory roles in Ca2+ handling or transcriptional control. NF-AT3 could also potentially activate some hypertrophic responsive genes, through mechanisms independent of GATA4. This model also predicts that phosphorylation of the N-terminal regulatory sites in NF-AT3 could be a key regulatory step in the hypertrophic signaling pathway. By inference, activation of the NF-AT kinase would be expected to interfere with hypertrophy, whereas its inhibition would lead to hypertrophy.
Figure 9. A Model for the Calcineurin-Dependent Transcriptional Pathway in Cardiac Hypertrophy.

AngII, PE, and possibly other hypertrophic stimuli acting at the cell membrane lead to elevation of intracellular Ca2+ and activation of calcineurin in the cytoplasm. Calcineurin dephosphorylates NF-AT3, resulting in its translocation to the nucleus, where it interacts with GATA4 to synergistically activate transcription. Whether all actions of NF-AT3 are mediated by its interaction with GATA4 or whether there are GATA4-independent pathways for activation of certain hypertrophic responses remains to be determined. Solid arrows denote pathways that are known. Dotted lines denote possible pathways that have not been demonstrated.
These studies also raise the possibility that an NF-AT-dependent mechanism could control normal cardiac growth during development. In this regard, NF-AT3 is expressed in cardiomyocytes within the embryonic heart (J. D. M. and E. N. O., unpublished data).
Calcineurin and NF-AT3 Transgenic Mice as Models for Dilated Cardiomyopathy and Heart Failure in Humans
The results of this study raise the possibility that calcineurin activation may regulate hypertrophy in response to a variety of pathologic stimuli and suggest a sensing mechanism for altered sarcomeric function. Of note, there are several familial hypertrophic cardiomyopathies (FHC) caused by mutations in contractile protein genes, which result in subtle disorganization in the fine crystalline-like structure of the sarcomere (Watkins et al., 1995; Vikstrom and Leinwand, 1996). It is unknown how sarcomeric disorganization is sensed by the cardiomyocyte, but it is apparent that this leads to altered Ca2+ handling (Lin et al., 1996; Botinelli et al., 1997; Palmiter and Solaro, 1997). Calcineurin could represent the sensing molecule that couples altered Ca2+ handling associated with FHC with cardiac hypertrophy and heart failure.
The α-MHC-calcineurin transgenic mice also represent a model in which to investigate the potential reversibility of cardiac hypertrophy and the architectural changes associated with heart failure because the hypertrophic effector molecule can be inhibited pharmacologically. Since these mice ultimately progress to dilated cardiomyopathy, treatment with CsA at these later stages will suggest the degree to which pathological conditions and structural alterations associated with the hypertrophic response are reversible.
These results also raise the question whether CsA or FK506 might be useful in the treatment of cardiac hypertrophy and heart failure in humans. These immunosuppressants are used routinely in transplant patients to prevent tissue rejection, but clinical data correlating CsA treatment with cardiac function in transplant patients are inconclusive (Haverich et al., 1994). However, it has been reported in a study of heart transplant patients that CsA increases cardiac function and left ventricular ejection fraction and results in fewer ischemic episodes (Reid and Yacoub, 1988). Given the remarkable similarity between the molecular and pathophysiological responses of the heart in calcineurin transgenic mice and in human heart failure, these transgenic mice should be a useful model for drug discovery and for identifying additional genes that influence the hypertrophic response.
Experimental Procedures
Two-Hybrid Screens
The GATA4 bait used for the yeast two-hybrid screen contained amino acids 130–409 fused in-frame with the GAL4 DNA-binding domain. This region of GATA4 encompasses the two zinc finger domains and was encoded within a PstI-NsiI fragment, which was cloned into a Pst I site in the pAS yeast expression vector. pAS-GATA4 was cotransformed into yeast with an embryonic 10.5 mouse cDNA library that contained the GAL4 activation domain fused to random cDNAs. From over 5 million primary colonies screened, approximately 100 positive colonies were identified. From each individual colony, the activating plasmid was rescued and the cDNA insert was sequenced. Clones containing cDNA inserts in the anti-sense orientation or out-of-frame were discarded. The remaining clones (approximately 21) were retransformed back into yeast to test for specificity. Three separate criteria were set for determination of specificity. First, the isolated clones had to recapitulate the interaction. Second, the isolated clones could not interact with a nonspecific bait, in this case a GAL4-E12 fusion. Third, we chose to focus initially on factors that could also interact with GATA5, since there is greater than 92% amino acid conservation within the zinc finger domains of GATA4 and GATA5. The NF-AT3 prey clone fulfilled these criteria.
