

Summary
Segmented negative-strand RNA virus (sNSV) polymerases transcribe and replicate the viral RNA (vRNA) within a ribonucleoprotein particle (RNP). We present cryo-EM and X-ray structures of, respectively, apo- and vRNA bound La Crosse orthobunyavirus (LACV) polymerase that give atomic-resolution insight into how such RNPs perform RNA synthesis. The complementary 3′ and 5′ vRNA extremities are sequence specifically bound in separate sites on the polymerase. The 5′ end binds as a stem-loop, allosterically structuring functionally important polymerase active site loops. Identification of distinct template and product exit tunnels allows proposal of a detailed model for template-directed replication with minimal disruption to the circularised RNP. The similar overall architecture and vRNA binding of monomeric LACV to heterotrimeric influenza polymerase, despite high sequence divergence, suggests that all sNSV polymerases have a common evolutionary origin and mechanism of RNA synthesis. These results will aid development of replication inhibitors of diverse, serious human pathogenic viruses.
Graphical Abstract
Highlights
-
•
Bunyavirus polymerase binds its complementary 3′ and 5′ vRNA ends in distinct sites
-
•
5′ vRNA binding allosterically structures and activates the polymerase catalytic site
-
•
Distinct template/product exit tunnels explain RNA synthesis in a circularized RNP
-
•
Monomeric bunyavirus and trimeric influenza polymerases are structurally similar
The structure of the monomeric bunyavirus polymerase reveals that divergent segmented negative-strand RNA virus polymerases have a common overall architecture, explains how viral RNA binding allosterically regulates polymerase activity, and suggests a replication model that could apply to all related RNA viruses.
Introduction
Arenaviridae, Bunyaviridae, and Orthomyxoviridae, the principal families of segmented negative single-stranded RNA viruses (sNSV), each include serious human pathogens such as Lassa fever, Crimean-Congo haemorrhagic fever, and influenza viruses, respectively. Orthomyxoviruses have six to eight genome segments, whereas Bunyaviridae (reviewed in Elliott, 2014) have three, and Arenaviridae two. For each segment, transcription, generating capped viral mRNAs, and replication, generating full-length genome or antigenome copies (vRNA and cRNA, respectively), are performed by the same virally encoded RNA-dependent RNA polymerase (RdRp). For arena- and bunyaviruses, which replicate in the cytoplasm, the RdRp is the single-chain L protein, whereas for orthomyxoviruses, which replicate in the nucleus, it is a hetero-trimeric complex, formed by the PA, PB1, and PB2 subunits (Fodor, 2013; Guu et al., 2012; Morin et al., 2013; te Velthuis, 2014). vRNA genome segments are always packaged by multiple copies of the viral nucleoprotein (NP) together with one copy of the RdRp into filamentous ribonucleoprotein particles (RNPs), which are the functional replication and transcription units (Reguera et al., 2014).
sNSV polymerases have two unique features. First, they perform transcription by the “cap-snatching” mechanism, whereby short 5′ capped RNA fragments are cleaved from host cell mRNA by an endonuclease intrinsic to the RdRp and then used to prime synthesis of viral mRNAs (Morin et al., 2010; Plotch et al., 1981; Reguera et al., 2010; Reich et al., 2014). Second, they recognize each genome segment via their highly conserved, quasi-complementary 3′ and 5′ extremities (over a length of 13–19 nucleotides), known as the promoter (Barr and Wertz, 2004). Correlated with this, sNSV RNPs are generally circularized, which is thought to occur by base pairing between the genome ends (forming a double-stranded “panhandle”) and/or the simultaneous binding of both ends to the polymerase (Reguera et al., 2014). vRNA promoter binding to influenza polymerase was visualized recently for the first time in a co-crystal structure. This revealed that each vRNA extremity binds sequence specifically as a single strand to distinct sites on the polymerase but then come together to form a short duplex of about four base pairs (Pflug et al., 2014). Furthermore, this mode of promoter binding is required for activation of diverse influenza polymerase functions (Fodor, 2013). For bunyaviruses, exact self-complementarity of the genome ends extends for 15–19 nts (except for only one G-U mismatch in the case of orthobunyaviruses [Barr and Wertz, 2004; Kohl et al., 2004]), potentially allowing formation of a much more stable panhandle than for influenza vRNA. However, the exact nature of the vRNA-vRNA and vRNA-L interactions that circularize bunyavirus RNPs are not known. Cross linking suggests that the vRNA ends within bunyavirus RNPs are base paired at least partially (Raju and Kolakofsky, 1989) and a distal duplex region is essential for RNA synthesis by bunyavirus (Barr and Wertz, 2004; Kohl et al., 2004) and arenavirus (Kranzusch et al., 2010) polymerases. The absence of significant sequence similarity, outside of the cap-snatching endonuclease (Reguera et al., 2010) and the conserved RdRp motifs (Müller et al., 1994), between Arenaviridae and Bunyaviridae L proteins and Orthomyxoviridae heteotrimeric polymerases also poses the question as to whether all sNSV have a structurally and evolutionary conserved architecture to match their functional similarity.
To answer this question and those related to promoter binding, we determined the crystal structure of 77% of the L protein from La Crosse orthobunyavirus (LACV) in complex with just the 3′ or both 3′ and 5′ conserved genomic RNA ends and the cryo-EM structure of the apo-form. LACV is a potentially serious but rare, mosquito-transmitted human pathogen that causes 50–100 cases of encephalitis per year in the USA (http://www.cdc.gov/lac/) (Elliott, 2014; Haddow and Odoi, 2009). The structure reveals high similarity, but also interesting differences, to the equivalent influenza complex (Pflug et al., 2014; Reich et al., 2014). In particular, it shows the structural basis of the specific recognition of the vRNA 3′ end, the allosteric regulation mediated by vRNA 5′ end binding, and the likely path of the template into the polymerase active site cavity and out again. These findings, combined with those gained from the influenza polymerase structures, provide new insight into the common mechanism of action, the conserved features, and the diversity among sNSV polymerases.
Results
Structure Determination of the LACV L1750 Protein
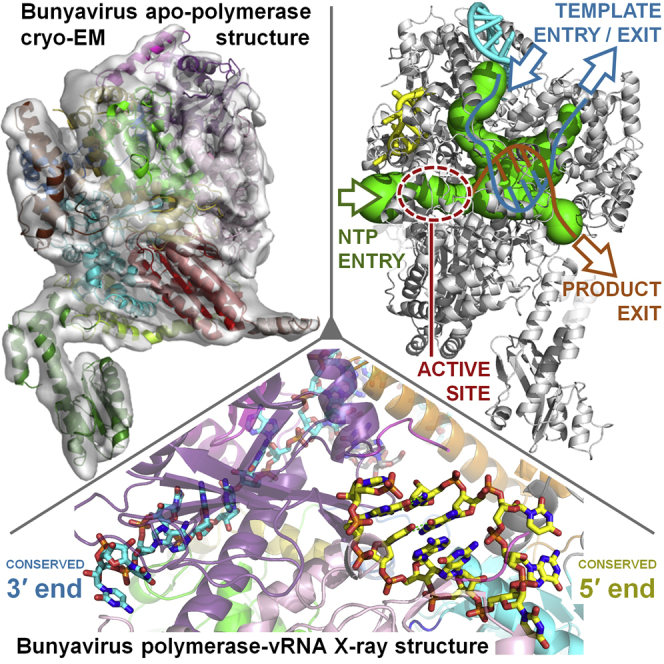
L1750, a construct comprising residues 1–1750 (out of 2263) of the LACV L protein, was expressed in insect cells and purified to homogeneity in milligram amounts (Figure S1A, Figures S2A and S2B). In vitro RNA-protein interaction experiments show that the separated single stranded LACV genomic extremities each bind with high affinity and specificity to the polymerase with dissociation constants of 13.8 ± 2.6 nM and 9.3 ± 1.6 nM for the 5′ and the 3′ ends, respectively, whereas the polymerase only has low affinity (∼1.5 μM) for the pre-annealed double stranded panhandle (Figure S3A). However, as shown by mobility shift assays, the polymerase still binds with high affinity to partially double stranded 3′ vRNA provided the first eight nucleotides from the 3′ end are single stranded (Figure S3B). Similar conclusions were previously found for Machupo arenavirus polymerase (Kranzusch et al., 2010). Using these results, co-crystals of L1750 diffracting up to 2.6 Å resolution were obtained with nucleotides 1–16 of the genomic 3′ vRNA (3′OH-UCAUCACAUGAUGGUU), to which was annealed a complementary 8-mer (5′OH-GCUACCAA), corresponding to nucleotides 9–16 from the 5′ vRNA extremity. The structure was solved by multiple isomorphous replacement with anomalous scattering (Figures S1B–S1D). Soaking the first 10 nucleotides of the 5′ vRNA (5′p-AGUAGUGUGC) into the crystals gave a new structure at 3.0 Å resolution that revealed the 5′ end in a distinct binding site. Crystallographic statistics are given in Table S1 and a sequence alignment of representative orthobunyavirus polymerases, annotated with the secondary structure, is shown in Data S1. The L1750 model contains 1652 residues (94.4% complete) with several connecting loops missing, some of which become ordered upon 5′ vRNA binding (Figure 1, Data S1). The structure of apo-L1750 was determined from cryo-EM images by single particle 3D reconstruction at 8.3 Å resolution and allows visualization of secondary structure elements in most of the protein (Figure 2, Figure S2).
Figure S1.

