

Abstract
Background
Recent advances in genetics are particularly relevant in the field of intellectual disability (ID), where sub-microscopic deletions or duplications of genetic material are increasingly implicated as known or suspected causal factors. Data-driven reports on the impact of providing an aetiological explanation in ID are needed to help justify widespread use of new and expensive genetic technologies.
Methods
We conducted a survey of caregivers on the value of a genetic/aetiologic diagnosis of 22q11.2 deletion syndrome (22q11.2DS), the most common microdeletion syndrome in ID. We also surveyed the opinion of a high-functioning subset of adults with 22q11.2DS themselves. We used standard quantitative and qualitative methods to analyse the responses.
Results
In total, 73 of 118 surveys were returned (61.9%). There was convergence of quantitative and qualitative results, and consistency between adult patient and caregiver responses. A definitive molecular diagnosis of 22q11.2DS was a critical event with diverse positive repercussions, even if occurring later in life. Frequently cited benefits included greater understanding and certainty, newfound sense of purpose and a platform for advocacy, and increased opportunities to optimise medical, social and educational needs.
Conclusions
This is the first study to characterise the impact of a diagnosis of this representative microdeletion syndrome on adult patients and their families. The results both validate and expand on the theoretical benefits proposed by clinicians and researchers. The use of genome-wide microarray technologies will provide an increasing number of molecular diagnoses. The importance of a diagnosis of 22q11.2DS demonstrated here therefore has implications for changing attitudes about molecular genetic diagnosis that could benefit individuals with ID of currently unknown cause and their families.
Keywords: 22q11 deletion syndrome, adult, caregivers, chromosome deletion, genetic services, intellectual disability
Introduction
In an era of advanced genetic technologies such as microarrays, anomalies of chromosome structure are an even more important part of clinical practice than before (Lee & Scherer 2010; D.T. Miller et al. 2010). Recent advances in genetics are particularly relevant in the field of intellectual disability (ID) (Raymond 2003), where sub-microscopic deletions or duplications of genetic material are increasingly implicated as known or suspected causal factors (Hochstenbach et al. 2009). Use of microarray technology doubles the rate of detection of likely causal genetic anomalies in ID over traditional genome-wide diagnostic tests (i.e. karyotype) (Hochstenbach et al. 2009), and the prevalence of children with ID who have a genetic diagnosis is increasing in clinical practice (Verri et al. 2004; Henderson & Dick 2005). A recent consensus statement proposed that microarray should now be considered the first-tier genetic diagnostic test for ID (D.T. Miller et al. 2010).
The most common microdeletion syndrome in ID (approximately 1 in 40) and in the general population (approximately 1 in 2000–4000) is 22q11.2 deletion syndrome (22q11.2DS) (MIM #188400/#192430) (Kobrynski & Sullivan 2007; Lopez-Rangel et al. 2008). Most individuals with 22q11.2DS have mild to borderline ID (Chow et al. 2006; De Smedt et al. 2007; Antshel et al. 2010); severe levels of ID occur but are uncommon (Bassett et al. 2005; Chow et al. 2006; De Smedt et al. 2007). Even the minority of individuals with intellect in the average range usually have learning difficulties of various sorts, most notably in arithmetic, abstraction and social cognition (Bassett et al. 2005; Chow et al. 2006; De Smedt et al. 2009). Other features frequently associated with 22q11.2DS include multisystem congenital and late-onset conditions [e.g. schizophrenia in 20–25%, hypocalcaemia in 60% and major congenital heart disease (CHD) in 30–40%] (McDonald-McGinn et al. 1999; Bassett et al. 2005; Fung et al. 2010). Testing for the associated 22q11.2 microdeletion, using fluorescence in situ hybridisation (FISH) and a specific 22q11.2 probe, has been available since about 1992, but this requires an increased index of suspicion for the syndrome and 22q11.2DS remains under-diagnosed. Routine use of microarray testing is expected to greatly increase identification of 22q11.2 deletions (Bassett et al. 2011). Recognising 22q11.2DS at any age has the potential to lead to significant changes in medical management, follow-up and genetic counselling that are helpful to the patient, family and clinicians (Kapadia & Bassett 2008; Bassett et al. 2011). A similar sentiment is expressed for genetic syndromes more generally, although also with limited empiric data (Griffiths & King 2004; Robin 2006; Shur & Abuelo 2009).
What is the current clinical reality with respect to genetic diagnosis? Data-driven reports on the impact of providing an aetiological explanation in ID are needed to help justify widespread use of new and expensive genetic technologies, and to convince clinicians of their usefulness in practice (Taylor et al. 2006; Ginsburg 2008). Testing uptake may be particularly low for adults (Taylor et al. 2006) and/or for those with milder forms of ID (the ‘forgotten generation’) (Tymchuk et al. 2001; Fujiura 2003), even though many could now receive a genetic/aetiologic diagnosis for the first time (Hochstenbach et al. 2009). Diagnosis of important aetiologies in mild and severe ID is already known to occur as a late event in a significant percentage of the population (Verri et al. 2004). However, this is typically conceptualised only as a missed opportunity with respect to therapeutic interventions and specific genetic counselling for parents (Verri et al. 2004; Henderson & Dick 2005). Benefits of diagnosis for adults and their families are particularly poorly characterised, and the opinions of adults with ID themselves on this topic are unknown. We surveyed adults with 22q11.2DS and their caregivers across Canada on the advantages and disadvantages of the molecular diagnosis of this representative microdeletion syndrome.