The rescued NF-AT3 cDNA fragment was also subcloned as a XhoI fragment into the SalI site of the mammalian GAL4 fusion plasmid pM1 and tested for activation of the GAL4-dependent reporter. Methods for culturing and transfection of 10T1/2 cells along with the analysis of CAT activity were described previously (Molkentin et al., 1996).
In Vitro Translation and Immunoprecipitation
The partial NF-AT3 cDNA region rescued from the two-hybrid prey plasmid was subcloned as an XhoI fragment into the SalI site of the pECE-Flag mammalian expression vector. To generate a vector suitable for in vitro translation, the NF-AT3 cDNA fragment along with the 5′ Flag epitope was excised from pECE-Flag-NF-AT3 as a NotI-XbaI fragment and cloned into the pCite2B T7 promoter-containing in vitro transcription vector (Invitrogen). This allowed for the generation of a 387 amino acid NF-AT3-Flag fusion protein. SalI-XbaI fragments corresponding to the denoted amino acids in GATA4 were subcloned to generate pCite2A-GATA4 80–441, pCite2A-GATA4 181–441, pCite2A-GATA4 239–441, pCite2A-GATA4 80–328, pCite-GATA4 181–328, and pCite2A 80–441/d265–294. A cDNA fragment encoding amino acids 130–350 of mouse GATA6 was also cloned as a SalI-XbaI fragment into pCite2A. In addition, a T7 promoter-directed construct encoding the entire Rel homology domain of the human NF-AT3 protein (amino acids 404–694) was used in these studies (gift from Dr. T. Hoey) (Hoey et al., 1995).
Coupled in vitro transcription and translation from the T7 promoter was performed in the presence of 35S-methionine according to the TNT kit protocol (Promega, Madison, WI). Immunoprecipitations were directed against the Flag epitope using Flag antibody (Kodak IBI, New Haven, CT) (Figures 2A and 2B) or against the Rel homology domain of NF-AT3 (antibody described in Lyakh et al., 1997) (Figure 2D). In vitro transcription-translation was performed in a reaction volume of 25 μl with 0.5 μg of each construct. Five microliters of this reaction mix was immunoprecipitated according to the manufacturer’s recommended conditions (Kodak IBI, New Haven, CT) in a total volume of 100 μl with 2 μl of anti-FLAG monoclonal antibody, or 5 μl of NF-AT3-specific antibody together with 25 μl of protein-A/G agarose. The precipitated products were analyzed by SDS-PAGE and autoradiography.
Preparation of Primary Rat Cardiomyocytes
Cardiomyocyte cultures were prepared by dissociation of 1-day-old neonatal rat hearts and were differentially plated to remove fibroblasts. To induce the hypertrophic response, AngII and PE were added to cardiomyocyte cultures at 10 nM and 10 μM, respectively, in serum-free M199 media. The culture media containing either agonist was changed every 12 hr for a period of 72 hr. CsA and FK 506 were present at 500 ng/ml and 150 ng/ml, respectively, over the entire 72 hr culturing period. To analyze effects of these agents on NF-AT3 activity, an NF-AT-dependent reporter was transfected into cardiomyocytes by Ca2+ phosphate transfection in M199 serum-free media. Cardiomyocytes were then cultured for 72 hr with the identified agent. The NF-AT-dependent reporter contained three NF-AT binding sites from the IL-2 promoter cloned upstream of the thymidine kinase minimal promoter and the luciferase gene (gift from T. Hoey). Methods of preparation for cellular extracts and luciferase assays have been described (Molkentin et al., 1994).
