

Abstract
Histone deacetylases (HDACs) catalyze removal of acetyl-groups from lysine residues within nucelosomal histone tails and thousands of non-histone proteins. The 18 mammalian HDACs are grouped into four classes. Class I, II and IV HDACs employ zinc as a co-factor for catalytic activity, while class III HDACs (also known as sirtuins) require NAD+ for enzymatic function. Small molecule inhibitors of zinc-dependent HDACs are efficacious in multiple pre-clinical models of pressure overload and ischemic cardiomyopathy, reducing pathological hypertrophy and fibrosis, and improving contractile function. Emerging data have revealed numerous mechanisms by which HDAC inhibitors benefit the heart, including suppression of oxidative stress and inflammation, inhibition of MAP kinase signaling, and enhancement of cardiac protein aggregate clearance and autophagic flux. Here, we summarize recent findings with zinc-dependent HDACs and HDAC inhibitors in the heart, focusing on newly described functions for distinct HDAC isoforms (e.g. HDAC2, HDAC3 and HDAC6). Potential for pharmacological HDAC inhibition as a means of treating age-related cardiac dysfunction is also discussed.
1. Introduction
Acetylation of nucleosomal histone tails provides a critical mechanism for epigenetic control of gene expression. Additionally, proteomic studies have revealed that thousands of non-histone proteins are also subject to reversible lysine acetylation [1, 2], further highlighting the biological significance of this post-translational modification. Acetyl groups are transferred to lysine residues by histone acetyltransferases (HATs) and removed by histone deacetylases (HDACs), which are often referred to as “writers” and “erasers”, respectively. Lysine acetylation also creates binding sites for bromodomain-containing “reader” proteins such as bromodomain and extraterminal (BET) proteins. Although HATs, HDACs and acetyl-lysine readers have all been shown to contribute to the pathogenesis of heart failure, this review specifically focuses on HDACs.
The 18 mammalian HDACs are encoded by distinct genes and are grouped into four classes on the basis of similarity to yeast transcriptional repressors. Class I HDACs (HDACs 1, 2, 3 and 8) are related to yeast RPD3, class II HDACs (HDACs 4, 5, 6, 9 and 10) to yeast HDA1, and class III HDACs (SirT1 – 7) to yeast Sir2. Class II HDACs are further divided into two subclasses, IIa (HDACs 4, 5, 7 and 9) and IIb (HDACs 6 and 10). HDAC11 falls into a fourth class [3]. Coordination of a zinc ion in the catalytic domains of class I, II and IV HDACs is required for catalysis (Fig. 1A). In contrast, class III HDACs (sirtuins) utilize nicotinamide adenine dinucleotide (NAD+) as a co-factor for catalytic activity. Class III HDACs are most commonly associated with aging (decreased activity and expression is thought to contribute to aging), and these HDACs clearly serve important roles in the heart. However, class III HDACs will not be discussed further in this review, since they are not inhibited by the small molecule HDAC inhibitors that were used in the pre-clinical models of heart failure described below.
Figure 1. Zinc-dependent HDACs and cardiac aging.

(A) Zinc-dependent HDACs fall into three classes, with class II being subdivided into IIa and IIb. Class III HDACs (sirtuins), which are NAD+-dependent, are not shown. (B) In response to hypertrophic stimuli, HDAC2 is acetylated by p300/CBP-associated factor (PCAF), which primes the protein for phosphorylation by casein kinase 2 (CK2). Acetylated and phosphorylated HDAC2 is more active, and thus has increased capacity to repress anti-hypertrophic gene expression. Hypertrophic signals also lead to HDAC3-mediated repression of the gene encoding dual-specificity phosphatase 5 (DUSP5). In HDAC inhibitor-treated cardiomyocytes, DUSP5 expression increases, creating a negative feedback loop that blocks pro-hypertrophic ERK signaling in the nucleus.
2. HDAC inhibitors in heart failure models
Positive effects of pan- and isoform-selective HDAC inhibitors in rodent models of heart failure have been reviewed extensively [4, 5]. Importantly, HDAC inhibition is capable of regressing established cardiac hypertrophy and systolic dysfunction in mice subjected to aortic constriction [6, 7]. Recently, a major advance in the field was provided by the discovery that SAHA (vorinostat), an FDA-approved pan-HDAC inhibitor, was efficacious in a rabbit model of cardiac ischemia-reperfusion (I/R) injury [8]. Delivery of SAHA before or during reperfusion resulted in a 40% decrease in infarct size and preservation of systolic function of the heart. Efficacy of SAHA in this model appeared to be due to enhancement of autophagic flux in the infarct border zone. It is thought that autophagy serves to protect cardiomyocytes during ischemia by resupplying energy, and by destroying damaged mitochondria [9]. This proof-of-concept study in a large animal model sets the stage for a clinical trial in humans to assess effects of HDAC inhibition on pathological cardiac remodeling post-myocardial infarction. Such a trial would be the first assessment of an HDAC inhibitor for a cardiovascular indication.
It will be interesting to determine whether isoform-selective HDAC inhibitors are efficacious in the rabbit I/R model. A recent evaluation of HDAC inhibitors in an ex vivo model of rat cardiac I/R injury demonstrated that MS-275, a class I HDAC (HDAC1, -2, -3)-selective inhibitor, preserved cardiac function and reduced infarct size [10]. These results suggest that class I HDAC activity contributes to ischemic cardiac damage.
Fibrosis is a hallmark of the aging heart, and pan-HDAC inhibitors have clearly been shown to reduce excess extracellular matrix (ECM) deposition in multiple models of cardiac disease [11]. We discovered that a small molecule inhibitor of class I HDACs, MGCD0103, blocks angiotensin II-mediated cardiac fibrosis [12]. A follow-up study with the same compound showed that class I HDAC inhibition reduces cardiac fibrosis and improves ventricular function in a chronic coronary artery ligation model in rats [13].
Multiple mechanisms account for class I HDAC inhibitor-mediated suppression of cardiac fibrosis. First, class I HDAC inhibition blocks cardiac fibroblasts in the G0/G1 phase of the cell cycle by preventing retinoblastoma (Rb) phosphorylation, which is required to stimulate downstream expression of target genes that drive the G1-to-S transition. Specifically, class I HDAC inhibitor treatment of cardiac fibroblasts results in upregulation of expression of p15 and p57, which are endogenous suppressors of the kinases that target Rb, cyclin-dependent kinases [12]. Through this mechanism, class I HDAC inhibitors prevent expansion of the pool of ECM-producing myofibroblasts in the myocardium in response to stress. Class I HDAC inhibition is also able to block cardiac myofibroblast activation [13], and promotes protein SUMOylation in cardiac fibroblasts [14]. Given the cardioprotection mediated by enhanced SUMOylation [15], it is possible that some of the anti-fibrotic action of class I HDAC inhibitors is governed by this post-translational modification.
