

Summary
Objective
Hyperactivation of the mechanistic target of rapamycin (mTOR) pathway has been demonstrated in human cortical dysplasia (CD) as well as in animal models of epilepsy. While inhibition of mTOR signaling early in epileptogenesis suppressed epileptiform activity in the neuron subset-specific Pten knockout (NS-Pten KO) mouse model of CD, the effects of mTOR inhibition after epilepsy is fully established were not previously examined in this model. Here, we investigated whether mTOR inhibition suppresses epileptiform activity and other neuropathological correlates in adult NS-Pten KO mice with severe and well-established epilepsy.
Methods
The progression of epileptiform activity, mTOR pathway dysregulation, and associated neuropathology with age in NS-Pten KO mice were evaluated using video-electroencephalography (EEG) recordings, western blotting, and immunohistochemistry. A cohort of NS-Pten KO mice was treated with the mTOR inhibitor rapamycin (10 mg/kg i.p., five days/week) starting at postnatal week 9 and video-EEG monitored for epileptiform activity. Western blotting and immunohistochemistry were performed to evaluate the effects of rapamycin on the associated pathology.
Results
Epileptiform activity worsened with age in NS-Pten KO mice, with parallel increases in the extent of hippocampal mTORC1 and mTORC2 dysregulation and progressive astrogliosis and microgliosis. Rapamycin treatment suppressed epileptiform activity, improved baseline EEG activity, and increased survival in severely epileptic NS-Pten KO mice. At the molecular level, rapamycin treatment was associated with a reduction in both mTORC1 and mTORC2 signaling and decreased astrogliosis and microgliosis.
Significance
These findings reveal a wide temporal window for successful therapeutic intervention with rapamycin in the NS-Pten KO mouse model and support mTOR inhibition as a candidate therapy for established, late-stage epilepsy associated with CD and genetic dysregulation of the mTOR pathway.
Keywords: malformation of cortical development, seizures, rapamycin, astrogliosis, microgliosis
Introduction
Cortical dysplasia (CD) is a malformation of cortical development that is a prevalent cause of intractable pediatric epilepsy1. CD has been linked to mutations in the genes encoding known regulators of the mechanistic target of rapamycin (mTOR; also known as mammalian target of rapamycin2) signaling pathway, including TSC1/23, PI3K, mTOR, AKT34, and STRADα5; 6, suggesting an important role for this pathway in the pathophysiology of CD and the associated epilepsy phenotype.
The mTOR pathway regulates a number of important functions, including cell growth, proliferation, protein synthesis, neuronal morphology, and cortical development2; 7, and has recently gained attention in epilepsy research as a candidate novel therapeutic target. In genetic mouse models, hyperactivation of mTOR signaling due to loss of the upstream regulators phosphatase and tensin homolog (PTEN) or tuberous sclerosis complex (TSC) has been associated with cortical malformations and the development of epilepsy8. In some rodent models of acquired temporal lobe epilepsy (TLE), aberrant mTOR signaling has been shown to play crucial roles in epileptogenesis and the maintenance of epilepsy9; 10. Evidence for increased mTOR activation has also been demonstrated in human CD4; 11–13, TSC13; 14, and TLE15; 16 tissue.
mTOR is a serine/threonine kinase that exerts it functions through two distinct complexes, mTORC1 and mTORC22. Association of mTOR with Raptor characterizes mTORC1, a rapamycin-sensitive complex that mediates protein synthesis by activating the translational machinery. Association of mTOR with Rictor characterizes mTORC2, a rapamycin-insensitive complex involved in cell survival, metabolism, and actin dynamics. Although mTORC2 is insensitive to acute rapamycin exposure, it can be disrupted by prolonged rapamycin treatment17.
To investigate the role of mTOR signaling in epilepsy and CD, we previously characterized neuron subset-specific Pten knockout (NS-Pten KO) mice that exhibited hyperactive mTOR signaling and mimicked features of human CD, including neuronal hypertrophy, cortical and hippocampal disorganization, aberrant mossy fiber sprouting, and epilepsy18–21. These abnormalities were evident after four to six weeks of postnatal development and worsened with age. Interestingly, treatment with rapamycin, an mTOR inhibitor, during postnatal weeks 4 and 5 resulted in short-term suppression of epileptiform activity, neuronal hypertrophy, and mossy fiber sprouting in these mice20; 21. Long-lasting suppression of epileptiform activity was further achieved with intermittent treatment consisting of two weeks on and four weeks off the drug21. Without treatment, epilepsy exacerbated and these mice died prematurely18–21. In both studies, treatment was initiated early during epilepsy development, when epileptiform activity and many of the associated pathologies were not fully developed. Thus, one important question that remains unanswered is whether mTOR inhibition would be effective if it is started at late stages of the pathology, when the brain circuitry is aberrantly wired and epilepsy is well-established. Therefore, in the present study, we investigated the effects of mTOR inhibition in adult NS-Pten KO mice with severe, chronic epilepsy and fully developed neuropathological correlates.
Methods
Animals
NS-Pten KO mice have been previously described18–20. Littermate wildtype (WT) and NS-Pten KO mice were generated by breeding heterozygotes. All experiments utilized mice of both genders. Animal care and use were in compliance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee at Baylor College of Medicine.
Rapamycin treatment
Rapamycin (LC Laboratories, Woburn, MA, USA) was dissolved in a vehicle solution of 4% ethanol, 5% polyethylene glycol 400, and 5% Tween 8020. 13 NS-Pten KO and 11 WT mice were treated with rapamycin (10 mg/kg body weight) by intraperitoneal (i.p.) injections five days per week, starting at postnatal week 9. 14 NS-Pten KO and 10 WT mice were treated with vehicle. 15 NS-Pten KO and 11 WT mice that did not receive any treatment were included as naive controls. No significant differences were observed between naïve and vehicle-treated mice for each genotype (data not shown), and therefore, these were pooled into control groups consisting of roughly equal numbers of naïve and vehicle-treated animals (hereafter referred to as WT or NS-Pten KO controls). Approximately half of the mice from each treatment group were used for video-EEG while the other half was used for western blotting.