Gel Mobility Shift Assays and Mutagenesis
To identify potential NF-AT binding sites within the BNP promoter, gel mobility shift assays were performed with double-stranded oligonucleotides corresponding to putative sites located at −927, −327, and −27, relative to the transcription start site (Owaga et al., 1995), or the consensus site from the IL-2 promoter. Sequences of probes were as follows: BNP-927: 5′-CTATCCTTTTGTTTTCCATC CTG-3′; BNP-327: 5′-TCCCTGCCTTTTCCAGCAACGGT-3′; BNP-27: 5′-GCTCCAGGATAAAAGGCCACGGT-3′; IL-2: 5′-TACATTGGAAAA TTTTATTACAC-3′. For gel mobility shift assays utilizing the NF-AT3 Rel homology domain, 2 μl of a coupled in vitro transcription-translation reaction (TNT Kit, Promega, Madison, WI) was incubated with the indicated oligonucleotide probe (40,000 cpm of a 32P-labeled probe per reaction) in the presence of 1 μg of poly (dI-dC) for 20 min at room temperature, followed by nondenaturing electrophoresis. Unlabeled competitor oligonucleotides were added at a 100-fold molar excess, and 2 μl of NF-AT3-specific antiserum (Gift from N. Rice; Lyakh et al., 1997) was added for the supershift experiments. The gel mobility shift buffers and electrophoresis conditions are described elsewhere (Molkentin et al., 1994). Site-directed mutations were introduced into the 1800 bp BNP promoter (Ogawa et al., 1995) by rolling-circle polymerase chain reaction as described (Molkentin et al., 1994).
Immunocytochemistry
To visualize sarcomeric organization in primary cardiomyocytes, anti-α-actinin mouse monoclonal antibody was used (Sigma). Cells were washed in PBS, fixed in 3.7% paraformaldehyde for 5 min, washed three times with PBS, and then preblocked in PBS containing 2% horse serum, 2% BSA, and 0.1% NP40 for 30 min. Anti-α-actinin antibody was added at a dilution of 1:800 in fresh preblock solution and incubated for an additional 30 min. Alternatively, cells were incubated with anti-NF-AT3 polyclonal antiserum at a dilution of 1:400 (Lyakh et al., 1997). Subsequently, cells were washed three times in PBS with 0.1% NP40. Anti-mouse TRITC-conjugated secondary antibody was then added at a dilution of 1:400 for 30 min in preblock solution, and the cells were again washed three times in PBS containing 0.1% NP40. Nuclear staining for DNA was performed with 0.5 μg/ml of bis-benzimide in PBS for 15 min followed by three rinses with PBS.
Transgenic Mice
Transgenic mice expressing calcineurin and NF-AT3 in the heart were created as follows. A cDNA encoding a constitutively active form of the calcineurin A catalytic subunit (O’Keefe et al., 1992) was cloned by PCR with a 5′ SalI linker and 3′ HindIII linker into an expression vector containing the α-MHC promoter. The expression pattern and characteristics of this expression vector have been described (Jones et al., 1994). To generate transgenic mice expressing a constitutively nuclear form of the NF-AT3 protein in the heart, PCR primers were generated to allow specific amplification of a region of sequence encoding amino acids 317–902 of the human NF-AT3 protein, referred to as NF-AT3Δ317. XhoI linkers on the ends of these primers allowed cloning into the SalI site of the α-MHC expression vector. Both the calcineurin and NF-AT3Δ317-α-MHC vectors were digested with NotI, and the α-MHC-fusion cDNA fragment was purified and eluted in oocyte injection buffer (5 mM Tris-HCl [pH 7.4] and 0.2 mM EDTA). DNA was then injected into fertilized oocytes derived from FVB mice, and oocytes were transferred into the oviducts of pseudopregnant ICR mice.
