

Gliwa and Berkman (2013) elegantly outline many of the challenges posed by the concept of incidental genetic findings. However, there is another critical barrier to interpreting human genetic variation that is unrelated to the specific disease under study. Research study participants often lack extended phenotype and family history data. Regardless of the anticipated advances in sequencing technology, bioinformatics, and our understanding of the human genome, the clinical context will remain a key component in making sense of genomic data.
CASE
We provide an illustrative example that arose in the course of an ongoing genetic family study of schizophrenia (Brzustowicz et al. 2000). Using genome-wide microarray technology, we first noted a structural mutation (loss) disrupting several exons of the low-density lipoprotein receptor (LDLR) gene because of its rarity and incomplete co-segregation with schizophrenia in the family (Figure 1). The same mutation was noted independently in DNA samples from five study participants from a single family, suggesting high analytic validity. We were aware that thousands of individual mutations in this gene have previously been implicated in familial hypercholesterolemia (Leigh et al. 2008). Although the hemizygous deletion found in this family was apparently novel (Leigh et al. 2008), its (1) rarity, (2) predicted disruption of the coding sequence of the gene, and (3) overlap with previously reported deletions at LDLR in familial hypercholesterolemia supported potential clinical validity. Familial hypercholesterolemia may be asymptomatic at young ages, and is a significant risk factor for premature cardiovascular disease (Sullivan et al. 2012; Wierzbicki, Humphries, and Minhas 2008). Early screening and treatment is indicated to those at known risk, and may help to reduce morbidity and mortality (Sullivan et al. 2012; Wierzbicki et al. 2008).
Figure 1.
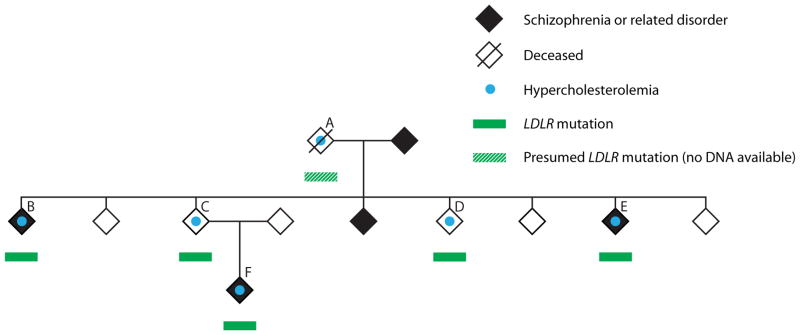
Discovery of an incidental genetic finding in the course of a family study of schizophrenia. The LDLR mutation (see text for details) was found to co-segregate perfectly with familial hypercholesterolemia in the family. Specifically, all five adult family members with the mutation, and none of the six adults without the mutation, met clinical criteria for a diagnosis of familial hypercholesterolemia. DNA was available for all members of the family except individual A (deceased). Individuals B, D, and E had no offspring (Color figure available online).
A singular feature of this longitudinal family study is, however, the “depth” (Beskow and Burke 2010) of our relationship with the study participants. The family mentioned has been participating in the study for more than 20 years. Regular updates and detailed lifetime medical records allowed us to better understand the clinical context for this variant. Three of four siblings and a nephew who harbored the LDLR mutation had already been clinically diagnosed with type IIa hyperlipoproteinemia and treated appropriately with statin medications (Figure 1, individuals B, D, E, F). The fourth sibling (Figure 1, individual C) had no family doctor. One of us (ASB) met with him on a clinical basis, ordered the standard cholesterol testing already indicated on the basis of his family history, made the clinical/laboratory diagnosis of hypercholesterolemia, ensured that treatment was initiated, and provided genetic counseling based on his family history. Perfect segregation with complete penetrance of the LDLR deletion with familial hypercholesterolemia in this family provided key further support for the clinical validity of this mutation.
Regarding disclosure of this incidental finding, we reasoned similarly to Gliwa and Berkman: (1) the additional benefit to participants would be low given their current clinical states and their already well-recognized family history, (2) the uniqueness of access was low, with clinical genetic testing of the LDLR gene available in many jurisdictions, and (3) considerable time, effort, and resources would realistically be required of both our research group and the public health care system to properly confirm and return molecular genetic findings to all of the geographically dispersed members of this family. After careful deliberation and informal consultation with an endocrinologist, we therefore decided against offering to return this incidental genetic finding to the family at this time. Notably, this decision was predicated on an in-depth understanding of the specific clinical context.
DISCUSSION
In clinically characterizing study participants, most researchers understandably maintain a narrow focus on the disease(s) under study and related inclusion/exclusion criteria. For other conditions, the typical “best case scenario” might involve acquisition at a single time point by a research assistant of a self-reported limited medical history. This then creates a significant barrier to the proper assessment of incidental genetic findings that are by definition unrelated to the primary research target. Interpretation of most mutations is probabilistic and conditional on the available data regarding the clinical context: the cross-sectional presence or absence of the specific disease in the individual, and his or her age, sex, and associated risk factors. A simplistic example is that an uncharacterized mutation computationally predicted to interfere with the function of a gene implicated in a potentially fatal childhood-onset disease would be interpreted very differently in (1) a seemingly healthy newborn enrolled in a population cohort study, (2) a child originally recruited for a study of asthma who has since died of this fatal disease, or (3) the now 84-year-old Nobel laureate James Watson. In our case, knowledge of the previous clinical diagnoses and treatment of familial hypercholesterolemia allowed us to better gauge the potential benefit to the participants of returning the incidental LDLR genetic finding. Many studies focused on interpreting incidental genetic findings have also implicitly or explicitly used detailed clinical (and family history) information (see, e.g., Johnston et al. 2012; Solomon et al. 2012), but to our knowledge this context would be practically unavailable to most current genetic researchers in most cases.