Video-electroencephalography (EEG)
Cortical EEG electrodes were implanted at postnatal week 3 or 5–7 as previously described20; 21. Mice were monitored with synchronous video-EEG recording using Stellate or NicoletOne systems (Natus, San Carlos, CA, USA) for 4–6 consecutive hours per week thereafter. EEG activity was analyzed as described in the supporting material.
Western blotting
Western blotting on whole hippocampal tissue was performed as previously described22. Optical densities of all immunoreactive bands were normalized to actin levels for loading control. Phosphorylated protein levels were subsequently normalized to total protein levels. Results were expressed as percentages of 8–9 week-old WT mice (Figs. 2, 3) or control WT mice (Fig. 6).
Figure 2. Aberrant mTORC1 and mTORC2 signaling increases with age in NS-Pten KO mice.

(A, B) Representative western blots and quantification of (A) p-S6 and (B) p-AKT protein levels in whole hippocampal tissue from 2, 4, 6, and 8–9 week-old NS-Pten KO and WT mice are shown. (A) p-S6 levels were significantly higher in NS-Pten KO compared to age-matched WT mice at postnatal weeks 6 and 8–9. Note that at postnatal week 2, both NS-Pten KO and WT mice displayed significantly higher levels of p-S6 compared to 8–9 week-old WT mice. (B) p-AKT levels were significantly higher in NS-Pten KO compared to age-matched WT mice at postnatal weeks 4, 6, and 8–9. (A, B) n= 6–15 mice/group; *p<0.05, ***p<0.001 (compared to age-matched WT), ‡p<0.05, ‡‡‡p<0.001 (compared to 8–9 week-old WT) by two-way ANOVA with Bonferroni post-hoc test; error bars = ± SEM. (C, D) Single confocal images of (C) p-S6- or (D) p-AKT-stained coronal sections of 6 week-old NS-Pten KO and WT hippocampi are shown. High magnifications of DG gcl showing co-labeling with NeuN are presented in the right panels. (C) p-S6 staining appeared more intense within DG gcl with weaker labeling in the CA3 area in NS-Pten KO compared to WT mice. No visible differences were found in the CA1 area between the two groups. (D) p-AKT staining appeared more intense within both DG gcl and DG ml with weaker labeling in the CA3 area in NS-Pten KO compared to WT mice. No visible differences were found in the CA1 area between the two groups. (C, D) n=3 mice/group. Abbreviations: DG, dentate gyrus; gcl, granule cell layer; ml, molecular layer. Dotted lines outline the hippocampal fissure.
Figure 3. Glial markers increase with age in NS-Pten KO mice.

(A, B) Representative western blots and quantification of (A) GFAP and (B) IBA1 protein levels in whole hippocampal tissue from 2, 4, 6, and 8–9 week-old NS-Pten KO and WT mice are shown. (A) GFAP levels were significantly higher in NS-Pten KO compared to age-matched WT mice at postnatal weeks 6 and 8–9. (B) IBA1 levels were significantly higher in NS-Pten KO compared to age-matched WT mice at postnatal weeks 8–9. (A, B) n=6–15 mice/group; *p<0.05, ***p<0.001 by two-way ANOVA with Bonferroni post-hoc test; error bars = ± SEM. Black vertical line on the blot denotes gap in loading order; all bands are from the same gel and exposure time. (C, D) Photomicrographs of (C) GFAP- or (D) IBA1-stained coronal sections of 7–9 week-old NS-Pten KO and WT hippocampi are shown. Nissl-stained cell nuclei are shown in purple. High magnifications of the DG, CA1, and CA3 areas are shown in the right panels. Staining for (C) GFAP and (D) IBA1 was more intense in NS-Pten KO compared to WT mice. Several of the IBA1-stained microglia in NS-Pten KO hippocampi were hypertrophied and ameboid compared to normal appearing, ramified microglia observed in WT mice (insets). (C, D) n= 2–4 mice/group (2 WT, 3–4 KO).
Figure 6. Rapamycin treatment suppresses mTORC1 and mTORC2 signaling and glial markers in severely epileptic NS-Pten KO mice.
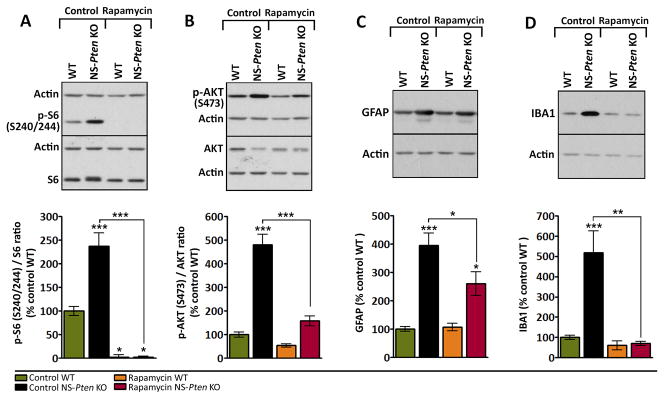
(A–D) Representative western blots and quantification of (A) p-S6, (B) p-AKT, (C) GFAP, and (D) IBA1 protein levels in whole hippocampal tissue from 11 week-old control and rapamycin-treated NS-Pten KO and WT mice are shown. (A) p-S6 levels were significantly higher in NS-Pten KO compared to WT mice in the control group. Rapamycin treatment suppressed p-S6 levels below control WT levels in both NS-Pten KO and WT mice. (B) p-AKT levels were significantly higher in NS-Pten KO compared to WT mice in the control group. Rapamycin treatment reduced p-AKT levels in NS-Pten KO mice to control WT levels but did not affect p-AKT levels in WT mice. (C) GFAP levels were significantly higher in NS-Pten KO compared to WT mice in the control group. Rapamycin treatment decreased GFAP levels in NS-Pten KO mice, however, the levels still remained higher than that of control WT mice. (D) IBA1 levels were significantly higher in NS-Pten KO compared to WT mice in the control group. Rapamycin treatment reduced IBA1 levels in NS-Pten KO mice to control WT levels. n=7–14 mice/group; *p<0.05, **p<0.01, ***p<0.001 (compared to control WT unless otherwise noted by connecting lines) by one-way ANOVA with Tukey’s post-hoc test; error bars = ± SEM.