Fibrocytes are another pro-fibrotic cell type that is targeted by class I HDAC inhibitors [12, 16]. Fibrocytes are a bone marrow-derived cell population defined by co-expression of CD34 (stem cell marker), CD45 (hematopoietic cell marker), a monocyte markers (e.g., CD11), and either collagen or α-smooth muscle actin (mesenchymal markers) [17]. In response to stress, monocytic fibrocyte precursors migrate to damaged tissue and differentiate into fibrocytes, which ultimately become myofibroblasts. We found that class I HDAC inhibitors potently block fibrocyte differentiation, providing another means of reducing the number of ECM producing cells in the heart [12]. Age-related cardiac fibrosis and diastolic dysfunction coincide with accumulation of fibrocytes in ventricular interstitial space [18]. As such, chronic HDAC inhibitor treatment has the potential to reduce age-dependent cardiac fibrosis.
3. Class I HDACs and cardiac signaling and gene expression
Class I HDACs, especially HDACs -1, -2 and -3, are thought to primarily reside in the nucleus, where they serve canonical roles in the control of gene expression through deacetylation of histone tails. These HDACs are present in large multi-protein complexes referred to as Sin3, NuRD, CoREST and NCoR/SMRT, which are recruited to gene regulatory elements by sequence-specific DNA binding transcription factors [19, 20]. In general, HDAC1 and HDAC2 are found together in Sin3, NuRD and CoREST complexes, while HDAC3 is a component of the NCoR/SMRT complex. However, it is important to emphasize that this is an oversimplification. For example, HDAC3 has also been found at the plasma membrane, where it regulates Src tyrosine kinase activity [21], and in cardiomyocytes HDAC3 has been shown to co-localize with sarcomeres [22].
Several studies have suggested that HDAC2 promotes cardiac hypertrophy [23]. Recently, HDAC2 was shown to be acetylated by p300/CBP-associated factor (PCAF) during cardiac hypertrophy [24]. Acetylation appears to prime HDAC2 for subsequent phosphorylation by casein kinase 2, leading to enhanced HDAC2-mediated gene repression. It is thought that HDAC2 promotes pathological growth of the heart by suppressing expression of anti-hypertrophic genes in cardiac myocytes (Fig. 1B).
Cardiac-specific deletion of HDAC3 in mice results in metabolic catastrophe in the heart, with massive cardiac hypertrophy and excessive myocardial lipid accumulation due to constitutive activation of peroxisome proliferator-activated receptor (PPAR)-mediated gene expression [25]. Cardiac deletion of HDAC3 in this study was mediated by Cre recombinase that was expressed under the control of the α-MyHC promoter, which is active during embryonic development. When HDAC3 was deleted from the heart at a later time point, using an MCK-Cre transgene, adult mice did not exhibit a phenotype until fed a high fat diet, whereupon they developed severe cardiomyopathy coincident with downregulation of genes involved in lipid metabolism [26].
In contrast to the deleterious effects of knocking out HDAC3 in the heart, knockdown of HDAC3 expression using a morpholino oligonucleotide led to reduced LV hypertrophy and fibrosis in a mouse pressure overload model [27]. When considering the different outcomes of HDAC3 knockout versus knockdown, it is important to also acknowledge that HDAC inhibitors such as SAHA and MGCD0103 potently inhibit HDAC3 catalytic activity, yet improve cardiac function (see above). The simplest interpretation of these collective findings is that HDAC3 has a non-catalytic function, presumably a scaffolding activity, which is required for cardiac homeostasis. As such, knocking out HDAC3 will exert a completely different effect on the heart than reducing HDAC3 expression or inhibiting its catalytic activity. This point was illustrated by the demonstration that HDAC3 depletion in mouse liver led to hepatosteatosis, which was rescued by catalytically inactive mutants of HDAC3 [28]. In order to advance the heart failure field, it will be essential to assess effects of newly developed HDAC3-selective inhibitors in models of pathological cardiac remodeling.
We recently described a novel function for HDAC3 in the regulation of cardiomyocyte mitogen-activated protein kinase (MAP kinase) signaling [29]. HDAC inhibitor treatment of cardiomyocytes led to reduced phosphorylation of ERK1/2 at two sites that are required for catalytic activity of the kinase. Remarkably, the upstream kinase for ERK, MEK1, remained active in the presence of HDAC inhibition. Instead, HDAC inhibitor treatment triggered a negative-feedback loop by stimulating expression of an ERK-specific phosphatase, termed dual-specificity phosphatase 5 (DUSP5). In response to hypertrophic stimuli, DUSP5 expression is downregulated through an HDAC3-dependent mechanism, and selective inhibition or knockdown of HDAC3 leads to depression of the dusp5 gene, with consequent suppression of cardiac ERK signaling (Fig. 1B). Interestingly, DUSP5 is a nuclear phosphatase, so HDAC inhibition specifically blocks ERK signaling in the nuclear compartment of cardiomyocytes [29]. This is significant because other work has suggested that phosphorylation of nuclear ERK substrates promotes pathological hypertrophy, while ERK signaling in the cytoplasm is cardioprotective [30, 31].
HDAC inhibitors have also been shown to influence p38 MAP kinase signaling in the heart [32, 33]. HDAC inhibitors enhance cardiac p38 signaling through a mechanism involving enhanced acetylation of a lysine residue in the ATP binding pocket of the kinase [32]. In this manner, HDAC inhibitors function non-genomically to alter signal transduction in the heart.
The catalytic activity of the fourth class I HDAC, HDAC8, was shown to be elevated in the heart in a model of hypertension-induced cardiac hypertrophy [34]. However, the function of HDAC8 in the heart remains unknown. In non-cardiac cells, HDAC8 interacts with numerous nuclear proteins that control gene expression [35], although clear demonstration of HDAC8-mediated deacetylation of histones is lacking [36]. HDAC8 can also control cytoplasmic functions. For example, HDAC8 interacts with α-smooth muscle actin and promotes smooth muscle cell contraction [37], and HDAC8 is able to deacetylate cytoplasmic heat shock protein 20 (HSP20) [38]. The availability of HDAC8-selective inhibitors that are stable in vivo, such as ITF3056 [39], provides an opportunity to assess the possible role of this class I HDAC in the pathogenesis of heart failure.