Purification and X-Ray Structure Solution, Related to Figure 1
(A) SDS PAGE of purified L1750 after a S200 gel filtration run.
(B) Scheme for X-ray crystal structure solution by the MIRAS methods using tantalum cluster, platinum, and selenomethionine derivatives.
(C) X-ray energy scan of selenomethionine derivative at Se absorption edge.
(D) X-ray energy scan of tantalum derivative at Ta absorption edge.
(E) Experimentally phased map at 2.85 Å resolution contoured at 1 σ (cyan mesh) with superposed final model showing a number of selenomethionines (anomalous difference peaks contoured at 4.0 σ, orange mesh) and one tantalum cluster position (anomalous difference peaks contoured at 4.0 σ, green mesh).
Figure S2.

Electron Microscopy of Apo-L1750, Related to Figure 2
(A) Negative stain EM micrograph of purified L1750. Scale bar is indicated.
(B) Representative cryo-EM micrograph collected at 80 kV on a Krios microscope using a Falcon II direct detector (defocus of the chosen micrograph: 1.4 μm).
(C) Comparison between ab initio 2D class averages (top) and reprojections of the 3D reconstruction (bottom), showing the reliability of the 3D reconstruction. Below each comparison, the corresponding view of the 3D reconstruction with the fitted pseudo-atomic model is displayed.
(D) Fourier Shell Correlation (FSC) based on the gold standard FSC = 0.143 criterion shows a resolution of 8.3 Å.
(E) Separated helices are clearly visible inside the map, consistent with the 8.3 Å resolution identified by the FSC.
(F) Local resolution identifies flexible parts of the polymerase. A cut-away view is displayed with the pseudo-atomic model on the left and the cryo-EM reconstruction colored by resolution on the right. A bar indicates the color code corresponding to the local resolution. The most flexible parts, which correspond to the vRBL domain and the α-ribbon, are highlighted with a dotted ellipse.
Figure S3.

Biophysical and Biochemical Analysis of vRNA Binding, Related to Figures 3 and 4
(A) Measurements of Kd by fluorescence anisotropy.
(B) Binding of duplexes with different length 3′ overhangs.
(C) Sequence analysis of 3′ end binding.
(D) Protection from trypsin cleavage by 3′ or 5′ end vRNA binding.
(A) 3′ and 5′ vRNA affinity to L1750 measured by fluorescence anisotropy. 5 nM of 25-mer RNAs corresponding to 3′ (red) or 5′ (blue) vRNA and labeled with fluorescein were titrated with L1750 over the protein concentration range from 3 nM to 1 μM (left). Comparison of ssRNA (red) and dsRNA (green) binding to L1750 upon titration with a broader protein concentration range (right). The calculated affinity values are indicated in the offset tables.
(B) Mobility shift assay of radiolabelled, artificial panhandle RNAs with different lengths of 3′ overhang bound to L1750. The RNAs used are schematically represented showing the overhangs and coloring the polymerase interacting nucleotides in red, the distal duplex nucleotides in green, the up/downstream nucleotides in black and guanine linker in white. The radioactive signals belonging to shifted bands were recorded with a Typhoon and quantified with ImageQuant. Amounts of bound RNAs were normalized against the amount of bound 22 nt long 3′ ssRNA used as a reference. The graphic plots the average values and Sd from four independent experiments. Radiography for one MSA experiment is shown in the offset maintaining the same order of RNAs as in the graphic but shifting the ssRNA control to the right lane.
(C) Sequence specificity of the 3′ vRNA end binding to L1750. Mobility shift assay of the wild-type radiolabelled 22 nt long 3′ vRNA (WT) (top-left, same as in B) and all possible 33 point mutants within the first 3′ 11 nucleotides. As in (B) amounts of bound RNAs were normalized against the amount of bound WT 3′ vRNA used as a reference. The graphic plots the average values and SD from four independent experiments. The RNA variants tested are loaded in the gel lanes maintaining the same order as in the graphic.
(D) Protection from trypsin cleavage by 3′ or 5′ end vRNA binding. Limited trypsination of L1750 without RNA, with 3′ vRNA and with 5′ vRNA was visualized by SDS-PAGE. The cleavage products were identified by ESI-TOF mass spectrometry to determine their molecular mass, tryptic MALDI-TOF to identify the tryptic peptides belonging to each, and Edman degradation to sequence the N-term of each band. The resulting molecular weight, peptide boundaries, and methods used to cross-validate the identification on each experiment are shown in the table. The results are schematically summarized on the bottom right of the panel showing the L1750 construct as a colored bar where the regions before and after the trypsin cleavage site are colored in red and blue, respectively, the RdRp region in green and the L1750 C-term region in yellow. The differentially cleaved region is represented by the dashed red lines box. The protein fragments belonging to each region are highlighted with dashed lines squares on the SDS-PAGE.
Figure 1.

Overall Structure of LACV Polymerase
(A) Schematic representation of the domain structure of the monomeric LACV polymerase (top) aligned to that of heterotrimeric (PA-PB1-PB2) influenza polymerase (bottom). Structurally or functionally equivalent domains are similarly colored. A notable difference with the influenza polymerase is the clamp (magenta), involved in 3′ vRNA end binding, which is inserted into the LACV PA-C like domain. The LACV α-ribbon (orange) is structurally equivalent to the influenza β-ribbon despite being inserted in a different loop of the fingers domain. The LACV palm domain has an insertion specific for the California serogroup of orthobunyaviruses (salmon). The LACV fingernode (gray) is functionally equivalent to the influenza β-hairpin. The PB1 C-ext/PB2-Nterm interface is replaced by the LACV bridge domain. The LACV thumb ring domain (yellow) is structurally homologous to the influenza PB2 N1 and N2 domains. L1750 lacks the last 518 residues of the L protein currently of unknown structure (black stripes).
(B) Illustrated representation of two views of the crystal structure of L1750 in complex with the 3′ (cyan) and 5′ vRNA (yellow). Protein domains are colored as in (A).
(C) Structural comparison between L1750 and influenza (FluB2 structure, PDB: 4WRT) polymerases with equivalent PA-like, PB1-like and PB2-N like regions colored green, blue, and red, respectively. The 3′ (cyan) and 5′ vRNA (yellow) vRNAs are indicated. A more detailed structural comparison is in Figure S4.
See also Figures S1 and S4.
Figure 2.

Cryo-EM Reconstruction of Apo-L1750
(A) 3D reconstruction of the apo-L1750 containing the entire dataset of cryo-EM imaged particles, determined at 8.3 Å resolution. The dataset can be separated into three distinct states:
(B) A 3D class displaying only partial density for the endonuclease (9.7 Å resolution).
(C) A 3D class displaying density for all regions of the polymerase (9.7 Å resolution).
(D) A 3D class lacking density for most of the vRBL domain and California insertion (9.3 Å resolution). Flexible regions are indicated with dotted lines. The domains are colored as in Figure 1.
See also Figure S2.
Overall Structure of LACV Polymerase
The RNA-bound and apo-LACV L1750 structures display the same overall shape with a large globular central core and a flexible protrusion (Figures 1 and 2). The overall structural organization is strikingly similar to that of the influenza polymerase (Pflug et al., 2014) despite the complete lack of extended sequence homology (Figures 1A and 1C, Figure S4). In fact L1750 corresponds precisely to PA, PB1, and PB2-N (residues 1–250 of PB2), confirming the linear, head-to-tail mapping of the influenza heterotrimeric polymerase onto the orthobunyavirus L protein, as previously proposed (Reguera et al., 2010). The central PB1-like RdRp region of L1750 (residues 758–1433) contains the canonical fingers, fingertips, palm, and thumb domains with the conserved polymerase motifs exposed into the internal RNA synthesis chamber. It is buttressed on one side by the PA-C like region, which also has distinct pockets for the 3′ and 5′ vRNA extremities, and on the other by the PB2-N like region.
Figure S4.