Immunohistochemistry
Immunohistochemistry on paraformaldehyde-fixed brain sections were performed as previously described22. Cell nuclei were visualized by standard Nissl staining. Images were captured using an LSM 710 confocal system (Zeiss, Thornwood, NY, USA) or BX51 microscope (Olympus, Center Valley, PA, USA). All images directly compared were uniformly processed.
Statistics
Statistics were performed using Prism 5 software (GraphPad, La Jolla, CA, USA). Parametric tests were applied to data that conformed to normal distribution. Nonparametric tests were used for non-normal data. Specific tests were used as noted in the figure legends. The significance level was set at p< 0.05. Data are presented as mean ± standard error of the mean (SEM).
Additional methods can be found in the supporting material.
Results
Epilepsy progressively worsens with age in NS-Pten KO mice
NS-Pten KO mice exhibit abnormal EEG activity and spontaneous seizures20; 21. To further characterize the progression of the epilepsy phenotype, we monitored NS-Pten KO and WT mice with video-EEG recordings starting at postnatal week 3 for several weeks. Epileptiform activity in the form of spikes, repetitive spike and polyspike activity, and seizures was present in NS-Pten KO mice early on and became more frequent with age. In comparison, no abnormal EEG activity was observed in WT mice at any age (Fig. 1A). Quantification of EEG activity revealed a significant increase in the time NS-Pten KO mice spent in epileptiform activity at postnatal week 9 compared to postnatal week 4 (p<0.001) (Fig. 1B). In 9 week and older NS-Pten KO mice, subclinical polyspike and seizure activity occurred almost continuously throughout the recording periods (Fig. 1C). Superimposed upon the subclinical epileptiform activity, NS-Pten KO mice also displayed intermittent motor seizures involving myoclonic jerks, overt tail and forelimb tonus-clonus, wild running, and loss of postural control. Electrographically, the motor seizures were typically associated with a characteristic increase in spike frequency and amplitude that progressed into spike-and-wave patterns lasting ≥10 sec in total and followed by post-ictal depression of EEG activity (Fig. 1D). These motor seizures occurred more frequently at later time points, during postnatal weeks 8–11, compared to earlier, during postnatal weeks 3–6 (p=0.045) (Fig. 1E). Taken together, these data indicate the presence of a progressively worsening epilepsy phenotype in NS-Pten KO mice.
Figure 1. Epilepsy progressively worsens with age in NS-Pten KO mice.

(A) Representative EEG traces from 9 week-old WT and 4, 6, and 9 week-old NS-Pten KO mice are shown. NS-Pten KO mice exhibit progressively worsening epileptiform activity with age, while no abnormal EEG activity was observed in WT mice at any age. (B) Quantification of EEG activity in NS-Pten KO mice revealed a significant increase in the time animals spent in epileptiform activity at postnatal week 9 compared to postnatal week 4. n=12–14 mice/group; ***p<0.001 by Kruskal-Wallis ANOVA with Dunn’s post-hoc test; error bars = ± SEM. (C) Compressed EEG traces from a 90-min epoch of a recording session illustrating normal background EEG activity in WT mice and continuous subclinical polyspike and seizure activity in NS-Pten KO mice at late pathological stages are shown. (D) Representative EEG activity during a motor seizure that was associated with myoclonic jerks, tail and forelimb tonus-clonus, and loss of postural control is shown. Motor seizures were superimposed upon the continuous subclinical polyspike and seizure activity and were typically associated with a characteristic increase in spike frequency and amplitude that progressed into spike-and wave patterns, followed by post-ictal depression of EEG activity. (E) The frequency of motor seizures was significantly higher during postnatal weeks 8–11 compared to postnatal weeks 3–6. n=26–34 mice/group; *p=0.045 by Mann Whitney test; error bars = ± SEM.
Aberrant mTORC1 and mTORC2 signaling increases with age in NS-Pten KO mice
Given that aberrant mTOR signaling plays a crucial role in the development of epilepsy in NS-Pten KO mice20; 21, we investigated whether the observed progressive epilepsy phenotype (Fig. 1) was associated with parallel changes in the extent of mTOR dysregulation. We narrowed our studies to the hippocampal formation as there is marked deletion of Pten in dentate gyrus (DG) granule cells18; 19 and seizures have been shown to originate in both the hippocampus and cortex in NS-Pten KO mice21. In addition, Pten loss and seizures in NS-Pten KO mice have been associated with hippocampal abnormalities, including pronounced disorganization, dysplasia, cytomegaly, and aberrant mossy fiber sprouting18–21. To evaluate the levels of mTOR activation in the hippocampus of NS-Pten KO and WT mice over time, western blotting was performed using whole hippocampal tissue collected at postnatal weeks 2, 4, 6, and 8–9. Phosphorylation levels of the S6 ribosomal protein [p-S6 (S240/244)]23 and AKT [p-AKT (S473)]24 were used as markers for mTORC1 and mTORC2 activation, respectively. We also compared the distribution of p-S6 and p-AKT in 6 week-old NS-Pten KO and WT hippocampi using immunohistochemistry.
Levels of p-S6 in WT and NS-Pten KO hippocampi at postnatal week 2 were significantly higher compared to adult WT levels at postnatal weeks 8–9 (p<0.001). In WT mice, p-S6 levels decreased with age, consistent with previous findings in rats25. However, in NS-Pten KO mice, p-S6 levels remained persistently elevated throughout all of the evaluated time points. By postnatal weeks 6 and 8–9, p-S6 levels were significantly higher in NS-Pten KO compared to age-matched WT mice (p<0.001) (Fig. 2A). Hippocampal levels of p-AKT remained unchanged between postnatal weeks 2 and 8–9 in WT mice but increased with age in NS-Pten KO mice. By postnatal week 4, p-AKT levels were significantly higher in NS-Pten KO compared to age-matched WT mice (p<0.05). This difference was larger by postnatal weeks 6 and 8–9 (p<0.001) (Fig. 2B).
