

Abstract
Behavioral disinhibition (BD) is a quantitative measure designed to capture the heritable variation encompassing risky and impulsive behaviors. As a result, BD represents an ideal target for discovering genetic loci that predispose individuals to a wide range of antisocial behaviors and substance misuse that together represent a large cost to society as a whole. Published genome-wide association studies (GWAS) have examined specific phenotypes that fall under the umbrella of BD (e.g. alcohol dependence, conduct disorder); however no GWAS has specifically examined the overall BD construct. We conducted a GWAS of BD using a sample of 1,901 adolescents over-selected for characteristics that define high BD, such as substance and antisocial behavior problems, finding no individual locus that surpassed genome-wide significance. Although no single SNP was significantly associated with BD, restricted maximum likelihood analysis estimated that 49.3% of the variance in BD within the Caucasian sub-sample was accounted for by the genotyped SNPs (p=0.06). Gene-based tests identified seven genes associated with BD (p≤2.0×10−6). Although the current study was unable to identify specific SNPs or pathways with replicable effects on BD, the substantial sample variance that could be explained by all genotyped SNPs suggests that larger studies could successfully identify common variants associated with BD.
Behavioral disinhibition (BD) is a latent quantitative measure designed to capture common variation shared across many harmful or dangerous behaviors including substance problems, antisocial or criminal behavior, and novelty seeking (Young et al. 2000). In addition, 60%–80% of variation in BD is attributed to additive genetic effects, making BD more heritable than many of the individual component behaviors used to formulate the latent BD construct (Young et al. 2000, Krueger et al. 2002, Hicks et al. 2013). To date, genome-wide association studies (GWAS) have been restricted to such individual component behaviors of BD (McGue et al. 2013), including use or abuse of various substances (e.g. alcohol (Bierut et al. 2010, Edenberg et al. 2010, Frank et al. 2012, Gelernter et al. 2014a, Kapoor et al. 2013, Schumann et al. 2011, Treutlein et al. 2009, Wang et al. 2012), tobacco (Bierut et al. 2007, Furberg et al. 2010, Liu et al. 2010, Thorgeirsson et al. 2010), cannabis (Agrawal et al. 2011, Verweij et al. 2013), methamphetamine (Uhl et al. 2008), opioids (Gelernter et al. 2014b, Nielsen et al. 2010), and cocaine (Gelernter et al. 2014c)), conduct disorder (Dick et al. 2011), adult antisocial behavior (Tielbeek et al. 2012), and related personality constructs such as excitement seeking (Terracciano et al. 2011). Although certain behaviors, most notably tobacco use (Bierut et al. 2007, Furberg et al. 2010, Liu et al. 2010, Thorgeirsson et al. 2010), have identified robust associations with specific variants, many GWAS fail to identify individual loci with genome-wide significant effects. This suggests that much of the heritability underlying each trait is unlikely the result of a small number of variants with large effects, and will require larger sample sizes in order to identify variants with small effects (Lee et al. 2011). GWAS of other phenotypes have identified significant replicated effects when large enough samples sizes have been amassed to provide adequate statistical power to identify variants despite very small effect sizes (e.g., accounting for 0.1% of the total variance, or less; Sullivan et al. 2011; Rietveld et al. 2013).
Increasing sample sizes is only one of a number of ways to increase statistical power. Improved phenotypic assessment and modeling could also provide increased statistical power for studies conducted in more moderately sized samples, and particularly for phenotypes that are presumed to be continuously distributed in the population (van der Sluis et al. 2012). BD is a prime example in this context, as relevant quantitative differences in phenotypic severity are maintained between individuals, whereas a case-control approach is fairly insensitive to these differences. However, one issue with searching for specific genetic influences on many continuous phenotypes, such as BD, is that the most severe, clinically significant levels are relatively rare in the general population, as they are located on the extreme ends of the distribution. Ascertaining samples specifically for individuals with extreme phenotypes may improve our ability to detect small genetic effects by increasing the sample variance. Therefore, an ideal sample might be considered one that is enriched (and well-measured) for extreme BD characteristics.