Methods
Participants
We have recruited Canadian adults (≥18 years) with 22q11.2DS through genetic, adult congenital cardiac and psychiatric services using active screening (Fung et al. 2008; Bassett et al. 2010a) and/or clinical referrals, and have (re)confirmed 22q11.2 deletions using standard methods (Bassett et al. 2008). Informed consent was obtained and the study was approved by local research ethics boards. Major CHD status and lifetime Diagnostic and Statistical Manual of Mental Disorders (fourth edition; DSM-IV) psychiatric diagnoses were determined as before (Bassett et al. 2009), and intellectual level was assessed using standard methods to obtain full-scale IQ (Chow et al. 2006). We used the American Association on Mental Retardation Classification for mild (IQ 50–75) and severe (IQ <50) ID. Data on adaptive functioning in this sample of adults with 22q11.2DS will be reported elsewhere (Butcher et al. unpublished data). All patients are followed at our clinic for adults with 22q11.2DS and receive treatment for associated conditions, including psychiatric illness, according to clinical practice guidelines (Bassett et al. 2011). Principal caregivers were self-identified at clinic visits. All known principal caregivers (n = 84), in addition to a higher-functioning subset of adults with 22q11.2DS, psychiatrically stable and capable of reading at a grade 4 level (n = 34), were included in the study.
Measures
We created a 28-item survey to determine the opinions of adults with 22q11.2DS and caregivers on several topics related to the syndrome. The current study focussed on the six closed-ended 5-point Likert scale items concerning the value of the definitive diagnosis (Appendix 1). Questions were informed by a review of the literature and clinical experience, and were developed with a participatory design. A comment section for each item offered respondents the opportunity to: (i) describe in greater detail any aspect of their experience with 22q11.2DS; and (ii) provide context for Likert scale responses. In this study, we retrospectively contacted previously identified patients and families seen through our clinic and associated research programme. The survey was distributed by surface mail, accompanied by a letter explaining the aim of the study and a postage-paid return envelope with a confidential ID number unique to each family. A reminder letter was sent after 8 weeks, and up to two reminder phone calls were made to non-respondents.
In a minority of cases, a clinical diagnosis of DiGeorge or velocardiofacial syndrome preceded availability of genetic testing for the 22q11.2 deletion. However, most clinical diagnoses were immediately followed by molecular confirmation of the deletion. Questions in the survey were specific to the molecular diagnosis.
Quantitative analysis
Descriptive statistics (frequencies, medians and modes) were used to characterise Likert scale responses. Non-parametric tests (Mann–Whitney U) were used to compare: (i) caregiver responses and adult patient responses; (ii) caregiver responses pertaining to adults with and without mild to severe ID; and (iii) caregiver responses pertaining to adults with and without schizophrenia. Analyses using SAS 9.1 determined statistical significance at the P < 0.05 level (two-sided) (SAS Institute Inc. 1997).
Qualitative analysis
Initial qualitative analysis of substantive comments was done as a paid service by an independent company (van Amsterdam Education and Research Inc.), blind to the identity of the respondents and to the corresponding quantitative responses. Using content analysis, the data were systematically coded by hand and then studied using QSR NVivo9 software (Gibbs 2009) to identify key themes. Identified themes were implicit or explicit in: (1) three or more caregiver responses; (2) two or more patient responses; or (3) one or more caregiver responses and one or more patient responses. The textual data were subsequently re-examined by the authors, guided by these preliminary themes and now within the appropriate clinical context. Specific subthemes relating to the impact of the 22q11.2DS diagnosis were isolated, including subthemes implicit or explicit in two or more caregiver or one or more patient responses and that supported a previously suggested benefit or disadvantage of diagnosis. Representative quotations included in the text are all from different individuals, and are therefore not labelled with anonymous individual-specific identification numbers.
Results
In total, 73 of 118 surveys were returned (61.9%): 53 from caregivers (63.1%) and 20 from adults with 22q11.2DS (58.8%) (Table 1). Respondents and non-respondents did not differ significantly on any demographic or clinical measure (Table 1). There was a non-significant trend (P = 0.090) towards a different rate of response from caregivers of individuals with, compared to those without, CHD (Table 1).
Table 1.