4. Class IIa HDACs and cardiac hypertrophy
Class IIa HDACs (HDACs -4, -5, -7 and -9) have several unique features [40]. First, these HDACs have long (~500 amino acid) amino-terminal extensions that harbor binding sites for transcription factors and cofactors. For example, this domain mediates binding to the myocyte enhancer factor 2 (MEF2) transcription factor, and results in suppression of MEF2 target genes that govern cardiac hypertrophy. Second, class IIa HDACs undergo signal-dependent nuclear export upon phosphorylation of two serine residues within their amino-terminal extensions. Phosphorylation of these sites by kinases such as protein kinase D (PKD) or calcium/calmodulin-dependent protein kinase (CaMK) results in class IIa HDAC nuclear export, freeing MEF2 to activate pro-hypertrophic target genes.
A third unique feature of class IIa HDACs is that, despite having conserved catalytic domains, these proteins are unable to deacetylate histones; class IIa HDAC catalytic activity is only detectable using an artificial substrate [41, 42]. The search for bona fide class IIa HDAC substrates continues. However, it has been proposed that class IIa HDACs primarily regulate gene expression in a non-catalytic manner, by functioning as scaffolds. Indeed, class IIa HDAC mutants that lack catalytic activity are still able to block pro-hypertrophic MEF2 target gene expression by recruiting co-repressors such as C-terminal-binding protein (CtBP), heterochromatin protein-1 (HP1) and the SUV39H1 histone methyltransferase [40, 43]. An elegant study, performed primarily with human heart samples, showed that HDAC4 represses pro-hypertrophic gene expression by recruiting a histone methyltransferase, SUV39H1, to regulatory elements for these genes [44]. In response to hypertrophic stimuli, HDAC4:SUV39H1 complexes are exported from the nucleus, freeing MEF2 to interact with histone acetyltransferases (HATs) and thereby replace repressive methyl-histone marks with gene stimulatory acetyl-histone marks (Fig. 2).
Figure 2. Newly described regulation and function of class II HDACs in the heart.
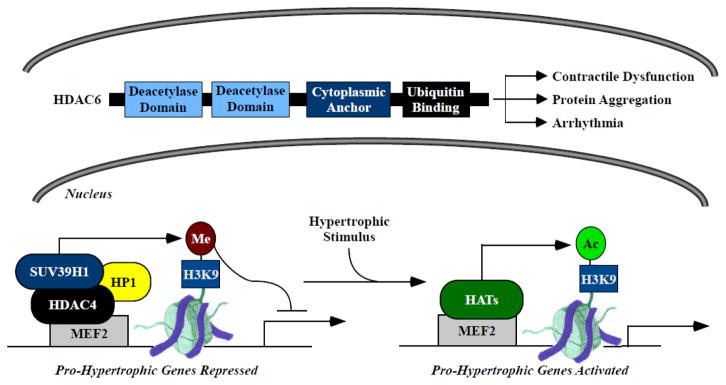
Class IIb HDAC6 contains tandem deacetylase domains, a cytoplasmic anchor and a ubiquitin binding motif. Roles of HDAC6 in cardiac disease have only recently been described, and include impairment of contractile function, promotion of protein aggregate formation, and atrial myocyte remodeling and atrial fibrillation propagation. Class IIa HDAC4 represses pro-hypertrophic gene expression in the heart by promoting formation of repressive methylation marks on histone H3 lysine-9 (H3K9). HDAC4 is recruited to pro-hypertrophic genes through association with the DNA binding transcription factor myocyte enhancer factor 2 (MEF2). This complex also contains the methyl-histone binding protein HP1 and the histone methyltransferase SUV39H1. In response to hypertrophic stimuli, HDAC4 is exported from the nucleus, freeing MEF2 to interact with histone acetyltransferases (HATs) and stimulate pro-hypertrophic gene expression.
5. Class IIb HDAC6 – regulator of cardiac contraction and protein aggregation
HDAC6 is also unique because it is not an epigenetic regulator. This class IIb HDAC contains two deacetylase domains and resides primarily in the cytoplasm, where it is known to deacetylate tubulin, heat shock protein 90 (HSP90) and the F-actin binding protein, cortactin [45] (Fig. 2). The function of HDAC6 in the heart has only recently been studied. Using cell-based assays, a dog pacing model, and human atrial tissue, Brundel and colleagues provided compelling evidence that HDAC6 contributes to structural and functional remodeling of atrial myocytes, thereby promoting atrial fibrillation progression [46]. We found that HDAC6 catalytic activity is elevated in multiple models of cardiac hypertrophy [47], suggesting a possible role for this HDAC isoform in the control of pathological ventricular remodeling. Indeed, subsequent studies with HDAC6 null mice and wild-type mice treated with a highly selective HDAC6 inhibitor, tubastatin A [48], revealed that HDAC6 contributes to systolic LV dysfunction in response to pressure overload or chronic angiotensin II signaling [49]. Interestingly, however, HDAC6 deletion does not block cardiac hypertrophy or fibrosis. Instead, in the absence of HDAC6, isolated cardiac myofibrils are able to produce more force. We have found that HDAC6 co-localizes with cardiac contractile machinery [49], and speculate that deacetylation of sarcomeric proteins by this HDAC isoform impairs systolic function of the heart.
HDAC6 inhibition was recently shown to be protective in a model of desmin-related cardiomyopathy in mice [50]. Expression of a mutant form of αB-crystallin in mouse heart causes accumulation of toxic, misfolded protein aggregates (aggresomes), which leads to heart failure. HDAC6 inhibition was found to reduce aggresome formation in cultured cardiomyocytes, and SAHA administration reduced cardiac aggregates and improved ventricular function in mutant αB-crystallin mice. Interestingly, the reduction in cardiac protein aggregates upon HDAC6 inhibition was associated with elevated autophagic flux. These data suggest that selective inhibition of HDAC6 has potential for the treatment of cardiac proteinopathies. Given the diminished capacity of the aged heart to remove protein aggregates via autophagy [51], it is intriguing to speculate that HDAC6-selective inhibitors will improve cardiac function in the elderly population.