We report here results and characterization of the initial GWAS from the Center on Antisocial Drug Dependence (CADD), an adolescent sample over-selected for severe BD characteristics. Any genetic effects on BD are potentially attributable to many (i.e., thousands of) variants, each with a very, very small effect. Incorporating methods for aggregating effects across multiple variants, such as gene- and pathway-based analyses, can identify promising causal biological systems beyond the significance of any single variant. In addition to SNP level association, the current study applied gene-based, pathway-based, and genome-wide approaches to characterize genetic influences on BD in a diverse, clinically-oversampled, thoroughly phenotyped sample. By supplementing a GWAS with several methods of aggregating genetic evidence across many potentially associated variants, we sought to generate novel insights into the potential genetic etiology of BD and identify promising candidates, either old or new, for future study.
Methods
Participants
Participants with genetic and relevant phenotypic data were ascertained from the CADD projects; full details of participant selection for inclusion in the GWAS sample are provided in the Supplemental materials. GWAS participants were drawn from several primary studies described elsewhere (Hartman et al. 2008, Petrill et al. 2003, Rhea et al. 2006, Stallings et al. 2005). The current sample of 1,901 unrelated adolescents was over-selected for adolescent BD, with half of the participants ascertained specifically from high-risk populations (i.e. recruited through substance abuse treatment, special schools, or involvement with the criminal justice system; see Supplement for additional criteria for clinical probands). CADD GWAS participants had an average age of 16.5 (SD=1.4, range=13.0–19.9), 28.9% were female, and 37.3% of participants reported non-Caucasian ancestry (primarily Hispanic or African; see Supplemental Table S1 for complete demographic statistics).
Phenotype
BD was defined as a composite measure of substance dependence vulnerability (assessed across 10 substances), novelty seeking, and conduct disorder symptoms. The BD phenotype has been previously examined within the CADD samples, including Young et al. (2000) demonstrating that the component measures have loadings ≥0.4 on a common, highly heritable BD latent factor, and linkage analyses by Stallings et al. (2003, 2005). A full description of construction of the BD phenotype is provided in the Supplement; Supplemental Figure S1 shows the distribution of BD in the CADD GWAS sample. Briefly, principal component scores were normed to community-representative samples in CADD and applied to all CADD GWAS participants from both the community-representative (48.2%) and high-risk samples (51.8%). Average scores on the BD composite measure were 0.19 (SD=1.2, range=−1.9–5.0) for the community-representative participants and 2.76 (SD=1.2, range=−0.3–6.7) for the high-risk participants.
Genotyping
All participants were genotyped on the Affymetrix 6.0 platform (Affymetrix, Inc., Santa Clara CA), with a total of 696,388 autosomal SNPs available for analysis after quality control. Full details on processing and cleaning genotypes for the CADD GWAS sample is provided in the Supplement. Population stratification was examined by performing multidimensional scaling in PLINK (Purcell et al. 2007), in which ten ancestry dimensions were estimated. The first three dimensions notably captured genetic variation among individuals of self-reported African, Hispanic, and Asian ancestry, compared to a central (majority) node of individuals of self-reported European ancestry. Supplemental Figure S2 illustrates the first three ancestry dimensions within the CADD GWAS sample (along with individuals’ self-reported ancestry).
Analyses
Genome-wide analysis was conducted as a linear regression of the additive effect of each SNP on BD in PLINK (Purcell et al. 2007). All autosomal SNPs that passed basic quality controls were tested for association with BD, and 10 ancestry dimensions were included as covariates. Age and sex were accounted for in the estimation of the BD phenotype. The criterion for individual SNP significance was set at p<5×10−8.
Genome-wide data from the CADD GWAS sample were further characterized using Genome-wide Complex Trait Analysis (GCTA; Yang et al. 2011). GCTA allows us to estimate the proportion of variance in the phenotype that may be explained using all of the genotyped SNPs using restricted maximum likelihood (REML) analysis. While this method does not specifically identify any causal variants, it does estimate the total proportion of sample variance that may be explained by all of the genotyped SNPs.
Gene-based tests were conducted using VEGAS (Liu et al. 2010), which aggregates evidence of association across all SNPs within a gene. A total of 16,094 autosomal genes were tested for association with BD in CADD, based on the primary GWAS results, with a multiple-testing-corrected significance threshold set at p<3.1×10−6.