In 6 week-old WT mice, basal staining for p-S6 was found in the CA1 and CA3 pyramidal cell layers as well as in the DG granule cell layer at relatively lower intensity (Fig. 2C top left panel). In contrast, markedly stronger staining for p-S6 was found in the DG granule cell layer in NS-Pten KO mice. Relatively less p-S6 staining was found in the CA3 area of NS-Pten KO compared to WT mice, while no visible differences were found in the CA1 area between the two groups (Fig. 2C bottom left panel). In both WT and NS-Pten KO mice, p-S6 was enriched within DG granule cells, as evidenced by co-localization of p-S6 with the neuronal marker NeuN. DG granule cells also appeared notably larger in NS-Pten KO mice, consistent with previous studies18–20 (Fig. 2C right panels). Basal staining for p-AKT in WT mice was found in the CA1, CA3, and DG cell layers, with signals being most prominent in the CA3 area. (Fig. 2D top left panel). In comparison, p-AKT staining in NS-Pten KO mice was stronger in the molecular and granule cell layers of the DG area (Fig. 2D bottom left panel). Less p-AKT staining was found in the CA3 area of NS-Pten KO compared to WT mice, while no visible differences were observed in the CA1 area between the two groups. In both WT and NS-Pten KO mice, p-AKT staining was found within DG granule cells (Fig. 2D right panels).
Taken together, these data reveal an age-dependent increase in mTORC1 and mTORC2 dysregulation in the hippocampus of NS-Pten KO mice in parallel with the development of a progressively worsening epilepsy phenotype. Furthermore, aberrant mTORC1 and mTORC2 activation was mainly distributed in the DG area, consistent with a region where substantial Pten loss has been reported in this model18; 19.
Glial markers increase with age in NS-Pten KO mice
As mTOR signaling has been shown to play a role in a number of glial properties26 and hippocampal gliosis has been widely associated with seizures and epilepsy27, we performed western blotting on whole hippocampal tissue from NS-Pten KO and WT mice collected at postnatal weeks 2, 4, 6, and 8–9 to evaluate the protein levels of the astrocyte and microglia markers GFAP and IBA1, respectively. We found no changes in the levels of GFAP between age-matched NS-Pten KO and WT mice at postnatal weeks 2 and 4. However, at postnatal weeks 6 and 8–9, GFAP levels in NS-Pten KO mice were significantly increased compared to age-matched WT mice (p<0.001) (Fig. 3A). Similarly, IBA1 levels were unchanged between age-matched NS-Pten KO and WT mice at postnatal weeks 2, 4, and 6, but were significantly increased in NS-Pten KO compared to WT mice at postnatal weeks 8–9 (p<0.05) (Fig. 3B).
Immunohistochemistry on 7–9 week-old brains revealed uniform distribution of GFAP-labeled astrocytes and IBA1-labeled microglia throughout the hippocampus in both NS-Pten KO and WT mice, however, the staining appeared more intense in NS-Pten KO hippocampi (Fig. 3C, D). Notably, many of the microglia in the CA1 area of NS-Pten KO mice were hypertrophied and ameboid compared to ramified microglia in WT mice (Fig. 3D insets).
Taken together, these data reveal increased levels of astroglial and microglial markers in older NS-Pten KO that were not found in younger mice, suggesting an age-dependent occurrence of gliosis in parallel with worsening epilepsy and mTOR dysregulation. Furthermore, changes in microglia morphology that are consistent with microglial activation28 were observed more frequently in NS-Pten KO hippocampi, suggesting the presence of reactive microgliosis.
Rapamycin treatment suppresses epileptiform activity and improves baseline EEG activity in severely epileptic NS-Pten KO mice
We previously demonstrated that early onset of mTOR inhibition with rapamycin treatment at postnatal week 4 suppressed epileptiform activity and prevented epilepsy progression in NS-Pten KO mice20; 21. To determine whether mTOR inhibition at late and advanced stages of the pathology also attenuated epileptiform activity in NS-Pten KO mice, we delayed rapamycin treatment until postnatal week 9 in the present study (Fig. 4A). By this age, the epilepsy phenotype was fully established and severe as evidenced by continuous subclinical epileptiform activity and superimposed recurrent motor seizures (Fig 1). While no abnormal EEG activity was observed in control and rapamycin-treated WT mice, we found marked epileptiform activity in control NS-Pten KO mice that was attenuated with rapamycin treatment (Fig. 4B). Quantification of EEG activity after 3–4 weeks of treatment, at postnatal weeks 12–13, revealed a significant reduction in the time rapamycin-treated NS-Pten KO mice spent in epileptiform activity compared to age-matched control NS-Pten KO mice (p=0.008) (Fig. 4C). Further analyses comparing epileptiform activity after 3–4 weeks of treatment to within-subject activity before treatment showed a significant decrease in epileptiform activity following treatment in NS-Pten KO mice (p=0.005). In comparison, no significant changes were found in control NS-Pten KO mice (Fig. 4D). In support of improved epileptiform activity, rapamycin-treated NS-Pten KO mice exhibited a significant decrease in spike (p=0.0043) (Fig. 4E) and motor seizure (p=0.026) (Fig. 4F) frequency compared to control NS-Pten KO mice. Taken together, these data suggest that rapamycin treatment at late stages of the pathology suppresses the epilepsy phenotype in NS-Pten KO mice.
Figure 4. Rapamycin treatment suppresses epileptiform activity and improves baseline EEG activity in severely epileptic NS-Pten KO mice.