RNA Analysis
Total RNA was collected and purified with Triazol reagent (GIBCO BRL) as recommended. RNA from wild-type and transgenic hearts, as well as from cultured cardiomyocytes, was subjected to dot blot hybridization against a panel of oligonucleotide probes as described previously (Jones et al., 1996).
Histology
Hearts from wild-type and transgenic mice were subjected to histological analysis. In brief, hearts were collected, fixed overnight in 10% formalin buffered with PBS, dehydrated in ethanol, transferred to xlyene, then into paraffin. Paraffin-embedded hearts were sectioned at 4 μm and subsequently stained with hematoxylin and eosin for routine histologic examination or with Masson trichrome for collagen (Woods and Ellis, 1994).
Acknowledgments
This work was supported by grants from NIH to E. N. O. and from the American Heart Association to S. R. G. J. D. M. was supported by an NIH postdoctoral fellowship at UT Southwestern. We are grateful to the following individuals for reagents: T. Hoey, Y. Ogawa, S. O’Keefe, and N. Rice. We also thank Chuanzhen Wu and Brian Mercer for generating transgenic mice, A. Tizenor for assistance with graphics, Robert Webb for histology, and our colleagues in the Olson lab for advice and encouragement.
References
- Botinelli R, Coviello DA, Redwood CS, Pellegrino MA, Maron BJ, Spirito P, Watkins H, Reggiani C. A mutant tropomyosin that causes hypertrophic cardiomyopathy is expressed in vivo and associated with an increased calcium sensitivity. Circ Res. 1997;82:106–115. doi: 10.1161/01.res.82.1.106. [DOI] [PubMed] [Google Scholar]
- Bustamante JO, Ruknudin A, Sachs F. Stretch-activated channels in heart cells: relevance to cardiac hypertrophy. J Cardiovasc Pharmacol. 1991;17:S110–S113. doi: 10.1097/00005344-199117002-00024. [DOI] [PubMed] [Google Scholar]
- Chien KR, Zhu H, Knowlton KU, Miller-Hance W, van Bilsen M, Obrein TX, Evans SM. Transcriptional regulation during cardiac growth and development. Annu Rev Physiol. 1993;55:77–95. doi: 10.1146/annurev.ph.55.030193.000453. [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature. 1997;386:855–858. doi: 10.1038/386855a0. [DOI] [PubMed] [Google Scholar]
- Emmel EA, Verweij CL, Durand DB, Higgins KM, Lacy E, Crabtree GR. Cyclosporin A specifically inhibits function of nuclear proteins involved in T cell activation. Science. 1989;246:1617–1620. doi: 10.1126/science.2595372. [DOI] [PubMed] [Google Scholar]
- Evans T. Regulation of cardiac gene expression by GATA4/5/6. Trends Cardiovasc Med. 1997;7:75–83. doi: 10.1016/S1050-1738(97)00010-8. [DOI] [PubMed] [Google Scholar]
- Flanagan WM, Corthesy B, Bram RJ, Crabtree GR. Nuclear association of a T-cell transcription factor blocked by FK-506 and cyclosporin A. Nature. 1991;352:803–807. doi: 10.1038/352803a0. [DOI] [PubMed] [Google Scholar]
- Force T, Pombo CM, Avruch JA, Bonventre JV, Kyriakis JM. Stress-activated protein kinases in cardiovascular disease. Circ Res. 1996;78:947–953. doi: 10.1161/01.res.78.6.947. [DOI] [PubMed] [Google Scholar]
- Grepin C, Dagnino LL, Robitaille L, Haberstroh LL, Antakly T, Nemer M. A hormone-encoding gene identifies a pathway for cardiac but not skeletal muscle gene transcription. Mol Cell Biol. 1994;14:3115–3129. doi: 10.1128/mcb.14.5.3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruver CL, DeMayo F, Goldstein MA, Means AR. Targeted developmental overexpression of calmodulin induces proliferative and hypertrophic growth of cardiomyocytes in transgenic mice. Endocrinology. 1993;133:376–388. doi: 10.1210/endo.133.1.8319584. [DOI] [PubMed] [Google Scholar]