The other class IIb HDAC, HDAC10, as well as the lone class IV HDAC, HDAC11, remain poorly characterized, in part due to the inability to effectively monitor their catalytic activity using in vitro assays [52]. HDAC10 is most closely related to HDAC6, and we have found knockdown of HDAC10 in cardiomyocytes leads to upregulation of HDAC6, and vice versa [47]. Thus, it is possible that these class IIb HDACs have redundant functions in the heart.
6. Cardiac aging and diastolic dysfunction
Heart failure is typically classified as either systolic, in which there is reduced ventricular pump function, or diastolic, which is characterized by impaired cardiac relaxation and abnormal ventricular filling. Cardiac aging is commonly associated with diastolic heart failure, which is also referred to as heart failure with preserved ejection fraction (HFpEF) [53]. Approximately half of the 5 million heart failure patients in the United States have been diagnosed with HFpEF, and more than 90% of HFpEF patients are over the age of 60 at the time of diagnosis [54, 55]. Large clinical trials using standard-of-care systolic heart failure medications have failed to demonstrate efficacy in patients with HFpEF [56–59], highlighting the need to better understand the molecular basis of HFpEF so that novel therapeutic interventions can be developed.
HFpEF is characterized by increased LV filling pressure, increased LV stiffness, and prolonged relaxation in the presence of normal systolic function. Reduced compliance of the LV in the setting of HFpEF is commonly attributed to cardiac fibrosis and hypertrophy, as well as altered calcium handling and elevated resting tension of myofibrils. Nothing is known about the roles of HDACs in the pathogenesis of HFpEF. However, given the ability of HDAC inhibitors to attenuate cardiac hypertrophy and fibrosis, and alter pathological signaling in cardiac myocytes, HDAC inhibition has potential to improve diastolic function of the heart (Fig. 3).
Figure 3. A model for suppression of age-related HFpEF by HDAC inhibitors.

Given the ability of HDAC inhibitors to block cardiac hypertrophy and fibrosis, as well as suppress oxidative stress and inflammation, it is hypothesized that these compounds will reduce age-depended cardiac dysfunction.
7. Oxidative stress, cardiac aging, and HDACs
Elevated levels of reactive oxygen species (ROS) are present in aged hearts, which may contribute to insidious inflammation in the myocardium [60–63]. HDAC inhibitors have both anti-oxidant and anti-inflammatory properties [64], further suggesting that this compound class has potential for the treatment of age-related cardiomyopathy. Treatment of spontaneously hypertensive (SHR) rats for 20 weeks with valproic acid (VPA) [65], a weak albeit selective inhibitor of class I HDACs [66], attenuated cardiac hypertrophy and fibrosis, which was associated with reduced levels of ROS and pro-inflammatory cytokines in ventricular tissue. VPA-mediated lowering of ROS in SHR hearts appeared to be due to diminished expression of gp91phox, a component of the superoxide-generating NADPH oxidase complex, which plays a critical role in age-associated cardiac remodeling [67]. In the rat cardiac I/R injury described above, it was found that cardioprotection mediated by the class I HDAC inhibitor, MS-275, was associated with increased expression of the free radical scavengers mitochondrial superoxide dismutase (SOD2) and catalase in the heart [10]. Importantly, myocardial SOD levels have been shown to be reduced with aging, which contributes to decreased protection against oxidative stress in aged hearts [68]. Furthermore, overexpression of mitochondria-targeted catalase in C57BL/6 mice prevents age-related diastolic dysfunction and associated cardiac hypertrophy and fibrosis [69, 70]. The impact of chronic class I HDAC inhibition on SOD and catalase expression in the heart has yet to be assessed.
Class II HDACs also likely play crucial roles in the control of cardiac redox control. For example, peroxiredoxin (Prx) I and Prx II, which are endogenous antioxidant enzymes, are negatively regulated by HDAC6 [71]. Prx proteins are direct substrates of HDAC6, and inhibition of HDAC6 leads to accumulation of acetylated Prx, which has increased reducing activity and is resistant to superoxidation. HDAC6 is also directly involved in inflammatory signaling. HDAC6 inhibition in macrophages amplifies p38 kinase signaling, leading to enhanced expression of the anti-inflammatory cytokine IL-10 [72]. In addition, HDAC6 inhibition enhances the anti-inflammatory action of regulatory T cells (Tregs), which are a major source of IL-10 [73]. The anti-oxidant and anti-inflammatory action of HDAC6 inhibitors provides further impetus for testing compounds that target this cytoplasmic deacetylase for efficacy in the setting of age-dependent cardiomyopathy.
8. On-target versus off-target actions of HDAC inhibitors
Studies with pharmacological inhibitors are often complemented with genetic loss-of-function to address whether a given action of a compound is due to on-target or off-target action. However, as discussed above using HDAC3 as an example, HDAC knockout/knockdown does not always recapitulate effects of HDAC inhibitors, presumably due to non-catalytic functions of HDACs. An alternative approach to address on/off-target effects of HDAC inhibitors is to employ multiple, structurally distinct compounds, since it is highly unlikely that all of the compounds will bind to a common off-target effector. In heart failure models, HDAC inhibitors from four broad chemical classes are efficacious: short chain fatty acids (e.g., valproic acid), hydroxamic acids (e.g., SAHA), aminobenzamides (e.g., MGCD0103), and cyclic peptides (e.g., apicidin) [74]. As such, it is likely that the beneficial effects of HDAC inhibitors in the heart are due to HDAC inhibition as opposed to off-target action.
9. Conclusions and next steps
Much of this review is framed in the context of HFpEF, which is a common outcome of aging, yet nothing is known about the roles of non-sirtuin HDACs in the control of diastolic heart failure. Rodent hearts develop many of the sequelae associated with aging in humans [75]. Experiments need to be performed to assess whether pan- and/or isoform-selective HDAC inhibitors alter the course of cardiac aging in mice and rats. For these studies it will be essential to formulate the inhibitors in chow or drinking water, and to longitudinally assess cardiac structure and function using echocardiography. The discovery of diverse and highly isoform-selective HDAC inhibitors provides an unprecedented opportunity to use these ‘chemical biological’ probes to dissect the roles of distinct HDAC isoforms in the pathogenesis of various diseases, including HFpEF [76]. These pharmacology studies should be complemented with assessment of mice in which genes for specific HDAC isoforms have been deleted. Given the points listed above, evaluation of age-dependent cardiac remodeling in HDAC6 null mice should be particularly enlightening. However, it is important to emphasize that knocking out an HDAC is not the same as inhibiting its activity, as illustrated by the work on HDAC3 [28].
