

Abstract
While hypertension has predominantly been attributed to perturbations of the vasculature, kidney, and central nervous system, research for almost 50 yr has shown that the immune system also contributes to this disease. Inflammatory cells accumulate in the kidneys and vasculature of humans and experimental animals with hypertension and likely contribute to end-organ damage. We and others have shown that mice lacking adaptive immune cells, including recombinase-activating gene-deficient mice and rats and mice with severe combined immunodeficiency have blunted hypertension to stimuli such as ANG II, high salt, and norepinephrine. Adoptive transfer of T cells restores the blood pressure response to these stimuli. Agonistic antibodies to the ANG II receptor, produced by B cells, contribute to hypertension in experimental models of preeclampsia. The central nervous system seems important in immune cell activation, because lesions in the anteroventral third ventricle block hypertension and T cell activation in response to ANG II. Likewise, genetic manipulation of reactive oxygen species in the subfornical organ modulates both hypertension and immune cell activation. Current evidence indicates that the production of cytokines, including tumor necrosis factor-α, interleukin-17, and interleukin-6, contribute to hypertension, likely via effects on both the kidney and vasculature. In addition, the innate immune system also appears to contribute to hypertension. We propose a working hypothesis linking the sympathetic nervous system, immune cells, production of cytokines, and, ultimately, vascular and renal dysfunction, leading to the augmentation of hypertension. Studies of immune cell activation will clearly be useful in understanding this common yet complex disease.
Keywords: T cell, cytokines, angiotensin, subfornical organ, costimulation
in the 1960s, a role for the immune system in the development of hypertension was first discovered. Several recent investigations have further defined the role of immune system, particularly the adaptive immune system, in hypertension, provided novel insights into the genesis of hypertension, and identified novel targets for the treatment of hypertension. The purpose of this review was to summarize recent discoveries by our laboratory and others on the role of the immune system in hypertension.
Early Studies in Immunity and Hypertension
The concept that the immune system contributes to hypertension had its genesis in the 1960s, when Grollman et al. (40, 56) demonstrated that immunosuppression blunted hypertension in a model of renal infarction and that transfer of lymphocytes from rats with renal infarction induced hypertension in previously nonhypertensive animals. Later, Svenson found that hypertension was not maintained in thymectiomized or athymic nude mice with renal infarction (46). In the 1980s, Ba et al. (2) found that transplanting the thymus from a Wistar-Kyoto (WKY) rat to a spontaneously hypertensive rat (SHR) resulted in a decrease in blood pressure in the SHR (2). Blood pressure was also lowered in SHRs with treatment by either anti-thymocyte serum or the immunosuppressive drug cyclophosphamide (5, 12). Nerve growth in the thymus of SHR was found to be greater than that of WKY rats, suggesting a neural component of immune cell activation in hypertension (42). Rodruiguez-Iturbe et al. (43) found that immunsupression with mycophenlate mofetil blunted salt-induced hypertension after ANG II infusion. In the early 2000s, Muller and Luft (35, 36, 49) conducted a series of investigations showing that NF-κB and ROS play roles in ANG II-induced end-organ damage. More recently, advances in genetic mouse models and knowledge of immunology prepared the way for discoveries that would lead to further understanding of the importance of the immune system in hypertension.
T Cells and Hypertension
In 2007, our laboratory (18) published a study demonstrating that T cells contribute to the development of hypertension. In this study, mice lacking recombinase-activating gene 1 (Rag-1−/− mice) were used as these mice cannot generate functional T cell receptors or B cell antibodies and thus lack both T and B lymphocytes. The increase in blood pressure caused by either ANG II or DOCA salt was significantly blunted in Rag-1−/− mice, suggesting that either T or B cells mediate overt hypertension. Rag-1−/− mice did not exhibit increased vascular superoxide production and endothelial dysfunction. The hypertensive response to ANG II was restored when Rag-1−/− mice received adoptive transfer of T cells but not B cells. In wild-type mice, ANG II increased circulating CD69+, CCR5+, and CD44high T cells, markers of effector memory T cells. In addition, T cells accumulated in the perivascular adipose tissue of the aorta. The results of this study indicate that T cells play a major role in hypertension. Supporting the role of T cells in hypertension, severe combined immunodeficieny mice have also been shown to be protected against hypertension and exhibit reduced albuminuria and renal damage (10). Recently, Mattson et al. (32) deleted the Rag1 gene in Dahl salt-sensitive rat using zinc finger nuclease technology and have shown that this attenuates blood pressure, albuminuria, and kidney damage. Thus, T cells seem to contribute to the development of various forms of hypertension in different strains of mice and in rats.