Demographic features of survey respondents and non-respondents, and clinical features of the corresponding adults with 22q11.2 deletion syndrome (22q11.2DS)
| Caregivers (n = 84)
|
Adults with 22q11.2DS (n = 34)
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Response (n = 53)
|
No response (n = 31)
|
Response (n = 20)
|
No response (n = 14)
|
|||||||
| Patient characteristics | n | (%) | n | (%) | P-value* | n | (%) | n | (%) | P-value† |
| Male sex | 28 | (52.8) | 15 | (48.4) | 0.694 | 8 | (40.0) | 7 | (50.0) | 0.728 |
| Mild or severe ID | 37 | (69.8) | 22 | (71.1) | 0.911 | 10 | (50.0) | 9 | (64.3) | 0.495 |
| Psychotic illness | 23 | (43.4) | 10 | (32.3) | 0.313 | 5 | (25.0) | 1 | (7.1) | 0.364 |
| Major CHD | 19 | (35.8) | 17 | (54.8) | 0.090 | 10 | (50.0) | 6 | (42.9) | 0.738 |
| Lives with parents | 28 | (52.8) | 19 | (61.3) | 0.451 | 11 | (55.0) | 7 | (50.0) | >0.999 |
| Lives in metropolitan area‡ | 42 | (79.2) | 24 | (77.4) | 0.844 | 14 | (70.0) | 11 | (78.6) | 0.704 |
| Parent of a child | 2 | (3.8) | 3 | (9.7) | 0.353† | 5 | (25.0) | 3 | (21.4) | >0.999 |
| Maternal caregiver | 36 | (67.9) | 25 | (80.6) | 0.207 | – | – | – | ||
|
| ||||||||||
| Mean | (SD) | Mean | (SD) | P-value§ | Mean | (SD) | Mean | (SD) | P-value§ | |
|
| ||||||||||
| Age (years) | 33.2 | (10.8) | 30.8 | (8.4) | 0.296 | 34.8 | (11.1) | 36.2 | (9.9) | 0.715 |
| Age at molecular diagnosis (years) | 23.2 | (12.3) | 21.5 | (10.1) | 0.519 | 27.2 | (12.8) | 26.5 | (12.5) | 0.884 |
| Full scale IQ | 70.3 | (12.7) | 70.7 | (11.3) | 0.889 | 76.1 | (11.4) | 73.9 | (8.6) | 0.549 |
| Age of caregiver (years) | 54.7 | (10.4) | 55.7 | (6.4) | 0.581 | – | – | – | ||
Pearson’s chi-square test.
Fisher’s exact test.
Defined as a very large urban area (‘urban core’; >100 000 people) together with adjacent urban and rural areas that have a high degree of social and economic integration with the urban core.
Two-tailed Student’s t-test.
CHD, congenital heart disease; ID, intellectual disability; IQ, intelligence quotient; SD, standard deviation.
Quantitative results
Over 50% of participants endorsed each of the five statements about positive effects of the diagnosis (Fig. 1a,b,d–f), that is, median and mode ‘Agree’ or ‘Strongly agree’. Responses to the statement about increasing worry about the future were more equivocal (Fig. 1c). There were no significant differences between caregiver and adult patient responses [Fig. 1: P = 0.925 (a), P = 0.836 (b), P = 0.652 (c), P = 0.592 (d), P = 0.417 (e), P = 0.063 (f)].
Figure 1.

Opinions of adults with 22q11.2 deletion syndrome (22q11.2DS) and their caregivers concerning the impact of the molecular diagnosis. The 100% stacked bar graphs display Likert scale responses of adults with 22q11.2DS (downward shading) and caregivers (no shading) to survey items concerning the diagnosis of the 22q11.2 deletion. Item-specific (a–f) response rates were 18, 19, 20, 19, 20 and 20 for adult patients, and 50, 51, 52, 51, 49 and 50 for caregivers, respectively. The greyscale coding key, from left to right, is: black = Strongly disagree, dark grey = Disagree, medium grey = Neither agree nor disagree, light grey = Agree (A), and off-white = Strongly agree (SA).
There were also no significant differences between responses of caregivers of those with and without mild to severe ID or schizophrenia (analyses not shown). The proportion of caregivers selecting ‘Agree’ or ‘Strongly agree’ for each of the six items (Appendix 1) for individuals with and without ID, respectively, was: (1) 22 of 34 vs. 11 of 16; (2) 23 of 35 vs. 10 of 16; (3) 13 of 36 vs. 9 of 16; (4) 21 of 35 vs. 11 of 16; (5) 24 of 33 vs. 13 of 16; and (6) 25 of 34 vs. 13 of 16, and for those with and without schizophrenia, respectively, was: (1) 17 of 22 vs. 16 of 28; (2) 15 of 22 vs. 18 of 29; (3) 8 of 23 vs. 14 of 29; (4) 16 of 23 vs. 16 of 28; (5) 20 of 23 vs. 17 of 26; and (6) 17 of 23 vs. 21 of 27.
Qualitative results
Substantive comments (>10 words) were recorded by 40 caregivers (75.5%; median = 87 words, range = 13–589 words) and 11 adults with 22q11.2DS (55.0%; median = 50 words, range = 10–236 words). Qualitative results were entirely consistent with the quantitative results and provided further insights into the patient and caregiver perspectives. Content analysis revealed several recurring themes concerning the impact of the genetic diagnosis.
Psychological benefits
Attaining a definitive diagnosis was cited often by respondents as a critical event in their lives. Many caregivers described how having this explanation for the adult’s lifelong history of serious intellectual, medical and behavioural issues gave a long-awaited justification for the individual’s struggles. In the words of one caregiver: ‘The diagnosis gives you a reason for understanding.’ The explanatory value of a genetic diagnosis was appreciated in part because of its relation to greater certainty, a sense of relief and better understanding of expectations and prognosis. This was particularly in relation to intellectual functioning. According to another mother: ‘[With the diagnosis], our daughter is relieved to know there is a reason she can’t do math, go to college or be like her school friends.’ An adult with 22q11.2DS also endorsed this viewpoint: ‘When I found out I had 22q it put me at a place where at least I knew why I had all this [sic] learning difficulties with school, work, etc.’ A brother of a patient stated: ‘I think that knowing the underlying genetic cause of the condition made it easier for us to understand and accept new diagnoses as they arose.’To some, the diagnosis engendered a newfound sense of purpose to participate in research and other endeavours to help similarly afflicted people. In one mother’s opinion: ‘[The diagnosis] gave us parents a platform.’