Structure Comparison between LACV and Influenza Polymerase, Related to Figure 1
In each panel equivalent structural features are highlighted for LACV (left) and influenza A (PDB: 4WSB) (right) polymerases after superposition. The root mean square structural similarity for Cα positions is 3.18 Å for PA, 3.24 Å for PB1 and 4.33 Å for the first third of PB2 (PB2-N).
(A) PA like region.
(B) PB1 like region.
(C) PB2-N like region.
The previously described N-terminal endonuclease domain (residues 1–184) (Reguera et al., 2010) is solvent exposed and differently orientated compared to influenza polymerase (Figure 1C, Figure S4A). However, it is clearly flexibly linked to the central polymerase core, as revealed by its lack of density in one 3D class of the EM map (Figure 2B). An extended linker (residues 185–270), analogous to the influenza PA-linker, that packs on and stabilizes the fingers and palm domains of the RdRp (Figure 1B, Figures S4A and S4B), connects the endonuclease to the PA-C like domain (residues 271–759), which is divided into two lobes (Figures 1A and 1B). The larger “core-lobe” is α-helical and buttresses the thumb and palm domains of the RdRp. The second lobe is mainly involved in vRNA promoter interactions and is therefore called the vRNA binding lobe (vRBL). It has a central β sheet with, on one side, a structure denoted the “clamp” (residues 355–400) that binds the 3′ of the vRNA (see below) and which has no equivalent in influenza PA. On the opposite side is a long loop (residues 420–440), analogous to the influenza PA-arch, which binds the vRNA 5′ end (Figure 1B, Figure S4). 3D classification of the apo L1750 cryo-EM data allows generation of EM maps corresponding to two states of the vRBL. In the first, the same conformation is observed as in the crystal structure, but the arch is invisible, whereas in the second state, the most of the vRBL is invisible, suggesting its enhanced flexibility in the absence of bound vRNA (Figures 2C and 2D). Two specific insertions emerge from the fingers domain, the partially ordered “α-ribbon” (residues 847–905), structurally equivalent to the influenza PB1 β-ribbon (but emerging from a different fingers domain loop, Figure S4B) and the “fingernode” (residues 1105–1135), functionally equivalent to the PB1 β-hairpin. The fingernode folds into two α helices (α43–44) linked by a flexible loop, and together with the vRBL arch, it plays a central role in sequence specific binding of the vRNA 5′ end (see below). The palm domain appears unusually elongated, partly due to the insertion of a solvent exposed helical hairpin of unknown function that is specific for the California orthobunyavirus serogroup including LACV (Figure 1B, Data S1). The thumb domain is surrounded by the PA-C like domain core lobe and a set of helices (α58–62 and α67) and strands (β31–34), denoted the thumb ring which is structurally homologous to the influenza PB2 N1 and N2 domains. A loop (residues 1402 to 1422) at the C-terminal end of the thumb is likely deployed into the polymerase cavity but lacks electron density map, indicating mobility. It is analogous to the influenza PB1 putative priming loop but is significantly shorter. The thumb domain is followed by a helical bundle called the bridge (residues 1433–1503), which replaces the helical PB1-PB2 interface and closes the circular architecture of the polymerase around its internal cavity. The highly conserved connection between the bridge and the thumb ring (residues 1498–1506) partly defines the template entry channel (see below). Inserted in the thumb ring is the lid (residues 1614–1703) which as in influenza PB2, borders the exit channel.
vRNA Promoter 3′ End Recognition
Nucleotides 1–8 from the vRNA 3′ end are bound in an extended, single-stranded configuration in a narrow cleft over which the clamp closes (Figure 3A). Diverse regions of the polymerase contribute 3′ end RNA binding loops, including both lobes of the PA-C like domain, the thumb (residues 1307–1315) and the thumb ring domain (residues 1513–1517). The protein-RNA interface buries a total of ∼3460 Å2 of surface area and includes >30 protein-RNA hydrogen bonds, indicating a high degree of sequence specificity (Figure S5A, Table S2). Nucleotides U1, C2, and A3 are orientated into individual pockets by an extensive hydrogen bond network with residues from helices α14 and α21 and two loops from the vRBL (residues 312–316 and 535–539) and another loop from the core lobe (residues 469–473) (Figure 3B). The very 3′ end is completely sequestered by the stacking of His312 onto the U1 ribose and hydrogen bonds from the 2′ and 3′ OH to Pro314 and Asn538 (Figure 3B). Nucleotides U4, C5, and A6 are stacked on each other with their bases facing the protein and their phosphates interacting with His1515 from the thumb ring and Arg372 from the clamp (Figures 3B and 3C). Nucleotides C7 and A8 are again orientated into separate pockets mainly formed by clamp residues, with C7 stacking between conserved Trp395 and Tyr524 as well as making base specific hydrogen bonds to residues Gln398 and Arg531. A8 stacks on Ile 378 and its N6 makes a base-specific hydrogen bond with the backbone of Lys381 (Figure 3C). These structural observations are consistent with RNA binding experiments with all possible single substitutions in 3′ end nucleotides 1–11, which show that nucleotides 6–8 are the most critical for sequence specific binding (Figure S3C). 3′ end nucleotides 9-UGAUGGUU-16 form a duplex with the co-crystallized complementary oligonucleotide 5′OH-GCUACCAA (Figure 3A) and the 3′ end backbone is neutralised by basic residues Lys859, Lys862, Arg869, and Lys870 from helix α33 of the α-ribbon (Figure S5A).
Figure 3.

3′ vRNA End Binding to LACV Polymerase
(A) Overview of the 3′ vRNA (cyan sticks) binding site showing the clamp (magenta) and other interacting loops colored as in Figure 1A. The distal short complementary strand is in gray sticks. The RNA electron density is from the final 2Fo-Fc map contoured at 1.5 σ. K368 on helix α16 is protected from trypsin cleavage upon 3′ end binding.
(B) Protein-RNA interactions of nucleotides 1–6 of the 3′ vRNA extremity. Hydrogen bonds are shown as green dotted lines.
(C) Protein-RNA interactions of the clamp with 3′ vRNA nucleotides 6–9.
See also Figures S3 and S5.
Figure S5.

Details of Protein-RNA Interactions, Related to Figures 3 and 4
(A) Residues (color coded as shown, according to Figure 1A) interacting with the 3′ vRNA and short duplex region. Based on data from CONTACT (Table S2)
(B) As (A) but for the 5′ vRNA.
vRNA Promoter 5′ End Recognition and Induced Structural Changes
The 10 first nucleotides of the 5′ vRNA, soaked into the crystal, binds as a stem-loop to the polymerase in a similar configuration and location to the ten nucleotide 5′ hook of influenza vRNA promoter (Pflug et al., 2014) (Figure 4, Figure S6A). However, LACV 5′ hook is less compact with only two base pairs between G2-C10 and U3-G9, compared to four in influenza. Bases G5 and A4 are consecutively stacked on U3, whereas U6, G7, and U8 are splayed out in the loop region, compared with only one in influenza. The 5′ vRNA interaction with the polymerase buries a total of ∼3030 Å2 of surface area and includes 37 protein-RNA hydrogen bonds (Figure S5B, Table S2). Upon RNA binding both the arch and the fingernode are structurally reconfigured to promote protein-RNA interactions (Figures S6B and S6C). An array of conserved, mainly positively charged, residues stabilizes the 5′ vRNA backbone phosphates (A1-Lys423, G2-Lys302/Arg592, U3-His306, A4-Arg600/Thr642, G5-Lys643/Tyr677, U6-Arg292, G7-Lys768, U8-His760/His761) (Figure S5B). Nucleotide A1 stacks onto the planar backbone of Cys419 and Gly420 and consecutive base stacking of nucleotides 1–5 is interrupted by conserved arch residue Pro440, which stacks on base G5 forcing base U6 to flip out (Figures 4A and 4B). Highly specific, induced fit interactions are made from residues 1116–1123 of the fingernode loop to flipped out bases G7 and U8. The loop structurally reconfigures to allow G7 to stack on Tyr1120 and make base specific interactions with Gln1116, Asp1123, and Lys768 (α30). U8 stacks on Gln1116 and makes three base specific main-chain interactions with the peptide 1118–1120 (Figure 4C, Figure S6C). Adjacent, conserved His760 and His761 on α30, further stabilize the RNA loop conformation by binding the U8 phosphate and by stacking onto the G9 and G5 ribose moieties, respectively (Figures 4B and 4C).
Figure 4.