(A) Timeline for video-EEG monitoring and rapamycin treatment. Mice were monitored weekly with video-EEG recordings between postnatal weeks 8 and 13. Rapamycin was administered at postnatal week 9 until death. All EEG analyses were performed at postnatal week 12–13, after 3–4 weeks of treatment, except for motor seizure frequency, which was evaluated during the entire period between postnatal weeks 9–13. (B) Representative EEG traces illustrating normal activity in control and rapamycin-treated WT mice, epileptiform activity in control NS-Pten KO mice, and attenuated epileptiform activity in rapamycin-treated NS-Pten KO mice are shown. (C) Time spent in epileptiform activity was significantly reduced in rapamycin-treated NS-Pten KO mice compared to control NS-Pten KO mice. n=5–6 mice/group; **p=0.008 by Mann-Whitney test. (D) Within-subject comparisons between postnatal week 8, before treatment, and postnatal weeks 12–13, after 3–4 weeks of treatment, revealed a significant decrease in epileptiform activity following rapamycin treatment in NS-Pten KO mice. No changes were observed in control NS-Pten KO mice. Data are presented as percentages of postnatal week 8. n=5–6 mice/group; **p=0.0043 (compared to age-matched WT) by Mann-Whitney test, ‡‡p=0.005 (compared to 100% at postnatal week 8) by one sample t-test. (E) Spike frequency was significantly decreased in rapamycin-treated NS-Pten KO mice compared to control NS-Pten KO mice. n=5–6 mice/group; **p=0.0043 by Mann-Whitney test. (F) Motor seizure frequency was significantly decreased in rapamycin-treated NS-Pten KO mice compared to control NS-Pten KO mice. n=6–11 mice/group; *p=0.026 by Mann-Whitney test. (G) Total power was significantly increased in control NS-Pten KO compared to control WT mice. This aberrant increase was significantly suppressed in the rapamycin-treated NS-Pten KO mice. n= 4–6 mice/group; *p<0.05 by one-way ANOVA with Tukey’s post-hoc test. For all graphs, error bars = ± SEM.
Total power analysis of baseline EEG activity revealed no significant differences between control WT and NS-Pten KO mice at postnatal week 8. However, by postnatal weeks 12–13, there was a significant decrease in WT (p<0.001) and an increase in NS-Pten KO (p<0.05) total power (Fig. S1). Rapamycin treatment significantly suppressed the aberrant total power increase in NS-Pten KO mice compared to control NS-Pten KO mice (p<0.05) such that no differences were observed between rapamycin-treated NS-Pten KO mice and control WT mice by postnatal weeks 12–13 (Fig. 4G). These data suggest that late rapamycin treatment also improves baseline EEG activity in NS-Pten KO mice.
Rapamycin treatment increases survival in severely epileptic NS-Pten KO mice
NS-Pten KO mice undergo premature death, with an average lifespan of 10.7 ± 1.9 weeks18; 19. Given that early exposure to rapamycin has been shown to increase the longevity of NS-Pten KO mice21, we determined whether late onset of rapamycin treatment also had an effect on their survival. Of the control NS-Pten KO mice in this study, only 33% (4 out of 12 mice) survived to postnatal 16. Rapamycin-treated NS-Pten KO mice lived significantly longer than control NS-Pten KO mice (p=0.034), with 86% (6 out of 7 mice) of these mice surviving to postnatal week 16. In comparison, all of the control and rapamycin-treated WT mice in this study lived beyond postnatal week 16 (Fig. 5). Taken together, these data suggest that late rapamycin treatment in severely epileptic NS-Pten KO mice effectively prolongs their survival.
Figure 5. Rapamycin treatment increases survival in severely epileptic NS-Pten KO mice.
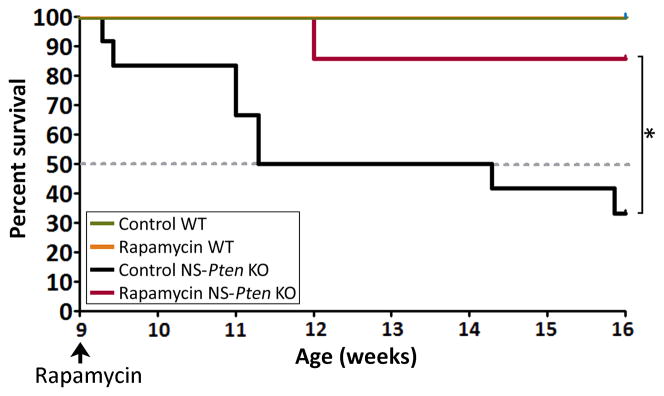
Kaplan-Meier survival curves for control and rapamycin-treated NS-Pten KO mice are shown. 86% of the rapamycin-treated NS-Pten KO mice (6 out of 7 mice) lived to postnatal week 16 compared to 33% of the control NS-Pten KO mice (4 out of 12 mice). Data from control and rapamycin-treated WT mice are plotted for comparison; all WT mice lived beyond postnatal week 16. n=3–12 mice per group; *p=0.034 by Kaplan-Meier log-rank test.
Rapamycin treatment suppresses mTORC1 and mTORC2 signaling and glial markers in severely epileptic NS-Pten KO mice
Given that long-term rapamycin treatment may affect both mTORC1 and mTORC2 signaling2; 17, we evaluated the effects of prolonged late rapamycin treatment on these pathways in NS-Pten KO and WT mice. Western blot analyses revealed a rapamycin-mediated reduction in p-S6 below control WT levels in NS-Pten KO and WT hippocampi after two weeks of treatment (p<0.05) (Fig. 6A). Hippocampal p-AKT levels were not significantly altered by rapamycin treatment in WT mice but were significantly reduced in rapamycin-treated NS-Pten KO mice compared to control NS-Pten KO mice (p<0.001) (Fig. 6B).