INRICH (Lee et al. 2012) was selected to conduct our pathway analyses as it is well-suited for testing both large (i.e., exploratory) and small (i.e., candidate) pathway sets. We took two, complementary approaches to pathway analysis: first, we sought to confirm previously proposed candidate gene pathways (Hodgkinson et al., 2008); second, we conducted an exploratory analysis aimed at identifying novel pathways involved in BD (The Gene Ontology Consortium, 2000). Additional details of the pathway analysis methods are discussed in the Supplemental Materials.
Promising results from the pathway analysis of the CADD sample were followed up in two additional samples: the Minnesota Center for Twin and Family Research (MCTFR; N=3,378), a community-based adolescent sample (McGue et al. 2013, Miller et al. 2012), and the Study of Addiction: Genes and Environment (SAGE; N=3,988), a clinically over-selected study of addiction (Bierut et al. 2010; dbGaP study accession: phs000092.v1.p1). A phenotype similar to BD as defined in the CADD sample was available in the MCTFR sample (Hicks et al. 2010; McGue et al. 2013). The phenotype analysed in the SAGE sample was the average number of dependence symptoms for substances that each participant used. Full description of the MCTFR and SAGE samples is provided in the Supplement.
Results
Figure 1 summarizes the GWAS results for BD in the over-selected CADD sample. No individual SNP reached genome-wide significance (p<5×10−8), nor did any SNP reach genome-wide significance in the MCFTR or SAGE samples (see Supplemental Figure S3 for QQ plots of the GWAS results from each study). Results from loci reaching p<5.0×10−5 in CADD are summarized in Table 1 (full GWAS results are available from the first author on request). The most significant SNP in the CADD GWAS was rs7104461 (p=5.8×10−6), an intergenic SNP on chromosome 11 for which there are no previously reported associated phenotypes. While this SNP was not genotyped in either the MCTFR or SAGE samples, it is in linkage disequilibium with rs341058 (r2=1.0 in 1000 Genomes Pilot 1 CEU sample, distance = 8721bp; Johnson et al. 2008), which was genotyped on both MCTFR and SAGE platforms and may serve as a proxy to compare results across samples. This proxy SNP was not associated with either adolescent BD in MCTFR (p=0.30) or adult substance dependence symptoms in SAGE (p=0.87).
Figure 1.
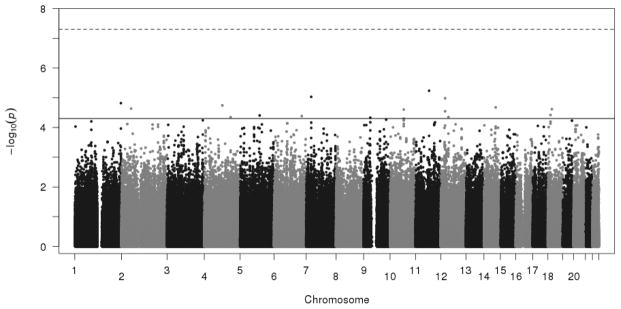
Plot of −log10(p) from the CADD GWAS, arranged by chromosomal location. The top (dashed) horizontal line indicates genome-wide significance at p=5×10−8; the lower (solid) line marks p=5×10−5 (loci described in Table 1).
Table 1.
Top associated loci from the CADD GWAS.