It will also be important to assess effects of inhibitors of zinc-dependent HDACs in other models of diastolic dysfunction, such as those in which the pathogenic stimulus is hypertension or diabetes. We have begun a preliminary investigation of pan-HDAC inhibition in a rat model of hypertension-induced diastolic dysfunction with preserved ejection fraction. Our initial findings indicate that HDAC inhibitors have remarkable ability to improve cardiac relaxation in male rats independently of effects on blood pressure. Future studies will need to address the mechanism(s) by which HDAC inhibitors alter disease progression in this model. Additionally, experiments will eventually need to be extended to female rodents, since HFpEF is more prevalent in women than men [55]. Pertaining to this point, 17-β-estradiol and estrogen receptor β agonists were shown to have a general suppressive effect on pro-hypertrophic HDACs in cardiomyocytes [77], suggesting the possibility that cardiac HDAC activity is augmented in post-menopausal women.
The therapeutic benefit of any pharmacological agent must be judiciously evaluated against the possible risk of toxicity, particularly for emerging classes of compounds such as HDAC inhibitors. In the context of cancer, HDAC inhibitors are generally well tolerated, with fatigue and thrombocytopenia being the most commonly reported dose-limiting toxicities [78]. We have found that HDAC inhibition improves diastolic cardiac function in rats at concentrations far below those that cause hematological toxicity. These findings suggest that it will be possible to establish a dosing regimen with HDAC inhibitors that provides a sufficient therapeutic index for the treatment of cardiac disease.
We discussed the involvement of zinc-dependent HDACs in cardiac hypertrophy, fibrosis, oxidative stress and inflammation. A multitude of additional processes contribute to age-related cardiac dysfunction, including titin-mediated cardiomyocyte stiffness [79], altered calcium reuptake into the sarcoplasmic reticulum by SERCA2a [80], as well as metabolic deficiencies in the cardiomyocyte due to impaired mitochondrial biogenesis and function [75]. Effects of HDAC inhibitors on these other pathogenic mechanisms have not been evaluated to date. Thus, despite the fact that investigative efforts into the roles of HDACs in the heart have been ongoing for over a decade, there is still a tremendous amount to be learned, especially regarding HDAC function in the context of aging and HFpEF. With the availability of novel chemical probes to target zinc-dependent HDACs, as well as technological advancements for genetic manipulation, progress in this field promises to be rapidly forthcoming, and has great potential to translate into innovative therapies for the treatment of our rapidly aging population.
Highlights.
HDAC inhibitors are efficacious in animal models of heart failure
HDAC inhibitors have multiple mechanisms of action in the heart
HDAC inhibitors have potential to treat age-related diastolic dysfunction
Acknowledgments
We are grateful to Dr. Mark Jeong (UC Denver) for critical discussions about HFpEF. B.S.F. was funded by fellowships from the American Heart Association (12POST10680000) and NIH (1F32HL124893-01). T.A.M. was supported by NIH (HL116848, AG043822 and HL114887) and the American Heart Association (Grant-in-Aid, 14510001).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–40. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 2.Lundby A, Lage K, Weinert BT, Bekker-Jensen DB, Secher A, Skovgaard T, et al. Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2012;2:419–31. doi: 10.1016/j.celrep.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gregoretti IV, Lee YM, Goodson HV. Molecular evolution of the histone deacetylase family: functional implications of phylogenetic analysis. J Mol Biol. 2004;338:17–31. doi: 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 4.Berry JM, Cao DJ, Rothermel BA, Hill JA. Histone deacetylase inhibition in the treatment of heart disease. Expert Opin Drug Saf. 2008;7:53–67. doi: 10.1517/14740338.7.1.53. [DOI] [PubMed] [Google Scholar]
- 5.McKinsey TA. Therapeutic potential for HDAC inhibitors in the heart. Annu Rev Pharmacol Toxicol. 2012;52:303–19. doi: 10.1146/annurev-pharmtox-010611-134712. [DOI] [PubMed] [Google Scholar]
- 6.Cao DJ, Wang ZV, Battiprolu PK, Jiang N, Morales CR, Kong Y, et al. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci U S A. 2011;108:4123–8. doi: 10.1073/pnas.1015081108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kee HJ, Sohn IS, Nam KI, Park JE, Qian YR, Yin Z, et al. Inhibition of histone deacetylation blocks cardiac hypertrophy induced by angiotensin II infusion and aortic banding. Circulation. 2006;113:51–9. doi: 10.1161/CIRCULATIONAHA.105.559724. [DOI] [PubMed] [Google Scholar]
- 8.Xie M, Kong Y, Tan W, May H, Battiprolu PK, Pedrozo Z, et al. Histone deacetylase inhibition blunts ischemia/reperfusion injury by inducing cardiomyocyte autophagy. Circulation. 2014;129:1139–51. doi: 10.1161/CIRCULATIONAHA.113.002416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Przyklenk K, Dong Y, Undyala VV, Whittaker P. Autophagy as a therapeutic target for ischaemia/reperfusion injury? Concepts, controversies, and challenges. Cardiovasc Res. 2012;94:197–205. doi: 10.1093/cvr/cvr358. [DOI] [PubMed] [Google Scholar]