Clinical benefits
In addition to providing a unifying explanation for a complex history, a definitive diagnosis sometimes facilitated access to special programmes, new funding opportunities (e.g. extended dental coverage) and more support. As a result of the sampling strategy, all adult patients and caregivers also reported having had some access to the resources of a specialised clinic (including medical consultations and genetic counselling) since receiving the genetic diagnosis. One adult with 22q11.2DS commented that specialised clinics and expert consultation were key to attaining significant clinical benefits from the diagnosis: ‘The only improvement that I received was from specialised Drs that did help me.’ Some respondents experienced a change in individual attitudes and resulting medical care when able to inform healthcare professionals about the known implications and possible trajectories of the syndrome. One mother stated: ‘The diagnosis changed the way medical specialists interacted with me, the caregiver, and as a consequence, the interaction between them and my child was better.’
Lament for missed opportunities
Many respondents recalled feelings of frustration and powerlessness shaped by the uncertainty of years without a genetic diagnosis, and believed that an earlier diagnosis would have been helpful in predicting and preventing difficulties. Some parents reported that not having an accurate diagnosis led to mismanagement in many aspects of their child’s life, including medical conditions dealt with in isolation and misattribution of behavioural problems by family members and educators. For example, one mother believed that: ‘Not knowing the diagnosis until the patient was 27 led to a lot of frustration and mishandling by medical people ignorant as to how best to manage the care.’ Written comments in support of neonatal screening were many and reflected an appreciation for the variability of expression of the 22q11.2 deletion and the absence of curative measures.
Discussion
To our knowledge, this is the first study to investigate the impact of diagnosis of a microdeletion or microduplication syndrome on adult patients and their families. The survey results provide important confirmation of the proposed value of a diagnosis of 22q11.2DS at any life stage and for varying degrees of ID (Griffiths & King 2004; Robin 2006; Kapadia & Bassett 2008; Lopez-Rangel et al. 2008). The many ways in which a genetic diagnosis was important from the perspective of patients and their caregivers can be summarised for a clinician audience by placing the results within the familiar framework of Maslow’s hierarchy of needs (Fig. 2) (Maslow 1943).
Figure 2.
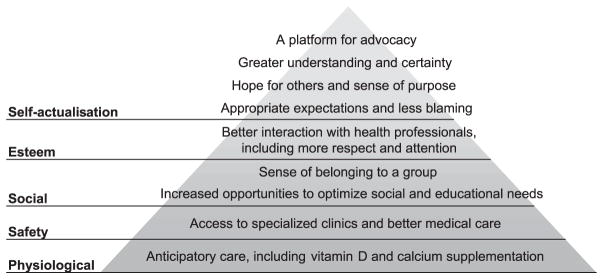
Why do individuals and their families value the diagnosis of 22q11.2 deletion syndrome? An outline of evidence-based benefits of a genetic diagnosis of 22q11.2 deletion syndrome to adult patients and their caregivers within the theoretical framework of Maslow’s hierarchy of needs.
The results may have implications for the increasing number of genetic diagnoses possible with new genomic analysis technologies (Hochstenbach et al. 2009; D.T. Miller et al. 2010) and the potential to change attitudes in the medical community for the benefit of families and individuals with currently unidentified causes of ID. A growing literature in prenatal testing, cancer predisposition and Huntington’s disease supports the value of genetic diagnosis in the absence of curative measures (Miller et al. 2006). Similar benefits are anticipated in ID (Raymond 2003; Lopez-Rangel et al. 2008), but there remain limited data to substantiate the importance of a genetic diagnosis to the care and well-being of children and adults with ID. Of the few other studies to date of single gene disorders and anomalies of chromosome number that are frequently associated with ID, Carmichael and colleagues used a postal questionnaire to catalogue the benefits and disadvantages of knowing the diagnosis for family members of a child with fragile X syndrome (Carmichael et al. 1999). Other studies reporting on interviews with parents and grandparents of children with fragile X, Down, Klinefelter and Turner syndromes have also delineated the primarily positive meanings that were attributed to these diagnostic labels (Starke et al. 2002; Lenhard et al. 2005; Poehlmann et al. 2005; Whitmarsh et al. 2007). The benefits of diagnosis of 22q11.2DS (Fig. 2) are consistent with those reported for these other conditions. The results of this study are also consistent with those of a preliminary study that explored the experiences with the diagnostic process of parents of 12 children with 22q11.2DS (Hallberg et al. 2010).
Advantages and limitations
A mixed-method design integrating quantitative and qualitative approaches was used to offset the limitations of each approach in isolation (Malterud 2001). In our sample, caregivers and patients had on average lived for two decades without a diagnosis and one decade with the diagnosis (Table 1), making them well suited to comment on before and after differences. Inclusion of a subset of patients themselves, which would not have been possible in a paediatric population, bolstered confidence in the presence of widespread benefits of 22q11.2DS identification (Fig. 2).
Our conclusions are dependent, however, on the diagnosis being accompanied by genetic counselling and provision of other key contextual information, which was true for the population surveyed but is not always the case in practice (Starke & Moller 2002). There may also be an age-related effect associated with reaction to diagnosis (Hallberg et al. 2010), suggesting caution in generalising our findings to neonatal diagnosis. Schizophrenia was overly represented in this sample of adults with 22q11.2DS (Table 1) (Fung et al. 2010), but there were no appreciable differences between responses from the schizophrenia and no psychosis groups.