5′ vRNA End Binding and Induced Structural Changes
(A) Overview of the 5′ vRNA stem-loop (yellow sticks) binding site with interacting loops colored as in Figure 1A. The RNA electron density is from the final 2Fo-Fc map contoured at 1.5 σ. K430 on the arch is protected from trypsin cleavage upon 5′ end binding.
(B) Protein-RNA interactions in the 5′ vRNA stem region with hydrogen bonds as green dotted lines.
(C) Protein-RNA interactions of the fingernode loop with 5′ vRNA loop bases G7 and U8.
(D) Superposition of the L1750 −3′ vRNA structure without (light green ribbons) and with (colored as in Figure 1A) soaking of nucleotides 1–10 of the 5′ vRNA. Upon 5′ vRNA binding (yellow) the backbone interaction with His760 and His761 pulls helix α30 up allowing stabilization of an ordered configuration of the fingertips residues 949–958 (blue sticks). Multiple new contacts are formed, including hydrophobic interactions with α30 residues V762 and L766 and hydrogen bonds (dashed green lines) with residues from the linker region between PA-C like domain α29 and fingers domain α30, notably His757. Hydrogen bonds between Arg958 and Glu959 to Gln1145 stabilize polymerase active site motif B (dark red).
See also Figures S3, S5 and S6.
Figure S6.

Comparison of 5′ vRNA Binding in LACV and Influenza and Induced Structural Changes in the Arch and Fingernode, Related to Figure 4
(A) Stick model of the LACV (left) and influenza (right) 5′ vRNA stem loops. The base paired residues are in pink, the additional stacked residues in gray and the flipped out bases of the loop in light cyan. Hydrogen bonds are shown as green dashed lines. Nucleotide U11, which is partially ordered in the LACV structure, is shown in yellow.
(B) The structure of L1750 before (light green) and after (colored as in Figure 1A) soaking with 10 nucleotides 5′ vRNA (yellow tube). 5′ vRNA binding prompts a reorientation of arch residues Val437 and Ser438 and induces a significant reconfiguration of the arch.
(C) The interaction with 5′ vRNA loop bases G7 and U8 radically changes the configuration of the fingernode loop (light green before to gray after) allowing the stacking of Tyr1120 onto G7 which makes base-specific hydrogen bonds with Gln1116 and Asp1123 and base-specific recognition of U8 by main-chain interactions.
(D) Influenza 5′ vRNA nucleotides 1–16 (red tube) and 3′ vRNA nucleotides 10–14 (blue tube) are superposed on the L1750 5′ vRNA soaked structure after superposition of bat influenza FluA structure (PDB: 4WSB) and LACV polymerases. LACV polymerase is shown as a gray cartoon with 5′ vRNA nucleotides 1–10 in yellow. Partially ordered LACV U11 is highlighted in yellow sticks and corresponds roughly to A10 in influenza. The LACV α-ribbon (orange) would clash with a distal, base paired region positioned as in influenza, but could rotate (arrow) to play a role similar to the PB1 β-ribbon (green) in binding the duplex region.
Biochemical evidence for the involvement of the clamp and arch in, respectively, 3′ and 5′ end binding comes from proteolysis experiments that show clamp residue Lys368 and arch residue Lys430 are protected from trypsination by 3′ and 5′ end binding, respectively (Figures 3A and 4A, Figure S3D). However, 5′ vRNA binding not only induces conformational changes in RNA binding loops but also structures elements of the polymerase active site (Figure 4D). Most significant is the complete ordering of the fingertips loop (residues 950–958), which contains motif F. The interactions of His760 and His761 pull helix α30 toward the 5′ vRNA, and the consequent displacements of Val762 and Leu766 make room for the fingertips to order into a structured active form through multiple contacts with residues from the PA-C like domain α29, fingers domain α30 and the linker joining them (Figure 4D). Conserved His757 plays a key role in stabilizing the fingertips β-turn through multiple hydrogen bonds to backbone carbonyl groups (Figure 4D). Fingertips ordering has a knock-on effect on stabilization of motif B notably through the interaction of conserved motif B Gln1145 with Arg958 and Glu959 (Figure 4D). This is the first observation of functionally important allosteric effects associated with 5′ vRNA binding and is only observed when 10–11 5′ end nucleotides are bound, but not eight (data not shown). Interestingly, soaking in 5′ cRNA nucleotides 1–10 (which differ from vRNA only in the substitution G9 to A) shows that the wobble base pair becomes canonical U3-A9 with no discernible difference on the induced polymerase rearrangements compared to vRNA (data not shown). This is consistent with only the identity of 3′ position 9 leading to a significant difference in propensity for transcription between vRNA and cRNA (Barr and Wertz, 2005). In the 5′ vRNA end 1–11 soaked structure, nucleotide U11 is only partially ordered and cannot base pair with A1 without displacement of conserved Arg595 which hydrogen bonds to N1 of A1 (Figure S6D). Furthermore soaking 5′ RNAs longer than 11 nucleotides resulted in no binding in the crystal, suggesting that further rearrangements of the polymerase (notably the α-ribbon) and probably a sharp turn in the RNA (as observed in influenza 5′ end between nucleotides 10–11) may be required to allow binding of the complete 5′ end, but which are incompatible with the crystal packing (Figure S6D).
The Active Site Cavity and Its Entrance and Exit Tunnels
The L protein internal active site chamber, where nucleotide addition occurs, is formed by the conserved polymerase motifs A-F and configured like other RNA polymerases, notably influenza polymerase(Pflug et al., 2014; te Velthuis, 2014) (Figure 5A). Motif F forms part of the fingertips, the flexible loop between fingers strands β20 and β21 that is only fully ordered when the 5′ vRNA is bound (see above). The other conserved polymerase motifs are all in the palm domain: motif A (1060-DMSKWS) between palm β22 and α41 with divalent cation binding D1060; motif B (1145-QGNFNYTSSY) between β24 and the long α45 with conserved N1149; the catalytic motif C (1186-SDD) in the turn between strands β25 and β26; motif D (1223-QANMKKTY) just before β27 and motif E (1236-KEFVSLFN) forming the tight loop between β28 and β29. Interestingly, structural alignment with influenza polymerase allows identification of two new active site motifs, denoted G and H, which appear to be conserved specifically in sNSV polymerases (Figure 5A, Figures S7A and S7B). Motif G (653-RYMI in LACV, 658-RKLL in influenza PA) is in helix α24 in the core-lobe of the PA-C like domain (Figure S7A), the conserved arginine being positioned to interact with the priming NTP (Figure 5A). Motif H (1101-KELIL in LACV and 347-KVARL in influenza PB1) forms a β strand (β23 in LACV) with the conserved lysine stabilizing the motif B backbone conformation by hydrogen bonding to multiple carbonyl-oxygens (Figure S7B).
Figure 5.

The LACV Polymerase Active Site and Entrance and Exit Tunnels.
(A) The arrangement of the conserved RdRp motifs in the LACV active site colored gold, light blue, green, red, brown, and blue for motifs A–F, respectively. Additional sNSV specific motifs G (from the PA-C like domain) and H are shown in pink and gray (see Figure S7). Superposition of the polio virus elongation complex structure (PDB: 3OLB, 3OL8) shows the positions of the catalytic divalent cations (black spheres), the priming nucleotide (N+1, gray) and incoming NTP (N+2, magenta) and template strand (light gray sticks).
(B) The LACV polymerase structure (gray cartoon) with the 5′ and 3′ vRNA in, respectively, yellow and cyan is shown with the tunnels (green) marked with arrows as template entry, NTP entry, product, and template exit, as calculated with MOLE 2.0 (Sehnal et al., 2013). The endonuclease, bridge, thumb-ring and lid are, respectively, in forest green, blue, gold, and brown.
(C) The same representation and orientation as (B) for the influenza A polymerase structure (PDB: 4WSB) with additionally the PB2 cap-binding domain in orange, the putative priming loop in magenta and the PB1 C-extension in dark gray.
(D) Diagram showing the conserved residues forming the template entrance in LACV polymerase which is partially occluded by the flexible α-ribbon (orange). Colors are as in Figure 1A.
(E) As (D) but showing the putative template exit channel in LACV polymerase.
See also Figure S7.
Figure S7.