| Index SNP | Index SNP description
|
Association
|
Locus description
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A1 | A2 | MAF | HWE P | Beta | P | Chr | Start | Stop | N | Kb | Genes | |
| rs4654186 | C | T | 0.467 | 5.7E-3 | −0.248 | 1.5E-5 | 1 | 246168017 | 246230729 | 6 | 62.7 | SMYD3 |
| rs11562945 | A | G | 0.029 | 2.1E-1 | 0.720 | 2.3E-5 | 2 | 51671180 | 51671180 | 1 | 0.0 | |
| rs2114532 | A | C | 0.025 | 2.8E-2 | 0.780 | 1.8E-5 | 4 | 96129959 | 96130145 | 2 | 0.2 | UNC5C |
| rs17050678 | A | G | 0.202 | 6.2E-1 | −0.289 | 4.5E-5 | 4 | 139945108 | 140143272 | 9 | 198.2 | PPP1R14BP3,ELF2,CCRN4L |
| rs10464002 | T | C | 0.164 | 1.8E-1 | 0.317 | 3.9E-5 | 5 | 103549477 | 103657217 | 4 | 107.7 | |
| rs10485364 | T | G | 0.011 | 2.0E-1 | 1.183 | 4.1E-5 | 6 | 147106285 | 147106285 | 1 | 0.0 | LOC729176,ADGB |
| rs12666574 | C | T | 0.326 | 7.9E-1 | −0.272 | 9.3E-6 | 7 | 26491618 | 26526960 | 2 | 35.3 | LOC441204 |
| rs10972581 | A | G | 0.407 | 2.7E-1 | −0.238 | 4.7E-5 | 9 | 35779571 | 35870001 | 6 | 90.4 | TMEM8B,SPAG8,OR13J1,OR13E1P,NPR2,LOC100128136,HINT2,FP588,FAM221B |
| rs10509330 | T | C | 0.289 | 6.2E-1 | −0.265 | 2.5E-5 | 10 | 72898795 | 72944032 | 12 | 45.2 | |
| rs7104461 | A | C | 0.132 | 1.6E-1 | −0.392 | 5.8E-6 | 11 | 72394446 | 72404958 | 2 | 10.5 | RPS12P20,PDE2A,ARAP1 |
| rs901625 | G | T | 0.419 | 5.7E-1 | 0.251 | 1.0E-5 | 12 | 22787796 | 22857819 | 10 | 70.0 | RPS27P22,ETNK1 |
| rs11175260 | C | T | 0.070 | 2.9E-1 | −0.459 | 4.4E-5 | 12 | 40387225 | 40601708 | 7 | 214.5 | SLC2A13,RPL30P13,LRRK2 |
| rs1652591 | T | C | 0.417 | 4.8E-1 | −0.250 | 2.1E-5 | 14 | 82542483 | 82559572 | 3 | 17.1 | |
| rs7233911 | A | G | 0.293 | 8.2E-1 | 0.261 | 3.8E-5 | 18 | 13927552 | 13931827 | 3 | 4.3 | RPL36AP49,MC2R |
| rs16940157 | A | C | 0.004 | 1.0E+0 | 1.849 | 2.4E-5 | 18 | 21149803 | 21149803 | 1 | 0.0 | NPC1 |
Note. Index SNP = most significant SNP tagging the LD block; A1 = tested minor allele; A2 = alternate major allele; MAF = minor allele frequency; HWE P = Hardy-Weinberg Equilibrium p-value; Beta = linear regression coefficient; P = association p-value; Chr, Start, Stop = location of the LD block tagged by the Index SNP; N = number of tested SNPs in the LD block; Kb = size of the LD block; Genes = genes overlapping the LD block.
Whole-genome SNP-heritability was estimated with GCTA in the CADD sample. SNPs genotyped in the current study explained 27.8% of the CADD sample variance in BD (SE=0.15, p=0.03). The point estimate of heritability remained fairly stable when the sample was restricted to individuals estimated to be <2.5% identical-by-state (N=1148, V(G)/Vp=30.9%, SE=0.28, p=0.10) or those individuals with only Caucasian ancestry (as determined by an examination of ancestry component plots, N=1031, V(G)/Vp=49.3%, SE=0.31, p=0.06).
Gene-based association tests identified seven genes as significant after Bonferroni correction for testing >16,000 genes: MAGI2 (p<1.0×10−6), NAV2 (p<1.0×10−6), CACNA1C (p=1.0×10−6), PCDH9 (p=1.0×10−6), MYO16 (p=1.0×10−6), IQCH (p=2.0×10−6), DLGAP1 (p<1.0×10−6). We examined overlap of these novel “candidate” genes derived from the CADD GWAS with results from MCTFR and SAGE as a single “pathway” (i.e., gene set) in INRICH (Lee 2012). This allowed us to estimate whether specific genes identified in the CADD results overlapped with the low p-value genomic regions (i.e., loci tagged at r2>0.5 by a SNP reaching GWAS p<5×10−3) in the MCTFR and SAGE results more than expected by chance. The CADD-identified gene set was not significant in analysis of either the MCTFR (0 regions overlapped genes identified in CADD, p=1.0) or SAGE samples (6 regions overlapped genes identified in CADD, p=0.14).