Role of the Central Nervous System in Immune-Mediated Hypertension
The blood vessels, kidney, and central nervous system (CNS) have all been shown to contribute to the development of hypertension. Interestingly, T cells may represent a link between these tissues. Lymphoid tissues are rich in sympathetic nerves (14). Ganta et al. (17) have shown that intracerebroventricular infusion of ANG II increased sympathetic nerve activity to the spleen and increased expression of multiple cytokines in the spleen. Our laboratory has preformed a series of investigations on the role of the CNS in mediating T cell activation.
The circumventricular organs (CVO) are highly vascularized and have an incomplete blood-brain barrier and can therefore be influenced by circulating hormones like ANG II. In addition, the CVO, and in particular, the subfornical organ (SFO), are important in both sending and receiving central signals that regulate cardiovascular function and electrolyte balance. Deletion of CVO extracellular (ec)SOD, using Cre-lox technology, provides a model to determine the role of central oxidative stress in hypertension. ecSOD deletion increased ROS levels in the CVO, increased heart low-frequency to high-frequency heart rate variability (indicative of increased sympathetic nervous activity), and elevated blood pressure (25). In addition, when mice with ecSOD deleted in the CVO were infused with ANG II at a dose that does not cause hypertension in normal mice (140 ng·kg−1·min−1), blood pressure was significantly elevated and was accompanied by aortic T cell infiltration. Interestingly, in a separate investigation, when ecSOD was specifically deleted in vascular smooth muscle, despite increases in vascular ROS, blood pressure and T cell responses were not altered compared with controls (27).
NADPH oxidases are major sources of superoxide anion production in mammalian cells. The subunit p22phox mediates trafficking of NADPH oxidase catalytic subunits to the cell membrane and is required for enzyme complex assembly and, ultimately, superoxide production. Complementing the studies that used deletion of ecSOD, our laboratory (26) also deleted p22phox in the SFO in a similar manner. Deletion of p22phox in the SFO blunted the pressor response to ANG II and decreased sympathetic outflow as assessed by heart rate variability. In addition, p22phox deletion abolished ANG II-induced aortic T cell infiltration. This study is in keeping with findings that intracerebroventricular injections of a superoxide scavenger reduces sympathetic drive, blood pressure, and renal damage in salt-induced hypertension in rats (16).
Supporting the role of the central nervous system, lesions in the anteroventral third cerebral ventricle (AV3V), a region which includes the SFO, can prevent ANG II-induced hypertension (7, 30). In addition, AV3V lesions protect against T cell activation and aortic infiltration in response to ANG II (30). This is of particular importance as it demonstrates that ANG II-induced T cell activation is not due to direct actions of ANG II on T cells but rather that central signals are required for T cell activation. Interestingly, mice infused with norepinephrine become hypertensive and exhibit T cell activation and aortic infiltration even after AV3V lesions. This supports the concept that sympathetic drive, and its attendant release of norepinephrine, likely mediates T cell activation and hypertension. In addition to the important role of the CNS, peripheral mechanisms appear to also contribute to T cell activation and vascular inflammation. Treatment with the vasodilator hydralzine to normalize blood pressure prevents T cell activation and vascular inflammation induced by ANG II infusion (30). This suggests that T cells may respond to elevations in pressure and that afferent nerves may activate central mechanisms in a feedforward manner to induce CVO oxidative stress, sympathetic outflow, further T cell activation, and overt hypertension. More recently, T cells have been shown to contribute to stress-induced hypertension (31). We exposed mice to 7 days of stress using a combination of restraint and cage switching. This stress paradigm resulted in increased blood pressure, activation of circulating T cells, and aortic T cell infiltration. Rag-1−/− mice were protected from stress-induced hypertension, and adoptive transfer of T cells restored the hypertensive response. These findings underscore the crucial role of the CNS in orchestrating the T cell response leading to hypertension.