- 10.Aune SE, Herr DJ, Mani SK, Menick DR. Selective inhibition of class I but not class IIb histone deacetylases exerts cardiac protection from ischemia reperfusion. J Mol Cell Cardiol. 2014;72:138–45. doi: 10.1016/j.yjmcc.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schuetze KB, McKinsey TA, Long CS. Targeting cardiac fibroblasts to treat fibrosis of the heart: focus on HDACs. J Mol Cell Cardiol. 2014;70:100–7. doi: 10.1016/j.yjmcc.2014.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Williams SM, Golden-Mason L, Ferguson BS, Schuetze KB, Cavasin MA, Demos-Davies K, et al. Class I HDACs regulate angiotensin II-dependent cardiac fibrosis via fibroblasts and circulating fibrocytes. J Mol Cell Cardiol. 2014;67:112–25. doi: 10.1016/j.yjmcc.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nural-Guvener HF, Zakharova L, Nimlos J, Popovic S, Mastroeni D, Gaballa MA. HDAC class I inhibitor, Mocetinostat, reverses cardiac fibrosis in heart failure and diminishes CD90+ cardiac myofibroblast activation. Fibrogenesis Tissue Repair. 2014;7:10. doi: 10.1186/1755-1536-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blakeslee WW, Wysoczynski CL, Fritz KS, Nyborg JK, Churchill ME, McKinsey TA. Class I HDAC inhibition stimulates cardiac protein SUMOylation through a post-translational mechanism. Cell Signal. 2014;26:2912–20. doi: 10.1016/j.cellsig.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vejpongsa P, Yeh ET. Wrestling with heart failure: SUMO-1 to the rescue. Circ Res. 2014;114:1561–3. doi: 10.1161/CIRCRESAHA.114.304125. [DOI] [PubMed] [Google Scholar]
- 16.Lishnevsky M, Haudek SB. Epigenetic regulation of fibrocyte differentiation? J Mol Cell Cardiol. 2014;69:85–7. doi: 10.1016/j.yjmcc.2014.01.019. [DOI] [PubMed] [Google Scholar]
- 17.Herzog EL, Bucala R. Fibrocytes in health and disease. Exp Hematol. 2010;38:548–56. doi: 10.1016/j.exphem.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cieslik KA, Taffet GE, Carlson S, Hermosillo J, Trial J, Entman ML. Immune-inflammatory dysregulation modulates the incidence of progressive fibrosis and diastolic stiffness in the aging heart. J Mol Cell Cardiol. 2011;50:248–56. doi: 10.1016/j.yjmcc.2010.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cunliffe VT. Eloquent silence: developmental functions of Class I histone deacetylases. Curr Opin Genet Dev. 2008;18:404–10. doi: 10.1016/j.gde.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kelly RD, Cowley SM. The physiological roles of histone deacetylase (HDAC) 1 and 2: complex co-stars with multiple leading parts. Biochem Soc Trans. 2013;41:741–9. doi: 10.1042/BST20130010. [DOI] [PubMed] [Google Scholar]
- 21.Longworth MS, Laimins LA. Histone deacetylase 3 localizes to the plasma membrane and is a substrate of Src. Oncogene. 2006;25:4495–500. doi: 10.1038/sj.onc.1209473. [DOI] [PubMed] [Google Scholar]
- 22.Samant SA, Courson DS, Sundaresan NR, Pillai VB, Tan M, Zhao Y, et al. HDAC3-dependent reversible lysine acetylation of cardiac myosin heavy chain isoforms modulates their enzymatic and motor activity. J Biol Chem. 2011;286:5567–77. doi: 10.1074/jbc.M110.163865. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 23.Eom GH, Kook H. The role of histone deacetylase 2 and its posttranslational modifications in cardiac hypertrophy. BMB Rep. 2014 doi: 10.5483/BMBRep.2015.48.3.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eom GH, Nam YS, Oh JG, Choe N, Min HK, Yoo EK, et al. Regulation of acetylation of histone deacetylase 2 by p300/CBP-associated factor/histone deacetylase 5 in the development of cardiac hypertrophy. Circ Res. 2014;114:1133–43. doi: 10.1161/CIRCRESAHA.114.303429. [DOI] [PubMed] [Google Scholar]
- 25.Montgomery RL, Potthoff MJ, Haberland M, Qi X, Matsuzaki S, Humphries KM, et al. Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J Clin Invest. 2008;118:3588–97. doi: 10.1172/JCI35847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun Z, Singh N, Mullican SE, Everett LJ, Li L, Yuan L, et al. Diet-induced lethality due to deletion of the Hdac3 gene in heart and skeletal muscle. J Biol Chem. 2011;286:33301–9. doi: 10.1074/jbc.M111.277707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharifi-Sanjani M, Shoushtari AH, Quiroz M, Baust J, Sestito SF, Mosher M, et al. Cardiac CD47 drives left ventricular heart failure through Ca2+-CaMKII-regulated induction of HDAC3. J Am Heart Assoc. 2014;3:e000670. doi: 10.1161/JAHA.113.000670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun Z, Feng D, Fang B, Mullican SE, You SH, Lim HW, et al. Deacetylase-independent function of HDAC3 in transcription and metabolism requires nuclear receptor corepressor. Mol Cell. 2013;52:769–82. doi: 10.1016/j.molcel.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ferguson BS, Harrison BC, Jeong MY, Reid BG, Wempe MF, Wagner FF, et al. Signal-dependent repression of DUSP5 by class I HDACs controls nuclear ERK activity and cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A. 2013;110:9806–11. doi: 10.1073/pnas.1301509110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lorenz K, Schmitt JP, Schmitteckert EM, Lohse MJ. A new type of ERK1/2 autophosphorylation causes cardiac hypertrophy. Nat Med. 2009;15:75–83. doi: 10.1038/nm.1893. [DOI] [PubMed] [Google Scholar]
- 31.Ruppert C, Deiss K, Herrmann S, Vidal M, Oezkur M, Gorski A, et al. Interference with ERK(Thr188) phosphorylation impairs pathological but not physiological cardiac hypertrophy. Proc Natl Acad Sci U S A. 2013;110:7440–5. doi: 10.1073/pnas.1221999110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pillai VB, Sundaresan NR, Samant SA, Wolfgeher D, Trivedi CM, Gupta MP. Acetylation of a conserved lysine residue in the ATP binding pocket of p38 augments its kinase activity during hypertrophy of cardiomyocytes. Mol Cell Biol. 2011;31:2349–63. doi: 10.1128/MCB.01205-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao TC, Cheng G, Zhang LX, Tseng YT, Padbury JF. Inhibition of histone deacetylases triggers pharmacologic preconditioning effects against myocardial ischemic injury. Cardiovasc Res. 2007;76:473–81. doi: 10.1016/j.cardiores.2007.08.010. [DOI] [PubMed] [Google Scholar]