Supplemental Table S2 presents gene-based association test results for previously identified addiction candidate genes (Hodgkinson et al. 2008), none of which were significant after adjustment for multiple testing (minimum p=1.4×10−3). Supplemental Table S3 gives results for each of the addiction candidate gene sets tested in CADD. None of the addiction candidate gene sets showed evidence of greater-than-chance overlap with low p-value genomic regions in the CADD GWAS (minimum p=5.0×10−1).
Promising pathways emerging from our exploratory pathway analysis were defined as those meeting nominal significance before correcting for multiple testing in CADD and either MCTFR or SAGE samples (Empirical p<0.05). Two pathways met these criteria: visual perception (Empirical pCADD=0.038, pMCTFR=0.012, pSAGE=0.22) and phosphatidylcholine biosynthetic process (Empirical pCADD=0.039, pMCTFR=1.0, pSAGE=0.026). Neither pathway achieved marginal significance in any sample after correction for multiple testing (i.e., Corrected p<0.10). Supplemental Table S4 provides results from all 72 pathways meeting Empirical p<0.05 in CADD (from a total of 3440 pathways tested) that were subsequently tested in the MCTFR and SAGE samples.
Discussion
No SNP was significantly associated with BD in the CADD GWAS. This is not surprising, given the relatively small sample. GWAS of psychiatric and behavioral phenotypes that have successfully identified and replicated individual effects of common SNPs have relied on very large samples (Rietveld et al. 2013; Ripke et al. 2013). Despite the lack of significance of any individual SNP, GCTA REML analysis estimated that 49.3% (SE=0.31, p=0.06) of the Caucasian ancestry sub-sample variation in BD could be accounted for by all of the genotyped SNPs. Conversely, a similar study found no evidence of variance in early adolescent (12-year-old) non-substance behavioral problems being attributable to common variants (Trzaskowski et al. 2013). This may suggest qualitative differences between genetic effects on BD at different ages, an effect that has been reported from twin models of comorbidity between dependence on different substances (Vrieze et al. 2012), which is a marker of BD.
Gene-based tests identified seven genes associated with BD in the CADD sample. However, neither the genes nor pathways identified as marginally overrepresented in the CADD GWAS results showed evidence of replicable low-p-values in either the MCTFR or SAGE samples. Taken together, these findings suggest that discoverable effects of common SNPs underlie the genetic architecture of BD, although better-powered studies are required to identify the associated loci.
The comparisons made between datasets must be considered in light of several limitations of the current study. There are substantial differences among the examined samples in terms of age (CADD and MCTFR represent adolescent data, while SAGE was comprised of adults), sex composition (MCTFR and SAGE are split evenly by sex, while CADD has an overrepresentation of males due to the sampling scheme), and diversity of ancestry (MCTFR is less diverse than either CADD or SAGE, which each have different representations of non-Caucasian ancestry groups). The sampling schemes of CADD and SAGE aimed to increase power to detect effects by oversampling extreme phenotype individuals, whereas the MCTFR study is closer to community-representative.
We sought to identify genetic influences on adolescent BD through a multifaceted approach. We initially characterized results from a standard GWAS by estimating the variance explained by common SNPs, and used gene- and pathway-based tests to identify potential novel candidate genes and pathways. Results from the estimation of sample variance explained by all genotyped SNPs and significant gene-based tests suggest there is a real genetic signal to be detected within the noise. However, the current sample is likely underpowered to detect realistic effect sizes of individual SNPs. Further, the lack of correspondence between pathway analyses in the CADD and replication samples may be due to limited power, or qualitative differences in the genetic effects on BD across different ages (adolescent versus adult) or sampling distributions (over-sampled for BD versus community-representative). Key to the search for causal genetic pathways underlying BD will be the availability of increasingly large, thoroughly phenotyped samples. Although the current analyses did not identify specific loci associated with BD, we demonstrate substantial heritability due to effects of common SNPs. Larger studies with appropriate phenotypes could well allow successful identification of common variants associated with BD.
Supplementary Material
Acknowledgments
The Center on Antisocial Drug Dependence (CADD) data reported here were funded by grants from the National Institute on Drug Abuse (P60 DA011015, R01 DA012845, R01 DA021913, R01 DA021905).
The Minnesota Center for Twin and Family Research (MCTFR) was supported in part by USPHS Grants from the National Institute on Alcohol Abuse and Alcoholism (AA09367 and AA11886), the National Institute on Drug Abuse (DA05147, DA13240, and DA024417), and the National Institute of Mental Health (MH066140).