T Cell Subtypes, Cytokines, and Mechanisms of Activation
The above studies demonstrated that T cells contribute to the development of hypertension; however, they do not provide extensive insights into the subsets of T cells involved. CD4+ T cells have been generally classified as either T helper (Th)1 or Th2, depending on their activation markers and cytokine production (34). Th17 cells are a newly characterized subset of T cells; these cells produce the cytokine IL-17 and contribute to numerous autoimmune diseases, obesity, and cardiovascular disease (13, 48, 57). To investigate the role of IL-17 in hypertension, our group studied IL-17a−/− mice. These mice exhibited a similar initial increase in blood pressure as wild-type mice in response to ANG II; however, after 7 days, blood pressure dropped in IL-17a−/− mice (28). The ANG II-induced aortic T cell infiltration observed in wild-type mice was abolished in IL-17a−/− mice, as were increases in vascular oxidative stress and endothelial dysfunction. Recent reports have shown that direct infusion of IL-17a mediated hypertension and endothelial dysfunction in mice (37) and that IL-17 mediated placental oxidative stress, resulting in hypertension during pregnancy in rats (11).
In addition to IL-17, other cytokines have been implicated in the pathogenesis of hypertension. Etanrecept, a TNF-α antagonist, is effective in preventing hypertension (18, 50, 52). IL-6 knockout mice are also protected from ANG II-induced hypertension (6, 24, 45). Interferon (IFN)-γ is upregulated in the kidneys of hypertensive mice (10), and inhibition of IFN-γ prevents ANG II-induced end-organ damage (29). Taken together, these observations suggest that hypertension is mediated by multiple proinflammatory T cell subsets. In accordance with this concept, T regulatory (Tregs) cells, which act to restrain proinflammatory T cells, attenuate hypertension-induced end-organ damage in mice (22) and blunt hypertension in rats (53).
Classically, T cells require two signals for activation: 1) interaction of the T cell receptor with an antigen presented in the context of a major histocompatibilty complex and 2) stimulation of costimulatory molecules on the T cell by ligands on the antigen-presenting cell (1). A major costimulatory molecule on T cells is CD28, which is bound by the B7 ligands CD80 and CD86 of the antigen-presenting cell. Ligation of the T cell receptor in the absence of costimulation leads to T cell apoptosis (15). The pharmacological agent CTLA4-Ig inhibits costimulation by binding to B7 ligands on antigen-presenting cells. To determine whether costimulation plays a role in hypertension, our laboratory used both pharmacological inhibition of costimulation with CTLA4-Ig and a genetic approach with B7-deficient (B7−/−) mice. CTLA4-Ig treatment blunted blood pressure, T cell activation, and vascular infiltration in both ANG II- and DOCA salt-induced hypertension (54). CTLA4-Ig treatment also abolished T cell production of TNF-α and IFN-γ induced by ANG II. Similar results were observed in B7−/− mice, which lack B7 ligands. These observations suggest that T cell receptor ligation and costimulation are necessary for T cell activation in hypertension.
The Innate Immune System and Hypertension
The role of the adaptive immune system in experimental hypertension has been well characterized; however, less is known about the role of the innate immune system. Recently, Abboud et al. (19) demonstrated that, in WKY rats, the choleneric agonist nicotine results in an anti-inflammatory response in splenic macrophages; in contrast, nicotine induced a proinflammatory response in macrophages from SHRs and enhanced Toll-like receptor-mediated cytokine release. In addition, perivascular macrophage infiltration has been observed in experimental hypertension in mice (3). Inflammatory cytokines, IL-1β, and IL-6 are elevated in SHRs compared with WKY rats; this can be reversed by treatment with an angiotensin-converting enzyme inhibitor (33). Similarly, ANG II receptor blockade prevented lipopolysaccharide-induced inflammatory responses of innate immune cells in the rat spleen (44). In humans, white blood cells from essential hypertensive patients produced more IL-1β and IL-6 when stimulated with lipopolysaccharide compared with controls (41). These observations are consistent with monocyte activation in hypertension. How the innate and adaptive immune systems interact in the development of hypertension is not well understood and is an important topic for future study.