Given that hippocampal GFAP and IBA1 protein levels were altered in NS-Pten KO mice (Fig. 3) and mTOR activation has been shown to modulate glial activity26, we also examined the effects of rapamycin treatment on GFAP and IBA1 levels in NS-Pten KO and WT hippocampi. Rapamycin treatment had no significant effects on GFAP or IBA1 levels in WT mice. However, the aberrantly elevated levels of GFAP and IBA1 in NS-Pten KO mice were significantly reduced following rapamycin treatment (p<0.05 and p<0.01, respectively) (Fig. 6C, D). GFAP levels in rapamycin-treated NS-Pten KO mice were significantly reduced compared to control NS-Pten KO mice (p<0.05) but the levels remained significantly higher than that of control WT mice (p<0.05), thereby suggesting a partial effect (Fig. 6C). IBA1 levels in rapamycin-treated NS-Pten KO mice were suppressed to levels resembling that of control WT mice (Fig. 6D).
Taken together, these data confirm suppression of both mTORC1 and mTORC2 signaling in NS-Pten KO mice with the late rapamycin treatment paradigm. This treatment was further associated with a reduction in hippocampal GFAP and IBA1 levels, suggesting that mTOR inhibition decreases ongoing expression of glial markers in NS-Pten KO mice.
Discussion
mTOR inhibitors have emerged as promising novel therapies for epilepsy, and efforts are ongoing to better understand their clinical utility in mTORopathies (disorders of mTOR signaling) such as CD and TSC. In the present study, we evaluated whether late intervention with the mTOR inhibitor rapamycin would effectively suppress epilepsy in the NS-Pten KO mouse model of CD once the pathology has become established and severe. Here, we have shown that epileptiform activity worsens with age in NS-Pten KO mice with parallel increases in mTOR pathway dysregulation, astrogliosis, and microgliosis. Furthermore, we have demonstrated that rapamycin treatment at advanced stages of the pathology attenuates epileptiform activity, reduces aberrant levels of glial markers, and extends the lifespan of these mice. Together, these findings support rapamycin treatment as an effective therapy for established epilepsy in this model. Furthermore, our findings that demonstrate the presence of astrogliosis and microgliosis in NS-Pten KO mice, which can be suppressed by rapamycin treatment, add a novel aspect to the epilepsy phenotype that has not previously been described in this model.
NS-Pten KO mice demonstrate an epilepsy phenotype in association with neuronal Pten deletion and mTOR hyperactivation11; 18–20. We found that NS-Pten KO mice progressed from having occasional spike activity at postnatal week 4 to continuous polyspike and seizure activity later in adult life, suggesting the presence of an ongoing, progressive epileptogenic process in these animals. Consistent with worsening epilepsy, we also found an increase in the frequency of superimposed recurrent motor seizures in older NS-Pten KO mice. However, the sensitivity of this analysis may be limited due to intermittent recording time and it is possible that the actual frequency is higher. Given that seizures tend to be variable and occur in clusters29–32, longer recording periods may provide more definitive results.
Given the involvement of mTOR signaling in neuronal hyperexcitability and epilepsy development33, the worsening epilepsy phenotype in NS-Pten KO mice may be related to the persistent increases in mTOR dysregulation observed in these animals. As increased mTOR activation following seizure induction has been shown in models of TLE acquired secondary to chemoconvulsant stimulants9; 10; 22; 34, one possible explanation for why mTOR signaling continues to increase over time in NS-Pten KO mice may be that seizures per se induce more pathway dysregulation. This in turn could contribute to worsening of seizures, more mTOR dysregulation, and perpetuation of the cycle. As the majority of Pten loss in this model occurs in the DG, an area where adult neurogenesis has been extensively described35, an additional explanation may be that new postnatally born neurons add to the existing pool of Pten-negative neurons with aberrant mTOR activation and thereby exacerbate mTOR dysregulation and epilepsy. In support of this idea, mTOR hyperactivation due to Pten deletion in a small number of postnatally born DG granule cells has been shown to cause epilepsy36. Future studies are needed to evaluate whether postnatally born neurons in the current model are indeed Pten-negative and whether this correlates with the levels of mTOR hyperactivity and epilepsy severity.
Worsening of the epilepsy phenotype could also partly be due to the induction of secondary mechanisms that exacerbate seizures. As glial contributions to neuronal hyperexcitability and epileptogenesis have been implicated27; 37, the astrogliosis and microgliosis that was found in NS-Pten KO mice may contribute to worsening epilepsy. Although increases in the glial markers could arguably be an indirect effect of neuronal Pten loss, these events did not occur until seizures became significantly prominent, thus supporting these findings as effects secondary to seizures. Furthermore, while mTOR dysregulation was mainly observed in the DG and less so in the CA1 and CA3 areas, astrogliosis and microgliosis were rather diffuse in the hippocampus. Given the established role for astrocytes and microglia in brain inflammation and the involvement of inflammatory mediators in epilepsy27, it would be interesting to investigate whether glial-mediated brain inflammation occurs in NS-Pten KO mice and whether it plays a role in the epilepsy progression.
Intervention with mTOR inhibitors early in epilepsy development has successfully prevented epilepsy in this20; 21 and related genetic models8, and therefore, mTOR inhibitors have emerged as potential disease-modifying agents for human epilepsies associated with mutations in the mTOR pathway. In a recent clinical trial, everolimus (a rapamycin-derivative) treatment effectively reduced seizures by more than 50% in 12 out of 20 TSC patients with refractory epilepsy38. However, some patients did not respond to treatment, raising the question of whether the timing of intervention is critical for a positive outcome. Successful rapamycin-mediated suppression of epilepsy has previously been described in symptomatic TSC KO mice39 and in a different Pten KO mouse model40, but these treatments were started relatively soon after the onset of epilepsy. Thus, to determine whether rapamycin treatment suppresses advanced and fully established epilepsy, we waited longer before initiating treatment. By the time rapamycin was administered in the present study, NS-Pten KO mice exhibited severe mTOR pathway dysregulation and chronic epileptiform activity. We found that rapamycin treatment was effective in suppressing mTORC1 and mTORC2 hyperactivation and epilepsy even after the pathology had become well-established. This suggests, in agreement with other studies, that mTOR plays a critical role in the maintenance of epilepsy and serves as important antiepileptic target in some epilepsies. Interestingly, rapamycin treatment also reduced aberrant levels of glial drug26; 41. Thus, rapamycin-mediated suppression of these events may also contribute to the improvement of epilepsy in NS-Pten KO mice.