Newly Identified Conserved Motifs in all sNSV Polymerases and Details of the Polymerase Tunnels, Related to Figure 5
(A) The conservation of motif G (RYφφ, bold absolutely conserved, φ denoting a hydrophobic residue) is shown in an alignment (left) including two strains each from Orthomyxoviruses (Influenza A and B), Bunyaviruses (Ortho: LACV/Bunyamwera, Hanta: Hantaan/Imgin, Phlebo: Rift Valley fever/Uukuniemi, Nairo: Crimean-Congo haemorrhagic fever/Kupe, Tospo: Tomato spotted wilt/ Peanut bud necrosis) and Arenavirus (Lassa/Junin). The environment of motif G is shown for LACV (middle) and influenza (right) with the neighboring motifs A, C, D (including conserved Lys1228/481) and E. Structurally similar elements are colored according to Figure 1A.
(B) The same for motif H (K)xφxφ) showing for LACV (middle) and influenza (right) the interactions of the conserved lysine with the backbone of the motif B loop. Neighboring LACV fingernode (gray) is highlighted (middle) as well as the analogous β-hairpin (gray) in influenza polymerase (right). In both cases neighboring motifs A, B, and F are marked. Structurally similar elements are colored according to Figure 1A.
(C) Section through the L1750 polymerase structure in van der Waals surface representation colored according to surface electrostatic potential showing that the four conserved channels and central cavity are positively charged. The 3′ and 5′ RNAs are shown as blue and yellow ribbons, respectively. Green arrows indicate the NTP and RNA traffic following the homology with influenza and other similar RNA polymerases.
(D) As C but solvent accessible residues are colored according to their degree of conservation among orthobunyaviruses (violet > 90%, 90% < pink > 60%, white < 60%).
(E) The NTP entry channel is lined by conserved basic residues R287, K673 (PA-C like), K956, R958 (fingertips), K1063 (motif A), K1227, K1228 (motif D) some of which are only positioned correctly upon 5′ vRNA binding. Coloring of domains is as in Figure 1A.
(F) View into the product exit channel showing contributing conserved residues from the fingers, bridge thumb ring and lid (colored as in Figure 1A).
Four positively charged solvent accessible tunnels, visualized using MOLE 2.0 (Sehnal et al., 2013) converge into a central inner cavity where the RdRp motifs mediate template directed RNA synthesis (Figure 5B, Figure S7C). The tunnels are delimited by residues conserved among Orthobunyavirus polymerases (Figure S7D). The configuration of the template entry channel, the NTP entry channel and the nascent strand exit channel is similar to that described for influenza polymerase (Figure 5C) (Pflug et al., 2014). The template channel entrance is defined by the vRBL, fingers and bridge and is partially obscured by the α-ribbon (Figure 5D), which together with several loops of the vRBL β sheet that are deployed toward the entrance but disordered in the structure, may modulate access. The NTP entry channel is lined by conserved basic residues R287, K673 (PA-C like), K956, R958 (fingertips), K1063 (motif A), K1227, and K1228 (motif D), some of which are only positioned correctly upon 5′ vRNA binding (Figure S7E). The product strand exit tunnel is surrounded by the lid domain and the thumb ring mainly by the extended joining linkers and by fingers and palm opposite side of the NTP entry channel (Figure S7F). In a LACV L there is a more obvious extra channel that we postulate is for the template to exit. In influenza polymerase the equivalent channel is present, but narrowed by the presence of the putative priming loop (Figure 5C), which is 15 residues longer than in LACV L. The putative template exit channel is defined by the thumb, thumb ring, bridge, and lid domains and lined by conserved basic residues R1430, K1492, R1493, K1686, and R1690 (Figure 5E). As discussed below, the arrangement of the tunnels in LACV L protein suggests an elegant strategy for RNA synthesis whereby the polymerase forces separation of the template and product strands and directs each down distinct exit channels on opposite sides of the molecule.
Discussion
Initiation of RNA Synthesis Requires Significant Conformational Changes in Protein and vRNA
In the L1750-vRNA complex, the 3′ end of the template is tightly and specifically bound but not accessing the entry tunnel, corresponding perhaps to the highly stable state that occurs in inactive RNPs, for example within virions. For active RNA synthesis, the 3′ end clearly has to be relocated into the polymerase active site (Figures 6A and 6B). Furthermore it has been shown that complementarity and presumably base pairing between at least 3′ and 5′ nucleotides 12 to 16 are required for RNA synthesis by orthobunyavirus polymerases (Barr and Wertz, 2004; Kohl et al., 2004). A pre-initiation configuration, with the 3′ end specifically bound on the outside but not entering the polymerase active site, was also visualized for influenza polymerase (Pflug et al., 2014; Reich et al., 2014). However, whereas in the influenza structures, both 3′ and 5′ extremities of the promoter simultaneously bind their separate single-stranded binding sites and form a distal duplex region (with 11–14 of 5′ base pairing with 10–13 of the 3′ end), for LACV polymerase in the crystallized conformation, this appears to be impossible. First, nucleotides 11 of the 3′ and 5′ ends are around 25 Å apart, too far to see how 3′ and 5′ nucleotides 12 to 16 could base pair (Figure 6A). Second, in the all L1750 structures, bases 9–16 of the 3′ end are already base paired with the co-crystallized cRNA, corresponding in sequence to 5′ nucleotides 9–16. Thus when 5′ nucleotides 1–11 are soaked into the crystal, nucleotides 9–11 are present in two distinct locations greater than 20 Å apart, which obviously cannot happen physiologically (Figure 6A). Given the similar mode of 5′ end binding to both LACV and influenza polymerases (and that the 5′ end remains bound as observed during initiation; Reich et al., 2014) and the similar requirement of a short distal 3′–5′ duplex, we propose that an alternative configuration of the bound promoter likely exists more analogous to the influenza pre-initiation conformation (Figure 6B). Release and repositioning of the 3′ end could occur by swinging of the clamp, without necessarily letting go of the 3′ end RNA, with a concomitant reorientation of the α-ribbon into a position analogous to the β-ribbon in influenza polymerase (Reich et al., 2014), to stabilize duplex formation (Figure 6B, Figure S6D). The flexibility of the vRBL and α-ribbon as seen by 3D classification of the EM images, together with the lower local resolution of these regions in the cryo-EM map, shows that such movements are plausible (Figure 2, Figure S2F). In the case of influenza virus, extrapolation of the template from the observed duplex region, based on the poliovirus polymerase elongation complex model (Gong and Peersen, 2010), would result in the 3′ end overshooting the polymerase active site by three nucleotides as previously discussed (Reich et al., 2014). For LACV, similar modeling suggests that this overshoot is accentuated. Assuming that for LACV the duplex is from nucleotides 12–15 of both strands (Barr and Wertz, 2004; Kohl et al., 2004), based on the influenza/polio models, the template would overshoot the polymerase active site by 5 or 4 nucleotides, depending on whether the LACV 3′–5′ 12–12 base pair corresponds to influenza 3′-5′ 10–11 or 11–12 base pair, respectively (Figure 6B). In the case of cap-dependent transcription, this overhang could favor base pairing with the incoming capped primer. In the case of replication, this situation could be explained by (1) a different, less direct path of the single stranded template, so that the 3′ nucleotide 1 was placed directly in the polymerase active site (i.e., the modeling is misleading); (2) a mechanism of internal initiation followed by realignment, dependent on the triplet repeat at the beginning of the template (3′-UCAUCA), as has been described for some bunya- and arena-viruses (Guu et al., 2012) (i.e., internal initiation at position 4 followed by realignment of the AGU triplet); or (3) initiation starting at nucleotide 1 but duplex formation between 3′ and 5′ nucleotides 12 to 16 only occurring after 4–5 nucleotides have been synthesized and the template has translocated further into the active site cavity. This latter possibility would be consistent with the observed position of LACV 3′ end nucleotide 8 being close to that of the nucleotide 8 counting from the active site along the polio template (Figure 6B).
Figure 6.