The Immune System and Preeclampsia
Preeclampsia is characterized by the onset of hypertension during pregnancy accompanied by proteinuriea. Preeclampsia is associated with the production of autoantibodies that stimulate the ANG II type 1 (AT1) receptor (55), and infusion of these antibodies can induce preeclampsia-like symptomps in pregnant mice (60). More recently, it has been shown that a specific subset of B cells produces these antibodies (21). Depletion of B cells using the anti-CD20 antibody rituximab blunts the blood pressure response in the reduced uterine perfusion pressure rat model of preeclampsia (23). Adoptive transfer of CD4+ T cells from reduced uterine perfusion pressure rats to normal pregnant rats results in increased blood pressure (39). This response is blunted by either rituximab or AT1 antagonism, suggesting an important role of cross-talk between T and B cells in preeclampsia. Supporting the role of T cells in preeclampsia, mice deficient in the cytokines IL-4 or IL-10, which skew T cells to an anti-inflammatory phenotype, develop preeclampsia-like symptoms when pregnant (8, 9). The proinflammatory cytokine IL-17 mediates placental oxidative stress and increases in blood pressure in pregnant rats (11). Together, these observations support a role for the adaptive immune system in preeclampsia where T and B cells act in a synergistic manner.
Pulmonary Hypertension and the Immune System
It has long been postulated that autoimmunity and inflammation is involved in pulmonary hypertension (38). In addition, T cells, B cells, and macrophages are present in the lungs of patients with pulmonary hypertension (51). Recently, a role of anti-inflammatory Tregs cells has been indentified in experimental pulmonary hypertension (47). In this study, in response to VEGF receptor 2 antagonism, athymic rats, which lack T cells, developed perivascular inflammation, including infiltration of B cells and macrophages in the lung and pulmonary hypertension. The authors then reconstituted different T cell subsets in these animals and found that CD4+ Tregs cells acted to blunt pulmonary vascular inflammation and the development of pulmonary hypertension. In a similar manner, Tregs cells appear to act to restrain experimental systemic hypertension in both mice and rats (3, 22, 53).
The Immune System in Human Hypertension
Although the majority of studies implicating the immune system in hypertension have been performed in experimental animals, a limited number of investigations have examined the role of the immune system in human hypertension. IL-6 and TNF-α are positively correlated with blood pressure in humans (4). In a small study, patients with either rheumatoid arthritis or psoriasis who also had essential hypertension were treated with mycophenlate mofetil, resulting in significantly lowered systolic and diastolic blood pressure (20). Recently, circulating proinflammatory CD8+ T cells have been indentified in hypertensive patients (58). These cells generate IFN-γ and TNF-α and exhibit loss of CD28 and gain of CD57, which is consistent with a proinflammatory, senescent T cell phenotype. These patients also exhibited increased circulating chemokines, which serve as T cell attractants.
Conclusions and Future Directions
In summary, it has been known for almost 50 yr that immune cells contribute to hypertension; in the last several years, investigations from our group and others have demonstrated the importance of T cells in the development of hypertension. In light of the data discussed here, we have formulated a working hypothesis for the development of overt hypertension. As shown in Fig. 1, hypertensive stimuli, such as ANG II or salt, leads to an initial elevation in blood pressure, consistent with clinical “prehypertension,” which results in protein modifications, possibly due to oxidative modifications. These altered proteins serve as neoantigens that no longer recognized as self, are processed and presented by dendritic cells, and promote T cell activation. In concert, afferent signals to the CNS result in increased sympathetic outflow contributing to T cell activation. Activated T cells infiltrate the kidney and vasculature and produce cytokines that promote renal Na+ and water retention and, in the vasculature, vasoconstriction and remodeling. Together, these alterations result in overt hypertension. The observation that hydralizine treatment abolished the T cell response suggests that signals from the periphery operate in a feedforward manner to signal the CNS to increase central sympathetic drive and mediate overt hypertension. An important point is that the T cell response is independent of the model of experimental hypertension, T cell responses have been observed in mice in response to ANG II, DOCA salt, and norepinephrine. In rats, this response has been observed in both salt-sensitive and genetic models. Emphasizing this point is the recent study of Zhang et al. (59), where ANG II receptors were specifically deleted in mouse T cells using Cre-lox technology. These mice were not protected against ANG II-induced hypertension, and, surprisingly, kidney damage in response to ANG II was exacerbated (59). These observations indicate that the T cell response in hypertension is independent of direct actions of ANG II on T cells. We believe, based on the studies of the CNS, that central signals mediate T cell activation in a variety of hypertension models. Recent observations by Abboud et al. (19) have suggested that central signals regulate the innate immune system in hypertension as well. It should be noted that the model for the immue system in hypertension outlined here is a working hypothesis; the precise mechanisms, particularly the initiating factors in the development of hypertension, are as yet unknown. Our hypothesis will almost certainly require refinement as new information comes available.
Fig. 1.