NS-Pten KO mice die prematurely on average by postnatal weeks 10–1118; 19. In a previous study, an early single course of rapamycin treatment did not affect the survival of these mice. However, with additional intermittent treatment, their lifespan was significantly increased21. Our present findings that late rapamycin treatment also prolonged survival in NS-Pten KO mice further suggest that while repeated exposure to rapamycin may be necessary for these beneficial effects, early initiation is not required.
Traditionally, research in animal models of epilepsy has mainly focused on mTORC1 due to its sensitivity to rapamycin9; 10; 20–22; 39; 40. However, prolonged rapamycin treatment also perturbs mTORC2, as shown in this and previous studies17; 40. mTORC2 regulates actin cytoskeletal dynamics that are implicated in neuronal morphology and synapse functions2; 7, and dysregulation of mTORC2 signaling may therefore be an important contributor to epilepsy. Thus, it is possible that some of the effects observed with rapamycin treatment in NS-Pten KO mice are mediated, at least in part, through mTORC2 suppression. Additional studies focused on understanding the roles of mTORC1 versus mTORC2 in the pathogenesis of epilepsy are needed, however, this has been challenging due to the lack of mTORC2-specific inhibitors.
In conclusion, we have provided evidence for a wide temporal window for successful therapeutic intervention with rapamycin in the NS-Pten KO mouse model of CD. While additional studies are required to identify the molecular targets downstream of the mTOR pathway that are responsible for establishing epileptic networks, our findings provide preclinical support for mTOR inhibition as an effective treatment for established, late-stage epilepsy in a genetic model of CD.
Supplementary Material
Figure S1: Changes in total EEG power with age in NS-Pten KO and WT mice
Acknowledgments
This work was supported by National Institutes of Health (NIH) R01 NS81053; 39943 (AEA), Vivian L. Smith Foundation Research Grant (AEA), CURE Challenge Award (AEA, GD), Epilepsy Foundation Predoctoral (LHN, CNS, VPP) and Postdoctoral (ALB) Research and Training Fellowships, and in part by NIH P30 HD024064 and U54 HD083092 from the Eunice Kennedy Shriver National Institute of Child Health and Human Development to the Intellectual and Developmental Disabilities Research Center (IDDRC) at Baylor College of Medicine. We thank Dr. Genevera Allen for statistical consultation and Dr. Yichen Lai for insightful discussions. The funding entities had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Disclosure
None of the authors has any conflict of interest to disclose. We confirm that we have read the Journal’s position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding entities.
References
- 1.Crino PB, Chou K. Epilepsy and Cortical Dysplasias. Curr Treat Options Neurol. 2000;2:543–552. doi: 10.1007/s11940-000-0032-z. [DOI] [PubMed] [Google Scholar]
- 2.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–293. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Orlova KA, Crino PB. The tuberous sclerosis complex. Ann N Y Acad Sci. 2010;1184:87–105. doi: 10.1111/j.1749-6632.2009.05117.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee JH, Huynh M, Silhavy JL, et al. De novo somatic mutations in components of the PI3K-AKT3-mTOR pathway cause hemimegalencephaly. Nat Genet. 2012;44:941–945. doi: 10.1038/ng.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Orlova KA, Parker WE, Heuer GG, et al. STRADalpha deficiency results in aberrant mTORC1 signaling during corticogenesis in humans and mice. J Clin Invest. 2010;120:1591–1602. doi: 10.1172/JCI41592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Puffenberger EG, Strauss KA, Ramsey KE, et al. Polyhydramnios, megalencephaly and symptomatic epilepsy caused by a homozygous 7-kilobase deletion in LYK5. Brain. 2007;130:1929–1941. doi: 10.1093/brain/awm100. [DOI] [PubMed] [Google Scholar]
- 7.Wong M. Mammalian target of rapamycin (mTOR) pathways in neurological diseases. Biomed J. 2013;36:40–50. doi: 10.4103/2319-4170.110365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wong M, Crino PB. mTOR and Epileptogenesis in Developmental Brain Malformations. In: Noebels JL, Avoli M, Rogawski MA, et al., editors. Jasper’s Basic Mechanisms of the Epilepsies. Bethesda (MD): 2012. [PubMed] [Google Scholar]
- 9.Zeng LH, Rensing NR, Wong M. The mammalian target of rapamycin signaling pathway mediates epileptogenesis in a model of temporal lobe epilepsy. J Neurosci. 2009;29:6964–6972. doi: 10.1523/JNEUROSCI.0066-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang X, Zhang H, Yang J, et al. Pharmacological inhibition of the mammalian target of rapamycin pathway suppresses acquired epilepsy. Neurobiol Dis. 2010;40:193–199. doi: 10.1016/j.nbd.2010.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ljungberg MC, Bhattacharjee MB, Lu Y, et al. Activation of mammalian target of rapamycin in cytomegalic neurons of human cortical dysplasia. Ann Neurol. 2006;60:420–429. doi: 10.1002/ana.20949. [DOI] [PubMed] [Google Scholar]
- 12.Aronica E, Boer K, Baybis M, et al. Co-expression of cyclin D1 and phosphorylated ribosomal S6 proteins in hemimegalencephaly. Acta Neuropathol. 2007;114:287–293. doi: 10.1007/s00401-007-0225-6. [DOI] [PubMed] [Google Scholar]
- 13.Miyata H, Chiang AC, Vinters HV. Insulin signaling pathways in cortical dysplasia and TSC-tubers: tissue microarray analysis. Ann Neurol. 2004;56:510–519. doi: 10.1002/ana.20234. [DOI] [PubMed] [Google Scholar]
- 14.Baybis M, Yu J, Lee A, et al. mTOR cascade activation distinguishes tubers from focal cortical dysplasia. Ann Neurol. 2004;56:478–487. doi: 10.1002/ana.20211. [DOI] [PubMed] [Google Scholar]