Funding support for the Study of Addiction: Genetics and Environment (SAGE) was provided through the NIH Genes, Environment and Health Initiative [GEI] (U01 HG004422). SAGE is one of the genome-wide association studies funded as part of the Gene Environment Association Studies (GENEVA) under GEI. Assistance with phenotype harmonization and genotype cleaning, as well as with general study coordination, was provided by the GENEVA Coordinating Center (U01 HG004446). Assistance with data cleaning was provided by the National Center for Biotechnology Information. Support for collection of datasets and samples was provided by the Collaborative Study on the Genetics of Alcoholism (COGA; U10 AA008401), the Collaborative Genetic Study of Nicotine Dependence (COGEND; P01 CA089392), and the Family Study of Cocaine Dependence (FSCD; R01 DA013423). Funding support for genotyping, which was performed at the Johns Hopkins University Center for Inherited Disease Research, was provided by the NIH GEI (U01HG004438), the National Institute on Alcohol Abuse and Alcoholism, the National Institute on Drug Abuse, and the NIH contract “High throughput genotyping for studying the genetic contributions to human disease” (HHSN268200782096C). The datasets used for the analyses described in this manuscript were obtained from dbGaP at http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000092.v1.p1 through dbGaP accession number phs000092.v1.p.
Jaime Derringer was supported by the National Institute of Mental Health (T32 MH016880).
References
- Agrawal A, Lynskey MT, Hinrichs A, et al. A genome-wide association study of DSM-IV cannabis dependence. Addiction biology. 2011;16(3):514–518. doi: 10.1111/j.1369-1600.2010.00255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierut LJ, Agrawal A, Bucholz KK, et al. A genome-wide association study of alcohol dependence. Proceedings of the National Academy of Sciences. 2010;107(11):5082–5087. doi: 10.1073/pnas.0911109107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierut LJ, Madden PA, Breslau N, et al. Novel genes identified in a high-density genome wide association study for nicotine dependence. Human molecular genetics. 2007;16(1):24–35. doi: 10.1093/hmg/ddl441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierut LJ, Strickland JR, Thompson JR, et al. Drug use and dependence in cocaine dependent subjects, community-based individuals, and their siblings. Drug and alcohol dependence. 2008;95(1):14–22. doi: 10.1016/j.drugalcdep.2007.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins AL, Kim Y, Sklar P, et al. Hypothesis-driven candidate genes for schizophrenia compared to genome-wide association results. Psychological medicine. 2012;42(3):607. doi: 10.1017/S0033291711001607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick DM, Aliev F, Krueger RF, et al. Genome-wide association study of conduct disorder symptomatology. Molecular psychiatry. 2011;16(8):800–808. doi: 10.1038/mp.2010.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edenberg HJ. The collaborative study on the genetics of alcoholism: an update. Alcohol Research and Health. 2002;26(3):214–218. [PMC free article] [PubMed] [Google Scholar]
- Edenberg HJ, Koller DL, Xuei X, et al. Genome-Wide Association Study of Alcohol Dependence Implicates a Region on Chromosome 11. Alcoholism: Clinical and Experimental Research. 2010;34(5):840–852. doi: 10.1111/j.1530-0277.2010.01156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank J, Cichon S, Treutlein J, et al. Genome-wide significant association between alcohol dependence and a variant in the ADH gene cluster. Addiction Biology. 2012;17(1):171–180. doi: 10.1111/j.1369-1600.2011.00395.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furberg H, Kim Y, Dackor J, et al. Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nat Genet. 2010;42(5):441–U134. doi: 10.1038/ng.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelernter J, Kranzler HR, Sherva R, et al. Genome-wide association study of alcohol dependence: significant findings in African-and European-Americans including novel risk loci. Molecular Psychiatry. 2014a;19:41–49. doi: 10.1038/mp.2013.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelernter J, Kranzler HR, Sherva R, et al. Genome-wide association study of opioid dependence: multiple associations mapped to calcium and potassium pathways. Biological Psychiatry. 2014b;76(1):66–74. doi: 10.1016/j.biopsych.2013.