More data are needed to address other important questions related to receipt of a diagnosis of 22q11.2DS. This study did not evaluate the impact of a diagnosis on family processes (e.g. communication) (Miller et al. 2006). Our study was also underpowered to assess potential differences between responses pertaining to adults with 22q11.2DS with and without children affected with the condition, a topic deserving of future study. Finally, more research is needed to provide objective evidence that use of new genomic technologies in clinical practice improves health outcomes (Ginsburg 2008). Future studies of 22q11.2DS, and of other microdeletion and microduplication syndromes, may better focus on the patient perspective by employing research methods such as interviewing that are more accessible and inclusive to those adults with more severe ID and/or literacy issues.
Implications for patients, families, clinicians and the public
Patients and their relatives often regard the function and meaning of a genetic diagnosis differently than do medical professionals (Evans 2006), and so their opinions are best assessed directly. For example, diagnostic certainty alone appears closely linked to psychological benefit (Rosenthal et al. 2001; Lenhard et al. 2005; Graungaard & Skov 2007), even when uncertainty remains about the extent of expression of a particular genetic anomaly in any particular individual (Whitmarsh et al. 2007). This may be overlooked by clinicians who equate diagnostic benefit solely with improved medical management. Identification of a 22q11.2 deletion after early childhood provided many adults with 22q11.2DS and families the satisfaction of an explanation for their lifelong challenges, sometimes after years of fruitless investigations (the ‘diagnostic odyssey’) (Robin 2006; Lee & Scherer 2010; Lewis et al. 2010). As for autism (F.A. Miller et al. 2010), genetic findings that explain ID, schizophrenia or other stigmatised neurodevelopmental conditions may be particularly valued for their explanatory value. Notably, this study demonstrated that this and other benefits of diagnosis extend to the adult with ID and not just to family members.
While caregivers valued a better understanding of prognosis, the reality of the syndrome diagnosis had varying effects on worry about the future (Fig. 1c). For example, psychotic illness is one of the greatest concerns of parents of children with 22q11.2DS (Hercher & Bruenner 2008), but these concerns have the potential to be productive. Although most individuals with 22q11.2DS do not suffer from schizophrenia, accurate information and knowledge about the increased risk can help caregivers to be more alert to early signs of this and other psychiatric illness and thereby better able to secure necessary treatment and supports in a timely manner (Bassett & Chow 2008). The known association between 22q11.2DS and treatable psychiatric illnesses like schizophrenia and anxiety disorders (Fung et al. 2010) is particularly important in those individuals with ID, where clinical diagnosis of a mental illness may be more challenging (Kerker et al. 2004; Werner & Stawski 2011).
As predicted previously (Griffiths & King 2004), identification of the underlying syndrome led in some cases to better care that stemmed from improved communication within clinical and medical disciplines and across disciplines (Fig. 2). For 22q11.2DS, it is increasingly possible to anticipate future care and health needs to allow for healthier ageing. Specific anticipatory care and changes in management (e.g. calcium and vitamin D supplementation to offset propensity for hypocalcaemia) are relevant regardless of the age of the patient (Bassett et al. 2005; Bassett et al. 2011). The detailed cognitive profile of adults with 22q11.2DS can inform appropriate expectations surrounding educational and vocational achievement (Chow et al. 2006). Such recommendations are not yet possible for many microdeletion and microduplication syndromes, which await sufficient numbers of individuals identified and assessed at different life stages (Bassett et al. 2010b; Lee & Scherer 2010).
What are the other main implications of this study? Apprehension regarding syndrome diagnosis within the general medical community may be driven by the belief that a genetic diagnosis merely brings a new ‘label’ to what is already known, with potentially harmful consequences for the patient and/or family (Griffiths & King 2004; Robin 2006). Such concerns may contribute to delayed and missed diagnoses of 22q11.2DS (Kapadia & Bassett 2008). In this study, no deleterious effects of a genetic diagnosis were reported by any caregiver or patient. In fact, as expected (Anonymous 2010; Bales et al. 2010), nearly all respondents endorsed newborn screening for 22q11.2 deletions (Fig. 1f). The non-significant trend towards different degrees of support between the patient and caregiver groups may be explained by a more nuanced understanding of the variability of expression of the deletion among the latter. Our findings also suggest that there is a clinical duty to inform individuals identified to have a 22q11.2 deletion in large-scale genetic screening studies where these deletions are likely to be ascertained. In certain countries and jurisdictions, this evidence for benefits of a molecular diagnosis may have important consequences with respect to private and public financial support for genetic testing and counselling (Taylor et al. 2006; Ginsburg 2008). Finally, increasing patient and family advocacy for rare conditions can lead to an increase in impactful, clinically focussed research (Ingelfinger & Drazen 2011).
Many individuals with 22q11.2DS remain undiagnosed, and most have ID. Awareness of 22q11.2DS is low among clinicians and educators (Lee et al. 2005). The results of this study provide further clinical impetus to identify, molecularly diagnose and provide genetic counselling for 22q11.2DS and other microdeletion and microduplication syndromes in adolescents and adults with ID – populations that remains largely invisible in clinical practice (Taylor et al. 2006; Kapadia & Bassett 2008). There exist clinical screening criteria that have been successfully implemented in other disease populations like schizophrenia (Bassett et al. 2010a) and adult CHD (Fung et al. 2008). Also, traditional genetic counselling approaches for adults can be successfully modified to take into account the presence of ID and any associated behavioural problems or psychiatric illnesses (Farwig et al. 2010; Finucane 2010). Opportunities to optimise management as a result of identifying 22q11.2DS suggest that many individuals with ID could be at the forefront of personalised medicine if diagnosed. Given the increasing indications for clinical use of microarrays in children and adults with ID, the demonstrated importance of a diagnosis of 22q11.2DS has potential implications for the burgeoning field of clinical genetics in ID.