Model of RNA Synthesis by LACV Polymerase
(A) Illustrated representation of the LACV polymerase (gray) looking down the template entry channel showing the disposition of key structural elements (arch, clamp, α-ribbon, fingertips, fingernode) colored as in Figure 1A. The 5′ and 3′ vRNA extremities are, respectively, yellow and cyan tubes, except that nucleotide 11 in each case is in red highlighting their wide separation (>20 Å). The figure shows the impossibility of formation of a distal 5′ and 3′ duplex between nucleotides 12–15 of each strand, while maintaining the single-stranded ends bound as in the observed conformation.
(B) Model for the initiation conformation of LACV based on superposition with the influenza polymerase (PDB: 4WSB) and the poliovirus elongation complex (PDB: 3OLB, 3OL8) structures. The observed 5′ and 3′ vRNAs are, respectively, red and blue for influenza and yellow and cyan for LACV and numbered accordingly. The LACV clamp binding to the 3′ end is in magenta. The poliovirus template strand is in gray and the active site is indicated by motif C (green), the catalytic divalent cations (black) and the priming and incoming NTPs (gray and magenta, respectively). The influenza vRNA distal duplex starts with the 3′-5′ 10:11 base pair (labeled). The template nucleotide numbering in outline white counts back from the active site, assuming initiation at position 1. The template nucleotide numbering in black numbers counts along the LACV template assuming the first LACV 3′-5′ base pair 12:12 aligns with the influenza 10:11 base pair. This would allow for connectivity between the distal LACV duplex and the observed 5′ end hook binding but imply an overshoot of the active site by 5 nucleotides. This is discussed further in the text.
(C) Model of the elongation state showing trajectories of template RNA (cyan) and product RNA (orange) and NTPs through the polymerase tunnels (green). The observed positions of the 3′ and 5′ ends are shown as well as the position of the active site. After a short template-product duplex, which is accommodated in the interior cavity, each strand exits separately along different tunnels, the template back to the front of the polymerase where it can re-integrate into the RNP and the nascent strand to the rear where product processing occurs i.e., progeny cRNP assembly in the case of replication or mRNP assembly or translation coupling in the case of transcription.
The Template Pathway through the Polymerase
Whatever mode of initiation, during elongation, a duplex formed by template and nascent strands starts to grow in the active site cavity as visualized in the structure of the poliovirus polymerase elongation complex (Gong and Peersen, 2010) (PDB: 3OL7) (Figure 6C). However, after a complete double-helical turn, the nascent and emerging template strands would appear to clash with the thumb and C-terminal lid domains, respectively, as described for the influenza case (Reich et al., 2014). To resolve this situation, we propose that the strands are forced to separate and each is extruded along a separate exit tunnel (Figure 6C). The nascent strand would exit to the rear of the polymerase and, by analogy to the influenza case, the cap-snatched transcription primer would probably enter by the same route (Reich et al., 2014). The template would exit to the front of polymerase through the template exit channel on the same side and not far from the template entrance (Figure 6C). The proximity of the entry and exit channels would allow reintegration of the vRNA genome template into the RNP with minimal disruption (Figure 7). Modeling shows that the template path through the entry channel, cavity, and the exit channel would accommodate around 20 nucleotides. Given that ∼11 nucleotides of the 5′ and ∼9 of the 3′ ends directly bind the polymerase in the pre-initiation state, and also a single LACV NP can accommodate 11 nucleotides (Reguera et al., 2013), no free RNA needs to be exposed, nor NPs removed or added, during template reading. The mobile clamp together with the α-ribbon and proximal NPs could mediate the RNA template translocation driven by the polymerase motor.
Figure 7.