Family history remains the cornerstone for personalized risk prediction for a range of conditions (Guttmacher, Collins, and Carmona 2004). In our experience, however, it is rare for researchers conducting large-scale genetic studies of specific diseases to take detailed three-generation family histories. Family histories that are informed by multiple sources and/or updated periodically are even rarer. As in the case described here, knowledge of the family history may (1) demonstrate that a potential increased risk for a disease based on molecular findings is already known on the basis of a positive family history (Costain and Bassett 2012), (2) reveal information critical to the proper interpretation of an incidental finding in a study participant (Johnston et al. 2012), and/or (3) clarify potential responsibilities of the researchers to other family members. With the typical absence or incompleteness of these data in research studies, a duty to search for incidental findings in a research participant could conceivably lead to researchers investing significant resources in context-less follow-up and expensive though clinically needless molecular testing.
One of the greatest potential benefits of searching for incidental findings is that new spontaneous (de novo) clinically relevant mutations may be discovered that reveal a previously unsuspected risk for an important medical condition. For example, this could include a typical mutation in the BRCA1 gene known to predispose to breast and ovarian cancer in a young woman with no personal or family history of either cancer (Johnston et al. 2012). However, most rare variants of potential clinical import are private (seemingly unique to the individual or family) and ultimately found to be inherited. An expectation for researchers to routinely return to the family to study inheritance patterns and potential co-segregation with illness of various uncharacterized genetic variants may be unduly burdensome, or practically impossible, depending on the study protocol and consent process (e.g., if DNA samples were collected anonymously).
Incidental findings can arise in all medical contexts, and in clinical as well as research settings. In the case of research-based genetic investigations, Gliwa and Berkman (2013) have carefully articulated the current factors influencing the potential duty to search for incidental findings, as well as those pertinent for the foreseeable future, in molecular genetic studies. We provide further support for their primary conclusion by emphasizing that the typical lack of knowledge of the clinical context in most of these research studies will confound the confident interpretation of many incidental genetic findings. Attempts to systematically tie genomic research samples to medical records, such as the Electronic Medical Records and Genomics (eMERGE) Network (www.genome.gov/27540473), and a shift away from the current standard of large-scale case-control study design with no ability to return to subjects, could in the future have an impact on this contemporary barrier. For now, however, the increasing application of next-generation sequencing technologies to incompletely characterized study cohorts suggests a large burden of potential incidental findings whose significance cannot realistically be determined.
Acknowledgments
We thank the family discussed in this commentary for their continued participation in our research study; research collaborators including Drs. Linda M. Brzustowicz, Christian R. Marshall, Stephen W. Scherer, and Eva W. C. Chow; and Fiona Fu for helping to create the figure.
Contributor Information
Gregory Costain, Centre for Addiction and Mental Health and University of Toronto.
Anne S. Bassett, Centre for Addiction and Mental Health and University of Toronto
References
- Beskow LM, Burke W. Offering individual genetic research results: Context matters. Science Translational Medicine. 2010;2(38):38cm20. doi: 10.1126/scitranslmed.3000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzustowicz LM, Hodgkinson KA, Chow EW, Honer WG, Bassett AS. Location of a major susceptibility locus for familial schizophrenia on chromosome 1q21-q22. Science. 2000;288(5466):678–682. doi: 10.1126/science.288.5466.678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costain G, Bassett AS. The ever-evolving concept of clinical significance and the potential for sins of omission in genetic research. American Journal of Bioethics. 2012;12(10):22–24. doi: 10.1080/15265161.2012.699142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gliwa C, Berkman BE. Do researchers have an obligation to actively look for genetic incidental findings? American Journal of Bioethics. 2013;13(2):32–42. doi: 10.1080/15265161.2012.754062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttmacher AE, Collins FS, Carmona RH. The family history–more important than ever. New England Journal of Medicine. 2004;351(22):2333–2336. doi: 10.1056/NEJMsb042979. [DOI] [PubMed] [Google Scholar]
- Johnston JJ, Rubinstein WS, Facio FM, et al. Secondary variants in individuals undergoing exome sequencing: screening of 572 individuals identifies high-penetrance mutations in cancer-susceptibility genes. American Journal of Human Genetics. 2012;91(1):97–108. doi: 10.1016/j.ajhg.2012.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh SE, Foster AH, Whittall RA, Hubbart CS, Humphries SE. Update and analysis of the University College London low density lipoprotein receptor familial hypercholesterolemia database. Annals of Human Genetics. 2008;72(Pt 4):485–498. doi: 10.1111/j.1469-1809.2008.00436.x. [DOI] [PubMed] [Google Scholar]
- Solomon BD, Hadley DW, Pineda-Alvarez DE, et al. Incidental medical information in whole-exome sequencing. Pediatrics. 2012;129(6):e1605–1611. doi: 10.1542/peds.2011-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan DR, Hamilton-Craig I, van Bockxmeer F, Watts GF. INTERIM guidelines for the diagnosis and management of familial hypercholesterolaemia. Heart, Lung and Circulation. 2012;21(3):159–162. doi: 10.1016/j.hlc.2011.11.006. [DOI] [PubMed] [Google Scholar]
- Wierzbicki AS, Humphries SE, Minhas R. Familial hypercholesterolaemia: summary of NICE guidance. British Medical Journal. 2008;337:a1095. doi: 10.1136/bmj.a1095. [DOI] [PubMed] [Google Scholar]