Acknowledgments
The authors are grateful to the adults with 22q11.2 deletion syndrome and their caregivers for sharing their experiences. The authors thank Savithiri Ratnapalan and Ivan Silver for their expertise in survey methodology, Sean Bekeschus for aesthetic design of the survey, Denise van Amsterdam (van Amsterdam Education and Research Inc.) for assistance with the qualitative analysis and Gladys Wong for assistance with preparing the manuscript. This work was supported by Canadian Institutes of Health Research grants (MOP-97800 and MOP-89066), a Vanier Canada Graduate Scholarship (G. C.) and a Canada Research Chair in Schizophrenia Genetics and Genomic Disorders (A. S. B).
Appendix 1
Caregiver survey items
‘With regard to diagnosing the 22q11.2 deletion that explains many of that person’s lifetime health problems:
The diagnosis changed the way medical specialists interact with him/her for the better.
The diagnosis improved the quality of care that he/she receives.
The diagnosis made me more worried about his/her future.
The diagnosis made a positive difference in his/her life.
The diagnosis would have made a bigger difference had it occurred earlier in his/her life.
All newborn babies should be screened for 22q11DS.’
Patient survey items
‘Finding out I’ve got 22q:
Changed the way doctors treat me for the better.
Improved the care that I get.
Made me more worried about my future.
Was a good thing in my life.
Would have been better if I had been younger when I found out.
All newborn babies should be tested for 22q.’
Footnotes
Conflict of interest: There are no actual or potential conflicts of interest to disclose.
References
- Anonymous. A question of benefit. Nature Genetics. 2010;42:811. doi: 10.1038/ng1010-811. [DOI] [PubMed] [Google Scholar]
- Antshel KM, Shprintzen R, Fremont W, Higgins AM, Faraone SV, Kates WR. Cognitive and psychiatric predictors to psychosis in velocardiofacial syndrome: a 3-year follow-up study. Journal of the American Academy of Child and Adolescent Psychiatry. 2010;49:333–44. [PMC free article] [PubMed] [Google Scholar]
- Bales AM, Zaleski CA, McPherson EW. Newborn screening programs: should 22q11 deletion syndrome be added? Genetics in Medicine. 2010;12:135–44. doi: 10.1097/GIM.0b013e3181cdeb9a. [DOI] [PubMed] [Google Scholar]
- Bassett AS, Chow EW. Schizophrenia and 22q11.2 deletion syndrome. Current Psychiatry Reports. 2008;10:148–57. doi: 10.1007/s11920-008-0026-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, Chow EW, Husted J, Weksberg R, Caluseriu O, Webb GD, et al. Clinical features of 78 adults with 22q11 deletion syndrome. American Journal of Medical Genetics. Part A. 2005;138:307–13. doi: 10.1002/ajmg.a.30984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, Marshall CR, Lionel AC, Chow EW, Scherer SW. Copy number variations and risk for schizophrenia in 22q11.2 deletion syndrome. Human Molecular Genetics. 2008;17:4045–53. doi: 10.1093/hmg/ddn307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, Chow EW, Husted J, Hodgkinson K, Oechslin E, Harris L, et al. Premature death in adults with 22q11.2 deletion syndrome. Journal of Medical Genetics. 2009;46:324–30. doi: 10.1136/jmg.2008.063800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, Costain G, Fung WL, Russell KJ, Pierce L, Kapadia R, et al. Clinically detectable copy number variations in a Canadian catchment population of schizophrenia. Journal of Psychiatric Research. 2010a;44:1005–9. doi: 10.1016/j.jpsychires.2010.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, Scherer SW, Brzustowicz LM. Copy number variations in schizophrenia: critical review and new perspectives on concepts of genetics and disease. The American Journal of Psychiatry. 2010b;167:899–914. doi: 10.1176/appi.ajp.2009.09071016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, McDonald-McGinn DM, Devriendt K, Digilio MC, Goldenberg P, Habel A, et al. Practical guidelines for managing patients with 22q11.2 deletion syndrome. The Journal of Pediatrics. 2011;159:332–9. doi: 10.1016/j.jpeds.2011.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmichael B, Pembrey M, Turner G, Barnicoat A. Diagnosis of fragile-X syndrome: the experiences of parents. Journal of Intellectual Disability Research. 1999;43:47–53. doi: 10.1046/j.1365-2788.1999.43120157.x. [DOI] [PubMed] [Google Scholar]
- Chow EW, Watson M, Young DA, Bassett AS. Neurocognitive profile in 22q11 deletion syndrome and schizophrenia. Schizophrenia Research. 2006;87:270–8. doi: 10.1016/j.schres.2006.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Smedt B, Devriendt K, Fryns JP, Vogels A, Gewillig M, Swillen A. Intellectual abilities in a large sample of children with velo-cardio-facial syndrome: an update. Journal of Intellectual Disability Research. 2007;51:666–70. doi: 10.1111/j.1365-2788.2007.00955.x. [DOI] [PubMed] [Google Scholar]
- De Smedt B, Swillen A, Verschaffel L, Ghesquiere P. Mathematical learning disabilities in children with 22q11.2 deletion syndrome: a review. Developmental Disabilities Research Reviews. 2009;15:4–10. doi: 10.1002/ddrr.44. [DOI] [PubMed] [Google Scholar]