Schematic Model of vRNA Replication
An LACV RNP is schematically represented with the polymerase (purple or green), with template entrance (TEn), template exit (TEx), NTP entry and nascent RNA exit channels as marked, interacting with the viral RNA (black or yellow) and proximal NPs (ellipses colored with a blue-to-red gradient). The complementary 5′ and 3′ vRNA ends are, respectively, cyan and red. The NPs form a chain linked together by flexible NP-NP interactions involving the N-terminal arm (blue) and the C-terminal arm (red) and each NP sequesters 11 nucleotides RNA (Reguera et al., 2014; Reguera et al., 2013). Small circles mark consecutive 11 nucleotide segments of the vRNA. The polymerase itself can sequester around 20–22 template nucleotides.
(1) In the inactive state, whether after vRNP assembly or in virions, both ends of the genomic RNA are sequestered into the specific 5′ and 3′ RNA binding sites of the polymerase, thus circularizing the RNP.
(2) For de novo RNA synthesis or cap-dependent transcription (not shown) the 3′ end is relocated into the polymerase active site for initiation, by an unknown mechanism. Distal 3′-5′ duplex formation may occur before or after initiation depending on whether initiation is internal (followed by prime and align) or at position 1 (see Figure 6B and main text). Duplex formation could bring the NPs at the 5′ and 3′ (NPa and NPz) closer enhancing the circularization of the NP scaffold but would need to be dissociated to proceed with elongation.
(3) With the 5′ end bound to the allosteric site for the activation of the RNA synthesis, a nascent cRNA begins to be synthesized.
(4) As elongation proceeds, the template dissociates from the proximal NP and is channelled into the active site. Because of the proximity of the entrance and exit channels the disruption of the RNA-NP assembly may only affect one NP. Early on, the 5′ end is detached from its specific binding site on the polymerase and enters the RNP by loading onto NPz. As incoming template is released from NPy on one side, the outgoing 3′ end is loaded on it from the other side. More generally, the RNA being pulled into the cavity by the polymerase motor detaches from the proximal NP which is pulled to the left thus pushing the NP-RNA array in the direction of the arrow. This model would imply that 5′ end binding is only required to activate initiation. This would be a difference from the influenza situation where the maintenance of 5′ end binding is required, at least during transcription, for self-polyadenylation to occur.
(5) Once the nascent c5′ end emerges from the exit channel it can recruit an incoming apo-polymerase as the first step in encapsidating the progeny cRNP with incoming apo-NPs. This may be facilitated by polymerase dimer formation (see main text).
(6) Approaching termination the template 5′ end would be copied and the template 3′ end (bound to NPy) would approach its starting point.
(7a) At termination the template 3′ end rebinds to its specific binding site on the polymerase to avoid base pairing with the emerging template 5′ end which subsequently rebinds to its polymerase binding site, thus completing the replication cycle.
(7b) Due to polymerase dimer formation, the nascent c3′ end, which emerges last from the product exit channel, can easily find and bind to specific 3′ binding site on the green polymerase, thus completing progeny cRNP formation. Without polymerase dimer formation being maintained throughout replication (or other mechanism for keeping the polymerases in close proximity), it is unclear how the c3′ could find and bind to the correct polymerase which may have diffused far away.
Conclusion
Our structures of the LACV L protein in the apo-state and with one or both vRNA ends bound suggest that assembly of a functional initiation complex is a multistep process. The structures clearly show that the polymerase has highly specific and distinct sites for the single-stranded 3′ and 5′ vRNA ends, preventing them from forming an extended panhandle. The mode of 5′ end hook binding is similar to that observed for influenza polymerase, but here we directly observe the associated allosteric effects that are essential for structuring critical active site loops. On the other hand the 3′ end appears to be preferentially and tightly bound in a groove closed by a clamp on the side of the polymerase. Elucidation of the exact purpose of this binding site and the mechanism for 3′ end relocation into the template tunnel for the initiation of RNA synthesis are clear questions for future studies. Furthermore, the extended complementarity of the LACV 3′ and 5′ ends appears to be a major obstacle to reconstitute the LACV initiation complex in vitro, since incubation, even sequentially, of the complete 3′ and 5′ ends leads to the preferred formation of a long, stable duplex, which has low affinity for the polymerase. Probably for the same reason, we have not yet been able to demonstrate robust template directed RNA synthesis activity for either L1750 or full-length polymerase (see Supplemental Experimental Procedures). It is therefore likely that in the case of bunya-, and probably, arenavirus polymerases (Kranzusch et al., 2010), to avoid stable base pairing of the highly complementary vRNA promoter, the free ends are prevented from ever meeting each other by the sequential mode of assembly of nascent RNPs starting at one end of a growing replicate (Figure 7). Thus it is plausible that, unlike the situation for recombinant influenza polymerase, which is fully active when reconstituted only with the vRNA promoter (Reich et al., 2014), NP-L and NP-RNA interactions may be required for bunyavirus polymerase activity.
The striking structural similarities between the single-chain LACV and heterotrimeric influenza polymerases strongly support the idea of an evolutionary common ancestor. Indeed, it now seems plausible that all sNSV polymerases (i.e., from arena-, bunya-, and orthomyxovirus families) have a similar architecture, despite very low overall sequence homology, and this is supported by structure-based identification of new common motifs (Figures S7A and S7B). However, this does not mean that these polymerases will not have idiosyncratic family and sub-family differences. For example, whereas the arch and fingernode that bind the 5′ hook have structural and functional homologs in influenza, the LACV 3′ end binding site and the clamp structure that pins it in place has no such equivalent. Similarly, it is reasonable to suppose that each polymerase is adapted to its cognate nucleoprotein, whose size, structure, mode of RNA binding, and number of nucleotides bound (e.g., 11 for orthobunyaviruses, 7 for phleboviruses), are very different for each sNSV family (Reguera et al., 2014). In this context, it is intriguing that LACV and influenza contain, respectively, an α- or β-ribbon, equivalently located extended and flexibly hinged structures that could both play a role in both RNA and NP interactions and could be adapted to the respective NP structures (Figure S6D) (Reich et al., 2014). Finally, it is highly significant that the L1750 construct ends precisely at the same position as separates PB2-N and PB2-C in influenza polymerase (Figure 1). PB2-C, which includes the cap-binding domain and the C-terminal nuclear localization motif (not relevant for cytoplasmic LACV), has already been shown to be loosely associated with the rest of influenza polymerase (Reich et al., 2014). It remains open as to how much the C-terminal residues 1751–2263 of LACV L, missing in L1750, are structurally homologous to PB2-C. In particular it is still unknown whether there is a cap-binding domain, for which there is no direct evidence yet, but, in the case of Lassa arenavirus a specific requirement for C-terminal residues for mRNA transcription has been established (Lehmann et al., 2014).
Based on the L1750 structure, we propose a model for RNA replication in which there are clearly separated exit tunnels for the single-stranded template and product (Figure 6C, Figure 7). The proximity of the template entrance and exit, on one side of the polymerase, is compatible with processive template reading with minimal disruption of the RNP. Template RNA would progressively dissociate from proximal NP as it translocates through the entrance tunnel, into the polymerase internal chamber and out again, to be reincorporated into the RNA free NP that concomitantly translocates round the outside of the polymerase between the entrance and exit tunnels, held together by flexible NP-NP interactions (Reguera et al., 2013) (Figure 7). Meanwhile, the products exit to the other side of the polymerase, thus allowing spatial separation of template translocation and product processing. In the case of (anti)-genome replicates, product processing involves assembly into progeny RNPs, possibly first with an incoming apo-polymerase binding specifically the emerging, nascent 5′ end and subsequently progressive packaging by incoming NPs (Figure 7). We propose that robust polymerase dimerization is necessary to ensure efficient and correct circularisation of progeny RNP (Figure 7), and this is consistent with some observations concerning replication by influenza polymerase (Jorba et al., 2008; York et al., 2013), but other influenza replication models involve more complicated higher order polymerization (Chang et al., 2015; Jorba et al., 2009). In the case of transcription, as suggested by the structure of influenza polymerase, the cap-snatched primer would enter the internal cavity via the product exit channel and then, upon elongation, extrude out in the same direction, where it likely interacts with host translation factors (in the case of bunyaviruses, transcription is closely coupled to translation; Barr, 2007). As suggested for influenza polymerase (Reich et al., 2014), the main role of the PB2-C like region may be in these product processing mechanisms. We think these concepts are likely to be applicable to all sNSVs polymerase and possibly those of nsNSV as well, which also operate in an RNP context.
Experimental Procedures
Protein Production, Crystallization, and Structure Determination
Residues 1–1750 (L1750) of the polymerase (L protein) sequence of La Crosse virus (LACV) were expressed in insect cells from a synthetic gene inserted in a pFastBac vector. Purified protein at 5 mg/ml was crystallized with an equimolar mixture of nucleotides 1–16 from the 3′ and nucleotides 9–16 from the 5′ vRNA ends (Dharmacon). Crystals were improved by microseeding. Diffraction data were collected on beamlines ID23-1 or ID29 at the European Synchrotron Radiation Facility and integrated with XDS (Kabsch, 2010). The structure was solved by the multiple isomorphous replacement with anomalous signal method using selenium, platinum, and tantalum cluster derivatives with SHARP (Bricogne et al., 2003). LACV endonuclease (PDB: 2XI5) and influenza polymerase PA and PB1 subunits (PDB: 4WSB) were used as a guide to model building. Selenomethionine positions from the anomalous difference map helped align the sequence and autobuilding with BUCCANNEER (Cowtan, 2006) was useful to extend the model, which was refined with REFMAC (Murshudov et al., 1997) and PHENIX (Afonine et al., 2012). 5′ end nucleotides 1–10 or 1–11 were soaked into pre-grown crystals to reveal the 5′ end binding site.
Electron Microscopy