- Hasegawa K, Lee SJ, Jobe SM, Markham BE, Kitsis RN. Cis-acting sequences that mediate induction of the α-myosin heavy chain gene expression during left ventricular hypertrophy due to aortic constriction. Circulation. 1997;96:3943–3953. doi: 10.1161/01.cir.96.11.3943. [DOI] [PubMed] [Google Scholar]
- Haverich A, Costard-Jackle A, Cremer J, Herrmann G, Simon R. Cyclosporin A and transplant coronary disease after heart transplantation: facts and fiction. Transplant Proc. 1994;26:2713–2715. [PubMed] [Google Scholar]
- Herzig TC, Jobe SM, Aoki H, Molkentin JD, Cowley AW, Izumo S, Markham BE. Angiotensin II type 1a receptor gene expression in the heart: AP-1 and GATA-4 mediate the response to pressure overload. Proc Natl Acad Sci USA. 1997;94:7543–7548. doi: 10.1073/pnas.94.14.7543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho SN, Thomas DJ, Timmerman LA, Li X, Francke U, Crabtree GR. NF-ATc3, a lymphoid-specific NF-AT3 family member that is calcium-regulated and exhibits distinct DNA binding specificity. J Biol Chem. 1995;270:19898–19907. doi: 10.1074/jbc.270.34.19898. [DOI] [PubMed] [Google Scholar]
- Hoey T, Sun YL, Williamson K, Xu X. Isolation of two new members of the NF-AT gene family and functional characterization of the NF-AT proteins. Immunity. 1995;2:461–472. doi: 10.1016/1074-7613(95)90027-6. [DOI] [PubMed] [Google Scholar]
- Hongo K, White E, Gannier F, Argibay JA, Garnier D. Orchard CH effect of stretch on contraction and the Ca2+ transient ferret ventricular muscles during hypoxia and acidosis. Am J Physiol. 1995;269:C690–C697. doi: 10.1152/ajpcell.1995.269.3.C690. [DOI] [PubMed] [Google Scholar]
- Jones WK, Sanchez A, Robbins J. Murine pulmonary myocardium: developmental analysis of cardiac gene expression. Dev Dyn. 1994;200:117–128. doi: 10.1002/aja.1002000204. [DOI] [PubMed] [Google Scholar]
- Jones WK, Grupp IL, Doetschman T, Grupp G, Osinska H, Hewett TE, Boivin G, Gulick J, Ng WA, Robbins J. Ablation of the murine α myosin heavy chain gene leads to dosage effects and functional deficits in the heart. J Clin Invest. 1996;98:1906–1917. doi: 10.1172/JCI118992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariya K, Karns LR, Simpson PC. An enhancer core element mediates stimulation of the rat α-myosin heavy chain promoter by an α1-adrenergic agonist and activated protein kinase C in hypertrophy of cardiac myocytes. J Biol Chem. 1994;269:3775–3782. [PubMed] [Google Scholar]
- Karliner JS, Kariya T, Simpson PC. Effects of pertussis toxin on α1-agonist-mediated phosphatidylinositide turnover and myocardial cell hypertrophy in neonatal rat myocytes. Experientia. 1990;46:81–84. doi: 10.1007/BF01955423. [DOI] [PubMed] [Google Scholar]
- Karns LR, Kariya K, Simpson PC. M-CAT, CArG, and Sp1 elements are required for α1-adrenergic induction of the skeletal α-actin promoter during cardiac myocyte hypertrophy. J Biol Chem. 1995;270:410–417. doi: 10.1074/jbc.270.1.410. [DOI] [PubMed] [Google Scholar]
- Komuro I, Yazaki Y. Control of cardiac gene expression by mechanical stress. Annu Rev Physiol. 1993;55:55–75. doi: 10.1146/annurev.ph.55.030193.000415. [DOI] [PubMed] [Google Scholar]