- Evans C. Genetic Counselling: A Psychological Approach. Cambridge University Press; NewYork, NY: 2006. [Google Scholar]
- Farwig K, Harmon AG, Fontana KM, Mervis CB, Morris CA. Genetic counseling of adults with Williams syndrome: a first study. American Journal of Medical Genetics. Part C. 2010;154C:307–15. doi: 10.1002/ajmg.c.30264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finucane B. Genetic counseling for women with intellectual disabilities. In: Leroy BS, Veach PM, Bartels DM, editors. Genetic Counseling Practice: Advanced Concepts and Skills. Wiley-Blackwell; Hoboken, NJ: 2010. pp. 281–303. [Google Scholar]
- Fujiura GT. Continuum of intellectual disability: demographic evidence for the ‘forgotten generation. Mental Retardation. 2003;41:420–9. doi: 10.1352/0047-6765(2003)41<420:COIDDE>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Fung WL, Chow EW, Webb GD, Gatzoulis MA, Bassett AS. Extracardiac features predicting 22q11.2 deletion syndrome in adult congenital heart disease. International Journal of Cardiology. 2008;131:51–8. doi: 10.1016/j.ijcard.2007.08.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung WL, McEvilly R, Fong J, Silversides C, Chow E, Bassett A. Elevated prevalence of generalized anxiety disorder in adults with 22q11.2 deletion syndrome. The American Journal of Psychiatry. 2010;167:998. doi: 10.1176/appi.ajp.2010.09101463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs G. Analysing Qualitative Data. Sage; London: 2009. [Google Scholar]
- Ginsburg GS. Genomic Medicine: ‘grand challenges’ in the translation of genomics to human health. European Journal of Human Genetics. 2008;16:873–4. doi: 10.1038/ejhg.2008.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graungaard AH, Skov L. Why do we need a diagnosis? A qualitative study of parents’ experiences, coping and needs, when the newborn child is severely disabled. Child: Care, Health and Development. 2007;33:296–307. doi: 10.1111/j.1365-2214.2006.00666.x. [DOI] [PubMed] [Google Scholar]
- Griffiths DM, King R, editors. Demystifying Syndromes: Clinical and Educational Implications of Common Syndromes Associated with Persons with Intellectual Disabilities. National Association for the Dually Diagnosed; Kington, NY: 2004. [Google Scholar]
- Hallberg U, Oskarsdottir S, Klingberg G. 22q11 deletion syndrome – the meaning of a diagnosis. A qualitative study on parental perspectives. Child: Care, Health and Development. 2010;36:719–25. doi: 10.1111/j.1365-2214.2010.01108.x. [DOI] [PubMed] [Google Scholar]
- Henderson A, Dick M. Response to Verri et al (Journal of Intellectual Disability Research 48, 679–686): is there room for hope when considering the problem of late diagnosis in intellectual disability? Journal of Intellectual Disability Research. 2005;49:690–1. doi: 10.1111/j.1365-2788.2005.00712.x. [DOI] [PubMed] [Google Scholar]
- Hercher L, Bruenner G. Living with a child at risk for psychotic illness: the experience of parents coping with 22q11 deletion syndrome: an exploratory study. American Journal of Medical Genetics. Part A. 2008;146:2355–60. doi: 10.1002/ajmg.a.32466. [DOI] [PubMed] [Google Scholar]
- Hochstenbach R, van Binsbergen E, Engelen J, Nieuwint A, Polstra A, Poddighe P, et al. Array analysis and karyotyping: workflow consequences based on a retrospective study of 36,325 patients with idiopathic developmental delay in the Netherlands. European Journal of Medical Genetics. 2009;52:161–9. doi: 10.1016/j.ejmg.2009.03.015. [DOI] [PubMed] [Google Scholar]
- Ingelfinger JR, Drazen JM. Patient organizations and research on rare diseases. New England Journal of Medicine. 2011;364:1670–1. doi: 10.1056/NEJMe1102290. [DOI] [PubMed] [Google Scholar]
- Kapadia RK, Bassett AS. Recognizing a common genetic syndrome: 22q11.2 deletion syndrome. Canadian Medical Association Journal. 2008;178:391–3. doi: 10.1503/cmaj.071300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerker BD, Owens PL, Zigler E, Horwitz SM. Mental health disorders among individuals with mental retardation: challenges to accurate prevalence estimates. Public Health Reports. 2004;119:409–17. doi: 10.1016/j.phr.2004.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobrynski LJ, Sullivan KE. Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11.2 deletion syndromes. Lancet. 2007;370:1443–52. doi: 10.1016/S0140-6736(07)61601-8. [DOI] [PubMed] [Google Scholar]
- Lee C, Scherer SW. The clinical context of copy number variation in the human genome. Expert Reviews in Molecular Medicine. 2010;12:e8. doi: 10.1017/S1462399410001390. [DOI] [PubMed] [Google Scholar]