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelernter J, Sherva R, Koesterer R, et al. Genome-wide association study of cocaine dependence and related traits: FAM53B identified as a risk gene. Molecular Psychiatry. 2014c;19(6):717–723. doi: 10.1038/mp.2013.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman CA, Gelhorn H, Crowley TJ, et al. Item Response Theory Analysis of DSM-IV Cannabis Abuse and Dependence Criteria in Adolescents. Journal of the American Academy of Child & Adolescent Psychiatry. 2008;47(2):165–173. doi: 10.1097/chi.0b013e31815cd9f2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks BM, Foster KT, Iacono WG, et al. Genetic and Environmental Influences on the Familial Transmission of Externalizing Disorders in Adoptive and Twin Offspring. JAMA psychiatry. 2013;70(10):1076–1083. doi: 10.1001/jamapsychiatry.2013.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicks BM, Schalet BD, Malone SM, et al. Psychometric and genetic architecture of substance use disorder and behavioral disinhibition measures for gene association studies. Behav Genet. 2011;41(4):459–475. doi: 10.1007/s10519-010-9417-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkinson CA, Yuan Q, Xu K, et al. Addictions biology: haplotype-based analysis for 130 candidate genes on a single array. Alcohol and Alcoholism. 2008;43(5):505–515. doi: 10.1093/alcalc/agn032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson AD, Handsaker RE, Pulit SL, et al. SNAP: a web-based tool for identification and annotation of proxy SNPs using HapMap. Bioinformatics. 2008;24(24):2938–2939. doi: 10.1093/bioinformatics/btn564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor M, Wang JC, Wetherill L, et al. A meta-analysis of two genome-wide association studies to identify novel loci for maximum number of alcoholic drinks. Human Genetics. 2013;132(10):1141–1151. doi: 10.1007/s00439-013-1318-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger RF, Hicks BM, Patrick CJ, et al. Etiologic connections among substance dependence, antisocial behavior and personality: Modeling the externalizing spectrum. J Abnorm Psychol. 2002;111(3):411. [PubMed] [Google Scholar]
- Lee PH, O’Dushlaine C, Thomas B, et al. INRICH: interval-based enrichment analysis for genome-wide association studies. Bioinformatics. 2012;28(13):1797–1799. doi: 10.1093/bioinformatics/bts191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Wray NR, Goddard ME, et al. Estimating missing heritability for disease from genome-wide association studies. Am J Hum Genet. 2011;88(3):294–305. doi: 10.1016/j.ajhg.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Basu S, Miller MB, et al. A rapid generalized least squares model for a genome-wide quantitative trait association analysis in families. Human heredity. 2011;71(1):67–82. doi: 10.1159/000324839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JZ, Mcrae AF, Nyholt DR, et al. A versatile gene-based test for genome-wide association studies. Am J Hum Genet. 2010;87(1):139–145. doi: 10.1016/j.ajhg.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JZ, Tozzi F, Waterworth DM, et al. Meta-analysis and imputation refines the association of 15q25 with smoking quantity. Nature Genetics. 2010;42(5):436–440. doi: 10.1038/ng.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGue M, Zhang Y, Miller MB, et al. A genome-wide association study of behavioral disinhibition. Behav Genet. 2013;43(5):363–373. doi: 10.1007/s10519-013-9606-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller MB, Basu S, Cunningham, et al. The Minnesota Center for Twin and Family Research genome-wide association study. Twin research and human genetics. 2012;15(6):767–774. doi: 10.1017/thg.2012.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen DA, Ji F, Yuferov V, et al. Genome-wide association study identifies genes that may contribute to risk for developing heroin addiction. Psychiatric genetics. 2010;20(5):207–214. doi: 10.1097/YPG.0b013e32833a2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrill S, Plomin R, DeFries JC, Hewitt JK, editors. Nature, Nurture, and the Transition to Adolescence. New York: Oxford University Press; 2003. [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhea SA, Gross AA, Haberstick BC, et al. Colorado twin registry. Twin Research and Human Genetics. 2006;9(06):941–949. doi: 10.1375/183242706779462895. [DOI] [PubMed] [Google Scholar]