Cryo-EM grids were prepared by applying 4 μl of L1750 at 0.2 mg/ml to a quantifoil grid, blotting excess solution and then freezing in liquid ethane. Cryo-EM images of apo-L1750 were collected on a Krios microscope at 80 kV with a Falcon II direct detector (FEI) at magnification 138,129 times. FEI EPU automatisation software was used to collect 6,129 micrographs with a defocus between 0.5 and 2 μm, an exposure time of 0.5 s and a dose of 14e−/Å2. After contrast transfer function correction, 10,102 manually picked particles were used to derive initial class averages with IMAGIC (van Heel et al., 1996). These were then used to select ∼180,000 particles for input to 3D reconstruction and refinement with RELION 1.3 (Scheres, 2012), using as initial model the L1750-vRNA crystal structure filtered at 30 Å resolution, leading to a map at 8.3 Å resolution. The dataset was subsequently partitioned by 3D classification resulting into three structures which revealed the less well-defined, flexible regions.
Polymerase-vRNA Binding Studies
For electrophoretic mobility shift assays, radioactively labeled RNAs were produced by in vitro transcription with T7 polymerase. For binding assays, 10 μM of L1750 in 10 μl buffer was mixed with radiolabelled RNA and 1 μl of non-specific poly(U) RNA (Sigma). Mixtures were incubated at room temperature for several hours and resolved on native TG gels. Radioactive signal from shifted bands was recorded with a Typhoon and quantified with ImageQuant.
For fluorescence anisotropy measurements, 5 nM 25-nucleotide long RNA oligos corresponding to 3′ or 5′ vRNA (IBA), labeled with fluorescein on the appropriate non-interacting end, were titrated with L1750 in order to obtain 10–15 protein concentration points ranging from 3 nM to 1 μM. Fluorescence and fluorescence anisotropy measurements used 495 nm excitation and 515 nm emission wavelengths. KaleidaGraph (Synergy Software) was used to evaluate the data and derive dissociation constants.
For proteolysis protection experiments, L1750 was incubated at 1 mg/ml with 1:1 molar ratios of 25-nucleotide long 3′ or 5′ genomic ends and then digested for 1 hr at room temperature with trypsin (1:1,000 w/w). Products of digestion were analyzed by various techniques including: SDS-PAGE, western-blot, ESI-TOF-MS, MALDI-TOF-MS, MALDI-TOF-MS with N-terminal acetylation, and N-terminal sequencing of protein fragments by Edman degradation.
For more details see Supplemental Information.
Author Contributions
P.G. and J.R. did protein production, biochemistry, RNA binding studies, and X-ray data collection and structural analysis. P.G. crystallized the L1750-vRNA complex. J.R. identified the L1750 construct and solved the crystal structure. H.M. did EM analysis. S.C. and J.R. co-directed the project and wrote the paper with input from P.G. and H.M.
Acknowledgments
We thank Wim Hagen and John Briggs for cryo-EM data collection at EMBL Heidelberg, Leandro Estrozi, Irina Gutsche, and Guy Schoehn for advice on EM image analysis; the staff of the EMBL Eukaryotic Expression and High Throughput Crystallisation facilities, and the EMBL-ESRF JSBG staff for access to ESRF beamlines. Darren Hart helped with construct design and together with Philippe Mas and Damien Maurin with the ESPRIT analysis. Mass-spectroscopy was performed by Joanna Kirkpatrick (EMBL Proteomics Core Facility) and Luca Signor (IBS), Edman degradation by Jean-Pierre Andrieu (IBS) and Stefan Reich (EMBL) helped with RNA binding studies. This work used the platforms of the Grenoble Instruct Center (ISBG: UMS 3518 CNRS-CEA-UJF-EMBL) within the Grenoble Partnership for Structural Biology (PSB), with support from FRISBI (ANR-10-INSB-05-02), GRAL (ANR-10-LABX-49-01) and, for the EM facility, the Rhône-Alpes Region and the Fondation Recherche Medicale (FRM). H.M. and J.R. were supported by EMBO long-term Fellowships for some of this work. S.C. acknowledges support by ANR grant ArenaBunya-L and ERC Advanced Grant V-RNA (322586).
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
Contributor Information
Stephen Cusack, Email: cusack@embl.fr.
Juan Reguera, Email: jreguera@embl.fr.
Accession Numbers
The accession number for the 8.3 Å cryo-EM map of apo-LACV1750 reported in this paper is EMDB: EMD-2930. The accession numbers for the L1750-3′ end and L1750-3′/5′ ends are PDB: 5AMR and 5AMQ, respectively.
Supplemental Information
References
- Afonine P.V., Grosse-Kunstleve R.W., Echols N., Headd J.J., Moriarty N.W., Mustyakimov M., Terwilliger T.C., Urzhumtsev A., Zwart P.H., Adams P.D. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr J.N. Bunyavirus mRNA synthesis is coupled to translation to prevent premature transcription termination. RNA. 2007;13:731–736. doi: 10.1261/rna.436607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr J.N., Wertz G.W. Bunyamwera bunyavirus RNA synthesis requires cooperation of 3′- and 5′-terminal sequences. J. Virol. 2004;78:1129–1138. doi: 10.1128/JVI.78.3.1129-1138.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr J.N., Wertz G.W. Role of the conserved nucleotide mismatch within 3′- and 5′-terminal regions of Bunyamwera virus in signaling transcription. J. Virol. 2005;79:3586–3594. doi: 10.1128/JVI.79.6.3586-3594.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricogne G., Vonrhein C., Flensburg C., Schiltz M., Paciorek W. Generation, representation and flow of phase information in structure determination: recent developments in and around SHARP 2.0. Acta Crystallogr. D Biol. Crystallogr. 2003;59:2023–2030. doi: 10.1107/s0907444903017694. [DOI] [PubMed] [Google Scholar]
- Chang S., Sun D., Liang H., Wang J., Li J., Guo L., Wang X., Guan C., Boruah B.M., Yuan L. Cryo-EM structure of influenza virus RNA polymerase complex at 4.3 Å resolution. Mol. Cell. 2015;57:925–935. doi: 10.1016/j.molcel.2014.12.031. [DOI] [PubMed] [Google Scholar]
- Cowtan K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 2006;62:1002–1011. doi: 10.1107/S0907444906022116. [DOI] [PubMed] [Google Scholar]
- Elliott R.M. Orthobunyaviruses: recent genetic and structural insights. Nat. Rev. Microbiol. 2014;12:673–685. doi: 10.1038/nrmicro3332. [DOI] [PubMed] [Google Scholar]
- Fodor E. The RNA polymerase of influenza a virus: mechanisms of viral transcription and replication. Acta Virol. 2013;57:113–122. doi: 10.4149/av_2013_02_113. [DOI] [PubMed] [Google Scholar]
- Gong P., Peersen O.B. Structural basis for active site closure by the poliovirus RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. USA. 2010;107:22505–22510. doi: 10.1073/pnas.1007626107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guu T.S., Zheng W., Tao Y.J. Bunyavirus: structure and replication. Adv. Exp. Med. Biol. 2012;726:245–266. doi: 10.1007/978-1-4614-0980-9_11. [DOI] [PubMed] [Google Scholar]
- Haddow A.D., Odoi A. The incidence risk, clustering, and clinical presentation of La Crosse virus infections in the eastern United States, 2003-2007. PLoS ONE. 2009;4:e6145. doi: 10.1371/journal.pone.0006145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorba N., Area E., Ortín J. Oligomerization of the influenza virus polymerase complex in vivo. J. Gen. Virol. 2008;89:520–524. doi: 10.1099/vir.0.83387-0. [DOI] [PubMed] [Google Scholar]
- Jorba N., Coloma R., Ortín J. Genetic trans-complementation establishes a new model for influenza virus RNA transcription and replication. PLoS Pathog. 2009;5:e1000462. doi: 10.1371/journal.ppat.1000462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabsch W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 2010;66:133–144. doi: 10.1107/S0907444909047374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl A., Dunn E.F., Lowen A.C., Elliott R.M. Complementarity, sequence and structural elements within the 3′ and 5′ non-coding regions of the Bunyamwera orthobunyavirus S segment determine promoter strength. J. Gen. Virol. 2004;85:3269–3278. doi: 10.1099/vir.0.80407-0. [DOI] [PubMed] [Google Scholar]
- Kranzusch P.J., Schenk A.D., Rahmeh A.A., Radoshitzky S.R., Bavari S., Walz T., Whelan S.P. Assembly of a functional Machupo virus polymerase complex. Proc. Natl. Acad. Sci. USA. 2010;107:20069–20074. doi: 10.1073/pnas.1007152107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann M., Pahlmann M., Jérôme H., Busch C., Lelke M., Günther S. Role of the C terminus of Lassa virus L protein in viral mRNA synthesis. J. Virol. 2014;88:8713–8717. doi: 10.1128/JVI.00652-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin B., Coutard B., Lelke M., Ferron F., Kerber R., Jamal S., Frangeul A., Baronti C., Charrel R., de Lamballerie X. The N-terminal domain of the arenavirus L protein is an RNA endonuclease essential in mRNA transcription. PLoS Pathog. 2010;6:e1001038. doi: 10.1371/journal.ppat.1001038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin B., Kranzusch P.J., Rahmeh A.A., Whelan S.P. The polymerase of negative-stranded RNA viruses. Curr Opin Virol. 2013;3:103–110. doi: 10.1016/j.coviro.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller R., Poch O., Delarue M., Bishop D.H., Bouloy M. Rift Valley fever virus L segment: correction of the sequence and possible functional role of newly identified regions conserved in RNA-dependent polymerases. J. Gen. Virol. 1994;75:1345–1352. doi: 10.1099/0022-1317-75-6-1345. [DOI] [PubMed] [Google Scholar]
- Murshudov G.N., Vagin A.A., Dodson E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Pflug A., Guilligay D., Reich S., Cusack S. Structure of influenza A polymerase bound to the viral RNA promoter. Nature. 2014;516:355–360. doi: 10.1038/nature14008. [DOI] [PubMed] [Google Scholar]
- Plotch S.J., Bouloy M., Ulmanen I., Krug R.M. A unique cap(m7GpppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell. 1981;23:847–858. doi: 10.1016/0092-8674(81)90449-9. [DOI] [PubMed] [Google Scholar]
- Raju R., Kolakofsky D. The ends of La Crosse virus genome and antigenome RNAs within nucleocapsids are base paired. J. Virol. 1989;63:122–128. doi: 10.1128/jvi.63.1.122-128.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reguera J., Weber F., Cusack S. Bunyaviridae RNA polymerases (L-protein) have an N-terminal, influenza-like endonuclease domain, essential for viral cap-dependent transcription. PLoS Pathog. 2010;6:e1001101. doi: 10.1371/journal.ppat.1001101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reguera J., Malet H., Weber F., Cusack S. Structural basis for encapsidation of genomic RNA by La Crosse Orthobunyavirus nucleoprotein. Proc. Natl. Acad. Sci. USA. 2013;110:7246–7251. doi: 10.1073/pnas.1302298110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reguera J., Cusack S., Kolakofsky D. Segmented negative strand RNA virus nucleoprotein structure. Curr Opin Virol. 2014;5:7–15. doi: 10.1016/j.coviro.2014.01.003. [DOI] [PubMed] [Google Scholar]
- Reich S., Guilligay D., Pflug A., Malet H., Berger I., Crépin T., Hart D., Lunardi T., Nanao M., Ruigrok R.W., Cusack S. Structural insight into cap-snatching and RNA synthesis by influenza polymerase. Nature. 2014;516:361–366. doi: 10.1038/nature14009. [DOI] [PubMed] [Google Scholar]
- Scheres S.H. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 2012;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sehnal D., Svobodova Varekova R., Berka K., Pravda L., Navratilova V., Banas P., Ionescu C.M., Otyepka M., Koca J. MOLE 2.0: advanced approach for analysis of biomacromolecular channels. J. cheminform. 2013;5:39. doi: 10.1186/1758-2946-5-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- te Velthuis A.J. Common and unique features of viral RNA-dependent polymerases. Cell. Mol. Life Sci. 2014;71:4403–4420. doi: 10.1007/s00018-014-1695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heel M., Harauz G., Orlova E.V., Schmidt R., Schatz M. A new generation of the IMAGIC image processing system. J. Struct. Biol. 1996;116:17–24. doi: 10.1006/jsbi.1996.0004. [DOI] [PubMed] [Google Scholar]
- York A., Hengrung N., Vreede F.T., Huiskonen J.T., Fodor E. Isolation and characterization of the positive-sense replicative intermediate of a negative-strand RNA virus. Proc. Natl. Acad. Sci. USA. 2013;110:E4238–E4245. doi: 10.1073/pnas.1315068110. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.