- 34.Kee HJ, Bae EH, Park S, Lee KE, Suh SH, Kim SW, et al. HDAC inhibition suppresses cardiac hypertrophy and fibrosis in DOCA-salt hypertensive rats via regulation of HDAC6/HDAC8 enzyme activity. Kidney Blood Press Res. 2013;37:229–39. doi: 10.1159/000350148. [DOI] [PubMed] [Google Scholar]
- 35.Olson DE, Udeshi ND, Wolfson NA, Pitcairn CA, Sullivan ED, Jaffe JD, et al. An unbiased approach to identify endogenous substrates of “histone” deacetylase 8. ACS Chem Biol. 2014;9:2210–6. doi: 10.1021/cb500492r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wolfson NA, Pitcairn CA, Fierke CA. HDAC8 substrates: Histones and beyond. Biopolymers. 2013;99:112–26. doi: 10.1002/bip.22135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Waltregny D, Glenisson W, Tran SL, North BJ, Verdin E, Colige A, et al. Histone deacetylase HDAC8 associates with smooth muscle alpha-actin and is essential for smooth muscle cell contractility. FASEB J. 2005;19:966–8. doi: 10.1096/fj.04-2303fje. [DOI] [PubMed] [Google Scholar]
- 38.Karolczak-Bayatti M, Sweeney M, Cheng J, Edey L, Robson SC, Ulrich SM, et al. Acetylation of heat shock protein 20 (Hsp20) regulates human myometrial activity. J Biol Chem. 2011;286:34346–55. doi: 10.1074/jbc.M111.278549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li S, Fossati G, Marchetti C, Modena D, Pozzi P, Reznikov LL, et al. Specific inhibition of histone deacetylase 8 reduces gene expression and production of proinflammatory cytokines in vitro and in vivo. J Biol Chem. 2014 doi: 10.1074/jbc.M114.618454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McKinsey TA. Derepression of pathological cardiac genes by members of the CaM kinase superfamily. Cardiovasc Res. 2007;73:667–77. doi: 10.1016/j.cardiores.2006.11.036. [DOI] [PubMed] [Google Scholar]
- 41.Jones P, Altamura S, De FR, Gallinari P, Lahm A, Neddermann P, et al. Probing the elusive catalytic activity of vertebrate class IIa histone deacetylases. Bioorg Med Chem Lett. 2008;18:1814–9. doi: 10.1016/j.bmcl.2008.02.025. [DOI] [PubMed] [Google Scholar]
- 42.Lahm A, Paolini C, Pallaoro M, Nardi MC, Jones P, Neddermann P, et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc Natl Acad Sci U S A. 2007;104:17335–40. doi: 10.1073/pnas.0706487104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang CL, McKinsey TA, Olson EN. Association of class II histone deacetylases with heterochromatin protein 1: potential role for histone methylation in control of muscle differentiation. Mol Cell Biol. 2002;22:7302–12. doi: 10.1128/MCB.22.20.7302-7312.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hohl M, Wagner M, Reil JC, Muller SA, Tauchnitz M, Zimmer AM, et al. HDAC4 controls histone methylation in response to elevated cardiac load. J Clin Invest. 2013;123:1359–70. doi: 10.1172/JCI61084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Valenzuela-Fernandez A, Cabrero JR, Serrador JM, Sanchez-Madrid F. HDAC6: a key regulator of cytoskeleton, cell migration and cell-cell interactions. Trends Cell Biol. 2008;18:291–7. doi: 10.1016/j.tcb.2008.04.003. [DOI] [PubMed] [Google Scholar]
- 46.Zhang D, Wu CT, Qi X, Meijering RA, Hoogstra-Berends F, Tadevosyan A, et al. Activation of histone deacetylase-6 induces contractile dysfunction through derailment of alpha-tubulin proteostasis in experimental and human atrial fibrillation. Circulation. 2014;129:346–58. doi: 10.1161/CIRCULATIONAHA.113.005300. [DOI] [PubMed] [Google Scholar]
- 47.Lemon DD, Horn TR, Cavasin MA, Jeong MY, Haubold KW, Long CS, et al. Cardiac HDAC6 catalytic activity is induced in response to chronic hypertension. J Mol Cell Cardiol. 2011;51:41–50. doi: 10.1016/j.yjmcc.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Butler KV, Kalin J, Brochier C, Vistoli G, Langley B, Kozikowski AP. Rational design and simple chemistry yield a superior, neuroprotective HDAC6 inhibitor, tubastatin A. J Am Chem Soc. 2010;132:10842–6. doi: 10.1021/ja102758v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Demos-Davies KM, Ferguson BS, Cavasin MA, Mahaffey JH, Williams SM, Spiltoir JI, et al. HDAC6 contributes to pathological responses of heart and skeletal muscle to chronic angiotensin-II signaling. Am J Physiol Heart Circ Physiol. 2014;307:H252–H258. doi: 10.1152/ajpheart.00149.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McLendon PM, Ferguson BS, Osinska H, Bhuiyan MS, James J, McKinsey TA, et al. Tubulin hyperacetylation is adaptive in cardiac proteotoxicity by promoting autophagy. Proc Natl Acad Sci U S A. 2014;111:E5178–E5186. doi: 10.1073/pnas.1415589111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Meyer GR, De Keulenaer GW, Martinet W. Role of autophagy in heart failure associated with aging. Heart Fail Rev. 2010;15:423–30. doi: 10.1007/s10741-010-9166-6. [DOI] [PubMed] [Google Scholar]
- 52.Bradner JE, West N, Grachan ML, Greenberg EF, Haggarty SJ, Warnow T, et al. Chemical phylogenetics of histone deacetylases. Nat Chem Biol. 2010;6:238–43. doi: 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sharma K, Kass DA. Heart failure with preserved ejection fraction: mechanisms, clinical features, and therapies. Circ Res. 2014;115:79–96. doi: 10.1161/CIRCRESAHA.115.302922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Borlaug BA, Paulus WJ. Heart failure with preserved ejection fraction: pathophysiology, diagnosis, and treatment. Eur Heart J. 2011;32:670–9. doi: 10.1093/eurheartj/ehq426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scantlebury DC, Borlaug BA. Why are women more likely than men to develop heart failure with preserved ejection fraction? Curr Opin Cardiol. 2011;26:562–8. doi: 10.1097/HCO.0b013e32834b7faf. [DOI] [PubMed] [Google Scholar]
- 56.Bhatia RS, Tu JV, Lee DS, Austin PC, Fang J, Haouzi A, et al. Outcome of heart failure with preserved ejection fraction in a population-based study. N Engl J Med. 2006;355:260–9. doi: 10.1056/NEJMoa051530. [DOI] [PubMed] [Google Scholar]