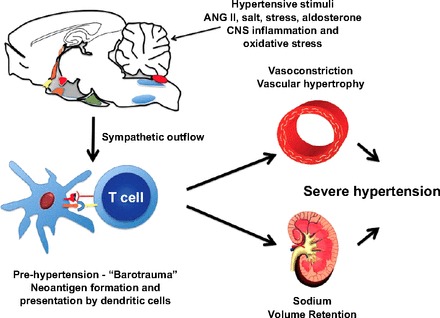
Working hypothesis describing the role of immune cells in hypertension. CNS, central nervous system.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: D.W.T. and D.G.H. conception and design of research; D.W.T. and D.G.H. prepared figures; D.W.T. and D.G.H. drafted manuscript; D.W.T. and D.G.H. approved final version of manuscript.
REFERENCES
- 1.Abbas AK, Lichtman AH, Pillai S. Cellular and Molecular Immunology. Philadelphia, PA: Saunders Elsevier, 2007, p. viii. [Google Scholar]
- 2.Ba D, Takeichi N, Kodama T, Kobayashi H. Restoration of T cell depression and suppression of blood pressure in spontaneously hypertensive rats (SHR) by thymus grafts or thymus extracts. J Immunol 128: 1211–1216, 1982. [PubMed] [Google Scholar]
- 3.Barhoumi T, Kasal DA, Li MW, Shbat L, Laurant P, Neves MF, Paradis P, Schiffrin EL. T regulatory lymphocytes prevent angiotensin II-induced hypertension and vascular injury. Hypertension 57: 469–476, 2011. [DOI] [PubMed] [Google Scholar]
- 4.Bautista LE, Vera LM, Arenas IA, Gamarra G. Independent association between inflammatory markers (C-reactive protein, interleukin-6, and TNF-α) and essential hypertension. J Hum Hypertens 19: 149–154, 2005. [DOI] [PubMed] [Google Scholar]
- 5.Bendich A, Belisle EH, Strausser HR. Immune system modulation and its effect on the blood pressure of the spontaneously hypertensive male and female rat. Biochem Biophys Res Commun 99: 600–607, 1981. [DOI] [PubMed] [Google Scholar]
- 6.Brands MW, Banes-Berceli AK, Inscho EW, Al-Azawi H, Allen AJ, Labazi H. Interleukin 6 knockout prevents angiotensin II hypertension: role of renal vasoconstriction and janus kinase 2/signal transducer and activator of transcription 3 activation. Hypertension 56: 879–884, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brody M, Fink G, Buggy J, Haywood J, Gordon F, Knuepfer M, Mow M, Mahoney L, Johnson A. Critical role of the anteroventral third ventricle (AV3V) region in development and maintenance of experimental hypertension. Perspect Nephrol Hypertens 6: 76–84, 1979. [Google Scholar]
- 8.Chatterjee P, Chiasson VL, Kopriva SE, Young KJ, Chatterjee V, Jones KA, Mitchell BM. Interleukin 10 deficiency exacerbates toll-like receptor 3-induced preeclampsia-like symptoms in mice. Hypertension 58: 489–496, 2011. [DOI] [PubMed] [Google Scholar]
- 9.Chatterjee P, Kopriva SE, Chiasson VL, Young KJ, Tobin RP, Newell-Rogers K, Mitchell BM. Interleukin-4 deficiency induces mild preeclampsia in mice. J Hypertens 31: 1414–1423, 2013. [DOI] [PubMed] [Google Scholar]
- 10.Crowley SD, Song YS, Lin EE, Griffiths R, Kim HS, Ruiz P. Lymphocyte responses exacerbate angiotensin II-dependent hypertension. Am J Physiol Regul Integr Comp Physiol 298: R1089–R1097, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dhillion P, Wallace K, Herse F, Scott J, Wallukat G, Heath J, Mosely J, Martin JN, Jr, Dechend R, LaMarca B. IL-17-mediated oxidative stress is an important stimulator of AT1-AA and hypertension during pregnancy. Am J Physiol Regul Integr Comp Physiol 303: R353–R358, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dzielak DJ. Immune mechanisms in experimental and essential hypertension. Am J Physiol Regul Integr Comp Physiol 260: R459–R467, 1991. [DOI] [PubMed] [Google Scholar]
- 13.Eid RE, Rao DA, Zhou J, Lo SF, Ranjbaran H, Gallo A, Sokol SI, Pfau S, Pober JS, Tellides G. Interleukin-17 and interferon-γ are produced concomitantly by human coronary artery-infiltrating T cells and act synergistically on vascular smooth muscle cells. Circulation 119: 1424–1432, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Felten DL, Livnat S, Felten SY, Carlson SL, Bellinger DL, Yeh P. Sympathetic innervation of lymph nodes in mice. Brain Res Bull 13: 693–699, 1984. [DOI] [PubMed] [Google Scholar]