- Kudoh S, Komuro I, Mizuno T, Yamazaki T, Zou Y, Shiojima I, Takekoshi N, Yazaki Y. Angiotensin II stimulates c-jun NH2-terminal kinase in cultured cardiac myocytes of neonatal rats. Circ Res. 1997;80:139–146. doi: 10.1161/01.res.80.1.139. [DOI] [PubMed] [Google Scholar]
- Kovacic-Milivojevic B, Wong VSH, Gardner DG. Selective regulation of the atrial natriuretic peptide gene by individual components of the activator protein-1 complex. Endocrinology. 1996;137:1108–1117. doi: 10.1210/endo.137.3.8603581. [DOI] [PubMed] [Google Scholar]
- Le Guennec JY, White E, Gannier F, Argibay JA, Garnier D. Stretch-induced increase of resting intracellular calcium concentration in single guinea-pig ventricular myocytes. Exp Physiol. 1991;6:975–978. doi: 10.1113/expphysiol.1991.sp003560. [DOI] [PubMed] [Google Scholar]
- Leite MF, Page E, Ambler SK. Regulation of ANP secretion by endothelin-1 in cultured atrial myocytes: desensitization and receptor subtype. Am J Physiol. 1994;267:H2193–H2203. doi: 10.1152/ajpheart.1994.267.6.H2193. [DOI] [PubMed] [Google Scholar]
- Lin D, Bobkova A, Homsher E, Tobacman LS. Altered cardiac troponin T in vitro function in the presence of a mutation implicated in familial hypertrophic cardiomyopathy. J Clin Invest. 1996;97:2842–2848. doi: 10.1172/JCI118740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh C, Carew JA, Kim J, Hogan PG, Rao A. T-cell receptor stimulation elicits an early phase of activation and a later phase of deactivation of the transcription factor NFAT1. Mol Cell Biol. 1996a;16:3945–3954. doi: 10.1128/mcb.16.7.3945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh C, Shaw KTY, Carew J, Viola JPB, Lou C, Perrino BA, Rao A. Calcineurin binds the transcription factor NFAT1 and reversibly regulates its activity. J Biol Chem. 1996b;271:10884–10891. doi: 10.1074/jbc.271.18.10884. [DOI] [PubMed] [Google Scholar]
- Lyakh L, Ghosh P, Rice NR. Expression of NF-AT-family proteins in normal human T cells. Mol Cell Biol. 1997;17:2475–2482. doi: 10.1128/mcb.17.5.2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marban E, Kitakaze M, Kusuoka H, Porterfield JK, Yue DT, Chacko VP. Intracellular free calcium concentrations measured with 19F NMR spectroscopy in intact ferret hearts. Proc Natl Acad Sci USA. 1987;84:6005–6009. doi: 10.1073/pnas.84.16.6005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda ES, Naito Y, Tokumitsu H, Campbell D, Saito F, Hannum C, Arai KI, Arai N. NF-ATx, a novel member of the nuclear factor of activated T cells family that is expressed predominantly in the thymus. Mol Cell Biol. 1995;15:2697–2706. doi: 10.1128/mcb.15.5.2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaffrey PG, Luo C, Kerppola TK, Jain J, Badalian TM, Ho AM, Burgeon E, Lane WS, Lambert JN, Curran T, et al. Isolation of the cyclosporin-sensitive T cell transcription factor NF-ATp. Science. 1993;262:750–754. doi: 10.1126/science.8235597. [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Olson EN. GATA4: a novel transcriptional regulator of cardiac hypertrophy? Circulation. 1997;96:3833–3835. [PubMed] [Google Scholar]