- Rietveld CA, Medland SE, Derringer J, Yang J, Esko T, Martin NW, McMahon G. GWAS of 126,559 individuals identifies genetic variants associated with educational attainment. science. 2013;340(6139):1467–1471. doi: 10.1126/science.1235488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke S, O’Dushlaine C, Chambert K, et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet. 2013;45(10):1150–1159. doi: 10.1038/ng.2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruano D, Abecasis GR, Glaser B, et al. Functional gene group analysis reveals a role of synaptic heterotrimeric G proteins in cognitive ability. Am J Hum Genet. 2010;86(2):113–125. doi: 10.1016/j.ajhg.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann G, Coin LJ, Lourdusamy A, et al. Genome-wide association and genetic functional studies identify autism susceptibility candidate 2 gene (AUTS2) in the regulation of alcohol consumption. Proceedings of the National Academy of Sciences. 2011;108(17):7119–7124. doi: 10.1073/pnas.1017288108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallings MC, Corley RP, Dennehey B, et al. A genome-wide search for quantitative trait loci that influence antisocial drug dependence in adolescence. Archives of General Psychiatry. 2005;62(9):1042–1051. doi: 10.1001/archpsyc.62.9.1042. [DOI] [PubMed] [Google Scholar]
- Sullivan P. Don’t give up on GWAS. Molecular psychiatry. 2011;17(1):2–3. doi: 10.1038/mp.2011.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terracciano A, Esko T, Sutin AR, et al. Meta-analysis of genome-wide association studies identifies common variants in CTNNA2 associated with excitement-seeking. Translational psychiatry. 2011;1(10):e49. doi: 10.1038/tp.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Gene Ontology Consortium. Gene Ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorgeirsson TE, Gudbjartsson DF, Surakka I, et al. Sequence variants at CHRNB3-CHRNA6 and CYP2A6 affect smoking behavior. Nat Genet. 2010;42(5):448–453. doi: 10.1038/ng.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tielbeek JJ, Medland SE, Benyamin B, et al. Unraveling the genetic etiology of adult antisocial behavior: A genome-wide association study. PloS one. 2012;7(10):e45086. doi: 10.1371/journal.pone.0045086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treutlein J, Cichon S, Ridinger M, et al. Genome-wide association study of alcohol dependence. Archives of general psychiatry. 2009;66(7):773–784. doi: 10.1001/archgenpsychiatry.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trzaskowski M, Dale PS, Plomin R. No genetic influence for childhood behavior problems from DNA analysis. Journal of the American Academy of Child & Adolescent Psychiatry. 2013;52(10):1048–1056. doi: 10.1016/j.jaac.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhl GR, Drgon T, Liu QR, et al. Genome-wide association for methamphetamine dependence: convergent results from 2 samples. Archives of general psychiatry. 2008;65(3):345–355. doi: 10.1001/archpsyc.65.3.345. [DOI] [PubMed] [Google Scholar]
- Van der Sluis S, Posthuma D, Nivard MG, et al. Power in GWAS: lifting the curse of the clinical cut-off. Molecular psychiatry. 2013;18:2–3. doi: 10.1038/mp.2012.65. [DOI] [PubMed] [Google Scholar]
- Verweij KJ, Vinkhuyzen AA, Benyamin B, et al. The genetic aetiology of cannabis use initiation: a meta-analysis of genome-wide association studies and a SNP-based heritability estimation. Addiction biology. 2013;18(5):846–850. doi: 10.1111/j.1369-1600.2012.00478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrieze SI, Hicks BM, Iacono WG, et al. Decline in genetic influence on the co-occurrence of alcohol, marijuana, and nicotine dependence symptoms from age 14 to 29. American Journal of Psychiatry. 2012;169(10):1073–1081. doi: 10.1176/appi.ajp.2012.11081268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JC, Foroud T, Hinrichs AL, et al. A genome-wide association study of alcohol-dependence symptom counts in extended pedigrees identifies C15orf53. Molecular Psychiatry. 2012;18(11):1218–1224. doi: 10.1038/mp.2012.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Lee SH, Goddard ME, et al. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88(1):76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young SE, Stallings MC, Corley RP, et al. Genetic and environmental influences on behavioral disinhibition. American journal of medical genetics. 2000;96(5):684–695. [PubMed] [Google Scholar]
- Yu K, Li Q, Bergen AW, et al. Pathway analysis by adaptive combination of P-values. Genetic epidemiology. 2009;33(8):700–709. doi: 10.1002/gepi.20422. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.