- 15.Frauwirth KA, Thompson CB. Activation and inhibition of lymphocytes by costimulation. J Clin Invest 109: 295–299, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fujita M, Ando K, Kawarazaki H, Kawarasaki C, Muraoka K, Ohtsu H, Shimizu H, Fujita T. Sympathoexcitation by brain oxidative stress mediates arterial pressure elevation in salt-induced chronic kidney disease. Hypertension 59: 105–112, 2012. [DOI] [PubMed] [Google Scholar]
- 17.Ganta CK, Lu N, Helwig BG, Blecha F, Ganta RR, Zheng L, Ross CR, Musch TI, Fels RJ, Kenney MJ. Central angiotensin II-enhanced splenic cytokine gene expression is mediated by the sympathetic nervous system. Am J Physiol Heart Circ Physiol 289: H1683–H1691, 2005. [DOI] [PubMed] [Google Scholar]
- 18.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204: 2449–2460, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harwani SC, Chapleau MW, Legge KL, Ballas ZK, Abboud FM. Neurohormonal modulation of the innate immune system is proinflammatory in the prehypertensive spontaneously hypertensive rat, a genetic model of essential hypertension. Circ Res 111: 1190–1202, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herrera J, Ferrebuz A, MacGregor EG, Rodriguez-Iturbe B. Mycophenolate mofetil treatment improves hypertension in patients with psoriasis and rheumatoid arthritis. J Am Soc Nephrol 17: S218–225, 2006. [DOI] [PubMed] [Google Scholar]
- 21.Jensen F, Wallukat G, Herse F, Budner O, El-Mousleh T, Costa SD, Dechend R, Zenclussen AC. CD19+CD5+ cells as indicators of preeclampsia. Hypertension 59: 861–868, 2012. [DOI] [PubMed] [Google Scholar]
- 22.Kvakan H, Kleinewietfeld M, Qadri F, Park JK, Fischer R, Schwarz I, Rahn HP, Plehm R, Wellner M, Elitok S, Gratze P, Dechend R, Luft FC, Muller DN. Regulatory T cells ameliorate angiotensin II-induced cardiac damage. Circulation 119: 2904–2912, 2009. [DOI] [PubMed] [Google Scholar]
- 23.LaMarca B, Wallace K, Herse F, Wallukat G, Martin JN, Jr, Weimer A, Dechend R. Hypertension in response to placental ischemia during pregnancy: role of B lymphocytes. Hypertension 57: 865–871, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee DL, Sturgis LC, Labazi H, Osborne JB, Jr, Fleming C, Pollock JS, Manhiani M, Imig JD, Brands MW. Angiotensin II hypertension is attenuated in interleukin-6 knockout mice. Am J Physiol Heart Circ Physiol 290: H935–H940, 2006. [DOI] [PubMed] [Google Scholar]
- 25.Lob HE, Marvar PJ, Guzik TJ, Sharma S, McCann LA, Weyand C, Gordon FJ, Harrison DG. Induction of hypertension and peripheral inflammation by reduction of extracellular superoxide dismutase in the central nervous system. Hypertension 55: 277–283, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lob HE, Schultz D, Marvar PJ, Davisson RL, Harrison DG. Role of the NADPH oxidases in the subfornical organ in angiotensin II-induced hypertension. Hypertension 61: 382–387, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lob HE, Vinh A, Li L, Blinder Y, Offermanns S, Harrison DG. Role of vascular extracellular superoxide dismutase in hypertension. Hypertension 58: 232–239, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, Harrison DG. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension 55: 500–507, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marko L, Kvakan H, Park JK, Qadri F, Spallek B, Binger KJ, Bowman EP, Kleinewietfeld M, Fokuhl V, Dechend R, Muller DN. Interferon-γ signaling inhibition ameliorates angiotensin II-induced cardiac damage. Hypertension 60: 1430–1436, 2012. [DOI] [PubMed] [Google Scholar]