- 57.Massie BM, Carson PE, McMurray JJ, Komajda M, McKelvie R, Zile MR, et al. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med. 2008;359:2456–67. doi: 10.1056/NEJMoa0805450. [DOI] [PubMed] [Google Scholar]
- 58.Pitt B, Pfeffer MA, Assmann SF, Boineau R, Anand IS, Claggett B, et al. Spironolactone for heart failure with preserved ejection fraction. N Engl J Med. 2014;370:1383–92. doi: 10.1056/NEJMoa1313731. [DOI] [PubMed] [Google Scholar]
- 59.Shah SJ, Gheorghiade M. Heart failure with preserved ejection fraction: treat now by treating comorbidities. JAMA. 2008;300:431–3. doi: 10.1001/jama.300.4.431. [DOI] [PubMed] [Google Scholar]
- 60.Dai DF, Rabinovitch PS. Cardiac aging in mice and humans: the role of mitochondrial oxidative stress. Trends Cardiovasc Med. 2009;19:213–20. doi: 10.1016/j.tcm.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Franceschi C, Bonafe M, Valensin S, Olivieri F, De LM, Ottaviani E, et al. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann N Y Acad Sci. 2000;908:244–54. doi: 10.1111/j.1749-6632.2000.tb06651.x. [DOI] [PubMed] [Google Scholar]
- 62.Judge S, Jang YM, Smith A, Hagen T, Leeuwenburgh C. Age-associated increases in oxidative stress and antioxidant enzyme activities in cardiac interfibrillar mitochondria: implications for the mitochondrial theory of aging. FASEB J. 2005;19:419–21. doi: 10.1096/fj.04-2622fje. [DOI] [PubMed] [Google Scholar]
- 63.Wu J, Xia S, Kalionis B, Wan W, Sun T. The role of oxidative stress and inflammation in cardiovascular aging. Biomed Res Int. 2014;2014:615312. doi: 10.1155/2014/615312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.McKinsey TA. Targeting inflammation in heart failure with histone deacetylase inhibitors. Mol Med. 2011;17:434–41. doi: 10.2119/molmed.2011.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cardinale JP, Sriramula S, Pariaut R, Guggilam A, Mariappan N, Elks CM, et al. HDAC inhibition attenuates inflammatory, hypertrophic, and hypertensive responses in spontaneously hypertensive rats. Hypertension. 2010;56:437–44. doi: 10.1161/HYPERTENSIONAHA.110.154567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fass DM, Shah R, Ghosh B, Hennig K, Norton S, Zhao WN, et al. Effect of Inhibiting Histone Deacetylase with Short-Chain Carboxylic Acids and Their Hydroxamic Acid Analogs on Vertebrate Development and Neuronal Chromatin. ACS Med Chem Lett. 2010;2:39–42. doi: 10.1021/ml1001954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang M, Zhang J, Walker SJ, Dworakowski R, Lakatta EG, Shah AM. Involvement of NADPH oxidase in age-associated cardiac remodeling. J Mol Cell Cardiol. 2010;48:765–72. doi: 10.1016/j.yjmcc.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Das KC, Muniyappa H. Age-dependent mitochondrial energy dynamics in the mice heart: role of superoxide dismutase-2. Exp Gerontol. 2013;48:947–59. doi: 10.1016/j.exger.2013.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Dai DF, Santana LF, Vermulst M, Tomazela DM, Emond MJ, MacCoss MJ, et al. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation. 2009;119:2789–97. doi: 10.1161/CIRCULATIONAHA.108.822403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Treuting PM, Linford NJ, Knoblaugh SE, Emond MJ, Morton JF, Martin GM, et al. Reduction of age-associated pathology in old mice by overexpression of catalase in mitochondria. J Gerontol A Biol Sci Med Sci. 2008;63:813–22. doi: 10.1093/gerona/63.8.813. [DOI] [PubMed] [Google Scholar]
- 71.Parmigiani RB, Xu WS, Venta-Perez G, Erdjument-Bromage H, Yaneva M, Tempst P, et al. HDAC6 is a specific deacetylase of peroxiredoxins and is involved in redox regulation. Proc Natl Acad Sci U S A. 2008;105:9633–8. doi: 10.1073/pnas.0803749105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang B, Rao YH, Inoue M, Hao R, Lai CH, Chen D, et al. Microtubule acetylation amplifies p38 kinase signalling and anti-inflammatory IL-10 production. Nat Commun. 2014;5:3479. doi: 10.1038/ncomms4479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.de Zoeten EF, Wang L, Butler K, Beier UH, Akimova T, Sai H, et al. Histone deacetylase 6 and heat shock protein 90 control the functions of Foxp3(+) T-regulatory cells. Mol Cell Biol. 2011;31:2066–78. doi: 10.1128/MCB.05155-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bush EW, McKinsey TA. Protein acetylation in the cardiorenal axis: the promise of histone deacetylase inhibitors. Circ Res. 2010;106:272–84. doi: 10.1161/CIRCRESAHA.109.209338. [DOI] [PubMed] [Google Scholar]
- 75.Loffredo FS, Nikolova AP, Pancoast JR, Lee RT. Heart failure with preserved ejection fraction: molecular pathways of the aging myocardium. Circ Res. 2014;115:97–107. doi: 10.1161/CIRCRESAHA.115.302929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.McKinsey TA. Isoform-selective HDAC inhibitors: closing in on translational medicine for the heart. J Mol Cell Cardiol. 2011;51:491–6. doi: 10.1016/j.yjmcc.2010.11.009. [DOI] [PubMed] [Google Scholar]
- 77.Pedram A, Razandi M, Narayanan R, Dalton JT, McKinsey TA, Levin ER. Estrogen regulates histone deacetylases to prevent cardiac hypertrophy. Mol Biol Cell. 2013;24:3805–18. doi: 10.1091/mbc.E13-08-0444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Fraczek J, Vanhaecke T, Rogiers V. Toxicological and metabolic considerations for histone deacetylase inhibitors. Expert Opin Drug Metab Toxicol. 2013;9:441–57. doi: 10.1517/17425255.2013.754011. [DOI] [PubMed] [Google Scholar]
- 79.LeWinter MM, Granzier HL. Cardiac titin and heart disease. J Cardiovasc Pharmacol. 2014;63:207–12. doi: 10.1097/FJC.0000000000000007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Janczewski AM, Lakatta EG. Modulation of sarcoplasmic reticulum Ca(2+) cycling in systolic and diastolic heart failure associated with aging. Heart Fail Rev. 2010;15:431–45. doi: 10.1007/s10741-010-9167-5. [DOI] [PMC free article] [PubMed] [Google Scholar]