- Lee TH, Blasey CM, Dyer-Friedman J, Glaser B, Reiss AL, Eliez S. From research to practice: teacher and pediatrician awareness of phenotypic traits in neurogenetic syndromes. American Journal on Mental Retardation. 2005;110:100–6. doi: 10.1352/0895-8017(2005)110<100:FRTPTA>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Lenhard W, Breitenbach E, Ebert H, Schindelhauer-Deutscher HJ, Henn W. Psychological benefit of diagnostic certainty for mothers of children with disabilities: lessons from Down syndrome. American Journal of Medical Genetics. Part A. 2005;133A:170–5. doi: 10.1002/ajmg.a.30571. [DOI] [PubMed] [Google Scholar]
- Lewis C, Skirton H, Jones R. Living without a diagnosis: the parental experience. Genetic Testing and Molecular Biomarkers. 2010;14:807–15. doi: 10.1089/gtmb.2010.0061. [DOI] [PubMed] [Google Scholar]
- Lopez-Rangel E, Mickelson ECR, Lewis MES. The value of a genetic diagnosis for individuals with intellectual disabilities: optimising healthcare and function across the lifespan. The British Journal of Developmental Disabilities. 2008;54:69–82. [Google Scholar]
- Malterud K. Qualitative research: standards, challenges, and guidelines. Lancet. 2001;358:483–8. doi: 10.1016/S0140-6736(01)05627-6. [DOI] [PubMed] [Google Scholar]
- Maslow AH. A theory of human motivation. Psychological Review. 1943;50:370–96. [Google Scholar]
- McDonald-McGinn DM, Kirschner R, Goldmuntz E, Sullivan K, Eicher P, Gerdes M, et al. The Philadelphia story: the 22q11.2 deletion: report on 250 patients. Genetic Counseling. 1999;10:11–24. [PubMed] [Google Scholar]
- Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, Carter NP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. American Journal of Human Genetics. 2010;86:749–64. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller FA, Hayeems RZ, Bytautas JP. What is a meaningful result? Disclosing the results of genomic research in autism to research participants. European Journal of Human Genetics. 2010;18:867–71. doi: 10.1038/ejhg.2010.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SM, McDaniel SH, Rolland JS, Feetham SL, editors. Individuals, Families, and the New Era of Genetics: Biopsychosocial Perspectives. W.W. Norton and Company; NewYork, NY: 2006. [Google Scholar]
- Poehlmann J, Clements M, Abbeduto L, Farsad V. Family experiences associated with a child’s diagnosis of fragile X or Down syndrome: evidence for disruption and resilience. Mental Retardation. 2005;43:255–67. doi: 10.1352/0047-6765(2005)43[255:FEAWAC]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Raymond FL. Genetic services for people with intellectual disability and their families. Journal of Intellectual Disability Research. 2003;47:509–14. doi: 10.1046/j.1365-2788.2003.00529.x. [DOI] [PubMed] [Google Scholar]
- Robin NH. It does matter: the importance of making the diagnosis of a genetic syndrome. Current Opinion in Pediatrics. 2006;18:595–7. doi: 10.1097/01.mop.0000247536.78273.78. [DOI] [PubMed] [Google Scholar]
- Rosenthal ET, Biesecker LG, Biesecker BB. Parental attitudes toward a diagnosis in children with unidentified multiple congenital anomaly syndromes. American Journal of Medical Genetics. 2001;103:106–14. doi: 10.1002/ajmg.1527. [DOI] [PubMed] [Google Scholar]
- SAS Institute Inc. SAS/STAT Software: Changes and Enhancements through Release 6.12. SAS Institute Inc; Cary, NC: 1997. [Google Scholar]
- Shur N, Abuelo D. Genetic syndromes: from clinical suspicion to referral to diagnosis. Pediatric Annals. 2009;38:419–25. doi: 10.3928/00904481-20090723-04. [DOI] [PubMed] [Google Scholar]
- Starke M, Moller A. Parents’ needs for knowledge concerning the medical diagnosis of their children. Journal of Child Health Care. 2002;6:245–57. doi: 10.1177/136749350200600402. [DOI] [PubMed] [Google Scholar]
- Starke M, Wikland KA, Moller A. Parents’ experiences of receiving the diagnosis of Turner syndrome: an explorative and retrospective study. Patient Education and Counseling. 2002;47:347–54. doi: 10.1016/s0738-3991(02)00010-1. [DOI] [PubMed] [Google Scholar]
- Taylor MR, Edwards JG, Ku L. Lost in transition: challenges in the expanding field of adult genetics. American Journal of Medical Genetics. Part C. 2006;142C:294–303. doi: 10.1002/ajmg.c.30105. [DOI] [PubMed] [Google Scholar]
- Tymchuk AJ, Lakin KC, Luckasson R, editors. The Forgotten Generation: the Status and Challenges of Adults with Mild Cognitive Limitations. Paul H. Brookes Publishing Company; Baltimore, MD: 2001. [Google Scholar]
- Verri AP, Maraschio P, Uggetti C, Pucci E, Ronchi G, Nespoli L, et al. Late diagnosis in severe and mild intellectual disability in adulthood. Journal of Intellectual Disability Research. 2004;48:679–86. doi: 10.1111/j.1365-2788.2003.00593.x. [DOI] [PubMed] [Google Scholar]
- Werner S, Stawski M. Knowledge, attitudes and training of professionals on dual diagnosis of intellectual disability and psychiatric disorder. Journal of Intellectual Disability Research. 2011 doi: 10.1111/j.1365-2788.2011.01429.x. [DOI] [PubMed] [Google Scholar]
- Whitmarsh I, Davis AM, Skinner D, Bailey DB., Jr A place for genetic uncertainty: parents valuing an unknown in the meaning of disease. Social Science and Medicine. 2007;65:1082–93. doi: 10.1016/j.socscimed.2007.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]