- Molkentin JD, Kalvakalanou D, Markham BE. GATA-4 positively regulates expression of the α-cardiac myosin heavy chain gene in vivo. Mol Cell Biol. 1994;14:4947–4957. doi: 10.1128/mcb.14.7.4947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molkentin JD, Black BL, Martin JF, Olson EN. Mutational analysis of the DNA binding, dimerization, and transcriptional activation domains of MEF2C. Mol Cell Biol. 1996;16:2627–2536. doi: 10.1128/mcb.16.6.2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northrop JP, Ho SN, Chen L, Thomas DJ, Timmerman LA, Nolan GP, Admon A, Crabtree GR. NF-AT components define a family of transcription factors targeted in T-cell activation. Nature. 1994;369:497–502. doi: 10.1038/369497a0. [DOI] [PubMed] [Google Scholar]
- Ogawa Y, Itoh H, Nakagawa O, Shirakami G, Tamura N, Yoshimasa T, Nagata K, Yoshida N, Nakao K. Characterization of the 5′-flanking region and chromosomal assignment of the human brain natriuretic peptide gene. J Mol Med. 1995;73:457–463. doi: 10.1007/BF00202264. [DOI] [PubMed] [Google Scholar]
- O’Keefe SJ, Tamura J, Kincaid RL, Tocci MJ, O’Neill EA. FK506- and CsA-sensitive activation of the interleukin-2 promoter by calcineurin. Nature. 1992;357:692–694. doi: 10.1038/357692a0. [DOI] [PubMed] [Google Scholar]
- Palmiter KA, Solaro RJ. Molecular mechanisms regulating the myofilament response to Ca2+: implications of mutations causal for familial hypertrophic cardiomyopathy. Basic Res Cardiol. 1997;92:63–74. doi: 10.1007/BF00794070. [DOI] [PubMed] [Google Scholar]
- Park J, Takeuchi A, Sharma S. Characterization of a new isoform of the NF-AT (nuclear family of activated T cells) gene family member NF-ATc. J Biol Chem. 1996;271:20914–20921. doi: 10.1074/jbc.271.34.20914. [DOI] [PubMed] [Google Scholar]
- Perreault CL, Shannon RP, Shen YT, Vatner SF, Morgan JP. Excitation-contraction coupling in isolated myocardium from dogs with compensated left ventricular hypertrophy. Am J Physiol. 1994;266:H2436–H2442. doi: 10.1152/ajpheart.1994.266.6.H2436. [DOI] [PubMed] [Google Scholar]
- Rao A, Luo C, Hogan PG. Transcription factors of the NF-AT family: regulation and function. Annu Rev Immunol. 1997;15:707–747. doi: 10.1146/annurev.immunol.15.1.707. [DOI] [PubMed] [Google Scholar]
- Reid CJ, Yacoub MH. Determinants of left ventricular function one year after cardiac transplantation. Br Heart J. 1988;59:397–402. doi: 10.1136/hrt.59.4.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney JW, Hodge MR, McCaffrey PG, Rao A, Glimcher LG. A common factor regulates both Th1- and The-specific cytokine gene expression. EMBO J. 1994;13:625–633. doi: 10.1002/j.1460-2075.1994.tb06300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadoshima J, Izumo S. Signal transduction pathways of angiotensin II-induced c-fos gene expression in cardiac myocytes in vitro. Circ Res. 1993;73:424–438. doi: 10.1161/01.res.73.3.424. [DOI] [PubMed] [Google Scholar]
- Sadoshima J, Izumo S. The cellular and molecular response of cardiac myocytes to mechanical stress. Annu Rev Physiol. 1997;59:551–571. doi: 10.1146/annurev.physiol.59.1.551. [DOI] [PubMed] [Google Scholar]
- Sadoshima J, Xu Y, Slayter HS, Izumo S. Autocrine release of angiotensin II mediates stretch-induced hypertrophy of cardiac myocytes in vitro. Cell. 1993;75:977–984. doi: 10.1016/0092-8674(93)90541-w. [DOI] [PubMed] [Google Scholar]
- Saeki Y, Kurihara S, Hongo K, Tanaka E. Tension and intracellular calcium transient of activated ferret ventricular muscle in response to step length changes. Adv Exp Med Biol. 1993;332:639–647. doi: 10.1007/978-1-4615-2872-2_57. [DOI] [PubMed] [Google Scholar]