- 30.Marvar PJ, Thabet SR, Guzik TJ, Lob HE, McCann LA, Weyand C, Gordon FJ, Harrison DG. Central and peripheral mechanisms of T-lymphocyte activation and vascular inflammation produced by angiotensin II-induced hypertension. Circ Res 107: 263–270, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marvar PJ, Vinh A, Thabet S, Lob HE, Geem D, Ressler KJ, Harrison DG. T lymphocytes and vascular inflammation contribute to stress-dependent hypertension. Biol Psychiatry 71: 774–782, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mattson DL, Lund H, Guo C, Rudemiller N, Geurts AM, Jacob H. Genetic mutation of recombination activating gene 1 in Dahl salt-sensitive rats attenuates hypertension and renal damage. Am J Physiol Regul Integr Comp Physiol 304: R407–R414, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miguel-Carrasco JL, Zambrano S, Blanca AJ, Mate A, Vazquez CM. Captopril reduces cardiac inflammatory markers in spontaneously hypertensive rats by inactivation of NF-κB. J Inflamm (Lond) 7: 21, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol 136: 2348–2357, 1986. [PubMed] [Google Scholar]
- 35.Muller DN, Dechend R, Mervaala EM, Park JK, Schmidt F, Fiebeler A, Theuer J, Breu V, Ganten D, Haller H, Luft FC. NF-κB inhibition ameliorates angiotensin II-induced inflammatory damage in rats. Hypertension 35: 193–201, 2000. [DOI] [PubMed] [Google Scholar]
- 36.Muller DN, Mervaala EM, Schmidt F, Park JK, Dechend R, Genersch E, Breu V, Loffler BM, Ganten D, Schneider W, Haller H, Luft FC. Effect of bosentan on NF-κB, inflammation, and tissue factor in angiotensin II-induced end-organ damage. Hypertension 36: 282–290, 2000. [DOI] [PubMed] [Google Scholar]
- 37.Nguyen H, Chiasson VL, Chatterjee P, Kopriva SE, Young KJ, Mitchell BM. Interleukin-17 causes Rho-kinase-mediated endothelial dysfunction and hypertension. Cardiovasc Res 97: 696–704, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nicolls MR, Taraseviciene-Stewart L, Rai PR, Badesch DB, Voelkel NF. Autoimmunity and pulmonary hypertension: a perspective. Eur Respir J 26: 1110–1118, 2005. [DOI] [PubMed] [Google Scholar]
- 39.Novotny SR, Wallace K, Heath J, Moseley J, Dhillon P, Weimer A, Wallukat G, Herse F, Wenzel K, Martin JN, Jr, Dechend R, Lamarca B. Activating autoantibodies to the angiotensin II type I receptor play an important role in mediating hypertension in response to adoptive transfer of CD4+ T lymphocytes from placental ischemic rats. Am J Physiol Regul Integr Comp Physiol 302: R1197–R1201, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Okuda T, Grollman A. Passive transfer of autoimmune induced hypertension in the rat by lymph node cells. Tex Rep Biol Med 25: 257–264, 1967. [PubMed] [Google Scholar]
- 41.Peeters AC, Netea MG, Janssen MC, Kullberg BJ, Van der Meer JW, Thien T. Pro-inflammatory cytokines in patients with essential hypertension. Eur J Clin Invest 31: 31–36, 2001. [DOI] [PubMed] [Google Scholar]
- 42.Purcell ES, Gattone VH., 2nd Immune system of the spontaneously hypertensive rat. I. Sympathetic innervation. Exp Neurol 117: 44–50, 1992. [DOI] [PubMed] [Google Scholar]
- 43.Rodriguez-Iturbe B, Pons H, Quiroz Y, Gordon K, Rincon J, Chavez M, Parra G, Herrera-Acosta J, Gomez-Garre D, Largo R, Egido J, Johnson RJ. Mycophenolate mofetil prevents salt-sensitive hypertension resulting from angiotensin II exposure. Kidney Int 59: 2222–2232, 2001. [DOI] [PubMed] [Google Scholar]
- 44.Sanchez-Lemus E, Benicky J, Pavel J, Larrayoz IM, Zhou J, Baliova M, Nishioku T, Saavedra JM. Angiotensin II AT1 blockade reduces the lipopolysaccharide-induced innate immune response in rat spleen. Am J Physiol Regul Integr Comp Physiol 296: R1376–R1384, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schrader LI, Kinzenbaw DA, Johnson AW, Faraci FM, Didion SP. IL-6 deficiency protects against angiotensin II induced endothelial dysfunction and hypertrophy. Arterioscler Thromb Vasc Biol 27: 2576–2581, 2007. [DOI] [PubMed] [Google Scholar]