- 15.Sha LZ, Xing XL, Zhang D, et al. Mapping the spatio-temporal pattern of the mammalian target of rapamycin (mTOR) activation in temporal lobe epilepsy. PLoS One. 2012;7:e39152. doi: 10.1371/journal.pone.0039152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sosunov AA, Wu X, McGovern RA, et al. The mTOR pathway is activated in glial cells in mesial temporal sclerosis. Epilepsia. 2012;53 (Suppl 1):78–86. doi: 10.1111/j.1528-1167.2012.03478.x. [DOI] [PubMed] [Google Scholar]
- 17.Sarbassov DD, Ali SM, Sengupta S, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 18.Backman SA, Stambolic V, Suzuki A, et al. Deletion of Pten in mouse brain causes seizures, ataxia and defects in soma size resembling Lhermitte-Duclos disease. Nat Genet. 2001;29:396–403. doi: 10.1038/ng782. [DOI] [PubMed] [Google Scholar]
- 19.Kwon CH, Zhu X, Zhang J, et al. Pten regulates neuronal soma size: a mouse model of Lhermitte-Duclos disease. Nat Genet. 2001;29:404–411. doi: 10.1038/ng781. [DOI] [PubMed] [Google Scholar]
- 20.Ljungberg MC, Sunnen CN, Lugo JN, et al. Rapamycin suppresses seizures and neuronal hypertrophy in a mouse model of cortical dysplasia. Dis Model Mech. 2009;2:389–398. doi: 10.1242/dmm.002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sunnen CN, Brewster AL, Lugo JN, et al. Inhibition of the mammalian target of rapamycin blocks epilepsy progression in NS-Pten conditional knockout mice. Epilepsia. 2011;52:2065–2075. doi: 10.1111/j.1528-1167.2011.03280.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brewster AL, Lugo JN, Patil VV, et al. Rapamycin reverses status epilepticus-induced memory deficits and dendritic damage. PLoS One. 2013;8:e57808. doi: 10.1371/journal.pone.0057808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hoeffer CA, Klann E. mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 2010;33:67–75. doi: 10.1016/j.tins.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 25.Talos DM, Sun H, Zhou X, et al. The interaction between early life epilepsy and autistic-like behavioral consequences: a role for the mammalian target of rapamycin (mTOR) pathway. PLoS One. 2012;7:e35885. doi: 10.1371/journal.pone.0035885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dello Russo C, Lisi L, Feinstein DL, et al. mTOR kinase, a key player in the regulation of glial functions: relevance for the therapy of multiple sclerosis. Glia. 2013;61:301–311. doi: 10.1002/glia.22433. [DOI] [PubMed] [Google Scholar]
- 27.Vezzani A, French J, Bartfai T, et al. The role of inflammation in epilepsy. Nat Rev Neurol. 2011;7:31–40. doi: 10.1038/nrneurol.2010.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kettenmann H, Hanisch UK, Noda M, et al. Physiology of microglia. Physiol Rev. 2011;91:461–553. doi: 10.1152/physrev.00011.2010. [DOI] [PubMed] [Google Scholar]
- 29.Bajorat R, Wilde M, Sellmann T, et al. Seizure frequency in pilocarpine-treated rats is independent of circadian rhythm. Epilepsia. 52:e118–122. doi: 10.1111/j.1528-1167.2011.03200.x. [DOI] [PubMed] [Google Scholar]
- 30.Goffin K, Nissinen J, Van Laere K, et al. Cyclicity of spontaneous recurrent seizures in pilocarpine model of temporal lobe epilepsy in rat. Exp Neurol. 2007;205:501–505. doi: 10.1016/j.expneurol.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 31.Williams PA, White AM, Clark S, et al. Development of spontaneous recurrent seizures after kainate-induced status epilepticus. J Neurosci. 2009;29:2103–2112. doi: 10.1523/JNEUROSCI.0980-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kadam SD, White AM, Staley KJ, et al. Continuous electroencephalographic monitoring with radio-telemetry in a rat model of perinatal hypoxia-ischemia reveals progressive post-stroke epilepsy. J Neurosci. 30:404–415. doi: 10.1523/JNEUROSCI.4093-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lasarge CL, Danzer SC. Mechanisms regulating neuronal excitability and seizure development following mTOR pathway hyperactivation. Front Mol Neurosci. 7:18. doi: 10.3389/fnmol.2014.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buckmaster PS, Ingram EA, Wen X. Inhibition of the mammalian target of rapamycin signaling pathway suppresses dentate granule cell axon sprouting in a rodent model of temporal lobe epilepsy. J Neurosci. 2009;29:8259–8269. doi: 10.1523/JNEUROSCI.4179-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ming GL, Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 2011;70:687–702. doi: 10.1016/j.neuron.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pun RY, Rolle IJ, Lasarge CL, et al. Excessive activation of mTOR in postnatally generated granule cells is sufficient to cause epilepsy. Neuron. 2012;75:1022–1034. doi: 10.1016/j.neuron.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Devinsky O, Vezzani A, Najjar S, et al. Glia and epilepsy: excitability and inflammation. Trends Neurosci. 2013;36:174–184. doi: 10.1016/j.tins.2012.11.008. [DOI] [PubMed] [Google Scholar]
- 38.Krueger DA, Wilfong AA, Holland-Bouley K, et al. Everolimus treatment of refractory epilepsy in tuberous sclerosis complex. Ann Neurol. 2013;74:679–687. doi: 10.1002/ana.23960. [DOI] [PubMed] [Google Scholar]
- 39.Zeng LH, Xu L, Gutmann DH, et al. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol. 2008;63:444–453. doi: 10.1002/ana.21331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou J, Blundell J, Ogawa S, et al. Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J Neurosci. 2009;29:1773–1783. doi: 10.1523/JNEUROSCI.5685-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hailer NP. Immunosuppression after traumatic or ischemic CNS damage: it is neuroprotective and illuminates the role of microglial cells. Prog Neurobiol. 2008;84:211–233. doi: 10.1016/j.pneurobio.2007.12.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Changes in total EEG power with age in NS-Pten KO and WT mice