- Schwinger RH, Bohm M, Schmidt U, Karczewski P, Bevendick U, Flesch M, Krause EG, Erdmann E. Unchanged protein levels of SERCA II and phospholamban but reduced Ca2+-ATPase activity of cardiac sarcoplasmic reticulum from dilated cardiomyopathy patients compared with patients with nonfailing hearts. Circulation. 1995;92:3220–3228. doi: 10.1161/01.cir.92.11.3220. [DOI] [PubMed] [Google Scholar]
- Shaw KTY, Ho AM, Raghavan A, Kim J, Jian J, Park J, Sharma S, Rao A, Hogan PG. Immunosuppressive drugs prevent a rapid dephosphorylation of transcription factor NFAT1 in stimulated immune cells. Proc Natl Acad Sci USA. 1995;92:11205–11209. doi: 10.1073/pnas.92.24.11205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemmer PM, Klee CB. Dual calcium ion regulation of calcineurin by calmodulin and calcineurin B. Biochemistry. 1994;33:6859–6866. doi: 10.1021/bi00188a015. [DOI] [PubMed] [Google Scholar]
- Su Q, Zhao M, Weber E, Eugster HP, Ryffel B. Distribution and activity of calcineurin in rat tissues. Evidence for post-transcriptional regulation of testis-specific calcineurin B. Eur J Biochem. 1995;230:469–474. doi: 10.1111/j.1432-1033.1995.0469h.x. [DOI] [PubMed] [Google Scholar]
- Thorburn A, Thorburn J, Chen SC, Powers S, Shubeita HE, Feramisco JR, Chien KR. HRas-dependent pathways can activate morphological and genetic markers of cardiac muscle cell hypertrophy. J Biol Chem. 1993;268:2244–2249. [PubMed] [Google Scholar]
- Vikstrom KL, Leinwand LA. Contractile protein mutations and heart disease. Curr Opin Cell Biol. 1996;8:97–105. doi: 10.1016/s0955-0674(96)80053-6. [DOI] [PubMed] [Google Scholar]
- Watkins H, Seidman JG, Seidman CE. Familial hypertrophic cardiomyopathy: a genetic model of cardiac hypertrophy. Hum Mol Genet. 1995;4:1721–1727. doi: 10.1093/hmg/4.suppl_1.1721. [DOI] [PubMed] [Google Scholar]
- Wolfe SA, Zhou P, Dotsch V, Chen L, You A, Ho SN, Crabtree GR, Wagner G, Verdine GL. Unusual Rel-like architecture in the DNA-binding domain of the transcription factor NFATc. Nature. 1997;385:172–176. doi: 10.1038/385172a0. [DOI] [PubMed] [Google Scholar]
- Woods AE, Ellis RC. Laboratory Histopathology: A Complete Reference. New York: Churchill Livingstone Publishers; 1994. pp. 7.1–13. [Google Scholar]
- Yamazaki T, Komuro I, Kudoh S, Zou Y, Shiojima I, Hiro Y, Mizuno T, Maemura K, Kurihara H, Aikawa R, Takano H, Yazaki Y. Endothelin-1 is involved in mechanical stress-induced cardiomyocyte hypertrophy. J Biol Chem. 1996;271:3221–3228. doi: 10.1074/jbc.271.6.3221. [DOI] [PubMed] [Google Scholar]
- Yamazaki T, Komuro I, Zou Y, Kudoh S, Shiojima I, Hiroi Y, Mizuno T, Aikawa R, Takano H, Yazaki Y. Norepinephrine induces the raf-1 kinase/mitogen-activated protein kinase cascade through both α1- and β-adrenoceptors. Circulation. 1997;95:1260–1268. doi: 10.1161/01.cir.95.5.1260. [DOI] [PubMed] [Google Scholar]
- Zou Y, Komuro I, Yamazaki T, Aikawa R, Kudoh S, Shiojima I, Hiroi Y, Mizuno T, Yazaki Y. Protein kinase C, but not tyrosine kinases or ras, plays a critical role in angiotensin II–induced activation of Raf-1 kinase and extracellular signal-regulated protein kinases in cardiac myocytes. J Biol Chem. 1996;271:33592–33597. doi: 10.1074/jbc.271.52.33592. [DOI] [PubMed] [Google Scholar]