- 46.Svendsen UG. The role of thymus for the development and prognosis of hypertension and hypertensive vascular disease in mice following renal infarction. Acta Pathol Microbiol Scand A 84: 235–243, 1976. [DOI] [PubMed] [Google Scholar]
- 47.Tamosiuniene R, Tian W, Dhillon G, Wang L, Sung YK, Gera L, Patterson AJ, Agrawal R, Rabinovitch M, Ambler K, Long CS, Voelkel NF, Nicolls MR. Regulatory T cells limit vascular endothelial injury and prevent pulmonary hypertension. Circ Res 109: 867–879, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tesmer LA, Lundy SK, Sarkar S, Fox DA. Th17 cells in human disease. Immunol Rev 223: 87–113, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Theuer J, Dechend R, Muller DN, Park JK, Fiebeler A, Barta P, Ganten D, Haller H, Dietz R, Luft FC. Angiotensin II induced inflammation in the kidney and in the heart of double transgenic rats. BMC Cardiovasc Disord 2: 3, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tran LT, MacLeod KM, McNeill JH. Chronic etanercept treatment prevents the development of hypertension in fructose-fed rats. Mol Cell Biochem 330: 219–228, 2009. [DOI] [PubMed] [Google Scholar]
- 51.Tuder RM, Groves B, Badesch DB, Voelkel NF. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am J Pathol 144: 275–285, 1994. [PMC free article] [PubMed] [Google Scholar]
- 52.Venegas-Pont M, Manigrasso MB, Grifoni SC, LaMarca BB, Maric C, Racusen LC, Glover PH, Jones AV, Drummond HA, Ryan MJ. Tumor necrosis factor-α antagonist etanercept decreases blood pressure and protects the kidney in a mouse model of systemic lupus erythematosus. Hypertension 56: 643–649, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Viel EC, Lemarie CA, Benkirane K, Paradis P, Schiffrin EL. Immune regulation and vascular inflammation in genetic hypertension. Am J Physiol Heart Circ Physiol 298: H938–H944, 2010. [DOI] [PubMed] [Google Scholar]
- 54.Vinh A, Chen W, Blinder Y, Weiss D, Taylor WR, Goronzy JJ, Weyand CM, Harrison DG, Guzik TJ. Inhibition and genetic ablation of the B7/CD28 T-cell costimulation axis prevents experimental hypertension. Circulation 122: 2529–2537, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wallukat G, Homuth V, Fischer T, Lindschau C, Horstkamp B, Jupner A, Baur E, Nissen E, Vetter K, Neichel D, Dudenhausen JW, Haller H, Luft FC. Patients with preeclampsia develop agonistic autoantibodies against the angiotensin AT1 receptor. J Clin Invest 103: 945–952, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.White FN, Grollman A. Autoimmune factors associated with infarction of the kidney. Nephron 1: 93–102, 1964. [DOI] [PubMed] [Google Scholar]
- 57.Winer S, Paltser G, Chan Y, Tsui H, Engleman E, Winer D, Dosch HM. Obesity predisposes to Th17 bias. Eur J Immunol 39: 2629–2635, 2009. [DOI] [PubMed] [Google Scholar]
- 58.Youn JC, Yu HT, Lim BJ, Koh MJ, Lee J, Chang DY, Choi YS, Lee SH, Kang SM, Jang Y, Yoo OJ, Shin EC, Park S. Immunosenescent CD8+ T cells and C-X-C chemokine receptor type 3 chemokines are increased in human hypertension. Hypertension 62: 126–133, 2013. [DOI] [PubMed] [Google Scholar]
- 59.Zhang JD, Patel MB, Song YS, Griffiths R, Burchette J, Ruiz P, Sparks MA, Yan M, Howell DN, Gomez JA, Spurney RF, Coffman TM, Crowley SD. A novel role for type 1 angiotensin receptors on T lymphocytes to limit target organ damage in hypertension. Circ Res 110: 1604–1617, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou CC, Zhang Y, Irani RA, Zhang H, Mi T, Popek EJ, Hicks MJ, Ramin SM, Kellems RE, Xia Y. Angiotensin receptor agonistic autoantibodies induce pre-eclampsia in pregnant mice. Nat Med 14: 855–862, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]